

Abstract
Lipoprotein particles accumulate in Bruch's membrane before the development of basal deposits and drusen, two histopathologic lesions that define age-related macular degeneration (AMD). We therefore, sought to determine which molecules could participate in lipoprotein retention. Wild-type or lipoprotein lipase–deficient mice were injected with low-dose d-galactose or PBS subcutaneously for 8 weeks to induce advanced glycation endproduct (AGE) formation. Some mice were also injected with the AGE breaker phenacylphiazolium bromide and d-galactose. Rhodamine-labeled low-density lipoproteins were injected into mice, and the fluorescence was measured up to 72 hours later. AGEs, proteoglycans, and other lipid-retaining molecules were evaluated by IHC. Lipoprotein lipase distribution was assessed in AMD samples by IHC. d-galactose–treated mice retained lipoproteins in the retinal pigment epithelial and Bruch's membrane to a greater extent than either PBS- or phenacylphiazolium bromide/d-galactose–treated mice at 24 and 72 hours after injection (P ≤ 0.04). Immunolabeling for carboxymethyllysine, biglycan, and lipoprotein lipase was found in d-galactose–treated mice only. Mice deficient for lipoprotein lipase treated with d-galactose did not retain lipoproteins to any measureable extent. Human AMD samples had lipoprotein lipase labeling within drusen, basal deposits, and the choroid. Mice treated with d-galactose to induce AGE formation in Bruch's membrane retain intravenously injected lipoproteins. Our results suggest that lipoprotein retention in Bruch's membrane is mediated by lipoprotein lipase.
Age-related macular degeneration (AMD) is the most common cause of vision loss among the elderly in the United States. Genetic studies have identified polymorphisms in complement factors C3, H, and I; HTRA1; ARMS2; and mitochondrial DNA polymorphism A4917G as risk factors and complement factor B and apolipoprotein E4 as protective factors.1–11 The influence of these genetic alterations on disease development remains unresolved at present. Bruch's membrane is a pentilaminar structure that is composed of the retinal pigment epithelial (RPE) basement membrane, inner collagenous layer, middle elastic layer, outer collagenous layer, and the choriocapillaris basement membrane. During aging, Bruch's membrane steadily accumulates neutral lipids contained within apolipoprotein B100 (apoB100) lipoprotein particles. These particles accumulate as a “wall” in the inner collagenous layer.12 Lipoprotein particles also accumulate in basal laminar deposits, basal linear deposits, and drusen, the pathognomonic histopathologic Bruch's membrane lesions of AMD.12–18 The identical laminar location of lipoprotein deposition within Bruch's membrane before the development of these hallmark lesions suggests that lipoproteins, in addition to the genetic and environmental influences, play a critical role in the pathophysiology of AMD.
The relative importance of lipoproteins in AMD pathogenesis is further suggested by studies that have identified polymorphisms in lipid-processing genes, including hepatic lipase and high-density lipoprotein c-associated alleles near lipoprotein lipase (LPL).19,20 Lipoproteins under normal conditions are designed to transport lipids to tissues around the body. ApoB100 lipoproteins, such as low-density lipoproteins (LDLs), appear to be particularly important for the retina. Tserentsoodol et al21 showed that LDLs are the preferred carrier of lipids into the retina and that they are internalized principally by the RPE through LDL receptors.
In atherosclerosis, lipoproteins in excess and their subsequent retention within the vessel wall initiates a chronic, pathologic inflammatory response from the oxidation of retained lipoproteins.22,23 Our laboratory has shown that oxidized lipoproteins accumulate in Bruch's membrane and basal deposits and drusen of patients with early AMD and that oxidized LDLs induce a pathologic phenotype of cultured RPE cells.24 This work suggests that lipoproteins that accumulate within Bruch's membrane are retained for a sufficient period to become oxidized and can induce a phenotypic change to the RPE.
Several changes to the subendothelial matrix promote lipoprotein retention during the development of atherosclerosis. These include an accumulation of certain matrix proteoglycans such as biglycan, decoran, and versican that retain lipoproteins due to their electronegative charge.22,25 Secreted sphingomyelinases and LPL, which are distinct from hepatic lipase, endothelial lipase, and pancreatic lipase (PL), in the subendothelial matrix promote lipoprotein retention.26–28 The formation of advanced glycation endproducts (AGEs) also promotes lipoprotein retention within the subendothelial matrix.29,30 Because of the cross-linking of AGEs, the supramolecular matrix architecture is modified so that the normal diffusion of metabolites or molecules such as lipoproteins might be impaired.
We previously demonstrated that AGEs develop within Bruch's membrane, including basal deposits and drusen during aging and AMD.31 In mice, we have also shown that low-dose d-galactose (D-gal) treatment promotes AGE formation in Bruch's membrane and induces an accelerated aging phenotype with features of AMD in Bruch's membrane.32,33 Given the accumulation of lipoproteins in Bruch's membrane, and their oxidation, we hypothesized that aging changes to Bruch's membrane encourages lipoprotein retention, which then leads to their oxidation. Oxidized lipoproteins are known to stimulate complement.34 Accumulation of oxidized lipoproteins, therefore, could induce complement-mediated inflammation, leading to either RPE cell injury and/or drusen formation within Bruch's membrane. We, therefore, treated mice with low-dose D-gal to induce AGE formation as an experimental model of Bruch's membrane aging to determine whether lipoproteins are retained.
Materials and Methods
Animals and Care
An equal number of male and female C57Bl6 (The Jackson Laboratories, Bar Harbor, ME), B6.129 (The Jackson Laboratories) or LPL-deficient mice (B6.129S4-Lpltm1Ijg/J; The Jackson Laboratories) were fed standard rodent chow and water ad libitum and kept in a 12-hour light-dark cycle. All experiments were conducted according to the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research, and the research was approved by the institutional research board at Johns Hopkins Medical Institutions.
Induction of AGE Changes
Five-month-old mice were divided into three groups. Group 1 mice were injected subcutaneously with D-gal (50 mg/kg; Sigma-Aldrich, St Louis, MO) daily for 8 weeks, following our previously published protocol.33 Group 2 mice were injected with the AGE breaker phenacylphiazolium bromide (PTB; 10 mg/kg; Prime Organics, Woburn, MA) and D-gal (50 mg/kg) for 8 weeks, and group 3 mice were injected subcutaneously with PBS for 8 weeks.
Injection of Labeled LDL
Human LDL or oxidized LDL was purchased from (Calbiochem, Gibbstown, NJ; Intracel, Frederick, MD, respectively). According to the manufacturer, the LDL preparations were purified by gradient-based ultracentrifugation. LDLs were verified by isoelectric focusing with the use of immunoelectrophoresis to obtain a single arc. LDL content was evaluated by staining for lipids by Fast Red 70 and proteins by Coomassie Blue staining. To avoid aggregation, LDL and oxidized LDL preparations were used within 1 week of purchase. LDLs were labeled with rhodamine with the use of the Fluororeporter Rhodamine Red Protein Labeling Kit (Invitrogen, Inc., Carlsbad, CA) according to the manufacturer's instructions. After the 8-week D-gal treatment, mice were injected intravenously via the tail vein with 500 μg of rhodamine-labeled LDL or oxidized LDL and sacrificed at 5 minutes, 24 hours, and 72 hours later to evaluate LDL retention in the fundus.
Tissue Processing
Mouse eyes were enucleated at the appropriate time points and lightly fixed in 2% paraformaldehyde (Sigma-Aldrich), cryopreserved, and sectioned for histochemical evaluation.
Human globes from 49- to 93-year-old donors were obtained from the National Disease Research Interchange (Philadelphia, PA). Eyes had been enucleated within 6 hours of death, were on life support for <24 hours, and were processed within 24 hours of death (Table 1). The protocol in this study adhered to the tenets of the Declaration of Helsinki for research involving human tissue. Globes were selected for early AMD and defined by clinical history and the presence of basal deposits thicker than the normal height of an RPE cell (ie, >8 μm), as defined by Sarks35 with the use of our previously described protocol.36 Macular calottes were fixed for 1 hour in 2% paraformaldehyde and then cryoprotected by progressive infiltration in 10% and 20% sucrose in PBS (w/v) before freezing in 2:1 sucrose 20% (w/v):OCT compound at −80°C.
Table 1.
Donor Characteristics
| Patient | AMD | Age (years) | Sex | Enucleation (hours) | Cause of death |
|---|---|---|---|---|---|
| 1 | No | 49 | F | 5:10 | Lung cancer |
| 2 | No | 91 | F | 5:25 | Lung cancer |
| 3 | No | 91 | F | 5:25 | Lung cancer |
| 4 | No | 91 | M | 2:20 | Subdural hematoma |
| 5 | No | 90 | F | 3:53 | Respiratory arrest |
| 6 | No | 90 | M | 2:45 | Stroke |
| 7 | Yes | 70 | F | 6:10 | Lung cancer |
| 8 | Yes | 72 | F | 6:11 | Cardiac arrest |
| 9 | Yes | 85 | F | 2:28 | Multiorgan failure |
| 10 | Yes | 90 | F | 3:53 | Respiratory arrest |
| 11 | Yes | 93 | F | 5:46 | Renal failure |
AMD, age-related macular degeneration; F, female; M, male.
IHC and Fluorescence IHC
For fluorescence immunohistochemistry (IHC), mouse cryosections (7 μm) were first blocked with 2% goat or donkey serum (Jackson ImmunoResearch, West Grove, PA) followed by blocking with avidin/biotin blocking reagent (Vector Labs, Burlingame, CA), and then treated with a mouse on mouse blocking reagent (anti-mouse IgG blocking reagent; Vector Labs) if necessary for 1 hour at room temperature. Sections were then incubated with the primary antibody: a monoclonal anti-carboxymethyl lysine (CML) monoclonal antibody (1:50; Wako, Richmond, VA), a rabbit polyclonal antibody to LPL (1:50; Santa Cruz Biotechnologies, Santa Cruz, CA), a rabbit polyclonal antibody to versican (1:50; Santa Cruz Biotechnologies), a goat polyclonal antibody to biglycan (1:50; Santa Cruz Biotechnologies), a rabbit polyclonal antibody to acid and neutral sphingomyelinase (1:50; Santa Cruz Biotechnologies), or the appropriate isotype IgG. The anti-LPL antibody was raised against amino acids 28 to 80, mapping near the N-terminus of LPL of human origin (Santa Cruz Biotechnologies). This antibody was Protein A purified and then glutathione S-transferase subtracted. It does not cross-react with other lipases and is suitable for use in human and mouse tissues.37–39 Sections were incubated with primary antibody overnight at 4°C, washed with PBS, and incubated with goat anti-rabbit and donkey anti-goat secondary antibodies, respectively (1:400; Santa Cruz Biotechnologies). Appropriate mouse, rabbit, and goat IgGs (Santa Cruz Biotechnologies) were used as isotype controls. Sections were imaged with a confocal microscope.
Human macular cryosections (7 μm) were blocked with avidin/biotin blocking reagent (Vector Labs) and then incubated with rabbit anti-LPL antibody (1:1000; Santa Cruz Biotechnologies). Sections were then incubated with biotinylated anti-rabbit IgG (Vector Labs) for 30 minutes at room temperature and then with alkaline phosphatase–conjugated avidin streptavidin (Vectastain ABC Kit; Vector Labs) and the appropriate substrate (Alkaline Phosphatase Substrate III; Vector Labs). Sections were counterstained with Nuclear Fast Red (Vector Labs). To permit interpretation of immunoreaction product in the RPE and choroid, some sections were fixed in 4% paraformaldehyde overnight at 4°C immediately after streptavidin APase IHC. Slides were washed in distilled water at room temperature, immersed in 0.05% potassium permanganate solution (Aldrich Chemical Co., Milwaukee, WI) for 25 minutes, and then rinsed in distilled water for 5 minutes. Sections were treated with 35% paracetic acid (Sigma-Aldrich) in a humidified chamber for 60 to 90 minutes at room temperature followed by washing in distilled water for 10 minutes and imaged in a axiovert 200M microscope (Carl Zeiss Micro Imaging, Inc., Thornwood, NY).
Confocal Microscopy
Confocal microscopy was performed on a laser scanning confocal microscope (Zeiss 510 META confocal microscope; Carl Zeiss Micro Imaging, Inc.). Rhodamine fluorescence was visualized with the use of an excitation wavelength of 543 nm and an emission wavelength between 560 and 585 nm. Green fluorescence staining was visualized by exciting with a 488-nm laser beam and collecting emissions between 500 and 552 nm. The fluorescence intensity was quantified with Zeiss software (AxioVision). Results were compared with animals that received an i.v. injection of PBS. Specifically, the measured fluorescence of the PBS-injected mice was subtracted from the fluorescence obtained from eyes in each of the experimental groups (D-gal, PTB/D-gal, and PBS). Separate intensities were calculated for the RPE, Bruch's membrane, and choroid. The fluorescence values were standardized by cross-sectional area of each tissue (n = 3 animals per time point per group).
Statistical Analysis
Statistical significance was determined with analysis of variance and Bonferroni procedure for post hoc analysis.
Results
D-Gal Treatment Induces AGE Formation in Bruch's Membrane
Our laboratory has previously shown that AGEs form in Bruch's membrane of mice treated with low-dose D-gal, coincident with the development of ultrastructural features of early AMD.32,33 To verify that this treatment induced AGE formation in Bruch's membrane, sections were evaluated for CML, a well-established AGE found to accumulate in basal deposits and drusen in AMD.31 Figure 1 (A and B) shows that mice treated with D-gal develop CML in Bruch's membrane, whereas PBS-treated mice did not develop CML (Figure 1, E and F). Mice treated with PTB, the AGE cross-link breaker, had reduced but detectable staining for CML (Figure 1, C and D). The RPE also showed CML labeling and an altered cell structure, as we have previously reported32 (Figure 1A).
Figure 1.
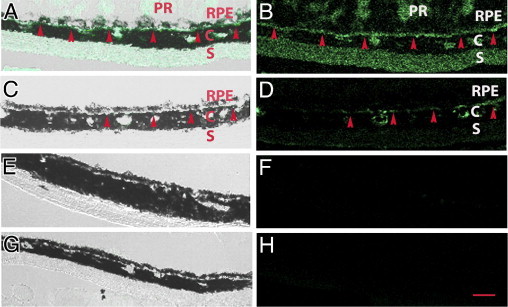
Immunofluorescence analysis of CML. Mice were treated with D-gal for 8 weeks to induce advanced glycation endproduct formation. Merged brightfield and fluorescence images (A, C, E, and G) and fluorescence image only (B, D, F, and H). A and B: Immunostaining for CML appears in the RPE, Bruch's membrane (red arrowheads), and choroidal vasculature (C) of D-gal–treated mice. Staining is also seen in the sclera (S) and photoreceptor segments (PR) when present. C and D: Mice treated with D-gal and phenacylphiazolium bromide, the advanced glycation endproduct cross-link breaker, had mild CML labeling. E and F: PBS-treated mice had no CML labeling. G and H: IgG control is also without labeling (n = 3 for each group). Scale bar = 10 μm.
LDLs Are Retained in Bruch's Membrane by AGE Formation
To show that AGE formation in Bruch's membrane is acutely associated with LDL retention, C57Bl6 mice treated with D-gal for 8 weeks were then injected intravenously with rhodamine-labeled LDLs. At 5 minutes, rhodamine-labeled LDL was found in the RPE, Bruch's membrane, and choroid of all three groups without a difference in fluorescence (data not shown). Although minimal labeling was observed in PTB/D-gal– and PBS-treated mice at 24 and 72 hours after injection, rhodamine-labeled LDL was more prominently retained in the RPE and Bruch's membrane, as shown in Figure 2A. As graphically quantified in Figure 2B, the RPE of D-gal mice retained LDLs 24 hours after injection significantly more than the PTB/D-gal– and PBS-injected mice (P < 0.05). LDLs were similarly retained in the RPE 72 hours after injection in the D-gal group relative to the PTB/D-gal and PBS controls (P < 0.01). In Bruch's membrane of the D-gal–treated mice, LDLs were increased at 24 and 72 hours after injection compared with the PTB/D-gal and PBS controls. At 24 hours, they were significantly higher in the D-gal group than in the PTB/D-gal and PBS controls (P ≤ 0.01). At 72 hours, the relative fluorescence remained higher in the D-gal group than in the PTB/D-gal and PBS groups (P < 0.01). A similar trend was seen with LDLs in the choroid, but the magnitude of retention was lower than the other tissues.
Figure 2.
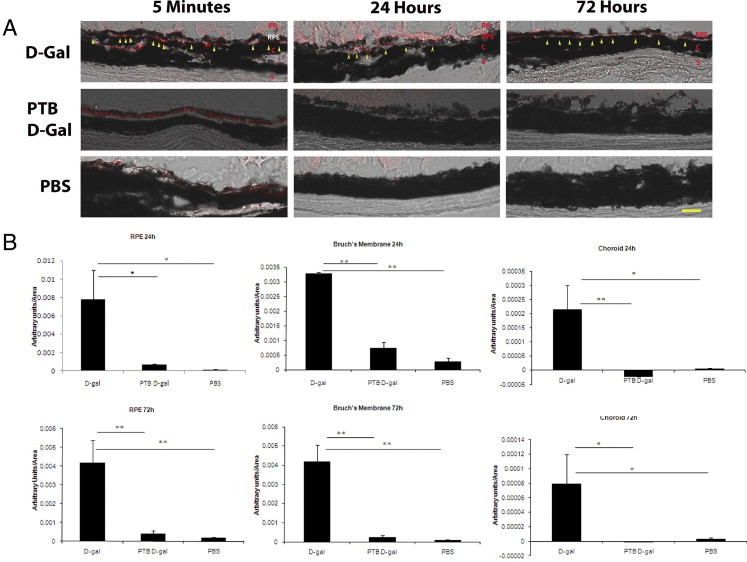
Rhodamine-labeled low-density lipoproteins (rho-LDLs) are retained in the RPE and Bruch's membrane of D-gal–treated mice. A: Five minutes after injection, rho-LDL is seen within Bruch's membrane (yellow arrowheads) and the choroid (C) in each of the three groups. Twenty-four and 72 hours after injection, rho-LDL appears in the RPE and Bruch's membrane (yellow arrowheads) mainly in the D-gal group. Occasional labeling of photoreceptors (PR) is seen when they are present. No labeling is observed in the phenacylphiazolium bromide (PTB)/D-gal or the PBS groups at these time points; n = 3 mice for each group. S, sclera. Scale bar = 25 μm. B: Quantification of LDL retention in the RPE, Bruch's membrane, and choroid at 24 and 72 hours after injection. Bars represent the mean ± SEM. *P < 0.05, **P < 0.01.
It is possible that oxidized LDLs from the systemic circulation can get retained in Bruch's membrane instead of LDLs being oxidized after being retained. To evaluate this possibility, rhodamine-labeled oxidized LDLs were injected with the use of the same protocol. Although labeling was seen at 5 minutes, confirming that oxidized LDLs transit to the RPE/choroidal complex, we did not see any measurable retention at 24 and 72 hours (data not shown). These results suggest that the oxidized LDLs that are identified in Bruch's membrane are probably formed after the retention of LDLs rather than systemically delivered as oxidized LDL.
Proteoglycans Accumulate in Bruch's Membrane after D-Gal Treatment
Several matrix proteoglycans, including decorin, versican, and biglycan, have been associated with LDL retention in the subendothelial cell matrix early in the course of atherosclerosis.22,25 We were particularly intrigued by biglycan because we previously identified a 1.4-fold increase in biglycan expression in the RPE/choroid of mice treated with D-gal.33 We, therefore, performed fluorescence IHC to determine whether these proteoglycans developed in Bruch's membrane after D-gal treatment. Although decorin and versican showed no staining, biglycan was detected in Bruch's membrane of D-gal–treated mice (n = 3) but not PTB/D-gal– or PBS-treated mice, as shown in Figure 3, A–F.
Figure 3.
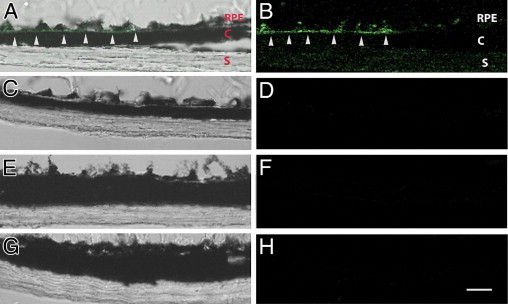
Immunofluorescence analysis of biglycan. Merged brightfield and fluorescence images (A, C, E, and G) and fluorescence image only to highlight green fluorescence image (B, D, F, and H). A and B: D-gal–injected mice have biglycan staining in Bruch's membrane (white arrowheads). C and D: Phenacylphiazolium bromide– and D-gal–injected mice. E and F: PBS-injected mice. G and H: IgG control. C, choroid; S, sclera; RPE, retinal pigment epithelial (n = 3 for each group). Scale bar = 25 μm.
LPL Also Accumulates in Bruch's Membrane after D-Gal Treatment
Because LPL and secretory sphingomyelinases also contribute to LDL retention in atherosclerosis,26–28 we performed fluorescence IHC experiments to determine whether these molecules accumulate in Bruch's membrane after D-gal treatment. We did not find IHC evidence of either secreted acid or neutral sphingomyelinase in any of our samples (data not shown). However, Figure 4 (A and B) shows that LPL localizes to the RPE, Bruch's membrane, and choroid of mice treated with D-gal. A similar, although more diffuse, staining pattern was seen in mice treated with D-gal and PTB (Figure 4, C and D), whereas mice treated with PBS showed no immunolabeling for LPL (Figure 4, E and F). These observations correlate with our previous work in which we found that LPL mRNA expression was increased in the RPE/choroid of mice treated with D-gal with the use of the same protocol as this study.33
Figure 4.
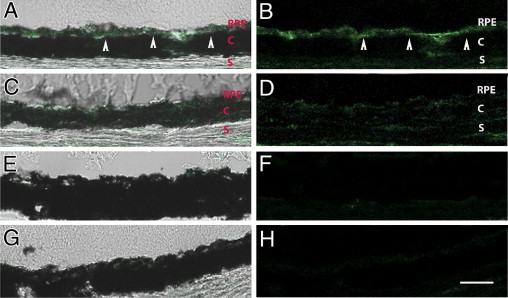
Immunofluorescence analysis of LPL. Merged brightfield and fluorescence images (A, C, E, and G) and fluorescence image only to highlight green fluorescence image (B, D, F, and H). A and B: D-gal–injected mice have LPL staining in the RPE, Bruch's membrane (arrowheads), and choroid (C). C and D: Phenacylphiazolium bromide– and D-gal–injected mice with mild LPL staining. E and F: PBS-injected mice have minimal LPL staining. G and H: IgG control; (n = 3 for each group). S, sclera. Scale bar = 25 μm.
LPL Deficiency Reduces D-gal–Associated LDL Retention
From this IHC survey, we determined that candidate molecules for retaining LDLs acutely in our model include AGEs, biglycan, and LPL. In mice overexpressing biglycan and human apoB100, Sallo et al,40 however, found that these mice did not have increased retention of lipid particles in Bruch's membrane over mice overexpressing only human apoB100. This observation suggests that molecules other than biglycan are important for lipoprotein binding in Bruch's membrane. We, therefore, obtained LPL-deficient mice and treated them with D-gal to induce AGEs to test whether LPL is involved in LDL retention. D-gal–treated LPL-deficient mice failed to retain LDLs to any measurable extent at 24 (n = 3) and 72 (n = 3) hours and showed no difference with mice that were injected with PBS for 8 weeks (n = 3; data not shown). These results suggest that LPL accumulation in Bruch's membrane contributes significantly to LDL retention.
LPL Also Accumulates in AMD
To further explore whether LPL could contribute to LDL retention in AMD, we evaluated human maculae for LPL. In all eyes (n = 11), the choroid was strongly labeled for LPL. In eyes without AMD (n = 6), we detected LPL immunolabeling along the choriocapillaris basement membrane but little to no staining within other areas of Bruch's membrane (Figure 5, A and B). However, in eyes with AMD, we identified discrete LPL labeling in basal deposits and drusen, two hallmark histopathologic lesions associated with AMD (n = 5; Figure 5, C and D). Figure 5C also shows a line of LPL staining within the inner Bruch's membrane that localizes to the inner collagenous layer.
Figure 5.
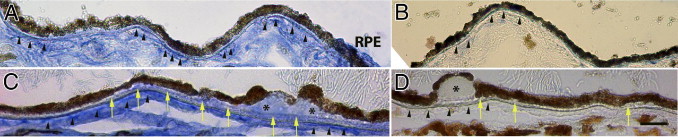
IHC evidence of LPL in AMD. A: A 49-year-old man without AMD with LPL labeling along the choriocapillaris basement membrane (arrowheads). Left side of image has mild basal deposit with diffuse immunolabeling for LPL. B: IgG control. Arrowheads point to choriocapillaris basement membrane. C: A 93-year-old man with non-neovascular AMD has LPL labeling along the choriocapillaris basement membrane (arrowheads) and diffuse LPL labeling in basal deposits and drusen (asterisk). Note line of LPL labeling at the inner collagenous layer of Bruch's membrane (yellow arrows). D: IgG control. Yellow arrows point to basal deposits. Scale bar = 25 μm.
Discussion
Our experiments show that age-related changes to Bruch's membrane through AGE formation can promote the acute retention of LDLs within the RPE and Bruch's membrane after systemic administration. To investigate whether molecules involved in LDL retention in atherosclerosis are similarly involved in retaining LDLs in Bruch's membrane, we identified in D-gal–treated mice the presence of the AGE product CML, biglycan, and LPL, but not versican, decorin, or sphingomyelinases. Because mice overexpressing biglycan and apoB100 did not have significant chronic lipid retention in Bruch's membrane,40 we instead focused on the role of LPL and tested in LPL-deficient mice whether D-gal treatment still caused LDL retention. We found that mice with LPL deficiency treated with D-gal did not retain LDLs to any measurable extent. These results suggest that acute LDL accumulation in Bruch's membrane of mice with AGE formation is attributed in large part by LPL.
Proteoglycans have a well-established role of retaining LDLs early in the pathogenesis of atherosclerosis.22,25 Gustaffson et al41 have shown in mice that LDL retention is initiated by direct binding to proteoglycans but shifts to binding with LPL later in the disease. Proteoglycans are glycoproteins that contain a core protein with covalently attached glycosaminoglycans. In atherosclerosis, the important and most abundant core proteins include biglycan, versican, and decorin.25 The most abundant glycosaminoglycan side chains are chondroitin sulfate and dermatan sulfate, both of which are increased in atherosclerosis, as well as heparan sulfate.42 Biglycan and versican have been extensively studied for their role in lipoprotein retention because they i) have strong affinity for chondroitin and dermatan sulfate proteoglycans,42 ii) are enriched in areas of atherosclerosis,42 and iii) bind to apoB100-containing lipoproteins.43 Biglycan has emerged as the most important core protein involved in the initial lipoprotein retention because it localizes to areas of lipoprotein retention before lesion development.44,45 Versican, however, appears to play a minor role in early lipoprotein retention because it often does not colocalize with lipoproteins in vivo.25 The role for decorin is less clear. It can bind lipoproteins in vitro, but it tends to localize to areas that are devoid of atherosclerotic lesions.42
Although it is well established that Bruch's membrane is a pentilaminar structure composed of the RPE basement membrane, the inner collagenous layer, middle elastic layer, outer collagenous layer, and choriocapillaris basement membrane, less is known about its proteoglycan content. Hewitt et al46 have identified a change in glycosaminoglycan content from its normal level of 77% chondroitin/dermatan sulfate and 23% heparan sulfate proteoglycans to 42% chondroitin/dermatan sulfate and 58% heparan sulfate with aging, and similarly 45% chondroitin/dermatan sulfate and the 55% heparan sulfate content in AMD. Kliffen et al47 reported diffuse immunostaining for chondroitin sulfate within basal deposits, whereas heparan sulfate was focally stained within basal deposits adjacent to the RPE in AMD samples. Preferential localization of heparan sulfate to basal deposits is particularly intriguing, given the recent observations by Kelly et al48 that heparan sulfate of Bruch's membrane accelerated complement activation. These observations, along with our identification of biglycan, suggest that biglycan is a potentially important molecule involved in lipoprotein retention within Bruch's membrane.
We also identified LPL in Bruch's membrane of D-gal–treated mice. LPL is a part of the nutritional system to deliver fatty acids to body tissues by lipoproteins.49 LPL appears to have both a protective and a pathologic role in atherosclerosis. When adjacent to endothelial cells, LPL is protective because it breaks down potentially unwanted lipids in lipoproteins.26 However, it becomes pro-atherosclerotic when it is located within the intra-arterial space because it acts non-enzymatically as a molecular bridge by binding to the glycosaminoglycan component of proteoglycans and LDLs.26,27 The presence of both biglycan and LPL, as in atherosclerosis, suggested to us that lipoproteins might be retained in Bruch's membrane by binding to both of these molecules. We tested this possibility by inducing AGE formation in LPL-deficient mice and found that lipoproteins were not retained after acute injection of labeled lipoproteins. This result suggests that LPL is critical for lipoprotein retention under our experimental conditions. Our results also suggest that AGE formation may serve instead to influence lipoprotein retention by inducing LPL expression rather than altering the supramolecular architecture of Bruch's membrane. Our previous work identified the transcriptional up-regulation of LPL in the RPE/choroid of mice treated with D-gal compared with PBS-treated mice.33 This scenario would be analogous of that seen in macrophages, whereby AGEs or oxidative stress stimulate LPL production.50,51 These findings have relevance to AMD because we have previously identified AGEs in basal deposits and drusen in AMD.31,52 In this study, we also identified LPL in human samples. Strong labeling was seen in the choroid and the choriocapillaris basement membrane of all eyes. This pattern is similar to what is seen in arteries without lipoprotein retention. However, in eyes with AMD, LPL was also found in drusen and basal deposits. As seen in Figure 5C, a line of LPL immunolabeling is seen in the inner collagenous layer, which corresponds in location to the “lipid wall” that Ruberti et al12 have observed. This raises the possibility that increased production of LPL results in its diffusion into Bruch's membrane into an area of known lipoprotein accumulation and suggests that LPL may participate in lipoprotein retention.
How do our findings fit in context with the genetic variations associated with the risk of AMD? We focused our studies on the influence of AGEs to Bruch's membrane as one change that develops during aging and early AMD, regardless of genetic profile. Clark et al53,54 have shown that the coding change in the Y402H single nucleotide polymorphism alters complement factor H (CFH) binding to sulfated polysaccharides such as heparin sulfate proteoglycans and Bruch's membrane. Recently, Kelly et al48 found that the full-length CFH, and not a recombinant fragment as in the studies by Clark et al,53,54 is not influenced by heparin sulfate content in binding to Bruch's membrane. Instead, heparin sulfate can modulate the function of full-length CFH. Therefore, changes in heparin sulfate content in Bruch's membrane with aging can influence the degree of complement activation. It is unclear what effect biglycan or LPL themselves, or the lipid wall that is known to be comprised of lipoproteins, might have on the regulatory effect of heparin sulfates on CFH function. Given that the mechanism of how the CFH variant causes AMD remains poorly understood, further work on the effect of lipoproteins on CFH modulation is a worthwhile avenue for future studies.
The experimental design that tested acute lipoprotein retention does not guarantee that biglycan and LPL participates in chronic lipoprotein retention in Bruch's membrane. Because we did not specifically test the role of biglycan in our model system, the specific role of biglycan remains unknown. The deficiency of LPL virtually eliminated any acute retention in D-gal–treated mice. Future work will hopefully determine whether LPL is sufficient to retain LDLs in Bruch's membrane or whether it is the combination of biglycan and LPL that is ultimately required. We suspect that LPL, as in atherosclerosis, is necessary for lipoprotein retention in Bruch's membrane because mice that overexpress biglycan and apoB100 did not have enhanced lipoprotein retention compared with mice expressing apoB100.40
At present, the origin of lipoproteins that get deposited in Bruch's membrane, whether from the RPE or systemically derived, remains unknown. Both our laboratory and the Curcio laboratory have provided evidence for local production of apoB100 lipoprotein particles.55,56 Our experiments tested the effect of aging-related changes to Bruch's membrane on lipoprotein retention after i.v. injection. Because material can transit through Bruch's membrane in a bidirectional manner, these results indicate that lipoproteins from either a local or systemic source, because of changes in Bruch's membrane, can become retained. Our work indicates that lipoproteins can get retained by age-related changes in Bruch's membrane resulting from AGE formation, proteoglycans such as those containing biglycan, and LPL (Figure 6, A–D). Although LPL is protective early when localized near to the vascular endothelium, increased production from inducers, such as AGEs or oxidative stress, promotes its more widespread deposition into Bruch's membrane. Along with proteoglycans, LPL then promotes lipoprotein retention. In the future, we will pursue experiments in chronic models of lipoprotein retention to determine the extent to which lipoproteins are retained, whether they become oxidized from this retention, and whether they stimulate an immune response that could promote basal deposit and drusen formation.
Figure 6.
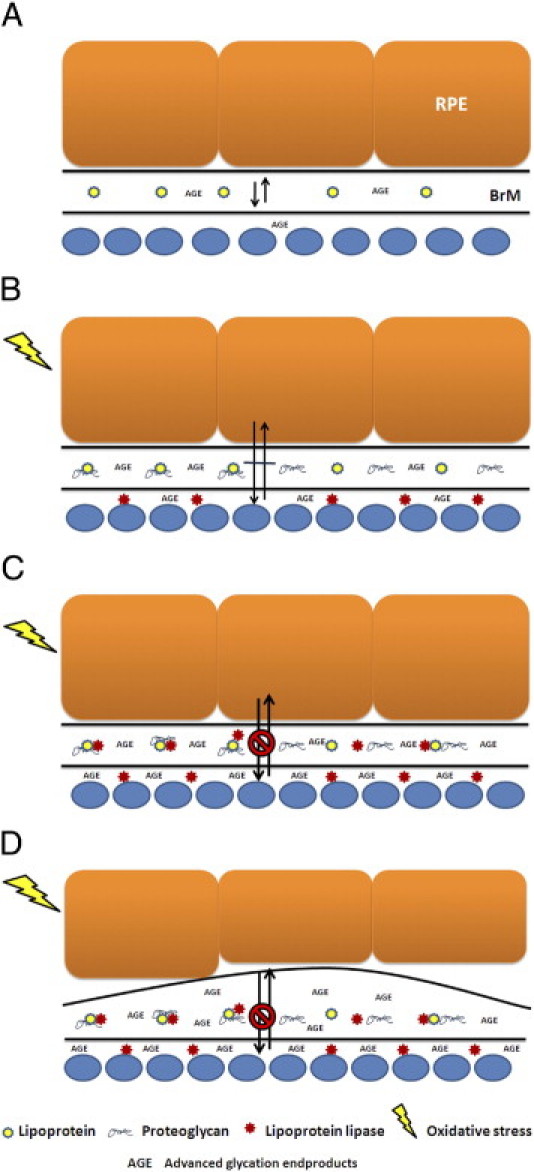
Proposed events that promote lipoprotein retention in Bruch's membrane. A: Free passage of lipoproteins through Bruch's membrane. RPE, retinal pigment epithelium; BrM, Bruch's membrane; CC, choriocapillaris. B: Biglycan deposition promotes lipoprotein retention. Oxidative stress induces protective lipoprotein lipase production with perivascular deposition. C: Continued production of lipoprotein lipase deposition within Bruch's membrane promotes lipoprotein retention. D: Retained lipoproteins become oxidized and stimulate an immune response with basal deposit formation.
Footnotes
Supported by grant EY14005 (J.T.H.), Robert Bond Welch Professorship (J.T.H.), an unrestricted grant from Research to Prevent Blindness (Wilmer Eye Institute), and generous gifts from Ric and Sandy Forsythe and the Merlau family.
References
- 1.Haines J.L., Hauser M.A., Schmidt S., Scott W.K., Olson L.M., Gallins P., Spencer K.L., Kwan S.Y., Noureddine M., Gilbert J.R., Schnetz-Boutaud N., Agarwal A., Postel E.A., Pericak-Vance M.A. Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005;308:419–421. doi: 10.1126/science.1110359. [DOI] [PubMed] [Google Scholar]
- 2.Klein R.J., Zeiss C., Chew E.Y., Tsai J.Y., Sackler R.S., Haynes C., Henning A.K., Sangiovanni J.P., Mane S.M., Mayne S.T., Bracken M.B., Ferris F.L., Ott J., Barnstable C., Hoh J. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308:385–389. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Souied E.H., Benlian P., Amouyel P., Feingold J., Lagarde J.P., Munnich A., Kaplan J., Coscas G., Soubrane G. The epsilon4 allele of the apolipoprotein E gene as a potential protective factor for exudative age-related macular degeneration. Am J Ophthalmol. 1998;125:353–359. doi: 10.1016/s0002-9394(99)80146-9. [DOI] [PubMed] [Google Scholar]
- 4.Hageman G.S., Anderson D.H., Johnson L.V., Hancox L.S., Taiber A.J., Hardisty L.I., Hageman J.L., Stockman H.A., Borchardt J.D., Gehrs K.M., Smith R.J., Silvestri G., Russell S.R., Klaver C.C., Barbazetto I., Chang S., Yannuzzi L.A., Barile G.R., Merriam J.C., Smith R.T., Olsh A.K., Bergeron J., Zernant J., Merriam J.E., Gold B., Dean M., Allikmets R. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci U S A. 2005;102:7227–7232. doi: 10.1073/pnas.0501536102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dewan A., Liu M., Hartman S., Zhang S., Liu D.T., Zhao C., Tam P.O., Chan W.M., Lam D.S., Snyder M., Barnstable C., Pang C.P., Hoh J. HTRA1 promoter polymorphism in wet age-related macular degeneration. Science. 2006;314:989–992. doi: 10.1126/science.1133807. [DOI] [PubMed] [Google Scholar]
- 6.Kanda A., Chen W., Othman M., Branham K.E., Brooks M., Khanna R., He S., Lyons R., Abecasis G.R., Swaroop A. A variant of mitochondrial protein LOC387715/ARMS2, not HTRA1, is strongly associated with age-related macular degeneration. Proc Natl Acad Sci U S A. 2007;104:16227–16232. doi: 10.1073/pnas.0703933104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gold B., Merriam J.E., Zernant J., Hancox L.S., Taiber A.J., Gehrs K., Cramer K., Neel J., Bergeron J., Barile G.R., Smith R.T., Hageman G.S., Dean M., Allikmets R. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat Genet. 2006;38:458–462. doi: 10.1038/ng1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yates J.R., Sepp T., Matharu B.K., Khan J.C., Thurlby D.A., Shahid H., Clayton D.G., Hayward C., Morgan J., Wright A.F., Armbrecht A.M., Dhillon B., Deary I.J., Redmond E., Bird A.C., Moore A.T. Complement C3 variant and the risk of age-related macular degeneration. N Engl J Med. 2007;357:553–561. doi: 10.1056/NEJMoa072618. [DOI] [PubMed] [Google Scholar]
- 9.Canter J.A., Olson L.M., Spencer K., Schnetz-Boutaud N., Anderson B., Hauser M.A., Schmidt S., Postel E.A., Agarwal A., Pericak-Vance M.A., Sternberg P., Jr., Haines J.L. Mitochondrial DNA polymorphism A4917G is independently associated with age-related macular degeneration. PLoS ONE. 2008;3:e2091. doi: 10.1371/journal.pone.0002091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fagerness J.A., Maller J.B., Neale B.M., Reynolds R.C., Daly M.J., Seddon J.M. Variation near complement factor I is associated with risk of advanced AMD. Eur J Hum Genet. 2009;17:100–104. doi: 10.1038/ejhg.2008.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Edwards A.O., Ritter Iii R., Abel K.J., Manning A., Panhuysen C., Farrer L.A. Complement factor H polymorphism and age-related macular degeneration. Science. 2005;308:421–424. doi: 10.1126/science.1110189. [DOI] [PubMed] [Google Scholar]
- 12.Ruberti J.W., Curcio C.A., Millican C.L., Menco B.P., Huang J.D., Johnson M. Quick-freeze/deep-etch visualization of age-related lipid accumulation in Bruch's membrane. Invest Ophthalmol Vis Sci. 2003;44:1753–1759. doi: 10.1167/iovs.02-0496. [DOI] [PubMed] [Google Scholar]
- 13.Green W.R., McDonnell P.J., Yeo J.H. Pathologic features of senile macular degeneration. Ophthalmology. 1985;92:615–627. [PubMed] [Google Scholar]
- 14.Curcio C.A., Millican C.L., Bailey T., Kruth H.S. Accumulation of cholesterol with age in human Bruch's membrane. Invest Ophthalmol Vis Sci. 2001;42:265–274. [PubMed] [Google Scholar]
- 15.Malek G., Li C.M., Guidry C., Medeiros N.E., Curcio C.A. Apolipoprotein B in cholesterol-containing drusen and basal deposits of human eyes with age-related maculopathy. Am J Pathol. 2003;162:413–425. doi: 10.1016/S0002-9440(10)63836-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Curcio C.A., Presley J.B., Millican C.L., Medeiros N.E. Basal deposits and drusen in eyes with age-related maculopathy: evidence for solid lipid particles. Exp Eye Res. 2005;80:761–775. doi: 10.1016/j.exer.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 17.Li C.M., Chung B.H., Presley J.B., Malek G., Zhang X., Dashti N., Li L., Chen J., Bradley K., Kruth H.S., Curcio C.A. Lipoprotein-like particles and cholesteryl esters in human Bruch's membrane: initial characterization. Invest Ophthalmol Vis Sci. 2005;46:2576–2586. doi: 10.1167/iovs.05-0034. [DOI] [PubMed] [Google Scholar]
- 18.Curcio C.A., Presley J.B., Malek G., Medeiros N.E., Avery D.V., Kruth H.S. Esterified and unesterified cholesterol in drusen and basal deposits of eyes with age-related maculopathy. Exp Eye Res. 2005;81:731–741. doi: 10.1016/j.exer.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 19.Chen W., Stambolian D., Edwards A.O., Branham K.E., Othman M., Jakobsdottir J. Genetic variants near TIMP3 and high-density lipoprotein-associated loci influence susceptibility to age-related macular degeneration. Proc Natl Acad Sci U S A. 2010;107:7401–7406. doi: 10.1073/pnas.0912702107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Neale BM, Fagerness J, Reynolds R, Sobrin L, Parker M, Raychaudhuri S, Tan PL, Oh EC, Merriam JE, Souied E, Bernstein PS, Li B, Frederick JM, Zhang K, Brantley MA Jr, Lee AY, Zack DJ, Campochiaro B, Campochiaro P, Ripke S, Smith RT, Barile GR, Katsanis N, Allikmets R, Daly MJ, Seddon JM: Genome-wide association study of advanced age-related macular degeneration identifies a role of the hepatic lipase gene (LIPC). Proc Natl Acad Sci U S A 107:7395–7400 [DOI] [PMC free article] [PubMed]
- 21.Tserentsoodol N., Sztein J., Campos M., Gordiyenko N.V., Fariss R.N., Lee J.W., Fliesler S.J., Rodriguez I.R. Uptake of cholesterol by the retina occurs primarily via a low density lipoprotein receptor-mediated process. Mol Vis. 2006;12:1306–1318. [PubMed] [Google Scholar]
- 22.Williams K.J., Tabas I. The response-to-retention hypothesis of early atherogenesis. Arterioscler Thromb Vasc Biol. 1995;15:551–561. doi: 10.1161/01.atv.15.5.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Patel S., Celermajer D.S., Bao S. Atherosclerosis-underlying inflammatory mechanisms and clinical implications. Int J Biochem Cell Biol. 2008;40:576–580. doi: 10.1016/j.biocel.2007.11.017. [DOI] [PubMed] [Google Scholar]
- 24.Yamada Y., Tian J., Yang Y., Cutler R.G., Wu T., Telljohann R.S., Mattson M.P., Handa J.T. Oxidized low density lipoproteins induce a pathologic response by retinal pigmented epithelial cells. J Neurochem. 2008;105:1187–1197. doi: 10.1111/j.1471-4159.2008.05211.x. [DOI] [PubMed] [Google Scholar]
- 25.Nakashima Y., Wight T.N., Sueishi K. Early atherosclerosis in humans: role of diffuse intimal thickening and extracellular matrix proteoglycans. Cardiovasc Res. 2008;79:14–23. doi: 10.1093/cvr/cvn099. [DOI] [PubMed] [Google Scholar]
- 26.Olin K.L., Potter-Perigo S., Barrett P.H., Wight T.N., Chait A. Lipoprotein lipase enhances the binding of native and oxidized low density lipoproteins to versican and biglycan synthesized by cultured arterial smooth muscle cells. J Biol Chem. 1999;274:34629–34636. doi: 10.1074/jbc.274.49.34629. [DOI] [PubMed] [Google Scholar]
- 27.Pentikainen M.O., Oksjoki R., Oorni K., Kovanen P.T. Lipoprotein lipase in the arterial wall: linking LDL to the arterial extracellular matrix and much more. Arterioscler Thromb Vasc Biol. 2002;22:211–217. doi: 10.1161/hq0102.101551. [DOI] [PubMed] [Google Scholar]
- 28.Devlin C.M., Leventhal A.R., Kuriakose G., Schuchman E.H., Williams K.J., Tabas I. Acid sphingomyelinase promotes lipoprotein retention within early atheromata and accelerates lesion progression. Arterioscler Thromb Vasc Biol. 2008;28:1723–1730. doi: 10.1161/ATVBAHA.108.173344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Khalil M.F., Wagner W.D., Goldberg I.J. Molecular interactions leading to lipoprotein retention and the initiation of atherosclerosis. Arterioscler Thromb Vasc Biol. 2004;24:2211–2218. doi: 10.1161/01.ATV.0000147163.54024.70. [DOI] [PubMed] [Google Scholar]
- 30.Pillarisetti S., Paka L., Obunike J.C., Berglund L., Goldberg I.J. Subendothelial retention of lipoprotein (a): Evidence that reduced heparan sulfate promotes lipoprotein binding to subendothelial matrix. J Clin Invest. 1997;100:867–874. doi: 10.1172/JCI119602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Farboud B., Aotaki-Keen A., Miyata T., Hjelmeland L.M., Handa J.T. Development of a polyclonal antibody with broad epitope specificity for advanced glycation endproducts and localization of these epitopes in Bruch's membrane of the aging eye. Mol Vis. 1999;5:11. [PubMed] [Google Scholar]
- 32.Ida H., Ishibashi K., Reiser K., Hjelmeland L.M., Handa J.T. Ultrastructural aging of the RPE-Bruch's membrane-choriocapillaris complex in the D-galactose-treated mouse. Invest Ophthalmol Vis Sci. 2004;45:2348–2354. doi: 10.1167/iovs.03-1337. [DOI] [PubMed] [Google Scholar]
- 33.Tian J., Ishibashi K., Ishibashi K., Reiser K., Grebe R., Biswal S., Gehlbach P., Handa J.T. Advanced glycation endproduct-induced aging of the retinal pigment epithelium and choroid: a comprehensive transcriptional response. Proc Natl Acad Sci U S A. 2005;102:11846–11851. doi: 10.1073/pnas.0504759102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miller Y.I., Chang M.K., Binder C.J., Shaw P.X., Witztum J.L. Oxidized low density lipoprotein and innate immune receptors. Curr Opin Lipidol. 2003;14:437–445. doi: 10.1097/00041433-200310000-00004. [DOI] [PubMed] [Google Scholar]
- 35.Sarks S.H. Ageing and degeneration in the macular region: a clinico-pathological study. Br J Ophthalmol. 1976;60:324–341. doi: 10.1136/bjo.60.5.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yamada Y., Ishibashi K., Ishibashi K., Bhutto I.A., Tian J., Lutty G.A., Handa J.T. The expression of advanced glycation endproduct receptors in RPE cells associated with basal deposits in human maculas. Exp Eye Res. 2006;82:840–848. doi: 10.1016/j.exer.2005.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fernandez-Veledo S., Nieto-Vazquez I., de Castro J., Ramos M.P., Bruderlein S., Moller P., Lorenzo M. Hyperinsulinemia induces insulin resistance on glucose and lipid metabolism in a human adipocytic cell line: paracrine interaction with myocytes. J Clin Endocrinol Metab. 2008;93:2866–2876. doi: 10.1210/jc.2007-2472. [DOI] [PubMed] [Google Scholar]
- 38.Li Y., Sugiyama E., Yokoyama S., Jiang L., Tanaka N., Aoyama T. Molecular mechanism of age-specific hepatic lipid accumulation in PPARalpha (+/−): LDLR (+/−) mice, an obese mouse model. Lipids. 2008;43:301–312. doi: 10.1007/s11745-008-3161-x. [DOI] [PubMed] [Google Scholar]
- 39.Kirpich IA, Gobejishvili LN, Bon Homme M, Waigel S, Cave M, Arteel G, Barve SS, McClain CJ, Deaciuc IV: Integrated hepatic transcriptome and proteome analysis of mice with high-fat diet-induced nonalcoholic fatty liver disease. J Nutr Biochem 22:38–45 [DOI] [PMC free article] [PubMed]
- 40.Sallo F.B., Bereczki E., Csont T., Luthert P.J., Munro P., Ferdinandy P., Santha M., Lengyel I. Bruch's membrane changes in transgenic mice overexpressing the human biglycan and apolipoprotein b-100 genes. Exp Eye Res. 2009;89:178–186. doi: 10.1016/j.exer.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 41.Gustafsson M., Levin M., Skalen K., Perman J., Friden V., Jirholt P., Olofsson S.O., Fazio S., Linton M.F., Semenkovich C.F., Olivecrona G., Boren J. Retention of low-density lipoprotein in atherosclerotic lesions of the mouse: evidence for a role of lipoprotein lipase. Circ Res. 2007;101:777–783. doi: 10.1161/CIRCRESAHA.107.149666. [DOI] [PubMed] [Google Scholar]
- 42.Hollmann J., Schmidt A., von Bassewitz D.B., Buddecke E. Relationship of sulfated glycosaminoglycans and cholesterol content in normal and arteriosclerotic human aorta. Arteriosclerosis. 1989;9:154–158. doi: 10.1161/01.atv.9.2.154. [DOI] [PubMed] [Google Scholar]
- 43.Boren J., Olin K., Lee I., Chait A., Wight T.N., Innerarity T.L. Identification of the principal proteoglycan-binding site in LDL: A single-point mutation in apo-B100 severely affects proteoglycan interaction without affecting LDL receptor binding. J Clin Invest. 1998;101:2658–2664. doi: 10.1172/JCI2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Olin-Lewis K., Krauss R.M., La Belle M., Blanche P.J., Barrett P.H., Wight T.N., Chait A. ApoC-III content of apoB-containing lipoproteins is associated with binding to the vascular proteoglycan biglycan. J Lipid Res. 2002;43:1969–1977. doi: 10.1194/jlr.m200322-jlr200. [DOI] [PubMed] [Google Scholar]
- 45.O'Brien K.D., Olin K.L., Alpers C.E., Chiu W., Ferguson M., Hudkins K., Wight T.N., Chait A. Comparison of apolipoprotein and proteoglycan deposits in human coronary atherosclerotic plaques: colocalization of biglycan with apolipoproteins. Circulation. 1998;98:519–527. doi: 10.1161/01.cir.98.6.519. [DOI] [PubMed] [Google Scholar]
- 46.Hewitt A.T., Nakazawa K., Newsome D.A. Analysis of newly synthesized Bruch's membrane proteoglycans. Invest Ophthalmol Vis Sci. 1989;30:478–486. [PubMed] [Google Scholar]
- 47.Kliffen M., Mooy C.M., Luider T.M., Huijmans J.G., Kerkvliet S., de Jong P.T. Identification of glycosaminoglycans in age-related macular deposits. Arch Ophthalmol. 1996;114:1009–1014. doi: 10.1001/archopht.1996.01100140217021. [DOI] [PubMed] [Google Scholar]
- 48.Kelly U., Yu L., Kumar P., Ding J.D., Jiang H., Hageman G.S., Arshavsky V.Y., Frank M.M., Hauser M.A., Rickman C.B. Heparan sulfate, including that in Bruch's membrane, inhibits the complement alternative pathway: implications for age-related macular degeneration. J Immunol. 2010;185:5486–5494. doi: 10.4049/jimmunol.0903596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang H., Eckel R.H. Lipoprotein lipase: from gene to obesity. Am J Physiol Endocrinol Metab. 2009;297:E271–E288. doi: 10.1152/ajpendo.90920.2008. [DOI] [PubMed] [Google Scholar]
- 50.Renier G., Desfaits A.C., Lambert A., Mikhail R. Role of oxidant injury on macrophage lipoprotein lipase (LPL) production and sensitivity to LPL. J Lipid Res. 1996;37:799–809. [PubMed] [Google Scholar]
- 51.Beauchamp M.C., Michaud S.E., Li L., Sartippour M.R., Renier G. Advanced glycation end products potentiate the stimulatory effect of glucose on macrophage lipoprotein lipase expression. J Lipid Res. 2004;45:1749–1757. doi: 10.1194/jlr.M400169-JLR200. [DOI] [PubMed] [Google Scholar]
- 52.Handa J.T., Verzijl N., Matsunaga H., Aotaki-Keen A., Lutty G.A., te Koppele J.M., Miyata T., Hjelmeland L.M. Increase in the advanced glycation end product pentosidine in Bruch's membrane with age. Invest Ophthalmol Vis Sci. 1999;40:775–779. [PubMed] [Google Scholar]
- 53.Clark SJ, Perveen R, Hakobyan S, Morgan BP, Sim RB, Bishop PN, Day AJ: Impaired binding of the age-related macular degeneration-associated complement factor H 402H allotype to Bruch's membrane in human retina. J Biol Chem 285:30192–30202 [DOI] [PMC free article] [PubMed]
- 54.Clark S.J., Higman V.A., Mulloy B., Perkins S.J., Lea S.M., Sim R.B., Day A.J. His-384 allotypic variant of factor H associated with age-related macular degeneration has different heparin binding properties from the non-disease-associated form. J Biol Chem. 2006;281:24713–24720. doi: 10.1074/jbc.M605083200. [DOI] [PubMed] [Google Scholar]
- 55.Wang L., Li C.M., Rudolf M., Belyaeva O.V., Chung B.H., Messinger J.D., Kedishvili N.Y., Curcio C.A. Lipoprotein particles of intraocular origin in human Bruch membrane: an unusual lipid profile. Invest Ophthalmol Vis Sci. 2009;50:870–877. doi: 10.1167/iovs.08-2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wu T., Tian J., Cutler R.G., Telljohann R.S., Bernlohr D.A., Mattson M.P., Handa J.T. Knockdown of FABP5 mRNA decreases cellular cholesterol levels and results in decreased apoB100 secretion and triglyceride accumulation in ARPE-19 cells. Lab Invest. 2010;90:963–965. doi: 10.1038/labinvest.2010.87. [DOI] [PubMed] [Google Scholar]