

Abstract
Reducing the concentration of circulating lipids leads to decreased cardiovascular morbidity and mortality, but the dynamic remodeling that established atherosclerotic lesions undergo upon lipid lowering is poorly understood. Early or advanced lesions in the aortic root were induced by feeding LDL receptor knockout mice a high-fat, high-cholesterol Western-type diet for 5 or 9 weeks, respectively. In the first week after switching to a chow diet, plasma total cholesterol levels dropped 70%, but both early and advanced lesions increased in size. Early lesions grew because of an increase in smooth muscle cells; advanced lesions had an enlargement of absolute macrophage area. From 1 to 3 weeks after the diet switch, plasma total cholesterol levels were completely normalized, but the size of early lesions remained stable; however, advanced lesions became smaller due to a reduction of the absolute macrophage area. From 3 to 6 weeks, both early and advanced lesions progressed further, as a result of expansion of the absolute collagen and necrotic core area. In contrast, early lesions became proinflammatory, as evidenced by the increased infiltration of neutrophils and increased oxidative stress, probably caused by the activation of mast cells in the adventitia. Thus, the severity of atherosclerotic lesions affects their dynamic response to lipid lowering, indicating the importance of establishing stage-specific therapeutic protocols for the treatment of atherosclerosis.
Atherosclerosis, a leading cause of morbidity and mortality in Westernized society, is a multifactorial complex disease with numerous etiologies that work synergistically to promote lesion development.1 Among the many cardiovascular risk factors, hypercholesterolemia, especially due to elevated levels of very low density lipoprotein cholesterol (VLDL-C) and low density lipoprotein cholesterol (LDL-C), is sufficient to initiate and promote atherogenesis even in the absence of other known risk factors.2 Clinical trials have shown that the extent of LDL-C lowering is negatively associated with the rate of atherosclerosis progression.3–5 Nonetheless, after normalization of hypercholesterolemia by lipid-lowering therapy, a considerable proportion of patients still show continued progression of atherosclerosis and the occurrence of clinical events such as myocardial infarction.6–7 In the ASTEROID trial (“A Study to Evaluate the Effect of Rosuvastatin on Intravascular Ultrasound-Derived Coronary Atheroma Burden”), after intensive LDL-C lowering by rosuvastatin, approximately 35% of patients still showed atherosclerotic lesion progression in the coronary artery.3,8 Also, the recent METEOR study (“Measuring Effects on Intima Media Thickness: An Evaluation of Rosuvastatin”) showed that intensive LDL-C lowering by rosuvastatin only reduced rather than stopped the progression of carotid atherosclerosis in asymptomatic subjects with low risk of cardiovascular disease.9 Thus, there is clearly a need for better insight into the mechanisms underlying the effects of plasma lipid lowering on atherogenesis.
The progression of an atherosclerotic lesion is a dynamic process, involving the influx and efflux of lipids, cell migration and emigration, cell proliferation and death, and matrix synthesis and degradation.10 Identification of lesion dynamics is expected to have important implications for the treatment of atherosclerosis, as it could allow establishment of the optimal timing for modulating specific cellular or acellular components of the lesion to induce lesion stabilization or regression. In the present study, we for the first time addressed the effects of plasma lipid lowering on the dynamic remodeling of atherosclerotic lesions with different degrees of severity in LDL receptor knockout mice by applying a dietary switch from a high-fat, high-cholesterol Western-type diet (WTD) to a regular chow diet. Evidence is provided that the complexity of the initial lesion determines its dynamic response to lipid lowering.
Materials and Methods
Mice
LDL receptor knockout (LDLr KO) mice obtained from the Jackson Laboratory (Bar Harbor, ME) were bred at the Gorlaeus Laboratories and were maintained on sterilized regular chow containing 4.3% (w/w) fat and no added cholesterol (RM3; Special Diets Services, Witham, UK). Mice at the age of 12 weeks received a Western-type diet containing 15% (w/w) total fat and 0.25% (w/w) cholesterol (Diet W; Special Diets Services) for 5 weeks (n = 32) or 9 weeks (n = 32) to induce the development of early and advanced atherosclerotic lesions, respectively. Subsequently, 8 of the 32 animals were euthanized (control group, C; week 0). The remaining animals were divided into three groups (n = 8 per group) and fed regular chow for 1 (W1 group), 3 (W3 group), and 6 (W6 group) weeks, respectively. Animal experiments were performed at the Gorlaeus Laboratories of the Leiden/Amsterdam Center for Drug Research in accordance with the National Laws. All experimental protocols were approved by the Ethics Committee for Animal Experiments of Leiden University.
Lipids Analysis
After an overnight fasting period, blood was collected by retro-orbital puncture under anesthesia. Hepatic lipids were extracted according to the method of Bligh and Dyer11 and dissolved in 2% Triton X-100. Triglycerides and phospholipids in serum and liver were determined using standard enzymatic colorimetric assays (for triglycerides, from Roche Diagnostics, Mannheim, Germany; for phospholipids, from Spinreact, Girona, Spain). The concentrations of cholesterol in serum and liver were determined by incubation with 0.025 U/mL cholesterol oxidase (Sigma-Aldrich, St. Louis, MO) and 0.065 U/mL peroxidase and 15 μg/mL cholesteryl esterase (Roche Diagnostics, Mannheim, Germany) in reaction buffer (1.0 KPi buffer; pH 7.7 containing 0.01 mol/L phenol, 1 mmol/L 4-amino-antipyrine, 1% polyoxyethylene-9-laurylether, and 7.5% methanol). Absorbance was read at 490 nm. The hepatic lipid levels were normalized to their protein concentrations determined using a BCA protein assay (Pierce; Thermo Fisher Scientific, Rockford, IL). The distribution of cholesterol over the different lipoproteins in serum was determined by fractionation of 30 μL of serum of individual mice using a Superpose 6 microchromatography column (3.2 × 300 mm, Smart system; Pharmacia, Uppsala, Sweden).
Histological Analysis
On sacrifice, the murine arterial tree was perfused in situ with PBS and the heart was excised and stored in 3.7% neutral-buffered formalin (Formal-fixx; Thermo Shandon, Cheshire, UK) until use. Sections (10-μm thick) were taken once the aortic root was identified by the appearance of aortic valve leaflets. The aortic arches were frozen, and longitudinal 8-μm cryosections were taken. Atherosclerotic lesion development was quantified in the aortic root and aortic arch from Oil Red O/hematoxylin-stained cryostat sections using a Leica image analysis system consisting of a Leica DMRE microscope coupled to a video camera and Leica Qwin imaging software version 3 (Leica Microsystems, Wetzlar, Germany). Mean lesion area (in μm2) was calculated from 10 Oil Red O/hematoxylin-stained sections in the aortic root and 15 sections with maximal lesion area in the aortic arch. Sections were immunolabeled against Moma-2 (monoclonal rat IgG2b, dilution 1:50; Research Diagnostics, Concord, MA), α-smooth muscle actin (monoclonal mouse IgG2a, dilution 1:500; Sigma-Aldrich), CD3 (polyclonal rabbit IgG, dilution 1:150; Neomarkers, Fremont, CA), and Ly6G (monoclonal rat IgG2b, dilution 1:100; eBioscience, San Diego, CA) for detection of monocytes/macrophages, smooth muscle cells, T lymphocytes, and neutrophils, respectively. Collagen content of the lesions was visualized with aniline blue by using Masson's trichrome Accustain according to the manufacturer's instructions (Sigma-Aldrich). TUNEL staining of lesions was performed to visualize apoptotic cells using an in situ cell death detection kit (POD kit, Roche Diagnostics). Mast cells were stained with naphthol AS-D chloroacetate esterase (Sigma-Aldrich). Histochemical stainings were subsequently quantified in five consecutive sections by computer-aided morphometric analysis using the Leica image analysis system. In addition, quantifications of necrotic core area were performed on Oil Red O/hematoxylin and Masson's trichrome stained sections. Oxidative stress was quantified by using α-nitrotyrosine (IgG2b, dilution 1:100; Abcam, Cambridge, UK) and α-8-hydroxy-2′-deoxyguanosine (8-OHdG) (IgG2a, dilution 1:100; QED Bioscience, San Diego, CA) monoclonal mouse antibodies. All analyses were performed blinded.
Circulating Leukocyte Analysis
On sacrifice, blood was collected by retro-orbital puncture under anesthesia. Total white blood cell count and neutrophil, lymphocyte, and monocyte counts in the blood were analyzed using an automated XT-2000iV veterinary hematology analyzer (Sysmex Corporation, Kobe, Japan).
Flow Cytometry
White blood cell suspensions from whole blood were prepared by lysis of red blood cells. Cell-surface immunolabeling of monocytes and neutrophils was performed according to the manufacturers' instructions (eBioscience; BD Biosciences, San Jose, CA). Briefly, fluorochrome-conjugated monoclonal antibodies to CD11b (eBioscience) and to Ly6G and Ly6C (BD Biosciences) were incubated with the white blood cell suspensions for 30 minutes at 4°C in labeling buffer (1% mouse serum in PBS). Flow cytometric analysis was performed with a BD Biosciences FACSCalibur system and then analyzed with the manufacturer's CellQuest Pro software, correcting for nonspecific staining with isotype antibody controls.
Statistical Analysis
Statistical analysis was performed using analysis of variance and the Student-Newman-Keuls post hoc test as implemented in InStat and Prism software (GraphPad software version 4.03, La Jolla, CA). Pearson's r test was used to perform correlation analysis. A level of P < 0.05 was considered significant.
Results
Dietary Lipid Lowering Leads to Decreased Lipid Levels in Plasma and Liver
LDLr KO mice were fed WTD for 5 weeks to induce atherosclerotic lesion formation. Thereafter the diet was switched to regular chow, to lower plasma cholesterol levels. Once the diet was switched to chow, plasma-free cholesterol and total cholesterol levels dropped approximately 60% (from 357 ± 36 mg/dL to 156 ± 2 mg/dL, P < 0.001) and 70% (from 1347 ± 78 mg/dL to 438 ± 13 mg/dL, P < 0.001) at 1 week and decreased further (to 84 ± 3 and 222 ± 6 mg/dL, respectively) at 6 weeks after the diet switch (Table 1). The reduced plasma total cholesterol levels were due mainly to decreased VLDL/LDL-C levels (see Supplemental Figure S1 at http://ajp.amjpathol.org). Moreover, an approximately 45% reduction in plasma triglycerides (P < 0.001) and phospholipids (P < 0.001) was observed at 1 week after the diet switch, and the levels remained low through the rest of the experimental period (Table 1). The reduction of plasma lipids thus occurred mainly in the first week after the dietary lipid withdrawal. In contrast, livers showed delayed cholesterol lowering on switch of WTD to chow, with the major effects seen at 3 weeks. After 3 weeks on regular chow, hepatic free and esterified cholesterol levels decreased approximately 35% (P < 0.001) and 75% (P < 0.001), respectively (Table 1). Dietary lipid lowering did not affect the hepatic triglyceride and phospholipid content (Table 1). Of note, dietary lipid lowering did normalize both plasma and hepatic lipid levels at 6 weeks after the diet switch, to levels similar to those in LDLr KO mice of the same age without WTD challenge (Table 1).
Table 1.
Effect of Dietary Cholesterol Lowering on Plasma and Hepatic Lipids
| Lipids | Group C | Group W1 | Group W3 | Group W6 | LDLr KO control |
|---|---|---|---|---|---|
| Plasma FC (mg/dL) | 357 ± 36 | 156 ± 2⁎⁎⁎ | 96 ± 4⁎⁎⁎† | 84 ± 3⁎⁎⁎† | 85 ± 3 |
| Plasma TC (mg/dL) | 1347 ± 78 | 438 ± 13⁎⁎⁎ | 232 ± 7⁎⁎⁎†† | 222 ± 6⁎⁎⁎†† | 237 ± 7 |
| Plasma TG (mg/dL) | 216 ± 21 | 104 ± 14⁎⁎⁎ | 120 ± 10⁎⁎⁎ | 115 ± 7⁎⁎⁎ | 106 ± 10 |
| Plasma PL (mg/dL) | 748 ± 50 | 489 ± 17⁎⁎⁎ | 447 ± 23⁎⁎⁎ | 405 ± 19⁎⁎⁎ | 425 ± 15 |
| Hepatic FC (μg/mg protein) | 26 ± 1 | 23 ± 1⁎ | 17 ± 1⁎⁎⁎†† | 17 ± 1⁎⁎⁎†† | 16 ± 1 |
| Hepatic CE (μg/mg protein) | 41 ± 3 | 31 ± 4⁎ | 11 ± 1⁎⁎⁎†† | 9 ± 1⁎⁎⁎†† | 8 ± 1 |
| Hepatic TG (μg/mg protein) | 67 ± 2 | 77 ± 7 | 73 ± 3 | 77 ± 4 | 64 ± 9 |
| Hepatic PL (μg/mg protein) | 74 ± 4 | 73 ± 4 | 65 ± 3 | 68 ± 2 | 75 ± 5 |
LDLr KO mice were fed a high-fat, high-cholesterol Western-type diet for 5 weeks. Thereafter, the animals were switched to regular chow and were euthanized at 0 (group C), 1 (group W1), 3 (group W3), and 6 (group W6) weeks after the diet switch. LDLr KO control mice were kept on chow diet at all times during the experiment. Data are reported as means ± SEM of 8 mice.
CE, cholesteryl ester; FC, free cholesterol; PL, phospholipids; TC, total cholesterol; TG, triglycerides.
P < 0.05 and
P < 0.001 versus group C;
P < 0.05 and
P < 0.01 versus group W1.
Decreased Circulating Neutrophils and Monocytes after Dietary Lipid Lowering
Next, we examined the effect of dietary lipid lowering on circulating leukocytes. The amount of total white blood cells was 4.0 ± 0.7 × 106/mL at baseline and had a tendency to decrease to 3.1 ± 0.5 × 106/mL at 3 weeks and remained low (3.3 ± 0.4 × 106/mL) until 6 weeks after the WTD withdrawal. Dietary lipid lowering did not affect the amount of lymphocytes in the circulation (approximately 2.5 × 106/mL). The tendency to a reduction in white blood cells by dietary lipid lowering can be attributed to reduced levels of circulating monocytes and neutrophils (Figure 1, A and B). Neutrophils in the circulation were reduced approximately twofold (P < 0.05) after 1 week on chow, but monocytes did not change at this time point. In contrast, at 3 weeks after dietary lipid withdrawal circulating monocytes were reduced approximately threefold (P < 0.05).
Figure 1.
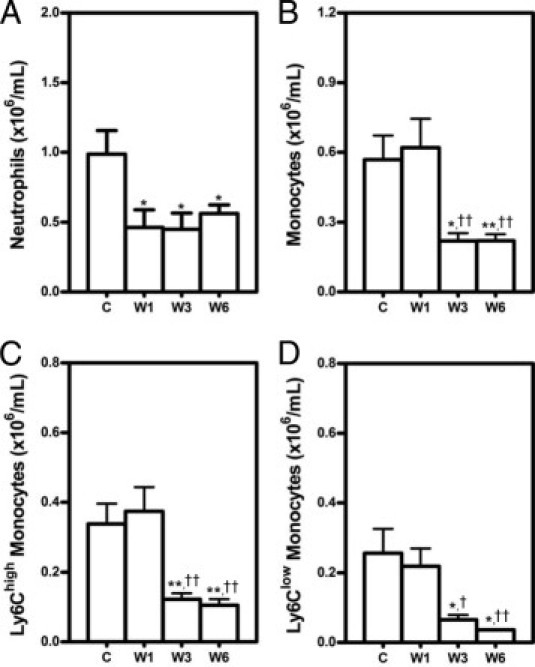
Decreased circulating neutrophils (A) and monocytes (B) on dietary lipid lowering. LDLr KO mice were fed Western-type diet (WTD) for 5 weeks and thereafter switched to regular chow for 0 (C), 1 (W1), 3 (W3), and 6 (W6) weeks. At each time point, circulating neutrophils and monocytes were analyzed by using flow cytometry. Neutrophils were defined as CD11b+Ly6G+ cells and monocytes as CD11b+Ly6G− cells. The CD11b+Ly6G− monocytes were separated further according to their Ly6C expression into monocyte subsets Ly6Chigh (C) and Ly6Clow (D). Data are reported as means ± SEM of eight mice. *P < 0.05 and **P < 0.01 versus group C; †P < 0.05 and ††P < 0.01 versus group W1.
Two specific monocyte subsets that vary in their capacity to infiltrate into atherosclerotic lesions are characterized by different expression levels of Ly6C. Ly6Chigh monocytes preferentially accumulate in the growing atheroma in hyperlipidemic mice, but Ly6Clow monocytes do not.12 Therefore, the Ly6Chigh and Ly6Clow monocyte subsets were further analyzed. Both subsets of monocytes were reduced after 3 weeks on chow, 3.1-fold for Ly6Chigh (P < 0.01) and 3.3-fold for Ly6Clow (P < 0.05) (Figure 1, C and D). Moreover, the percentage of Ly6Chigh monocytes decreased from 60 ± 2% at baseline to 47 ± 2% (1.3-fold, P < 0.001) at 6 weeks after the diet switch.
Dynamic Remodeling of Established Early Atherosclerotic Lesions upon Dietary Lipid Lowering
Feeding LDLr KO mice the WTD for 5 weeks induced development of early atherosclerotic lesions in the aortic root, characterized by fatty streaks and accumulation of macrophage foam cells (Figures 2C and 3A). The dynamic remodeling of these established early lesions after dietary lipid lowering was investigated by analyzing the effects on lesion size and lesion composition at 0 (group C), 1 (group W1), 3 (group W3), and 6 (group W6) weeks after the diet switch to regular chow.
Figure 2.
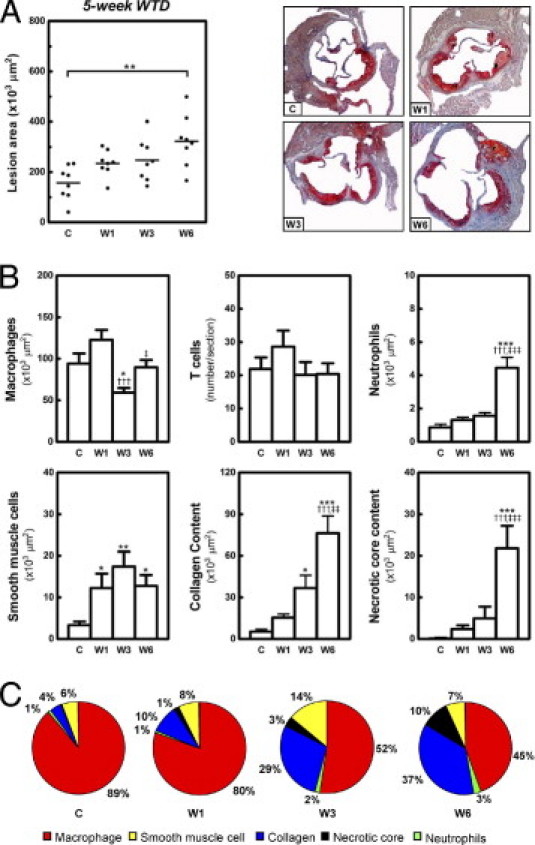
Dynamic remodeling of established early lesions on dietary lipid lowering. Feeding LDLr KO mice WTD for 5 weeks induced early atherosclerotic lesion formation in the aortic root. Atherosclerotic lesion development was analyzed at the aortic root at 0 (C), 1 (W1), 3 (W3), and 6 (W6) weeks after the switch of the mice to regular chow. A: Scatter dot plot of atherosclerotic lesion quantification, with photomicrographs of representative Oil Red O-stained sections. Each symbol represents the mean lesion area in a single mouse; horizontal bars indicate the mean value for each group. Original magnification, ×25. B: Bar graphs show the dynamic changes in macrophages, T cells, neutrophils, smooth muscle cells, collagen content, and necrotic core area of early lesions at and after the diet switch. *P < 0.05, **P < 0.01, and ***P < 0.001 versus group C; †††P < 0.001 versus group W1; and ‡P < 0.05, ‡‡P < 0.01, and ‡‡‡P < 0.001 versus group W3. C: Relative abundance of macrophages, smooth muscle cells, collagen, necrotic core, and neutrophils in early lesions during remodeling on dietary lipid lowering. The relative percentage of each lesion component was normalized to sum to 100%.
Figure 3.
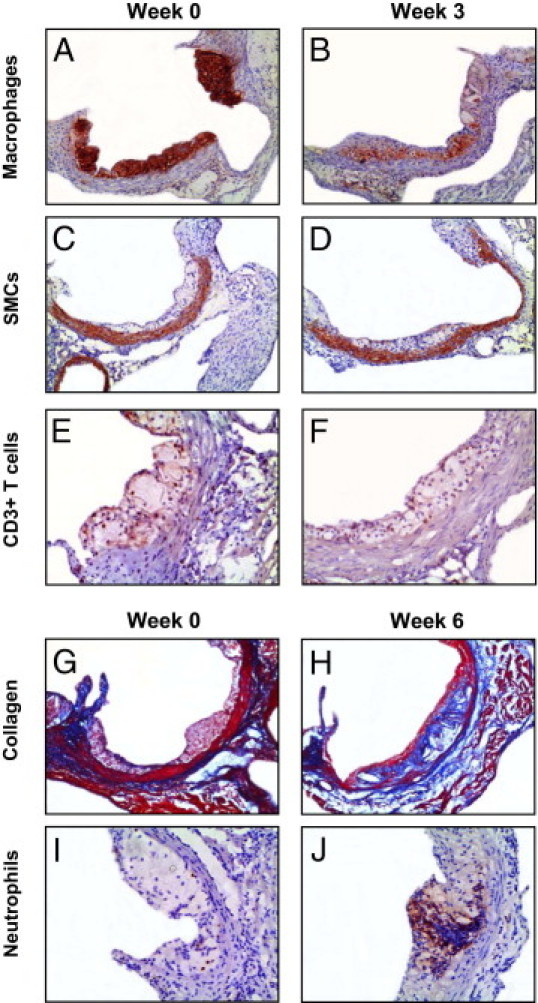
Morphometric changes of early lesions on dietary lipid lowering. Sections of the aortic roots were stained with antibodies against Moma-2, α-actin, CD3, and Ly6G to visualize macrophages (A and B), smooth muscle cells (SMCs) (C and D), T cells (E and F), and neutrophils (I and J), respectively. Atherosclerotic lesions in the aortic root were stained with Masson's trichrome Accustain, which stains cytoplasm and muscle fiber red and collagen blue (G and H). Original magnification: ×50 (A–D, G, and H); ×100 (E, F, I, and J).
In the initial phase (the first week after dietary lipid withdrawal), lesions still progressed, from 157 ± 24 × 103 μm2 to 235 ± 18 × 103 μm2, despite the 70% decrease in plasma total cholesterol levels to 438 ± 13 mg/dL (Figure 2A). Quantification of lesion composition indicated a 3.7-fold increase of smooth muscle cells (P < 0.05), but no significant change in macrophages, T cells, neutrophils, or collagen content of lesions (Figure 2B). The percentage of apoptotic cells in the lesions was only 1.3 ± 0.3% (see Supplemental Figure S2 at http://ajp.amjpathol.org). Morphological analysis of the lesions also revealed the appearance of a necrotic core at 1 week after the diet switch; however, the mean size of the necrotic core area was only 2.4 ± 0.9 × 103 μm2 (Figure 2B). After 1 week of chow diet feeding, the early lesions were still composed mainly of macrophage foam cells (Figure 2C).
In the second phase (from 1 to 3 weeks on chow), the plasma total cholesterol levels decreased further, to approximately 232 ± 7 mg/dL. Interestingly, no change in lesion size was detected, compared with 1 week of chow feeding (W3: 248 ± 18 × 103 μm2 versus W1: 235 ± 18 × 103 μm2) (Figure 2A). Of note, dietary lipid lowering for 3 weeks did lead to a reduction in macrophages (2.1-fold, P < 0.001 versus W1; 1.6-fold, P < 0.05 versus C) (Figures 2B and 3B), but the average size of macrophage foam cells remained unchanged (data not shown). There was further increase in smooth muscle cells (7.4-fold, P < 0.05) (Figure 3, C and D) and collagen content (sevenfold, P < 0.05), compared with baseline levels (Figure 2B), forming the subluminal fibrous cap (Figure 3B). The percentage of apoptotic cells increased approximately 2.3-fold (P < 0.05 versus C and W1), to 2.9 ± 0.4% (see Supplemental Figure S2 at http://ajp.amjpathol.org). Although the buildup of necrotic material inside the lesions was augmented (5.0 ± 2.8 × 103 μm2), the ratio of collagen to necrotic core was increased 3.3-fold, to 16.2 ± 5.8, indicating that dietary lipid lowering induced lesion stabilization. Moreover, T cells (Figure 3, E and F) and neutrophils of the lesions were not significantly changed (Figure 2, B and C). In accord, the lesion contains more collagen and smooth muscle cells and fewer macrophage foam cells after dietary lipid lowering for 3 weeks (Figure 2C).
In the third phase (from 3 to 6 weeks on chow), the size of the lesions reached 323 ± 36 × 103 μm2 (P < 0.01 versus C) (Figure 2A), although the plasma total cholesterol levels remained low (222 ± 6 mg/dL). The lesional macrophage area of mice kept on chow for 6 weeks increased 1.5-fold (P < 0.05), compared with the lesions of mice fed chow for 3 weeks, and became similar to baseline levels (Figure 2B). The amount of smooth muscle cells in the lesions remained relatively high, 3.9-fold (P < 0.05) higher than baseline levels. Moreover, the collagen content of the lesions was further increased (2.1-fold versus W3, 14.5-fold versus C, P < 0.001) (Figures 2B and 3H), indicating that the rate of collagen synthesis was still higher than that of its degradation. Importantly, in the 3 additional weeks of chow diet feeding, there was also a dramatic enlargement of necrotic core (4.4-fold, P < 0.001 versus W3) (Figure 2, B and C). The ratio of collagen to necrotic core was thus reduced 5.5-fold, to 2.9 ± 0.4 (P < 0.05 versus W3). A further increase in the percentage of apoptotic cells was also found (4.7 ± 0.6%, 1.6-fold versus W3, P < 0.05; 3.7-fold versus C and W1, P < 0.001) (see Supplemental Figure S2 at http://ajp.amjpathol.org). In addition, the neutrophil content of the lesions was increased 2.8-fold (P < 0.001 versus W3) (Figures 2B and 3J). The enhanced infiltration of neutrophils and a decreased ratio of collagen to necrotic core suggest a proinflammatory phenotype of the lesions. Strikingly, the T cell content still remained the same as at baseline (Figure 2B).
After 6 weeks of chow diet feeding, the early lesions consisted mainly of macrophages, collagen, and necrotic core (Figure 2C). Pearson's correlation analyses demonstrated that, after normalization of hypercholesterolemia, the size of early lesions was highly correlated with the macrophage (r = 0.67, P = 0.0048, n = 16), collagen (r = 0.85, P < 0.0001, n = 16), and necrotic core (r = 0.66, P = 0.0055, n = 16) areas.
Dynamic Remodeling of Established Advanced Atherosclerotic Lesions upon Dietary Lipid Lowering
We next examined whether the dynamic remodeling of established advanced atherosclerotic lesions differs from the response of early lesions on dietary lipid lowering. Advanced lesions in the aortic roots were induced by feeding LDLr KO mice WTD for 9 weeks. The lesion size and composition were again examined at 0, 1, 3, and 6 weeks after the diet switch to regular chow. When the mice were fed WTD for 9 weeks before the switch to chow diet, dietary lipid lowering led to a similar reduction in plasma and hepatic lipid levels, compared with 5 weeks of WTD feeding before the diet switch (data not shown). Compared with early lesions, advanced lesions displayed a markedly lower macrophage content, increased smooth muscle cell content and collagen deposition (Figures 2C and 4C), and a necrotic core and a fibrous cap could also be distinguished. In addition, advanced lesions contained twofold more T cells (P = 0.0013) and 49-fold more neutrophils (P = 0.0011), respectively, compared with early lesions. The percentage of apoptotic cells in advanced lesions was also largely increased (8. 5 ± 0.8%, 6.7-fold, P < 0.001) (see Supplemental Figure S2 at http://ajp.amjpathol.org). Of note, the neutrophil content of advanced lesions was approximately fivefold (P = 0.0003) higher than the level in early lesions after 6 weeks of WTD withdrawal (Figures 2B and 4B).
Figure 4.
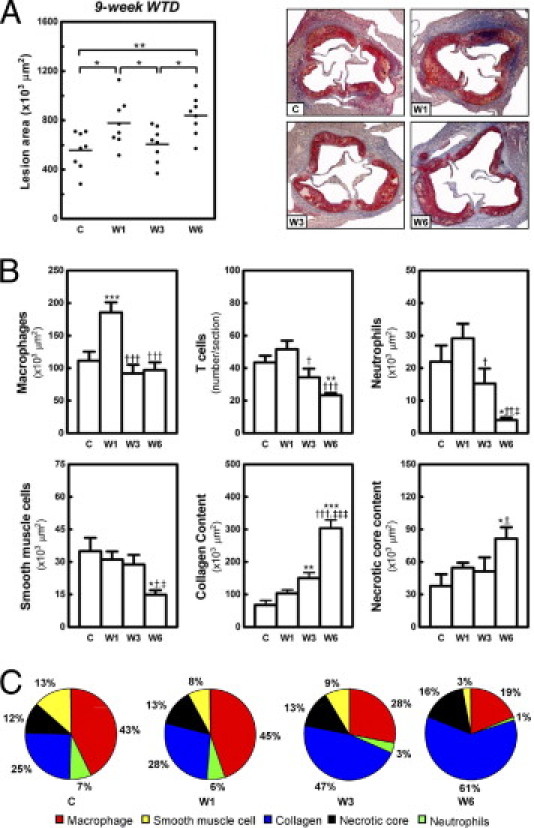
Dynamic remodeling of established advanced lesions on dietary lipid lowering. Feeding LDLr KO mice WTD for 9 weeks induced advanced atherosclerotic lesion formation in the aortic root. Atherosclerotic lesion development was analyzed at the aortic root at 0 (C), 1 (W1), 3 (W3), and 6 (W6) weeks after the switch to regular chow. A: Scatter dot plot of atherosclerotic lesion quantification, with photomicrographs of representative Oil Red O-stained sections (original magnification, ×25). Each symbol represents the mean lesion area in a single mouse; horizontal bars indicate the mean value for each group. B: Bar graphs show the dynamic changes in macrophages, T cells, neutrophils, smooth muscle cells, collagen content, and necrotic core area of advanced lesions at and after the diet switch. *P < 0.05, **P < 0.01, and ***P < 0.001 versus group C; †P < 0.05, ††P < 0.01, and †††P < 0.001 versus group W1; and ‡P < 0.05 and ‡‡‡P < 0.001 versus group W3. C: Relative abundance of macrophages, smooth muscle cells, collagen, necrotic core, and neutrophils in advanced lesions during remodeling on dietary lipid lowering. The relative percentage of each lesion component was normalized to sum to 100%.
In the initial phase of WTD withdrawal, advanced lesions increased 1.4-fold in size (778 ± 68 × 103 μm2 versus 557 ± 54 × 103 μm2, P < 0.05) (Figure 4A). In contrast to early lesions, this size increase was associated with a 1.7-fold (P < 0.001) increase of macrophage content in advanced lesions (Figure 4B). No significant change was observed in the other lesion components. Notably, in the second phase (from 1 to 3 weeks on chow), a significant 1.3-fold reduction in the size of advanced lesions was observed (606 ± 50 × 103 μm2, P < 0.05 versus W1) (Figure 3A), which was primarily the consequence of a twofold reduction in the macrophage content of the lesions (Figures 4B and 5B). In addition, T cells (1.5-fold, P < 0.05 versus W1) and neutrophils (1.9-fold, P < 0.05 versus W1) were decreased to baseline levels. Increased fibrous cap area in the subluminal Moma-2-negative region (Figure 5B) was correlated with increased collagen accumulation (2.2-fold, P < 0.01, compared with baseline), but smooth muscle cells and the necrotic core remained the same (Figure 4, B and C). The ratio of collagen to necrotic core was thus increased 1.8-fold, to 3.7 ± 0.6 (P < 0.05, versus C and W1). The percentage of apoptotic cells remained unchanged (8.6 ± 0.6%) (see Supplemental Figure S2 at http://ajp.amjpathol.org). All these changes indicate that advanced lesions became stabilized after dietary lipid lowering for 3 weeks. In accord, collagen became the major component of advanced lesions after 3 weeks of chow diet feeding (Figure 4C).
Figure 5.
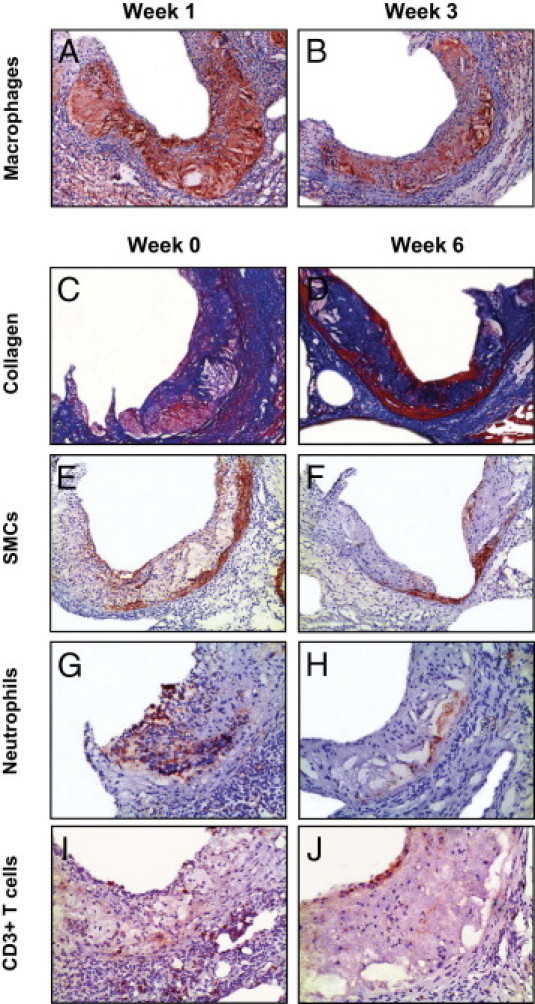
Photomicrographs showing morphometric changes of advanced lesions in response to dietary lipid lowering. Sections of the aortic roots were stained with antibodies against Moma-2, α-actin, Ly6G, and CD3 to visualize macrophages (A and B), smooth muscle cells (SMCs) (E and F), neutrophils (G and H), and T cells (I and J), respectively. Atherosclerotic lesions in the aortic root were stained with Masson's trichrome Accustain, which stains cytoplasm and muscle fiber red and collagen blue (C and D). Original magnification: ×50 (A–F); ×100 (G–J).
Like the early lesions, in the third phase the advanced lesions progressed, to 838 ± 56 × 103 μm2 (1.4-fold, P < 0.05 versus W3) (Figure 4A). This was primarily the consequence of an increase (twofold, P < 0.001) in collagen content of advanced lesions and to a lesser extent of an enhanced buildup of necrotic core material (1.7-fold, P < 0.001) (Figures 4B and 5D). The macrophage area of advanced lesions did not change, but smooth muscle cells (Figure 5, E and F) decreased 1.9-fold (P < 0.05 versus W3; 2.4-fold, P < 0.05 versus C) after the 3 additional weeks of chow diet feeding (Figure 4B). The ratio of collagen to necrotic core remained stably high (4.0 ± 0.4, P < 0.01 versus C). The percentage of apoptotic cells in the lesions was slightly decreased, to 7.1 ± 0.6% (P < 0.05 versus W1) (see Supplemental Figure S2 at http://ajp.amjpathol.org). Moreover, at 6 weeks neutrophils of advanced lesions (Figure 5, G and H) were dramatically reduced (3.7-fold, P < 0.05 versus W3, 5.4-fold, P < 0.05 versus C) (Figure 4B). T cells of advanced lesions (Figure 5, I and J) decreased further (1.9-fold, P < 0.01 versus C), reaching levels similar to those of early lesions (Figure 4B). In accord, advanced lesions became collagen-rich lesions after dietary lipid lowering for 6 weeks (Figure 4C). Of note, Pearson's correlation analyses demonstrated that, after normalization of hypercholesterolemia, the size of advanced lesions was also highly correlated with collagen (r = 0.91, P = 0.0002, n = 16) and necrotic core (r = 0.80, P = 0.0003, n = 16) content, but not with macrophage content (r = 0.39, P = 0.14, n = 16).
Effects of Dietary Lipid Lowering on Inflammatory Cells in the Adventitia of Mice with Early and Advanced Lesions
Adventitial inflammatory cells also play a critical role in the progression of atherosclerotic lesions.13 The response of inflammatory cells in the adventitia to dietary lipid lowering might thus influence the progression of established lesions. Therefore, total and degranulated mast cells, neutrophils, and T cells in the adventitia underlying the early and advanced lesions were analyzed after WTD withdrawal. On dietary lipid lowering, total and degranulated mast cells in the adventitia of early lesions increased 2.1-fold (P < 0.05 versus C and W1) and 3.8-fold (P < 0.05), respectively, at 3 weeks and remained stably high (total: 2.8-fold, P < 0.01 versus C; degranulated: 3.9-fold, P < 0.05 versus C) at 6 weeks (Figure 6, B and C). Degranulated mast cells can recruit neutrophils by secretion of KC, the murine ortholog of IL-8.14 In accord, a significant increase of neutrophils (1.9-fold, P < 0.01 versus C; 1.2-fold, P < 0.05 versus W1) was observed in the adventitia of early lesions at 3 weeks after WTD withdrawal. Unexpectedly, at 6 weeks, neutrophils declined twofold, to approximately baseline levels (P < 0.05 versus W3) (Figure 6D).
Figure 6.
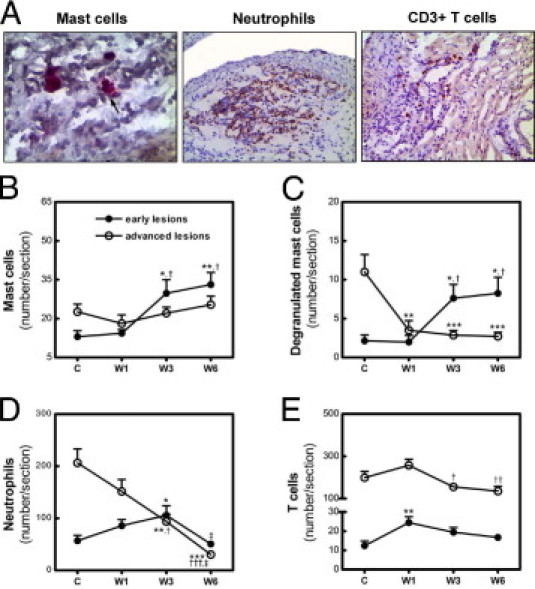
Effect of dietary lipid lowering on inflammatory cells in the adventitia of early and advanced lesions. A: Representative photomicrographs of mast cells (arrow indicates degranulated mast cells; original magnification, ×200), neutrophils (original magnification, ×100), and T cells (original magnification, ×100) in the adventitia. B–E: Dynamic changes of total mast cells (B) degranulated mast cells (C), neutrophils (D), and T cells (E) in the adventitia of early lesions (solid symbols) and advanced lesions (open symbols) after the switch from WTD to chow diet. Data are reported as means ± SEM (n = 8). *P < 0.05, **P < 0.01, and ***P < 0.001 versus group C; †P < 0.05, ††P < 0.01, and †††P < 0.01 versus group W1; and ‡P < 0.05 versus group W3.
Compared with early lesions, the adventitia of advanced lesions contained more total (1.7-fold, P = 0.0251) and degranulated (5.2-fold, P = 0.0072) mast cells and neutrophils (3.6-fold, P = 0.0008) before the diet switch. Although total mast cells did not change in the adventitia of advanced lesion after the WTD withdrawal, dietary lipid lowering did lead to a reduction of degranulated mast cells (3.1-fold, P < 0.001 versus C) after 1 week on chow, which remained stably low through the rest of the experimental period (Figure 6, B and C). Neutrophils in the adventitia of advanced lesions declined over time (6.8-fold, P < 0.001, W6 versus C), at 6 weeks reaching levels similar to the baseline for early lesions (Figure 6D).
As an important component of adaptive immunity, T cells are critical for the progression rather than initiation of atherosclerosis.15 In accord, 16-fold more T cells (P = 0.0004) were observed in the adventitia of advanced lesions, compared with early lesions. T cells in the adventitia of early lesions increased approximately twofold (P < 0.01 versus C) in the first week after the WTD withdrawal and did not significantly change afterward, but a 1.7-fold (P < 0.05 versus W1) reduction of T cells was detected in the adventitia of advanced lesions from 1 to 3 weeks, which remained low (1.9-fold, P < 0.01 versus W1) at 6 weeks on chow (Figure 6E). The amount of T cells in the adventitia of advanced lesions, however, remained 7.9-fold (P = 0.0003) higher than the levels in early lesions at 6 weeks on chow.
Effects of Dietary Lipid Lowering on Oxidative Stress in Early and Advanced Lesions
To study the effect of dietary lipid lowering on oxidative stress in established lesions, the amounts of nitrotyrosine and 8-OHdG in the lesions were quantified after the diet switch. Although plasma cholesterol decreased after 6 weeks on chow, the amount of nitrotyrosine and 8-OHdG in early lesions was significantly increased, 17-fold (P = 0.009) and 3.3-fold (P = 0.043), respectively (see Supplemental Figure S3A at http://ajp.amjpathol.org). In contrast, oxidative stress was reduced in advanced lesions, as evidenced by the reduction of nitrotyrosine (13-fold, P = 0.005) and 8-OHdG (fivefold, P = 0.0003) (see Supplemental Figure S3B at http://ajp.amjpathol.org).
Effects of Dietary Lipid Lowering on the Established Atherosclerotic Lesion Development in the Coronary Artery and Aortic Arch
Lesion development in the coronary artery and aortic arch was analyzed next. In accord with the fact that the aortic root is the most susceptible site for atherosclerosis in murine models, no lesion formation was visible in the coronary artery of LDLr KO mice after 5 weeks of WTD feeding. Even after 9 weeks on WTD, only two of the eight mice displayed lesions, and those were very small (5.6 × 103 μm2 and 7.2 × 103 μm2). Moreover, LDLr KO mice did not develop any atherosclerotic lesions in the aortic arch after 5 weeks on WTD (data not shown). After 9 weeks of WTD feeding, macrophage-rich fatty streak lesions had formed in the aortic arch, with a mean lesion area of 86.5 ± 16.4 × 103 μm2 (see Supplemental Figure S4A at http://ajp.amjpathol.org). The ratio of collagen to macrophages in the aortic arch after 9 weeks on WTD (0.053 ± 0.021) was similar to that of established early lesions in the aortic root (0.051 ± 0.015) after 5 weeks of WTD feeding. No necrotic core was visible in these established lesions of the aortic arch (see Supplemental Figure S4D at http://ajp.amjpathol.org). After the first week on the chow, no significant change was found in lesion size (97.8 ± 13.4 × 103 μm2) and composition (see Supplemental Figure S4 at http://ajp.amjpathol.org). Three weeks of chow diet feeding led to an approximately twofold (P < 0.05 versus C and W1) and approximately 7.5-fold (P < 0.01 versus C and W1) increase in size and collagen content, respectively (see Supplemental Figure S4, A and C, at http://ajp.amjpathol.org). In addition, a small necrotic core became evident; however, the mean size was only 2.5 × 103 μm2 (P < 0.05 versus C and W1) (see Supplemental Figure S4D at http://ajp.amjpathol.org). The lesion increased further in size (1.5-fold, P < 0.05 versus W3; approximately threefold, P < 0.001 versus C and W1) and collagen content (2.4-fold, P < 0.001 versus W3; approximately 20-fold versus C and W1) after an additional 3 weeks on chow, for a total of 6 weeks on chow (see Supplemental Figure S4, A and C, at http://ajp.amjpathol.org). An enlargement of the necrotic core was also observed (5.5-fold, P < 0.001 versus W3). The ratio of collagen to necrotic core was thereby reduced 5.2-fold to 5.8 ± 1.3 (P < 0.05 versus W3) (see Supplemental Figure S4D at http://ajp.amjpathol.org). Dietary lipid lowering did not significantly affect the macrophage content, although a tendency was observed toward a profile similar to that of established early lesions in the aortic root (see Supplemental Figure S4B at http://ajp.amjpathol.org). Taken together, dietary lipid lowering did lead to similar changes on early lesions in both aortic root and aortic arch.
Discussion
The present study for the first time investigated the dynamic remodeling of established early and advanced atherosclerotic lesions on dietary lipid lowering. Strikingly, both established early and advanced atherosclerotic lesions continued to progress after the withdrawal of dietary lipids, which was strongly correlated with the induction of collagen and necrotic core content of the lesions. After normalization of hypercholesterolemia, however, early lesions exhibited a proinflammatory phenotype, whereas advanced lesions became less inflammatory. The clear differences in the response of early and advanced lesions to dietary lipid lowering indicate that the severity of the lesions affected their dynamic response to lipid lowering.
Dietary lipid lowering, as expected, induced a decrease in plasma and hepatic cholesterol levels; however, both established early and advanced lesions in LDLr KO mice could further increase in size when the plasma cholesterol levels of the animals was normalized to approximately 220 mg/dL (ie, lower than the 300 mg/dL that is prerequisite for initiation of lesion development).16 This finding indicates that the plasma cholesterol levels required for lesion progression are lower than those for lesion initiation. This might also explain why progression of atherosclerotic lesions can occur in patients with normal LDL-C levels (<90 mg/dL) after lipid-lowering treatment with statins.3 In accord, a robust reduction of the plasma LDL-C levels (<70 mg/dL) is required to halt the progression of established atherosclerotic lesions in humans.3,4 Nonetheless, it should be noted that some patients cannot reach this threshold level of LDL-C even after intensive lipid-lowering treatment. Combined therapy that targets multiple pathogenic mechanisms involved in lesion progression is thus needed for efficient treatment of this group of patients.
Consistent with previous findings in rabbits17 and in miniature pigs,18 dietary lipid lowering also led to massive collagen accumulation in the lesion of LDLr KO mice. Importantly, we observed that collagen content was highly correlated with the size of both early and advanced lesions from 3 to 6 weeks after normalization of hypercholesterolemia. Despite the importance of collagen in providing mechanical stability to the lesion, excessive collagen may also stimulate lesion development by serving as a depot for proatherogenic molecules (modified lipoproteins, growth factors, and glycation end-products) and by promoting foam cell formation.19 Moreover, massive collagen accumulation might obstruct the access of healthy phagocytotic cells for clearing apoptotic cells in the lesions, thereby leading to the increased necrotic core formation. Necrotic core areas are sites of active inflammation and oxidative stress,20,21 which could further promote the progression of lesions. In accord with this notion, we found that necrotic core formation increased over time on dietary lipid lowering. It should therefore be carefully investigated whether inhibition of collagen synthesis in combination with lipid lowering might be a potential therapeutic strategy for the treatment of atherosclerosis.
Recruitment of monocytes into atherogenic foci is central to the initiation and progression of atherosclerosis. Hypercholesterolemia induces Ly6Chigh monocytosis,22,23 subsequently contributing to atherogenesis.12 Reduction of plasma cholesterol levels by statins could reduce Ly6Chigh monocytosis.22 However, the anti-inflammatory effects of statins could also contribute to these alterations, beyond the cholesterol-lowering effect. In the present study, we showed that cholesterol lowering alone by dietary lipid withdrawal could not only decrease circulating monocytes, but also normalize the increased levels of Ly6Chigh monocytes in LDLr KO mice. Of note, these effects were observed mainly at 3 weeks after the diet switch. Therefore, both early and advanced lesions continued to accumulate macrophages in the first week after dietary lipid lowering, probably as a result of monocyte recruitment from the circulation. Swirski et al24 demonstrated that, in addition to bone marrow, spleen is another reservoir for a large number of monocytes. These splenic monocytes can be recruited to inflammatory sites. Reduced monocytes in the subcapsular red pulp of spleen was also observed after 1 week on chow (unpublished data), indicating that splenic monocytes might account for the increased macrophage accumulation in lesions within the first week after the diet switch. More experiments would be needed to test this hypothesis.
From 1 to 3 weeks after the diet switch, dietary lipid lowering reduced the macrophage content of both early and advanced lesions; however, the size of macrophage foam cells and necrotic core was not significantly changed. Moreover, the rate of apoptosis was either very low in the early lesions or remained stable in advanced lesions. We speculate that the reduction of macrophages is a consequence mainly of their emigration. Emigration of monocyte-derived foam cells from atherosclerotic lesions and regression of established atherosclerotic lesions has been elegantly shown by transplantation of a region of the aorta with atherosclerotic lesions from hypercholesterolemic apoE KO mice to normocholesterolemic wild-type mice.25
The lesion size was not increased in early lesions, and was even reduced in advanced lesions from 1 to 3 weeks after WTD withdrawal, indicating that dietary lipid lowering can inhibit the progression and/or induce the regression of established lesions. Nonetheless, despite the reduced Ly6Chigh monocytosis on dietary lipid lowering, macrophage accumulation in the early lesions was enhanced again from 3 to 6 weeks after the diet switch. Of note, the macrophage area was highly correlated with the lesion size of early lesions but not of advanced lesions at this time point after WTD withdrawal. In accord, using bone marrow transplantation, we recently demonstrated that the growth of established early lesions under hypercholesterolemia is caused mainly by the continuous recruitment of monocyte-derived cells, although this process is largely impaired in established advanced lesions.26 Thus, inhibition of monocyte infiltration combined with lipid lowering might be a more efficient therapeutic strategy for the treatment of established early lesions than of advanced lesions.
Another important finding is that early and advanced lesions showed a different inflammatory phenotype after normalization of hypercholesterolemia. Early lesions became proinflammatory (as evidenced by a decreased ratio of collagen to necrotic core, an elevated neutrophil content, and increased oxidative stress), but advanced lesions became less inflammatory. Zernecke et al27 demonstrated that an expansion of circulating neutrophils leads to more neutrophil infiltration into lesions. However, dietary lipid lowering reduced the levels of circulating neutrophils, and so could not have contributed to increased neutrophil infiltration in the lesions. Moreover, in accord with a previous report,28 the area of necrosis did not correlate with neutrophil accumulation. Actually, almost no neutrophils were found in close proximity to the necrotic core in the atherosclerotic lesion.
Notably, quantification of inflammatory cells in the adventitia of early lesions revealed increased numbers of degranulated mast cells. Activation of mast cells not only recruits monocytes, neutrophils, and other mast cells by secreting their respective chemoattractant MCP-1, KC, and eotaxin,14,29 but also promotes macrophage apoptosis, probably thereby leading to augmented buildup of necrotic core material.30 In accord, the enhanced infiltration of monocytes and neutrophils into the lesions, the increased recruitment of mast cells into the adventitia, and a dramatic enlargement of necrotic core were observed on progression of the early lesions after normalization of hypercholesterolemia. Increased macrophages and neutrophils in early lesions could generate more reactive oxygen species through NADPH oxidase and myeloperoxidase,31 thereby leading to enhanced oxidative stress. Thus, the proinflammatory phenotype of early lesions might be the consequence of mast cell activation in the adventitia. Importantly, Sun et al32 recently demonstrated that mast cells promote atherogenesis by promoting the secretion of the proinflammatory cytokine IL-6 and IFN-γ. Also, the activation of mast cells is involved in induction of rupture of advanced lesions.30 Targeting mast cell activation might thus be a promising strategy for combination with lipid-lowering therapies for treatment of established atherosclerotic lesions.
We recognize the limitations of the present study. Dietary lipid lowering might not be as effective as the use of statins in the treatment of atherosclerosis, given the many pleiotropic effects beyond their cholesterol-lowering capacity.33 Also, mice have no cholesterol ester transport protein (CETP), but humans do.34 Moreover, in contrast to humans, the LDLr KO mice used in the present study developed atherosclerosis at the aortic root and descending aorta, rather than the coronary artery. In addition, mice seldom show signs of myocardial infarction or stroke,35 because murine atherosclerotic lesions are not prone to plaque rupture. Clearly, findings based on mouse models must be carefully evaluated in larger animal models and in humans.
In summary, the present study provides important insights into the dynamic remodeling of established atherosclerotic lesions on dietary lipid lowering. Progression of established atherosclerotic lesions after dietary lipid lowering indicates that more effective therapeutic strategies are needed for the treatment of atherosclerosis. The dynamics of lesion remodeling on dietary lipid lowering are complex, and depend on the stage of atherosclerotic lesion development. Establishment of stage-specific therapeutic protocols might thus improve the eventual outcome; however, the requirement of stage-specific therapy for atherosclerosis calls for the development of better molecular imaging tools to characterize lesion composition in vivo.
Acknowledgment
We thank Dr. Theo Reijmers (Division of Analytical Biosciences, Leiden/Amsterdam Center for Drug Research, Leiden University) for his help with statistical analysis.
Footnotes
Supported by grants from the Netherlands Heart Foundation (grant 2001T4101 to M.V.E. and Y.Z.; Established Investigator grant 2007T056 to M.V.E.), the Catalan University and Research Grants Management Agency (Beatriu de Pinós postdoctoral grant to L.C.-B.), the Top Institute Pharma (TI Pharma project T2-110, to Y.Z. and L.C.-B.), and the Netherlands Organization for Scientific Research (VIDI grant 917.66.301 to M.V.E.).
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.05.020.
Supplementary data
Effect of dietary cholesterol lowering on lipoprotein distribution of total cholesterol. LDLrKO mice were fed WTD for 5 weeks to induce lesion formation. Thereafter, the animals were switched to regular chow and were euthanized at 0 (C), 1 (W1), 3 (W3), and 6 (W6) weeks after the diet switch. A 30-μL aliquot of serum was fractionated in a Pharmacia Smart microchromatography column and the total cholesterol of each fraction was determined. Fractions 3 to 6 represent VLDL, fractions 8 to 12 represent LDL, and fractions 16 to 20 represent high density lipoprotein (HDL). Data are reported as means ± SEM of eight mice.
Effect of dietary cholesterol lowering on the cell apoptosis in established lesions. LDLrKO mice were fed WTD for 5 or 9 weeks to induce lesion formation. Thereafter, the animals were switched to regular chow and were euthanized at 0 (C), 1 (W1), 3 (W3), and 6 (W6) weeks after the diet switch. Apoptosis is expressed as the percentage TUNEL-positive relative to total nuclei in the atherosclerotic lesions. Data are reported as means ± SEM of eight mice. *P < 0.05 and ***P < 0.001 versus group C; #P < 0.05 and ###P < 0.001 versus group W1; and $P < 0.05 versus group W3.
Effect of dietary cholesterol lowering on oxidative stress in established lesions. LDLrKO mice were fed WTD for 5 or 9 weeks to induce lesion formation. Thereafter, the animals were switched to regular chow and were euthanized at 0 (C), 1 (W1), 3 (W3), and 6 (W6) weeks after the diet switch. Oxidative stress is expressed as the amount of nitrotyrosine and α-8-hydroxy-2′-deoxyguanosine (8-OHdG) in the atherosclerotic lesions. Data are reported as means ± SEM of five mice. *P < 0.05, **P < 0.01, and ***P < 0.001 versus group C.
Effect of dietary cholesterol lowering on the development of established lesions in the aortic arch. LDLrKO mice were fed WTD for 9 weeks to induce lesion formation. Thereafter, the animals were switched to regular chow and were euthanized at 0 (C), 1 (W1), 3 (W3), and 6 weeks after the diet switch. Mean lesion area (A) and macrophage (B), collagen (C), and necrotic core (D) content were quantified as described in Materials and Methods. Data are reported as means ± SEM of eight mice. *P < 0.05, **P < 0.01, and ***P < 0.001 versus group C; #P < 0.05, ##P < 0.01, and ###P < 0.001 versus group W1; and $P < 0.05 and $$$P < 0.001 versus group W3.
References
- 1.Zadelaar S., Kleemann R., Verschuren L., de Vries-Van der Weij J., van der Hoorn J., Princen H.M., Kooistra T. Mouse models for atherosclerosis and pharmaceutical modifiers. Arterioscler Thromb Vasc Biol. 2007;27:1706–1721. doi: 10.1161/ATVBAHA.107.142570. [DOI] [PubMed] [Google Scholar]
- 2.Falk E. Pathogenesis of atherosclerosis. J Am Coll Cardiol. 2006;47(8 Suppl):C7–C12. doi: 10.1016/j.jacc.2005.09.068. [DOI] [PubMed] [Google Scholar]
- 3.Nissen S.E., Tuzcu E.M., Schoenhagen P., Brown B.G., Ganz P., Vogel R.A., Crowe T., Howard G., Cooper C.J., Brodie B., Grines C.L., DeMaria A.N., REVERSAL Investigators Effect of intensive compared with moderate lipid-lowering therapy on progression of coronary atherosclerosis: a randomized controlled trial. JAMA. 2004;291:1071–1080. doi: 10.1001/jama.291.9.1071. [DOI] [PubMed] [Google Scholar]
- 4.Sipahi I., Nicholls S.J., Tuzcu E.M., Nissen S.E. Coronary atherosclerosis can regress with very intensive statin therapy. Cleve Clin J Med. 2006;73:937–944. doi: 10.3949/ccjm.73.10.937. [DOI] [PubMed] [Google Scholar]
- 5.Chhatriwalla A.K., Nicholls S.J., Wang T.H., Wolski K., Sipahi I., Crowe T., Schoenhagen P., Kapadia S., Tuzcu E.M., Nissen S.E. Low levels of low-density lipoprotein cholesterol and blood pressure and progression of coronary atherosclerosis. J Am Coll Cardiol. 2009;53:1110–1115. doi: 10.1016/j.jacc.2008.09.065. [DOI] [PubMed] [Google Scholar]
- 6.Brown B.G., Zhao X.Q., Sacco D.E., Albers J.J. Lipid lowering and plaque regression: New insights into prevention of plaque disruption and clinical events in coronary disease. Circulation. 1993;87:1781–1791. doi: 10.1161/01.cir.87.6.1781. [DOI] [PubMed] [Google Scholar]
- 7.Bucher H.C., Griffith L.E., Guyatt G.H. Systematic review on the risk and benefit of different cholesterol-lowering interventions. Arterioscler Thromb Vasc Biol. 1999;19:187–195. doi: 10.1161/01.atv.19.2.187. [DOI] [PubMed] [Google Scholar]
- 8.Nissen S.E., Nicholls S.J., Sipahi I., Libby P., Raichlen J.S., Ballantyne C.M., Davignon J., Erbel R., Fruchart J.C., Tardif J.C., Schoenhagen P., Crowe T., Cain V., Wolski K., Goormastic M., Tuzcu E.M., ASTEROID Investigators Effect of very high-intensity statin therapy on regression of coronary atherosclerosis: the ASTEROID trial. JAMA. 2006;295:1556–1565. doi: 10.1001/jama.295.13.jpc60002. [DOI] [PubMed] [Google Scholar]
- 9.Bots M.L., Palmer M.K., Dogan S., Plantinga Y., Raichlen J.S., Evans G.W., O'Leary D.H., Grobbee D.E., Crouse J.R., 3rd, METEOR Study Group Intensive lipid lowering may reduce progression of carotid atherosclerosis within 12 months of treatment: the METEOR study. J Intern Med. 2009;265:698–707. doi: 10.1111/j.1365-2796.2009.02073.x. [DOI] [PubMed] [Google Scholar]
- 10.Bennett M.R. The atherosclerotic plaque was not built in a day: the dynamic nature of plaque progression and instability. Heart Metab. 2007;36:5–7. [Google Scholar]
- 11.Bligh E.G., Dyer W.J. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 12.Tacke F., Alvarez D., Kaplan T.J., Jakubzick C., Spanbroek R., Llodra J., Garin A., Liu J., Mack M., van Rooijen N., Lira S.A., Habenicht A.J., Randolph G.J. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J Clin Invest. 2007;117:185–194. doi: 10.1172/JCI28549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maiellaro K., Taylor W.R. The role of the adventitia in vascular inflammation. Cardiovasc Res. 2007;75:640–648. doi: 10.1016/j.cardiores.2007.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bot I., de Jager S.C., Zernecke A., Weber C., van Berkel T.J., Biessen E.A. Mast cell activation promotes leukocyte recruitment and adhesion to the atherosclerotic plaque. Circulation. 2007;116(Suppl 1):II_242. [Google Scholar]
- 15.Weber C., Zernecke A., Libby P. The multifaceted contributions of leukocyte subsets to atherosclerosis: lessons from mouse models. Nat Rev Immunol. 2008;8:802–815. doi: 10.1038/nri2415. [DOI] [PubMed] [Google Scholar]
- 16.Getz G.S., Reardon C.A. Diet and murine atherosclerosis. Arterioscler Thromb Vasc Biol. 2006;26:242–249. doi: 10.1161/01.ATV.0000201071.49029.17. [DOI] [PubMed] [Google Scholar]
- 17.Aikawa M., Rabkin E., Okada Y., Voglic S.J., Clinton S.K., Brinckerhoff C.E., Sukhova G.K., Libby P. Lipid lowering by diet reduces matrix metalloproteinase activity and increases collagen content of rabbit atheroma: a potential mechanism of lesion stabilization. Circulation. 1998;97:2433–2444. doi: 10.1161/01.cir.97.24.2433. [DOI] [PubMed] [Google Scholar]
- 18.Verhamme P., Quarck R., Hao H., Knaapen M., Dymarkowski S., Bernar H., Van Cleemput J., Janssens S., Vermylen J., Gabbiani G., Kockx M., Holvoet P. Dietary cholesterol withdrawal reduces vascular inflammation and induces coronary plaque stabilization in miniature pigs. Cardiovasc Res. 2002;56:135–144. doi: 10.1016/s0008-6363(02)00515-1. [DOI] [PubMed] [Google Scholar]
- 19.Rekhter M.D. Collagen synthesis in atherosclerosis: too much and not enough. Cardiovasc Res. 1999;41:376–384. doi: 10.1016/s0008-6363(98)00321-6. [DOI] [PubMed] [Google Scholar]
- 20.Schrijvers D.M., De Meyer G.R., Kockx M.M., Herman A.G., Martinet W. Phagocytosis of apoptotic cells by macrophages is impaired in atherosclerosis. Arterioscler Thromb Vasc Biol. 2005;25:1256–1261. doi: 10.1161/01.ATV.0000166517.18801.a7. [DOI] [PubMed] [Google Scholar]
- 21.Clarke M.C., Talib S., Figg N.L., Bennett M.R. Vascular smooth muscle cell apoptosis induces interleukin-1-directed inflammation: effects of hyperlipidemia-mediated inhibition of phagocytosis. Circ Res. 2010;106:363–372. doi: 10.1161/CIRCRESAHA.109.208389. [DOI] [PubMed] [Google Scholar]
- 22.Swirski F.K., Libby P., Aikawa E., Alcaide P., Luscinskas F.W., Weissleder R., Pittet M.J. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. 2007;117:195–205. doi: 10.1172/JCI29950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Combadière C., Potteaux S., Rodero M., Simon T., Pezard A., Esposito B., Merval R., Proudfoot A., Tedgui A., Mallat Z. Combined inhibition of CCL2, CX3CR1, and CCR5 abrogates Ly6C(hi) and Ly6C(lo) monocytosis and almost abolishes atherosclerosis in hypercholesterolemic mice. Circulation. 2008;117:1649–1657. doi: 10.1161/CIRCULATIONAHA.107.745091. [DOI] [PubMed] [Google Scholar]
- 24.Swirski F.K., Nahrendorf M., Etzrodt M., Wildgruber M., Cortez-Retamozo V., Panizzi P., Figueiredo J.L., Kohler R.H., Chudnovskiy A., Waterman P., Aikawa E., Mempel T.R., Libby P., Weissleder R., Pittet M.J. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325:612–616. doi: 10.1126/science.1175202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Llodrá J., Angeli V., Liu J., Trogan E., Fisher E.A., Randolph G.J. Emigration of monocyte-derived cells from atherosclerotic lesions characterizes regressive, but not progressive, plaques. Proc Natl Acad Sci USA. 2004;101:11779–11784. doi: 10.1073/pnas.0403259101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ye D., Zhao Y., Hildebrand R.B., Singaraja R.R., Hayden M.R., Van Berkel T.J.C., Van Eck M. The dynamics of macrophage infiltration into the arterial wall during atherosclerotic lesion development in LDL receptor knockout mice. Am J Pathol. 2011;178:413–422. doi: 10.1016/j.ajpath.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zernecke A., Bot I., Djalali-Talab Y., Shagdarsuren E., Bidzhekov K., Meiler S., Krohn R., Schober A., Sperandio M., Soehnlein O., Bornemann J., Tacke F., Biessen E.A., Weber C. Protective role of CXC receptor 4/CXC ligand 12 unveils the importance of neutrophils in atherosclerosis. Circ Res. 2008;102:209–217. doi: 10.1161/CIRCRESAHA.107.160697. [DOI] [PubMed] [Google Scholar]
- 28.van Leeuwen M., Gijbels M.J., Duijvestijn A., Smook M., van de Gaar M.J., Heeringa P., de Winther M.P., Tervaert J.W. Accumulation of myeloperoxidase-positive neutrophils in atherosclerotic lesions in LDLR−/− mice. Arterioscler Thromb Vasc Biol. 2008;28:84–89. doi: 10.1161/ATVBAHA.107.154807. [DOI] [PubMed] [Google Scholar]
- 29.Libby P., Shi G.P. Mast cells as mediators and modulators of atherogenesis. Circulation. 2007;115:2471–2473. doi: 10.1161/CIRCULATIONAHA.107.698480. [DOI] [PubMed] [Google Scholar]
- 30.Bot I., de Jager S.C., Zernecke A., Lindstedt K.A., van Berkel T.J., Weber C., Biessen E.A. Perivascular mast cells promote atherogenesis and induce plaque destabilization in apolipoprotein E-deficient mice. Circulation. 2007;115:2516–2525. doi: 10.1161/CIRCULATIONAHA.106.660472. [DOI] [PubMed] [Google Scholar]
- 31.Parthasarathy S., Santanam N. Mechanisms of oxidation, antioxidants, and atherosclerosis. Curr Opin Lipidol. 1994;5:371–375. doi: 10.1097/00041433-199410000-00009. [DOI] [PubMed] [Google Scholar]
- 32.Sun J., Sukhova G.K., Wolters P.J., Yang M., Kitamoto S., Libby P., MacFarlane L.A., Mallen-St Clair J., Shi G.P. Mast cells promote atherosclerosis by releasing proinflammatory cytokines. Nat Med. 2007;13:719–724. doi: 10.1038/nm1601. [DOI] [PubMed] [Google Scholar]
- 33.Blum A., Shamburek R. The pleiotropic effects of statins on endothelial function, vascular inflammation, immunomodulation and thrombogenesis. Atherosclerosis. 2009;203:325–330. doi: 10.1016/j.atherosclerosis.2008.08.022. [DOI] [PubMed] [Google Scholar]
- 34.Breslow J.L. Transgenic mouse models of lipoprotein metabolism and atherosclerosis. Proc Natl Acad Sci USA. 1993;90:8314–8318. doi: 10.1073/pnas.90.18.8314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cullen P., Baetta R., Bellosta S., Bernini F., Chinetti G., Cignarella A., von Eckardstein A., Exley A., Goddard M., Hofker M., Hurt-Camejo E., Kanters E., Kovanen P., Lorkowski S., McPheat W., Pentikäinen M., Rauterberg J., Ritchie A., Staels B., Weitkamp B., de Winther M., MAFAPS Consortium Rupture of the atherosclerotic plaque: does a good animal model exist? Arterioscler Thromb Vasc Biol. 2003;23:535–542. doi: 10.1161/01.ATV.0000060200.73623.F8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Effect of dietary cholesterol lowering on lipoprotein distribution of total cholesterol. LDLrKO mice were fed WTD for 5 weeks to induce lesion formation. Thereafter, the animals were switched to regular chow and were euthanized at 0 (C), 1 (W1), 3 (W3), and 6 (W6) weeks after the diet switch. A 30-μL aliquot of serum was fractionated in a Pharmacia Smart microchromatography column and the total cholesterol of each fraction was determined. Fractions 3 to 6 represent VLDL, fractions 8 to 12 represent LDL, and fractions 16 to 20 represent high density lipoprotein (HDL). Data are reported as means ± SEM of eight mice.
Effect of dietary cholesterol lowering on the cell apoptosis in established lesions. LDLrKO mice were fed WTD for 5 or 9 weeks to induce lesion formation. Thereafter, the animals were switched to regular chow and were euthanized at 0 (C), 1 (W1), 3 (W3), and 6 (W6) weeks after the diet switch. Apoptosis is expressed as the percentage TUNEL-positive relative to total nuclei in the atherosclerotic lesions. Data are reported as means ± SEM of eight mice. *P < 0.05 and ***P < 0.001 versus group C; #P < 0.05 and ###P < 0.001 versus group W1; and $P < 0.05 versus group W3.
Effect of dietary cholesterol lowering on oxidative stress in established lesions. LDLrKO mice were fed WTD for 5 or 9 weeks to induce lesion formation. Thereafter, the animals were switched to regular chow and were euthanized at 0 (C), 1 (W1), 3 (W3), and 6 (W6) weeks after the diet switch. Oxidative stress is expressed as the amount of nitrotyrosine and α-8-hydroxy-2′-deoxyguanosine (8-OHdG) in the atherosclerotic lesions. Data are reported as means ± SEM of five mice. *P < 0.05, **P < 0.01, and ***P < 0.001 versus group C.
Effect of dietary cholesterol lowering on the development of established lesions in the aortic arch. LDLrKO mice were fed WTD for 9 weeks to induce lesion formation. Thereafter, the animals were switched to regular chow and were euthanized at 0 (C), 1 (W1), 3 (W3), and 6 weeks after the diet switch. Mean lesion area (A) and macrophage (B), collagen (C), and necrotic core (D) content were quantified as described in Materials and Methods. Data are reported as means ± SEM of eight mice. *P < 0.05, **P < 0.01, and ***P < 0.001 versus group C; #P < 0.05, ##P < 0.01, and ###P < 0.001 versus group W1; and $P < 0.05 and $$$P < 0.001 versus group W3.