

Abstract
The hyaluronan (HA) receptor CD44 plays an essential role in cell–cell or cell–extracellular matrix communications and is a bioactive signal transmitter. Although a number of studies have described the function of CD44 in breast cancer (BC) metastasis, the underlying mechanisms have yet to be determined. By using a validated tetracycline-off-regulated CD44 expression system in the MCF-7 cell line combined with microarray analysis, we identified survivin (SVV) as a potential downstream transcriptional target of CD44. To test the hypothesis that SVV underpins CD44-promoted BC cell invasion, we combined molecular and pharmacologic approaches and showed that CD44 induction increased SVV expression levels, which in turn promotes BC cell invasion. Further, clinical analysis of breast tissue samples showed that SVV expression patterns paralleled those of the standard form of CD44 during breast tumor progression. More interestingly, we identified the PI3K/E2F1 pathway as a potential molecular link between HA/CD44 activation and SVV transcription. In addition to identifying SVV as a target for HA/CD44 signaling, this investigation provides a better understanding of the molecular mechanisms that underpin the novel function of SVV in breast cancer metastasis.
Breast cancer (BC) is the most common cancer and the second most common cause of cancer-related deaths in women in the United States, with more than 175,000 women being diagnosed annually.1,2 In the later stages of progression, BC cells metastasize from the original tumor site and travel through the vasculature to distant organs such as liver, lungs, brain, and bone.2–5 Although the involvement of cell adhesion molecules in cancer development, progression, and metastasis has been established and discussed extensively in the literature, the mechanisms underlying their implication is still nascent.6–9 The hyaluronan (HA) receptor CD44, a multistructural and multifunctional cell adhesion molecule involved in cell–cell and cell–extracellular matrix interactions, functions as a bioactive signaling transmitter involved in a variety of cellular responses, including lymphocyte homing, hematopoiesis, inflammation, tumorigenesis, angiogenesis, and metastasis.10–13 The CD44–HA complex initiates a series of intracellular signaling events that lead to migration, adhesion, invasion, proliferation, and differentiation of a variety of cells. The transduction of HA/CD44 signaling can occur through various mechanisms including the following: i) HA binding to CD44 can initiate the extracellular clustering of CD44, resulting in the activation of downstream kinases,14 ii) CD44 can serve as a co-receptor physically associated with other cell signaling receptors,15–18 iii) CD44 can serve as a docking protein for pericellular or cytoplasmic proteins,19,20 and iv) the trans-membrane domain of CD44 can be cleaved, allowing the translocation of the cytoplasmic domain to the nucleus, where it functions as a transcription factor regulating the expression of target genes such as CD44 itself.21,22 CD44 and its variants can induce chemoresistance and invasion of human BC cell lines via different mechanisms.23–27
Despite knowing the role that CD44–HA plays in promoting BC invasion and metastasis, the underlying downstream signaling mechanism is nascent. In an attempt to elucidate these downstream signaling mechanisms, we generated a tetracycline (tet)-off–inducible system of CD44 expression both in vitro14 and in vivo.16 These studies showed that induction of CD44 potentiated migration and invasion of BC cells14 and promoted BC metastasis to the liver16 through the CD44–HA/NF-κB/cortactin signaling pathway.14 In the same study,14 we identified the anti-apoptotic protein survivin (SVV) as a potential transcriptional CD44 downstream target gene involved in BC invasion and metastasis. Several lines of evidence support the importance of SVV in BC metastasis. In a murine BC model, the treatment of mice with a plasmid encoding a construct of mouse SVV phosphorylation threonine 34 to alanine (T34A) mutant inhibited BC metastasis to the lung.28 Also, siRNA inhibition of SVV markedly decreased the growth of colorectal cancer cells and sensitized the cells toward radiotherapy.29 On the other hand, the expression of a dominant-negative form of SVV inhibited the growth as well as BC angiogenesis.30 In the present report we show a novel relationship between CD44–HA signaling and the transcription of SVV in BC cells. By using molecular and pharmacologic approaches, we have identified a novel signaling pathway and its molecular components linking HA/CD44 activation to SVV transcription that underpins CD44-promoted BC invasion.
Materials and Methods
Cell Culture
The MCF7, MDA-MB-231 (BC cell lines), were purchased from American Type Culture Collection (Manassas, VA). All cells were cultured in Dulbecco's modified Eagle's medium containing 10% (v/v) fetal bovine serum, 2 mmol/L l-glutamine, and 1 mmol/L sodium pyruvate (Gibco, Gaithersburg, MD). The MCF7-CD44 tet-off (MCF7-B5) BC cell line with the inducible expression of the CD44 standard isoform, previously established in our laboratory,14 was maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 2.5 μg/mL doxycycline (a tet analog that has a greater chemical stability and therefore a longer duration in the media), 100 μg/mL G418, and 1 μg/mL puromycin.
Antibodies, Chemicals, and Reagents
The following antibodies were used: mouse monoclonal anti-CD44 (R&D, Minneapolis, MN), rabbit polyclonal anti-SVV (Santa Cruz Biotechnology, Santa Cruz, CA), rabbit polyclonal anti-E2F1 (Santa Cruz Biotechnology), and goat anti-actin antibodies (Santa Cruz Biotechnology), goat anti-rabbit IgG horseradish peroxidase, donkey anti-mouse IgG horseradish peroxidase, and donkey anti-goat IgG–horseradish peroxidase secondary antibodies (Santa Cruz Biotechnology). Doxycycline (Sigma, St. Louis, MO), BD Matrigel Matrix (BD Biosciences, San Jose, CA), Puromycin and G418 (InvivoGen, San Diego, CA), and citrate buffer (pH 6) (Invitrogen, Carlsbad, CA), were all purchased from the indicated vendors.
Preparation of RNA Samples
RNA was harvested from MCF7-B5 cells cultured in the absence (CD44 expression) and presence (no CD44 expression) of doxycycline for 12 and 24 hours using the RNeasy Mini Kit (Qiagen, Valencia, CA), according to the manufacturer's protocol. Harvested RNA was assessed for quantity and purity, then aliquoted and stored at −80°C. Validation of the differential tet-regulated CD44 expression within the MCF7-B5 cells at the time of RNA sample extraction was performed in parallel immunoblotting experiments.
RT-PCR Analysis
Total RNA was isolated from MCF7–CD44 cells using the RNeasy Mini kit (Qiagen), according to the manufacturer's protocol. For RT-PCR analysis, 1.0 μg of total RNA was reverse-transcribed using standard reagents. Samples were incubated in the PCR machine for reverse transcription at 50°C for 30 minutes, initial PCR activation step (95°C for 5 minutes), followed by 27 PCR cycles. Each cycle consisted of 95°C for 30 seconds, 55°C for CD44 and E2F1, 54°C for SVV and STAT3, 56°C for specificity protein 1 (SP1) and 58°C for glyceraldehyde-3-phosphate dehydrogenase for 30 seconds each, and 72°C for 1 minute. Final extension was at 72°C for 10 minutes. The oligonucleotide primers used were CD44, 5′-TTTGCATTGCAGTCAACAGTC-3′ (sense) and 5′-TTACACCCCAATCTTCATGTCCAC-3′ (antisense); SVV, 5′-AGCCCTTTCTCAAGGACCA-3′ (sense) and 5′-TCAATCCATGGCAGCCAG-3′ (antisense); E2F1, 5′-TGCCCTGAGGAGACCGTAG-3′ (sense) and 5′-CTCCAAGCCCTGTCAGAAATC-3′ (antisense); SP1, 5′-ATGGGGGCAATGGTAATGGTGG-3′ (sense) and 5′-TCAGAACTTGCTGGTTCTGTAAG-3′ (antisense); STAT3, 5′-ATTGACCAGCAGTATAGCCG-3′ (sense) and 5′-TTCCAGCTGCTGCATCTTCT-3′ (antisense); and glyceraldehyde-3-phosphate dehydrogenase, 5′-ACCACAGTCCATGCCATCAC-3′ (sense) and 5′-TCCACCACCCTGTTGCTGTA-3′ (antisense).
The RT-PCR products were examined by electrophoresis in 1% agarose gel containing 0.2 μg/mL ethidium bromides.
Western Blot Analysis
Forty micrograms of total cell extracts, prepared as previously described,16 were boiled for 5 minutes in SDS-PAGE loading buffer, resolved by 12% SDS-PAGE, and transferred to nitrocellulose membranes. Membranes were probed with mouse monoclonal anti-CD44 (1:1000 dilution), rabbit polyclonal anti-SVV (1:500 dilution), rabbit polyclonal anti-E2F1 (1:500 dilution), or goat anti-actin antibodies (1:500 dilution), followed by an incubation with goat anti-rabbit (1:2000 dilution), donkey anti-mouse (1:2000 dilution), or donkey anti-goat IgG–horseradish peroxidase (1:2000 dilution) secondary antibodies, respectively. The presence of the protein then was detected using the West Femto Supersignal chemiluminescence kit (Thermo Scientific, Rockford, IL).
siRNA-Mediated Depletion of CD44, SVV, and E2F1
Oligonucleotides specific for human CD44, human SVV, and human E2F1, along with a Silencer Negative Control #1 small interfering RNA (siRNA), were synthesized commercially (Ambion, Austin, TX) for use in the siRNA knockdown of CD44, SVV, and E2F1. The sequences used were as follows: CD44, 5′-GGAAAUGGUGCAUUUGGUGTT-3′ (sense) and 5′-TTCCUUUACCACGUAAACCAC-3′ (antisense); SVV, 5′-GGACCACCGCAUCUCUACTT-3′ (sense) and 5′-TTCCUGGUGGCGUAGAUGU-3′ (antisense); and E2F1, 5′-GGACCUUCGUAGCAUUGCATT-3′ (sense) and 5′-UGCAAUGCUACGAAGGUCCTG-3′.
Cells were seeded and grown to 50% confluency, washed twice with sterile PBS, and then incubated with a transfection cocktail comprising OPTI-MEM1, Lipofectamine-2000 (Invitrogen), and the siRNA or scrambled oligonucleotides at a final concentration of 50 nmol/L for CD44 and E2F1, and 25 nmol/L for SVV at 37°C for 6 hours. After incubation, the media was replaced with 20% (v/v) fetal bovine serum–enriched growth media and subsequently incubated overnight at 37°C. Transfected cells then were transfected a second time with the CD44 or SVV siRNA transfection cocktail, as described previously. Total RNA and protein lysates were harvested within 24 to 48 hours after the second hit, respectively. Depletion of protein expression was confirmed by immunoblotting and RT-PCR analysis.
Cell Invasion Assay
MCF7-B5 cells were cultured in the absence of doxycycline and the presence of HA (100 μg/mL) for 24 hours to continuously induce CD44 expression. Cells then were treated with SVV-specific siRNA, described earlier, to inhibit SVV expression. Twenty-four hours after siRNA treatment, the cells were washed twice with sterile PBS and harvested by trypsinization. The harvested cells then were resuspended in Dulbecco's modified Eagle's medium supplemented with 0.5% bovine serum albumin and plated in Millicell culture inserts (12-μm pore size; Millipore, Billerica, MA), which previously were coated with a thin layer of 200 μg/mL of Matrigel (BD Biosciences). The inserts containing the cells were placed into a tissue culture dish (lower chamber) with the attracting medium, which consisted of MCF7-B5 special medium supplemented with 200 μg/mL HA. Cells were incubated for 22 hours at 37°C, after which the Millicell culture insert was removed and the upper surface of the insert was wiped quickly with a cotton swab to remove noninvasive cells. The cell culture inserts were dried under a laminar flow hood for 4 hours, the cells present on the bottom of the filter (the invasive cells) were stained using the Diff-Quick staining kit (Dade Behring, Inc., Newark, DE), according to the manufacturer's protocol, and the stained cells were counted under a phase-contrast microscope equipped with ocular grids. By using the same experimental strategy described earlier (except for HA sensitization), SVV expression levels were inhibited in the invasive BC cell line MDA-MB-231 (high endogenous level of CD44). Cells were collected after the second treatment with human-specific SVV siRNA and cultured in the cell inserts as described earlier (8-μm pore size Boyden chamber; Millipore). The lower chamber attracting media consisted of 10% fetal bovine serum–enriched Dulbecco's modified Eagle's medium supplemented with 100 μg/mL HA.
Inhibition of Survivin Pathways
MCF7-B5 cells, treated without doxycycline for 24 hours to induce CD44 expression as described earlier, were treated independently for either 1 hour or 24 hours with inhibitors specific for a variety of signal transduction kinases as follows: IKK2 (NF-κB kinase 2) inhibitor (SC-514, 20 μmol/L), phosphatidylinositol 3-kinase (PI3K) inhibitor (LY294002, 10 μmol/L), Src-kinase inhibitor (PP2, 10 nmol/L), mitogen-activated protein kinase kinase 1 (MEK1) inhibitor (U0126, 5 μmol/L), and protein kinase C inhibitor (bisindolemaleimide I, 10 nmol/L). CD44 was activated with 100 μg/mL HA, in the presence of inhibitors as described earlier (the cells were treated with 100 μg/mL HA in the presence of inhibitors, first for 1-hour sensitization and then at the start of the 24-hour cell treatment with inhibitors), and the expression levels of SVV were determined by Western blot analysis. The effects of the various inhibitors on SVV level were compared against a vehicle control using 0.1% (v/v) dimethyl sulfoxide and HA untreated cells as a negative control.
IHC
Twenty-four paraffin blocks including normal and tumor breast tissue were obtained from the archives of the Department of Pathology at Louisiana State University Health Sciences Center (New Orleans, LA). Ethical approval was obtained from the Louisiana State University Health Sciences Center Ethical Committee. Immunohistochemical (IHC) assays were performed as we have described previously, with slight modifications.16,31 Briefly, adjacent sections were examined for both CD44 and SVV expression using mouse anti-human CD44 antibody (1:100 dilution; R&D) and rabbit polyclonal anti-SVV antibody (1:100 dilution; Santa Cruz Biotechnology) after antigen retrieval performed by boiling the samples in 500 mL of 9 mmol/L citrate buffer (pH 6) (Invitrogen) for 25 minutes. Staining was performed using the Ventana Nexus immunostainer and Ventana detection kit system (Ventana Medical Systems, Inc., Tucson, AZ). Adjacent sections also were stained with H&E for histology. For the intensity of immunostaining, we adopted a simple comparison of the intensity of immunostaining using 1+ for low expression, 2+ for intermediate expression, and 3+ for high expression.
Statistical Analysis
Differences between mean values were assessed for statistical significance using the two-tailed Student's t-test comparisons (GraphPad Prism 4.0 software, La Jolla, CA). P values < 0.05 were considered statistically significant.
Results
Survivin Expression Is Dependent on HA–CD44 Signaling
Several reports have implicated SVV as a potential target for cancer therapy because its expression is restricted to cancer cells and absent from normal postmitotic adult cells. Further, as its name suggests, SVV has anti-apoptotic survival effects on cancer cells and is implicated in resistance of tumor cells to chemotherapy and radiotherapy.32,33 Despite this information, the mechanism by which SVV expression is induced and regulated in cancer cells is still unclear.34–37 Therefore, we used our previously described tet-controlled system (tet-off) to regulate CD44 expression. In this system, the weakly invasive breast adenocarcinoma cell line MCF7 was engineered to contain the tet-inducible expression of CD44, in which the removal of the drug regulates the expression of CD44. The tet-off cell line, called MCF7F-B5, allowed us to examine the ability of HA–CD44 signaling to regulate the transcription of SVV in a controlled manner.
To examine the effect of CD44 on SVV expression levels, MCF7F-B5 cells were cultured in the presence or absence of the tet-related drug doxycycline for 24 hours to repress or induce CD44 expression, respectively. The cells subsequently were stimulated with the CD44 ligand HA (100 μg/mL) for 18 and 24 hours, we isolated and used the mRNA samples for microarray analysis. This analysis showed a 3.2-fold increase in SVV mRNA levels as a consequence of CD44 induction. To further investigate these results, MCF7F-B5 cells were cultured in the absence (induction of CD44) or the presence (no CD44) of doxycycline after stimulation with HA. Total mRNA samples and protein lysates were collected at various time points after HA stimulation, and the levels of CD44 and SVV were determined by RT-PCR or Western blot analysis, respectively. Consistent with previous results,14 we observed a significant increase in CD44 expression in the absence of doxycycline at all time points of the experiment (Figure 1A). To determine whether there was a correlation between CD44 levels and expression of SVV, we performed time-course RT-PCR using specific primers for SVV. We observed a nearly threefold to fourfold increase in SVV mRNA levels at 9 and 12 hours (Figure 1B). Further, Western blot analysis confirmed the increase in SVV protein levels in the presence of tet 18 and 24 hours after HA stimulation, consistent with the increased levels of CD44 expression (Figure 1C).
Figure 1.
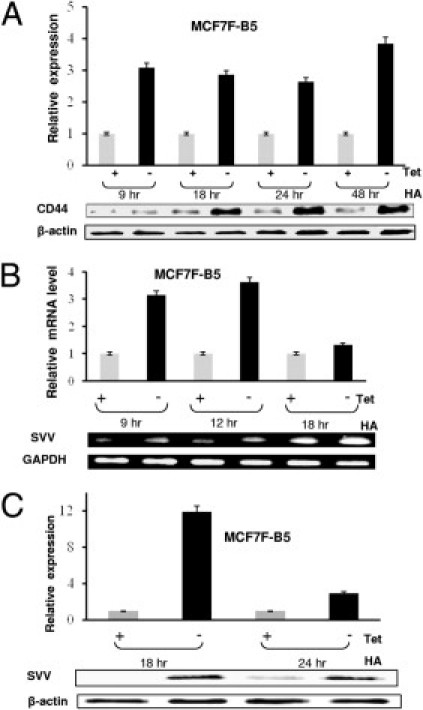
Validation of SVV as a downstream target for HA–CD44 signaling in MCF7-B5 cells. A: Western blot analysis showing time-dependent CD44 expression in the presence and absence of doxycycline and stimulation with HA (100 μg/mL). B: Semiquantitative RT-PCR showing regulation of SVV mRNA transcription after the removal of doxycycline from the culture media. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. C: Western blot analysis illustrating the relationship between HA–CD44 induction and SVV expression increase. Total cell extracts were performed under the indicated conditions and SVV protein was detected by Western blot analysis using antibodies specific for SVV. In all graphs the error bars represent the SD from the mean of at least three independent determinations and the expression levels of CD44 or SVV were normalized for the β-actin loading control. Expression of either CD44 or SVV in the presence of doxycycline (inhibition of expression) was given a relative expression level of 1 and induction is described relative to this negative control.
To further investigate the relationship between HA–CD44 interaction and regulation of SVV expression in BC cell lines, we inhibited the expression of CD44 using the RNAi method and determined the effect of this inhibition on the expression levels of SVV on HA stimulation. The presence of CD44 and SVV were determined by Western blot analysis. A nearly 80% decrease in the expression of CD44 was observed in MCF7-B5 cells after transfection with 25 nmol/L of CD44-specific siRNA oligonucleotides (Figure 2A). A parallel decrease in the expression levels of SVV was observed (Figure 2A), suggesting that SVV is a potential transcriptional target of HA–CD44 signaling.
Figure 2.
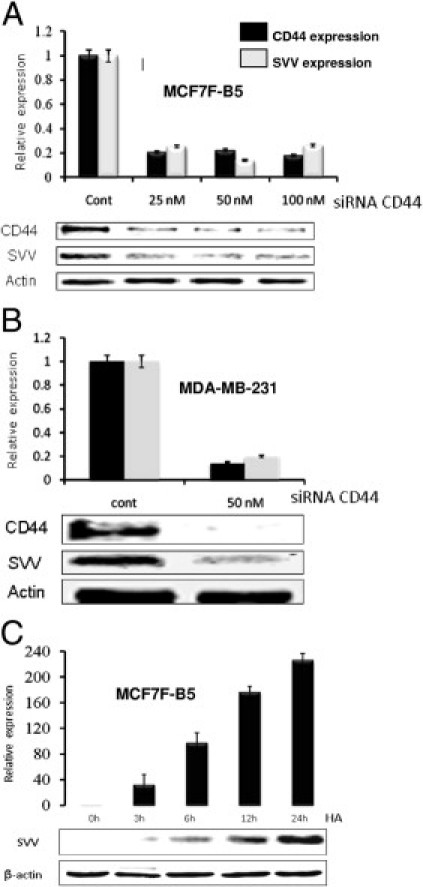
Validation of SVV as a transcriptional target for HA–CD44. siRNA oligonucleotides specific for CD44 were transfected into MCF7F-B5 (A) and MDA-MB-231 (B) cells and CD44 (black bars) and SVV (gray bars) protein expression were determined by Western blot analysis using antibodies specific for each protein. C: CD44 induction of SVV expression is HA-dependent in MCF7F-B5 cells. Cells were cultured for 24 hours in media without doxicycline to induce the expression of CD44, after which the cells were stimulated with HA (100 μg/mL). At the indicated time points total cell extracts were performed and the expression levels of SVV were determined by Western blot analysis. In all graphs the error bars represent the SD from the mean of at least three independent determinations and the expression levels of CD44 or SVV were normalized for the β-actin loading control. Expression of either CD44 or SVV in the control sample was given a relative expression level of 1 and all other expression levels are described relative to this negative control.
The cell-line specificity of the relationship between CD44 and the regulation of SVV transcription/expression also was examined using the CD44-expressing MDA-MB-231 BC cell line. As expected, quantitative PCR analysis confirmed that inhibition of CD44 in the MDA-MB-231 cells in the presence of 100 mg/mL HA resulted in a nearly 90% decrease in CD44 and SVV protein levels (Figure 2B), further validating the relationship between CD44 and SVV expression, independent of BC cell type.
The temporal relationship of HA-induced CD44 signaling to SVV gene regulation was investigated by analysis of SVV protein levels by Western blot analysis in MCF7F-B5 cells after exposure to 100 μg/mL HA. Increased SVV expression levels were detected as early as 3 hours after HA stimulation, reaching a fourfold peak induction at the 12-hour time point (Figure 2C).
Survivin Underpins CD44-Regulated Invasiveness of BC Cells
The functional validation of HA/CD44-regulated SVV expression in BC cell invasion was studied using two BC cell lines, MCF7F-B5 clone expressing CD44 (tet-free media) and MDA-MB-231 cell line that expresses high endogenous levels of CD44. Transfection of the cells with SVV-specific siRNA oligonucleotide (50 nmol/L) significantly reduced the endogenous levels of SVV expression in each of the cell lines (data not shown). In comparison, the transfection of the cells with scrambled RNAi oligonucleotides as negative control had nearly no effect on SVV expression in either of the two cell lines (data not shown). Subsequently, using an in vitro invasion assay, the HA/CD44-promoted invasiveness of MCF7F-B5 cells was attenuated markedly (∼70% reduction) after RNAi-mediated depletion of SVV expression in these cells in comparison with scrambled oligonucleotides. Similarly, abrogation of SVV expression in MDA-MB-231 cells attenuated HA/CD44-promoted invasiveness of these cells (∼75% reduction), whereas scrambled oligonucleotides had no effect on cell invasion (Figure 3, A and B).
Figure 3.
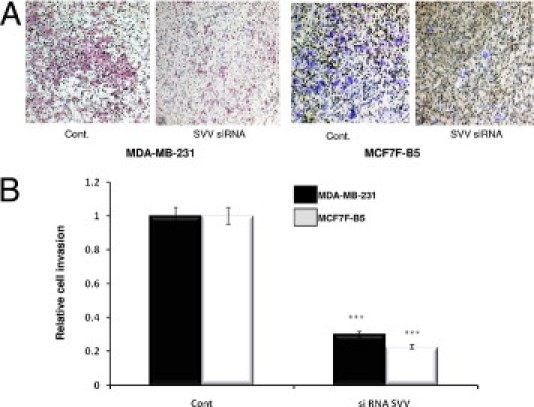
Functional validation of SVV expression in BC cell lines. A: Invasion of both MDA-MB-231 and MCF7F-B5 breast cancer cells as shown by a Boyden chamber invasion assay. Images of Boyden chamber membranes represent the number of captured cells during the invasion (original magnification, ×4), illustrating the difference in the number of invasive cells. B: Graphic representation of the quantitative determination of the number of invasive cells in MDA-MB-231 cells (black bars) and the inducible MCF7F-B5 cells (gray bars) (two-tailed Student's t-test, ***P < 0.001).
Expression of Both SVV and CD44 in Normal Versus Late-Stage Aggressive Breast Tumor Tissues
Our results showing that HA/CD44-dependent SVV expression contributes to BC invasiveness would suggest a significant increase of the expression of both CD44 and SVV in late-stage, metastatic BC samples. Therefore, to determine whether CD44 and SVV expression are increased during late stages of BC, we performed IHC analysis on breast tissues derived from highly aggressive stage 3 breast adenocarcinoma and the neighboring normal human breast tissue from the same patient. A minimal staining of both CD44 and SVV was observed in the normal human breast tissue (Figure 4, C and E). In contrast, high expression of CD44 and SVV were detected in the stage 3 breast adenocarcinoma tissues (Figure 4, D and F), which suggests a correlation between CD44 and SVV expression. Furthermore, these results indicate that the high expression levels of both CD44 and SVV, known to promote invasiveness in vitro, may contribute to the metastatic potential of late-stage BC cells.
Figure 4.
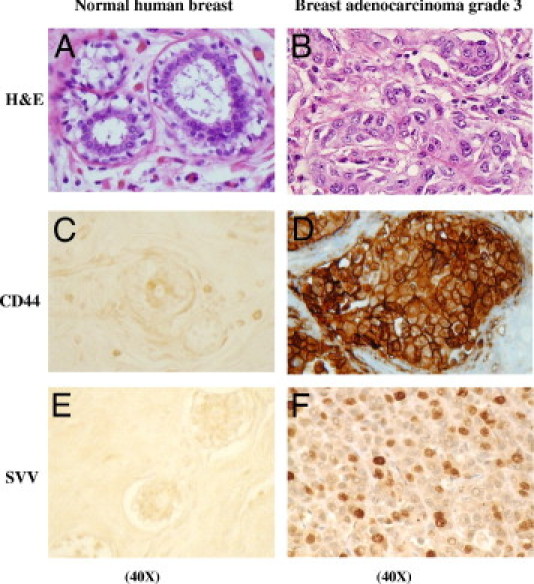
Protein expression levels of CD44 and SVV in normal human breast tissue and highly aggressive stage 3 breast adenocarcinoma tumor cells. H&E-stained normal human breast tissue (A) and stage 3 breast adenocarcinoma tissue (B). IHC analysis of CD44 expression in normal human breast tissue (C) and breast adenocarcinoma tumor tissues (D). SVV protein expression in normal human breast tissue (E) and in breast adenocarcinoma tumor tissue (F).
Elucidation of HA-CD44/SVV Signaling Pathway
To elucidate the signal transduction pathways coupling CD44 to the regulation of SVV transcription, MCF7F-B5 cells were induced to express CD44 for 24 hours, after which the cells were stimulated with 100 μg/mL HA in the presence or absence of known and well-characterized signal transduction: PI3K inhibitor LY294002 (10 μmol/L); MEK1 inhibitor U0126 (10 μmol/L); IKK2 inhibitor SC-514 (20 μmol/L); protein kinase C inhibitor bisindolemaleimide (10 nmol/L); and the Src inhibitor PP2 (5 nmol/L). Cells also independently were treated with dimethyl sulfoxide alone as a negative control. A nearly 80% decrease in SVV expression was observed in the absence of HA stimulation; a stimulation that was not effected by the presence of dimethyl sulfoxide (Figure 5A). Furthermore, no significant effects of the inhibitors specific for Src, MEK1, κ-kinase-2, or protein kinase C were observed (Figure 5A). In contrast, a significant decrease of HA–CD44–dependent SVV expression (∼65%) was observed in the presence of the PI3K inhibitor LY294002 (Figure 5A). This result implicates a PI3K-dependent signal transduction pathway in HA–CD44–dependent expression of SVV.
Figure 5.
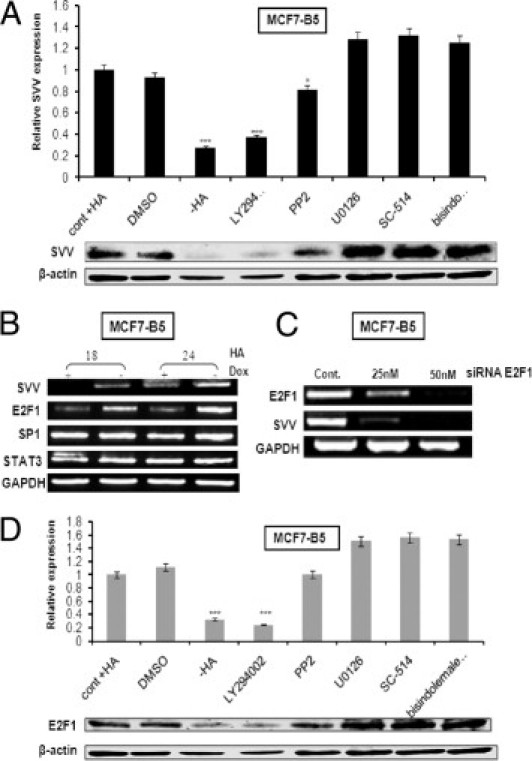
Involvement of PI3K and E2F1 in the CD44-dependent induction of SVV expression. A: MCF7-B5 cells induced to express CD44 were pretreated for 1 hour with different inhibitors specific for kinases involved in different signal transduction pathways (PI3K inhibitor LY294002, 10 μmol/L; MEK1 inhibitor U0126, 10 μmol/L; IKK2 inhibitor SC-514, 20 μmol/L; protein kinase C inhibitor bisindolemaleimide, 10 nmol/L; and the Src inhibitor PP2, 5 nmol/L), after which the cells were stimulated with HA (100 μg/mL) for 24 hours in the presence of the same inhibitors. SVV protein levels were determined by Western blot analysis and SVV expression in the presence of HA and induced CD44 was given the relative value of one (cont + HA). All values were normalized against β-actin. The error bars represent the SD from at least three independent experiments. DMSO, dimethyl sulfoxide. B: MCF7-B5 cells were induced to express CD44 and stimulated with HA. At the indicated periods of time mRNA was isolated from the cells and the expression levels of SVV, E2F1, SP1, and STAT3 were determined by semiquantitative RT-PCR. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. C: MCF7-B5 cells, induced to express CD44 and stimulated with HA, were transfected with increasing concentrations of siRNA oligonucleotides specific for E2F1. The expression levels of E2F1 and SVV were determined by semiquantitative RT-PCR analysis on mRNA isolated from the cells. D: MCF7-B5 cells induced to express CD44 were pretreated for 1 hour with the earlier-mentioned kinase inhibitors, after which the cells were stimulated with HA (100 μg/mL) for 24 hours, in the presence of the same inhibitors. E2F1 protein levels were determined by Western blot analysis and E2F1 expression in the presence of HA and induced CD44 were given the relative value of one (cont + HA). All values were normalized against β-actin. The error bars represent the SD from at least three independent determinations.
The PI3K signal transduction pathway is known to regulate many transcription factors, including E2F1, SP1, and STAT3. To determine which of these factors may play a role in HA–CD44–dependent expression of SVV, MCF7F-B5 cells induced to express CD44 were stimulated with 100 μg HA for 18 or 24 hours. The expression levels of SVV, E2F1, SP1, and STAT3 were determined by RT-PCR analysis (Figure 5B). The results showed no differences in the expression levels of both SP1 and STAT3 either in the presence or absence of CD44 (Figure 5B). In addition, Western blot analysis for phospho-STAT3 (the activated form of STAT3) revealed no significant changes (data not shown). However, on induction and activation of CD44, a time-dependent increase in the expression of both E2F1 and SVV was observed (Figure 5B), suggesting a relationship between HA stimulation of CD44, activation of E2F transcription factor, and induction of SVV transcription. To further support this finding, the expression of E2F1 was inhibited using the RNAi method and its effect on SVV expression levels was examined by RT-PCR (Figure 5C). The specific siRNA oligonucleotide was effective in decreasing E2F1 expression with a concomitant decrease in SVV expression (Figure 5C). Finally, similar experiments using signal transduction inhibitors as described in Figure 5A were performed, and E2F1 expression levels were examined by Western blot analysis (Figure 5D). The pattern of expression of E2F1 was nearly identical to that observed for SVV (Figure 5, A and D). Inhibitors specific for Src, MEK1, IKK2, and protein kinase C had minimal effect on E2F1 expression, similar to that seen in the absence of HA stimulation (Figure 5D). However, significant inhibition of E2F1 was observed in CD44-induced BC cells in the presence of the PI3K inhibitor LY294002 (Figure 5D). Thus, taken together, these results support our hypothesis that PI3K signal transduction and the E2F1 transcription factor assist in regulating the HA–CD44–dependent expression of SVV.
Discussion
The controversial function of CD44 in BC progression and the impact of its signaling on the cellular transcriptome initially prompted us to develop and validate a tet-off CD44-regulated CD44 expression in the weakly invasive MCF-7 B5 cell line.14 By using this system we showed that the standard form of CD44 promotes breast tumor cell invasion14 and metastasis to the liver.16 Our findings indicated that the underlying mechanism involves an interaction of CD44 with HA, leading to an induction and nuclear localization of NF-κB, which subsequently binds to the promoter of cortactin (CTN) and activates its expression, thereby promoting breast tumor cell invasion.14 The aim of the present study was to expand our knowledge of the role that CD44 plays in BC tumor progression. By using similar strategies, we identified SVV as a novel downstream target of CD44 signaling. Further, we provide compelling evidence to support the hypothesis that CD44 uses the PI3K signaling pathway and the transcription factor E2F1 to induce the expression of SVV leading to breast tumor invasion.
We initially were drawn to SVV as a potential target of HA–CD44 signaling for a possible role in BC tumor development because of its crucial role in anti-apoptosis, contributions to cell survival, and resistance to chemotherapies.32–39 SVV is unique relative to other inhibitor of apoptosis proteins because of three distinct features (reviewed by Ouhtit et al32) as follows: i) SVV expression is virtually absent in a majority of normal adult tissues, however, it is overexpressed in many human tumors, ii) SVV contains a baculovirus inhibitor of apoptosis proteins repeat domain that interacts with and inhibits the pro-apoptotic functions of caspases, and iii) SVV is the only inhibitor of apoptosis protein tightly regulated throughout cell-cycle progression. The dual role in inhibiting apoptosis and contributing to cellular proliferation makes SVV an attractive molecular target and a promising lead for BC therapy.35 However, despite these correlations the role that SVV plays in BC development and the upstream factors regulating its expression are not known.
In this report we present compelling evidence that supports our conclusion that HA–CD44 signaling induces SVV expression, which in turn contributes to BC cell invasiveness. This conclusion is based on our data showing that SVV is a transcriptional target of HA–CD44 signaling, as determined by microarray analysis (data not shown) and RT-PCR analysis on mRNA samples isolated from cells, specifically induced to express CD44 (Figures 1 and 2). Further, the specific inhibition of CD44 expression by siRNA, of either induced or endogenous CD44, resulted in a specific decrease in SVV expression that correlated with a significant loss of BC cell invasiveness (Figure 3). Finally, an analysis of tissue samples derived from normal human breast and from highly aggressive stage 3 breast adenocarcinoma confirmed simultaneous increased expression of both CD44 and SVV in late-stage metastatic BC cells, expression that was absent in normal breast tissue samples (Figure 4). Therefore, taken together, our results support our hypothesis that HA–CD44–regulated expression of SVV promotes BC cell invasiveness and may contribute to BC metastasis, a result consistent with previous studies describing the role of SVV in promoting BC cell motility.40,41
In addition to showing a link between HA–CD44 activation, SVV expression, and BC cell invasiveness, we also have provided evidence to support a role of the PI3K signaling pathway and the expression of E2F1 in the regulation of SVV. By using a panel of inhibitors specific for a variety of kinases involved in different signal transduction pathways, we determined that the inhibitor specific for PI3K was able to reduce the expression of SVV and E2F1 to background levels (Figure 5, A and D), suggesting the involvement of PI3K in SVV regulation. Further, siRNA reduction of E2F1 resulted in a concomitant reduction in SVV levels (Figure 5C), supporting the role of E2F1 in regulating the expression of SVV. Taken together, our results allow us to propose a hypothetical model (Figure 6) in which the binding of HA to CD44 activates a signal transduction pathway involving PI3K. This pathway subsequently activates E2F1, which then up-regulates the expression of SVV, most likely through a previously reported direct interaction with the SVV promoter,42,43 thereby contributing to the invasive potential of BC cells.
Figure 6.
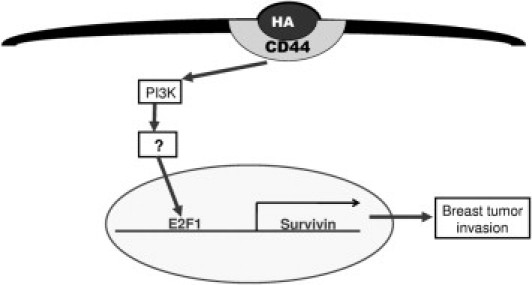
The proposed signal transduction pathway describing the HACD44-dependent activation SVV transcription and BC invasion. HA binding to CD44 activates a signal transduction cascade involving PI3K and E2F1, which subsequently activates SVV, leading to BC cell invasion and metastasis.
In summary, we used our previously described tet-regulated standard form of CD44 expression system to identify SVV as a transcriptional target for HA–CD44 signal transduction in BC cells. We validated SVV as a novel downstream transcriptional target of HA–CD44 signaling, and showed that SVV is functionally relevant in underpinning CD44-promoted breast tumor cell invasion. This investigation shows that HA–CD44 signaling can increase SVV expression in BC cells, showing for the first time that an extracellular stimulus may influence the regulation of SVV expression. Although the present investigation strongly supports our proposed novel pathway linking CD44 activation to SVV transcription and BC cell invasiveness, the molecular basis of additional downstream effector(s) and mechanism(s) by which SVV ultimately modulates cell migration and invasion remains to be determined. Therefore, SVV may play an important role in communicating cell motility signals initiated by CD44 signaling in BC cells to other cell-surface receptors involved in regulating cell migration and invasion.
Acknowledgment
We thank Dr. David J. Waugh for supplying the CD44-B5 clone.
Footnotes
Supported by an Egyptian government scholarship (A.M.E.).
None of the authors disclosed any relevant financial relationships.
References
- 1.Chu D., Lu J. Novel therapies in breast cancer: what is new from ASCO 2008? J Hematol Oncol. 2008;1:16. doi: 10.1186/1756-8722-1-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Greenlee H., Hershman D.L., Jacobson J.S. Use of antioxidant supplements during breast cancer treatment: a comprehensive review. Breast Cancer Res Treat. 2009;115:437–452. doi: 10.1007/s10549-008-0193-0. [DOI] [PubMed] [Google Scholar]
- 3.Diamond J.R., Finlayson C.A., Borges V.F. Hepatic complications of breast cancer. Lancet Oncol. 2009;10:615–621. doi: 10.1016/S1470-2045(09)70029-4. [DOI] [PubMed] [Google Scholar]
- 4.Vollmer R.T. Primary lung cancer vs metastatic breast cancer: a probabilistic approach. Am J Clin Pathol. 2009;132:391–395. doi: 10.1309/AJCPDIP12IUGVRQR. [DOI] [PubMed] [Google Scholar]
- 5.Lorger M., Felding-Habermann B. Capturing changes in the brain microenvironment during initial steps of breast cancer brain metastasis. Am J Pathol. 2010;176:2958–2971. doi: 10.2353/ajpath.2010.090838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baranwal S., Alahari S.K. Molecular mechanisms controlling E-cadherin expression in breast cancer. Biochem Biophys Res Commun. 2009;384:6–11. doi: 10.1016/j.bbrc.2009.04.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ouhtit A., Gaur R.L., Abd Elmageed Z.Y., Fernando A., Thouta R., Trappey A.K., Abdraboh M.E., El-Sayyad H.I., Rao P., Raj M.G. Towards understanding the mode of action of the multifaceted cell adhesion receptor CD146. Biochim Biophys Acta. 2009;1795:130–136. doi: 10.1016/j.bbcan.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 8.Zhang Y., Ma B., Fan Q. Mechanisms of breast cancer bone metastasis. Cancer Lett. 2010;292:1–7. doi: 10.1016/j.canlet.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 9.Oliveira L.R., Jeffrey S.S., Ribeiro-Silva A. Stem cells in human breast cancer. Histol Histopathol. 2010;25:371–385. doi: 10.14670/HH-25.371. [DOI] [PubMed] [Google Scholar]
- 10.Celebiler Cavusoglu A., Kilic Y., Saydam S., Canda T., Baskan Z., Sevinc A.I., Sakizli M. Predicting invasive phenotype with CDH1. CDH13, CD44, and TIMP3 gene expression in primary breast cancer. Cancer Sci. 2009;100:2341–2345. doi: 10.1111/j.1349-7006.2009.01333.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sherman L., Sleeman J., Herrlich P., Ponta H. Hyaluronate receptors: key players in growth, differentiation, migration and tumor progression. Curr Opin Cell Biol. 1994;6:726–733. doi: 10.1016/0955-0674(94)90100-7. [DOI] [PubMed] [Google Scholar]
- 12.Moll J., Khaldoyanidi S., Sleeman J.P., Achtnich M., Preuss I., Ponta H., Herrlich P. Two different functions for CD44 proteins in human myelopoiesis. J Clin Invest. 1998;102:1024–1034. doi: 10.1172/JCI2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gee K., Kryworuchko M., Kumar A. Recent advances in the regulation of CD44 expression and its role in inflammation and autoimmune diseases. Arch Immunol Ther Exp (Warsz) 2004;52:13–26. [PubMed] [Google Scholar]
- 14.Hill A., McFarlane S., Mulligan K., Gillespie H., Draffin J.E., Trimble A., Ouhtit A., Johnston P.G., Harkin D.P., McCormick D., Waugh D.J. Cortactin underpins CD44-promoted invasion and adhesion of breast cancer cells to bone marrow endothelial cells. Oncogene. 2006;25:6079–6091. doi: 10.1038/sj.onc.1209628. [DOI] [PubMed] [Google Scholar]
- 15.Wang S.J., Bourguignon L.Y. Hyaluronan and the interaction between CD44 and epidermal growth factor receptor in oncogenic signaling and chemotherapy resistance in head and neck cancer. Arch Otolaryngol Head Neck Surg. 2006;132:771–778. doi: 10.1001/archotol.132.7.771. [DOI] [PubMed] [Google Scholar]
- 16.Ouhtit A., Abd Elmageed Z.Y., Abdraboh M.E., Lioe T.F., Raj M.H. In vivo evidence for the role of CD44s in promoting breast cancer metastasis to the liver. Am J Pathol. 2007;171:2033–2039. doi: 10.2353/ajpath.2007.070535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tremmel M., Matzke A., Albrecht I., Laib A.M., Olaku V., Ballmer-Hofer K., Christofori G., Heroult M., Augustin H.G., Ponta H., Orian-Rousseau V. A CD44v6 peptide reveals a role of CD44 in VEGFR-2 signaling and angiogenesis. Blood. 2009;114:5236–5244. doi: 10.1182/blood-2009-04-219204. [DOI] [PubMed] [Google Scholar]
- 18.van der Voort R., Taher T.E., Wielenga V.J., Spaargaren M., Prevo R., Smit L., David G., Hartmann G., Gherardi E., Pals S.T. Heparan sulfate-modified CD44 promotes hepatocyte growth factor/scatter factor-induced signal transduction through the receptor tyrosine kinase c-Met. J Biol Chem. 1999;274:6499–6506. doi: 10.1074/jbc.274.10.6499. [DOI] [PubMed] [Google Scholar]
- 19.Yu Q., Stamenkovic I. Localization of matrix metalloproteinase 9 to the cell surface provides a mechanism for CD44-mediated tumor invasion. Genes Dev. 1999;13:35–48. doi: 10.1101/gad.13.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mori H., Tomari T., Koshikawa N., Kajita M., Itoh Y., Sato H., Tojo H., Yana I., Seiki M. CD44 directs membrane-type 1 matrix metalloproteinase to lamellipodia by associating with its hemopexin-like domain. EMBO J. 2002;21:3949–3959. doi: 10.1093/emboj/cdf411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Okamoto I., Kawano Y., Murakami D., Sasayama T., Araki N., Miki T., Wong A.J., Saya H. Proteolytic release of CD44 intracellular domain and its role in the CD44 signaling pathway. J Cell Biol. 2001;155:755–762. doi: 10.1083/jcb.200108159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Murakami D., Okamoto I., Nagano O., Kawano Y., Tomita T., Iwatsubo T., De Strooper B., Yumoto E., Saya H. Presenilin-dependent gamma-secretase activity mediates the intramembranous cleavage of CD44. Oncogene. 2003;22:1511–1516. doi: 10.1038/sj.onc.1206298. [DOI] [PubMed] [Google Scholar]
- 23.Fang X.J., Xu W.L., Gong J.L., Chen C., Fang L.L., Chen Q.Y. CD44 variant increases the invasive ability of human breast cancer cell line MCF-7 cells. Zhonghua Zhong Liu Za Zhi. 2010;32:22–28. [PubMed] [Google Scholar]
- 24.To K., Fotovati A., Reipas K.M., Law J.H., Hu K., Wang J., Astanehe A., Davies A.H., Lee L., Stratford A.L., Raouf A., Johnson P., Berquin I.M., Royer H.D., Eaves C.J., Dunn S.E. Y-box binding protein-1 induces the expression of CD44 and CD49f leading to enhanced self-renewal, mammosphere growth, and drug resistance. Cancer Res. 2010;70:2840–2851. doi: 10.1158/0008-5472.CAN-09-3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bourguignon L.Y., Xia W., Wong G. Hyaluronan-mediated CD44 interaction with p300 and SIRT1 regulates beta-catenin signaling and NFkappaB-specific transcription activity leading to MDR1 and Bcl-xL gene expression and chemoresistance in breast tumor cells. J Biol Chem. 2009;284:2657–2671. doi: 10.1074/jbc.M806708200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bourguignon L.Y., Spevak C.C., Wong G., Xia W., Gilad E. Hyaluronan-CD44 interaction with protein kinase C(epsilon) promotes oncogenic signaling by the stem cell marker Nanog and the production of microRNA-21, leading to down-regulation of the tumor suppressor protein PDCD4, anti-apoptosis, and chemotherapy resistance in breast tumor cells. J Biol Chem. 2009;284:26533–26546. doi: 10.1074/jbc.M109.027466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marangoni E., Lecomte N., Durand L., de Pinieux G., Decaudin D., Chomienne C., Smadja-Joffe F., Poupon M.F. CD44 targeting reduces tumour growth and prevents post-chemotherapy relapse of human breast cancers xenografts. Br J Cancer. 2009;100:918–922. doi: 10.1038/sj.bjc.6604953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peng X.C., Yang L., Yang L.P., Mao Y.Q., Yang H.S., Liu J.Y., Zhang D.M., Chen L.J., Wei Y.Q. Efficient inhibition of murine breast cancer growth and metastasis by gene transferred mouse survivin Thr34–>Ala mutant. J Exp Clin Cancer Res. 2008;27:46. doi: 10.1186/1756-9966-27-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rodel F., Frey B., Leitmann W., Capalbo G., Weiss C., Rodel C. Survivin antisense oligonucleotides effectively radiosensitize colorectal cancer cells in both tissue culture and murine xenograft models. Int J Radiat Oncol Biol Phys. 2008;71:247–255. doi: 10.1016/j.ijrobp.2008.02.011. [DOI] [PubMed] [Google Scholar]
- 30.Blanc-Brude O.P., Mesri M., Wall N.R., Plescia J., Dohi T., Altieri D.C. Therapeutic targeting of the survivin pathway in cancer: initiation of mitochondrial apoptosis and suppression of tumor-associated angiogenesis. Clin Cancer Res. 2003;9:2683–2692. [PubMed] [Google Scholar]
- 31.Lee C.W., Simin K., Liu Q., Plescia J., Guha M., Khan A., Hsieh C.C., Altieri D.C. A functional Notch-survivin gene signature in basal breast cancer. Breast Cancer Res. 2008;10:R97. doi: 10.1186/bcr2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ouhtit A., Matrougui K., Bengrine A., Koochekpour S., Zerfaoui M., Yousief Z. Survivin is not only a death encounter but also a survival protein for invading tumor cells. Front Biosci. 2007;12:1260–1270. doi: 10.2741/2144. [DOI] [PubMed] [Google Scholar]
- 33.Yamamoto H., Ngan C.Y., Monden M. Cancer cells survive with survivin. Cancer Sci. 2008;99:1709–1714. doi: 10.1111/j.1349-7006.2008.00870.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Duffy M.J., O'Donovan N., Brennan D.J., Gallagher W.M., Ryan B.M. Survivin: a promising tumor biomarker. Cancer Lett. 2007;249:49–60. doi: 10.1016/j.canlet.2006.12.020. [DOI] [PubMed] [Google Scholar]
- 35.Ryan B.M., O'Donovan N., Duffy M.J. Survivin: a new target for anti-cancer therapy. Cancer Treat Rev. 2009;35:553–562. doi: 10.1016/j.ctrv.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 36.Guha M., Altieri D.C. Survivin as a global target of intrinsic tumor suppression networks. Cell Cycle. 2009;8:2708–2710. doi: 10.4161/cc.8.17.9457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu J.J., Liu W.D., Yang H.Z., Zhang Y., Fang Z.G., Liu P.Q., Lin D.J., Xiao R.Z., Hu Y., Wang C.Z., Li X.D., He Y., Huang R.W. Inactivation of PI3k/Akt signaling pathway and activation of caspase-3 are involved in tanshinone I-induced apoptosis in myeloid leukemia cells in vitro. Ann Hematol. 2010;89:1089–1097. doi: 10.1007/s00277-010-0996-z. [DOI] [PubMed] [Google Scholar]
- 38.Liu F., Liu S., He S., Xie Z., Zu X., Jiang Y. Survivin transcription is associated with P-glycoprotein/MDR1 overexpression in the multidrug resistance of MCF-7 breast cancer cells. Oncol Rep. 2010;23:1469–1475. doi: 10.3892/or_00000786. [DOI] [PubMed] [Google Scholar]
- 39.Zhao P., Meng Q., Liu L.Z., You Y.P., Liu N., Jiang B.H. Regulation of survivin by PI3K/Akt/p70S6K1 pathway. Biochem Biophys Res Commun. 2010;395:219–224. doi: 10.1016/j.bbrc.2010.03.165. [DOI] [PubMed] [Google Scholar]
- 40.Khan S., Aspe J.R., Asumen M.G., Almaguel F., Odumosu O., Acevedo-Martinez S., De Leon M., Langridge W.H., Wall N.R. Extracellular, cell-permeable survivin inhibits apoptosis while promoting proliferative and metastatic potential. Br J Cancer. 2009;100:1073–1086. doi: 10.1038/sj.bjc.6604978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liang X., Da M., Zhuang Z., Wu W., Wu Z., Wu Y., Shen H. Effects of survivin on cell proliferation and apoptosis in MG-63 cells in vitro. Cell Biol Int. 2009;33:119–124. doi: 10.1016/j.cellbi.2008.10.013. [DOI] [PubMed] [Google Scholar]
- 42.Jiang Y., Saavedra H.I., Holloway M.P., Leone G., Altura R.A. Aberrant regulation of survivin by the RB/E2F family of proteins. J Biol Chem. 2004;279:40511–40520. doi: 10.1074/jbc.M404496200. [DOI] [PubMed] [Google Scholar]
- 43.Hallstrom T.C., Mori S., Nevins J.R. An E2F1-dependent gene expression program that determines the balance between proliferation and cell death. Cancer Cell. 2008;13:11–22. doi: 10.1016/j.ccr.2007.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]