

Abstract
Obesity is a major risk factor for the development and progression of breast cancer. Leptin, a cytokine mainly produced by adipocytes, plays a crucial role in mammary carcinogenesis and is elevated in hyperinsulinemia and insulin resistance. The antidiabetic thiazolidinediones inhibit leptin gene expression through ligand activation of the peroxisome proliferator-activated receptor-γ (PPARγ) and exert antiproliferative and apoptotic effects on breast carcinoma. In this study, we investigated the ability of PPARγ ligands to counteract leptin stimulatory effects on breast cancer growth in either in vivo or in vitro models. The results show that activation of PPARγ prevented the development of leptin-induced MCF-7 tumor xenografts and inhibited the increased cell-cell aggregation and proliferation observed on leptin exposure. PPARγ ligands abrogated the leptin-induced up-regulation of leptin gene expression and its receptors in breast cancer. PPARγ-mediated repression of leptin gene involved the recruitment of nuclear receptor corepressor protein and silencing mediator of retinoid and thyroid hormone receptors corepressors on the glucocorticoid responsive element site in the leptin gene expression regulatory region in the presence of glucocorticoid receptor and PPARγ. In addition, PPARγ ligands inhibited leptin signaling mediated by MAPK/STAT3/Akt phosphorylation and counteracted leptin stimulatory effect on estrogen signaling. These findings suggest that PPARγ ligands may have potential therapeutic benefits in the treatment of breast cancer.
Several epidemiologic findings have established that obesity is a risk factor for human breast cancer.1 Indeed, increased body weight has been associated with shorter disease-free and overall survival in patients with breast cancer.2
Leptin, a peptide hormone mainly secreted by adipocytes, is a pleiotropic molecule that regulates food intake, hematopoiesis, inflammation, immunity, cell differentiation, and proliferation.3 More recently, leptin has been found to be involved in neoplastic processes, particularly in mammary tumorigenesis.4,5 Specifically, in vitro and in vivo studies have shown that leptin stimulates tumor growth, cell survival, and transformation4,6,7 and amplifies estrogen signaling, contributing to hormone-dependent breast cancer growth and progression.6,8
Peroxisome proliferator-activated receptor-γ (PPARγ) is a member of the nuclear receptor family of ligand-dependent transcription factors, which is best known for its differentiating effects on adipocytes and insulin-mediated metabolic functions.9 Activators of PPARγ include thiazolidinediones, a new class of antidiabetic drugs, such as rosiglitazone (BRL), that rather than reducing hyperglycemia and hyperinsulinemia in insulin-resistant states10 inhibit leptin expression and its signal transduction in different cell and animal models.11–14 PPARγ is also involved in cell-cycle control, inflammation, atherosclerosis, apoptosis, and carcinogenesis.15 The controversial role of PPARγ ligands in carcinogenesis has been reported, although the precise mechanisms responsible for differential effects (ie, proapoptotic versus proliferative) remain incompletely clarified. Some studies have demonstrated that activation of PPARγ increases tumor cell growth.16–18 However, most published studies imply the inhibitory effects of PPARγ ligands on the tumor growth of several carcinomas, including breast cancer.19,20 In the past few years, we have investigated different molecular mechanisms through which PPARγ may induce antiproliferative effects, cell-cycle arrest, and apoptosis in human MCF-7 breast cancer cells.21–23
In this study, we evaluated the ability of PPARγ ligands to counteract leptin stimulatory effects on breast cancer growth in either in vivo or in vitro models. These results have shown that PPARγ ligands reverse the enhanced expression of leptin gene (Lep; formerly Ob) and its receptor (Lepr; formerly ObR), inhibiting the downstream signaling pathways induced by leptin. Transcriptional repression of Ob gene by PPARγ seems to be consequent to the recruitment of nuclear receptor corepressor protein (NCoR) and silencing mediator of retinoid and thyroid hormone receptors (SMRT) corepressors involving the participation of glucocorticoid receptor (GR).
Materials and Methods
Plasmids
The pHEGO plasmid, encoding the full length of estrogen receptor α (ERα) cDNA, and the reporter plasmid XETL, a construct containing an estrogen-responsive element, were gifts from Dr. Didier Picard (University of Geneva, Geneva, Switzerland). The plasmids containing the full-length human leptin promoter or its deletions were gifts from Dr. Marc Reitman (NIH, Bethesda, MD).
Site-Directed Mutagenesis
The leptin promoter plasmid-bearing glucocorticoid responsive element–mutated site (p1775 GRE mut) was created by site-directed mutagenesis using a QuikChange kit (Stratagene, La Jolla, CA) and as template the human leptin promoter p1775. The mutagenic primers were 5′-CCAGGCTGTAGTGCAATGGTCTtgcCTTGGCTCACTGCAACC-3′ and 5′-GGTTGCAGTGAGCCAAGgcaAGACCATTGCACTACAGCCTGG-3′. Mutations are shown as italic and lower case letters. The constructed reporter vector was confirmed by DNA sequencing.
Cell Cultures
MCF-7, BT-20, and CHO cells were obtained from the American Type Culture Collection (Manassas, VA), were maintained in Dulbecco's modified Eagle's medium/Ham's F-12 containing 5% fetal bovine serum (Sigma, Milan, Italy), and were cultured in serum-free medium for at least 24 hours before treatments. All the media were supplemented with 1% L-glutamine and 1% penicillin/streptomycin (Sigma).
In Vivo Experiments
Female 45-day-old athymic nude mice (nu/nu Swiss; Charles River Laboratories, Milan, Italy) were maintained in a sterile environment. At day 0, estradiol pellets (1.7 mg per pellet, 90-day release; Innovative Research of America, Sarasota, FL) were subcutaneously implanted into the intrascapular region of the mice. The next day, MCF-7 cells (5.0 × 106 cells per mouse) were inoculated subcutaneously in 0.1 mL of Matrigel (BD Biosciences, Bedford, MA). Leptin treatment was performed as previously described.7 When the tumors reached ∼0.2 cm3 (ie, in 4 weeks), the animals received rosiglitazone (10 mg/kg/day) in drinking tap water (Avandia, GlaxoSmith Kline, Middlesex, UK) for 8 weeks. MCF-7 xenograft tumor growth was monitored twice a week by caliper measurements, and tumor volumes (in cubic centimeters) were estimated by the following formula: TV = a × (b2)/2, where a and b are tumor length and width, respectively, in centimeters. At week 12, blood samples were collected from the mice, and the animals were sacrificed following standard protocols; the tumors were dissected from the neighboring connective tissue, frozen in nitrogen, and stored at −80°C for further analyses. After blood centrifugation, plasma was collected and kept at −80°C for analyses. Plasma leptin concentration was measured using a commercially available mouse and rat leptin enzyme-linked immunosorbent assay kit (BioVendor, Heidelberg, Germany). All the procedures involving animals and their care were conducted in accordance with the institutional guidelines and regulations at the Laboratory of Molecular Oncogenesis, Regina Elena Cancer Institute, Rome, Italy.
Histologic Analysis
Tumors, livers, lungs, spleens, and kidneys were fixed in 4% formalin, sectioned at 5 μm, and stained with hematoxylin and eosin Y (Bio-Optica, Milan, Italy). The epithelial nature of the tumors was verified by immunostaining with mouse monoclonal antibody directed against human cytokeratin 18 (Santa Cruz Biotechnology, Milan, Italy), and nuclei were counterstained with hematoxylin. For negative controls, nonimmune serum replaced the primary antibody.
Three-Dimensional Spheroid Culture and Cell Growth Assays
For three-dimensional cultures, MCF-7 and BT-20 cells plated on 2% agar–coated plates were treated with 1000 ng/mL of leptin (Invitrogen, Carlsbad, CA) and/or 10 μmol/L BRL (Alexis, San Diego, CA) and 10 μmol/L 15-deoxy-Δ12,14-prostaglandin J2 (PGJ2) (Sigma). After 48 hours, three-dimensional cultures were photographed using a phase-contrast microscope (Olympus, Milan, Italy). The extent of aggregation and cell numbers were evaluated as reported.7
[3H]Thymidine Incorporation Assays
MCF-7 and BT-20 cells were treated with leptin (1000 ng/mL) and/or BRL (10 μmol/L) and PGJ2 (10 μmol/L) for 48 hours and were pretreated for 2 hours with GW9662 (10 μmol/L; Sigma) or transfected with RNA interference (RNAi) for PPARγ where necessary. The assay was performed as previously reported.6
RT-PCR Assays
Total RNA was extracted using TRIzol reagent (Invitrogen). Reverse transcription was performed using a RETROscript kit (Ambion, Austin, TX). The cDNAs were amplified by PCR using the following primers: long isoform of ObR (ObRL): 5′-CGAGAAACGTTTCAGCATCT-3′ and 5′-CAAAAGCACACCACTCTCTC-3′; short isoform of ObR (ObRS): 5′-GAAGGAGTCGGAAAACCAAAG-3′ and 5′-CCACCATATGTTAACTCTCAG-3′; Ob: 5′-AGAGCCTTTGGATGACCAGAACAAGGTTCCCT-3′ and 5′-TTACGAGAGAACTAACTGGAGAGCGACCTTT-3′; cathepsin D: 5′-AACAACAGGGTGGGCTTC-3′ and 5′-ATGCACGAAACAGATCTGTGCT-3′; pS2 5′-TTCTATCCTAATACCATCGACG-3′ and 5′-TTTGAGTAGTCAAAGTCAGAGC-3′; cyclin D1: 5′-TCTAAGATGAAGGAGACCATC-3′ and 5′-GCGGTAGTAGGACAGGAAGTTGTT-3′; P450arom: 5′-CAAGGTTATTTTGATGCATGG-3′ and 5′-TTCTAAGGTTTGCGCATGA-3′; and 36B4: 5′-CTCAACATCTCCCCCTTCTC-3′ and 5′-CAAATCCCATATCCTCGT-3′.
The PCR was performed for 30 cycles for cathepsin D and cyclin D1; for 35 cycles for ObRL, ObRS, Ob, and P450arom; and for 15 cycles to amplify pS2 and 36B4 in the presence of 1 μL of first-strand cDNA, 1 μmol/L each primer, 0.5 mmol/L dNTP, and TaqDNA polymerase (2 U per tube) (Promega Corp, Madison, WI) in a final volume of 25 μL.
Western Blot Analysis and Immunoprecipitation Assays
The phosphorylated forms of MAPK, STAT3, Akt, and GR S211 were identified by Western blot analysis in 50 μg of whole lysate. The immunoblots were stripped and reprobed to determine total levels of MAPK, STAT3, Akt (Cell Signaling Technology, Milan, Italy), and GR (Santa Cruz Biotechnology). Immunoprecipitation was performed in 300 μg of nuclear extracts in the presence of appropriate antibodies (Santa Cruz Biotechnology). Proteins were resolved on 8% to 10% SDS-polyacrylamide gels, transferred to a nitrocellulose membrane, and probed with specific antibodies (Novus Biologicals, Milan, Italy). The antigen-antibody complex was detected by incubation of the membrane at room temperature with a peroxidase-coupled goat anti-mouse or anti-rabbit IgG and revealed using the ECL system (Amersham, Buckinghamshire, UK).
Transfection Assays
MCF-7 cells were transfected using the FuGENE 6 reagent (Roche Diagnostics, Mannheim, Germany) with the mixture containing 0.5 μg of luciferase-reporter plasmid and 5 ng of pRL-CMV. CHO cells were transfected with XETL (0.5 μg per well) in the presence of HEGO (0.2 μg per well). After 24 hours of transfection, treatments were added and cells were incubated for 24 hours. The firefly luciferase values of each sample were normalized by Renilla luciferase activity, and the data are reported in relative light units.
Electrophoretic Mobility Shift Assays
Nuclear extracts were prepared from MCF-7 cells as previously described.24 The probe was generated by annealing single-stranded oligonucleotides labeled with [γ32P]ATP and tyrosine polynucleotide kinase and then purified using Sephadex G-50 spin columns (Sigma). The DNA sequences used as probe or as cold competitors were as follows: GRE: 5′-ATGGTCTGATCTTGGCTCAC-3′ and 5′-GTGAGCCAAGATCAGACCAT-3′; CRE: 5′-CACCGACGTCATTTGCAGTTCC-3′ and 5′-CACCGACAGCTTTTGCAGTTCC-3′; and Sp1: 5′GAAAAACTCCGCCCTGGTAAAT-3′ and 5′-ATTTACCAGGCGCGGAGTTTTTC-3′. The protein-binding reactions were performed as reported.7 The specificity of the binding was tested by adding to the mixture the reaction-specific antibodies anti-GR and anti-PPARγ. The entire reaction mixture was electrophoresed through a 6% polyacrylamide gel in 0.25x Tris-borate-EDTA for 3 hours at 150 V.
Chromatin Immunoprecipitation and Re–Chromatin Immunoprecipitation Assays
MCF-7 cells were cross-linked with 1% formaldehyde and sonicated. Supernatants were immunocleared with salmon sperm DNA/protein A agarose for 1 hour at 4°C. The precleared chromatin was immunoprecipitated with specific anti-GR (E-20), PPARγ (H-100), or anti-polymerase II (RNA-PoII H-224) antibodies (Santa Cruz Biotechnology) and were reimmunoprecipitated with anti-PPARγ, anti-NCoR (NB120-2781), or anti-SMRT (NB300-732) antibodies (Novus Biologicals).
Normal mouse serum IgG was used as negative control. Pellets were washed as reported, eluted with elution buffer (1% SDS, 0.1 mol/L NaHCO3), and digested with proteinase K.25 DNA was obtained by phenol/chloroform/isoamyl alcohol extractions and were precipitated with ethanol; 5 μL of each sample and input were used for real-time PCR with the primers flanking GRE sequence present in the leptin promoter region: 5′-GCCCAGGCTGTAGTGCAAT-3′ and 5′-TAGCCAGGTGTGGTGG-3′. PCR reactions were performed as reported.26
Immunocytochemical Staining
Cells were fixed in 4% paraformaldehyde, and hydrogen peroxide was used to inhibit endogenous peroxidase activity. Cells were incubated with 10% normal horse serum to block nonspecific binding sites. Immunocytochemical staining was performed using specific primary antibodies (Santa Cruz Biotechnology). A biotinylated universal antibody made in horse was applied as secondary antibody (Vector Laboratories, Burlingame, CA). Amplification of avidin-biotin–horseradish peroxidase complex was performed, and 3,3′-diaminobenzidine tetrachloride dihydrate was used as a detection system. In control experiments (negative control), cells were processed, replacing the primary antibody with nonimmune rabbit IgG (Vector Laboratories).
RNAi
MCF-7 cells were transfected with 25-bp RNA duplex of validated RNAi-targeted human PPARγ mRNA sequence 5′-AGAAUAAUAAGGUGGAGAUGCAGGC-3′ or with a stealth RNAi-negative control low GC (Invitrogen) to a final concentration of 100 nmol/L using Lipofectamine 2000 (Invitrogen), as recommended by the manufacturer. After 5 hours, the transfection medium was changed with serum-free medium to avoid Lipofectamine 2000 toxicity, and then cells were exposed to treatments.
Statistical Analysis
Each data point represents the mean ± SE of at least three different experiments. Statistical analysis was performed using analysis of variance followed by Newman-Keuls testing to determine differences in means. Statistical comparisons for in vivo studies were made using the Wilcoxon-Mann-Whitney test. P < 0.05 was considered statistically significant.
Results
PPARγ Ligands Reverse Leptin-Induced Tumor Cell Growth in Vivo and in Vitro
First we explored the ability of BRL to inhibit leptin-induced breast tumor growth in vivo. To this aim, female nude mice, bearing into the intrascapular region of MCF-7 cell tumor xenografts, were treated with leptin and/or BRL. This administration was well tolerated because no change in body weight (see Supplemental Table S1 at http://ajp.amjpathol.org) or in food and water consumption was observed together with no evidence of reduced motor function. In addition, no significant difference in the mean weights or histologic features of the major organs (liver, lung, spleen, and kidney) after sacrifice was observed between vehicle-treated mice and those that received treatment, indicating a lack of toxic effects at the dose given.
Histologic examination of MCF-7 xenografts revealed that tumors were primarily composed of tumor epithelial cells (see Supplemental Figure S1 at http://ajp.amjpathol.org). Our results showed that leptin treatment induced tumor growth in mice, as we previously demonstrated,7 whereas a significant reduction in tumor volume was observed in the animal group receiving leptin plus BRL (Figure 1A). At week 12, tumor sizes were markedly smaller in animals treated with leptin plus BRL than with leptin alone (Figure 1B). Mean ± SE plasma leptin levels measured at week 12 were significantly higher in leptin-treated mice (2.05 ± 0.087 ng/mL, P < 0.01) than in vehicle-treated mice (1.50 ± 0.05 ng/mL); in contrast, in BRL- (1.13 ± 0.06 ng/mL, P < 0.01) and BRL + leptin– (1.36 ± 0.078 ng/mL, P < 0.01) treated mice, leptin concentrations were decreased compared with those in the control and leptin groups.
Figure 1.
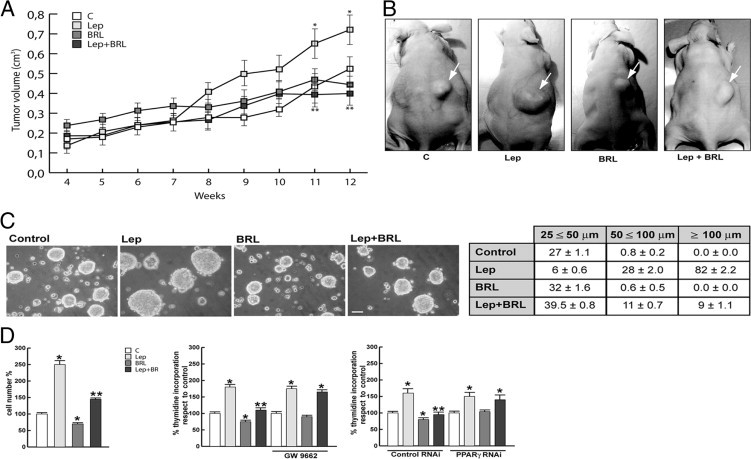
BRL reverses leptin-induced tumor cell growth. A: Tumor volume from MCF-7 xenografts implanted with estradiol pellets in female nude mice at weeks 4 and 12. The animals were treated with 230 μg/kg/day leptin (Lep) (n = 8), 10 mg/kg/day BRL (n = 8), Lep and BRL (n = 8), or vehicle as control (n = 8). *P < 0.05, leptin-treated group versus control group; **P < 0.05, leptin plus BRL–treated group versus leptin-treated group. B: Representative images of experimental tumors at week 12 (arrows). C: MCF-7 three-dimensional cultures were untreated or treated as indicated for 48 hours (top). Scale bar = 50 μm. The extent of aggregation was scored by measuring the spheroid diameters. The values represent the sum of spheroids in 10 optical fields under ×10 magnification (bottom). D: Cell numbers obtained from three-dimensional spheroids and proliferation determined by [3H]thymidine incorporation in MCF-7 cells transfected and treated as indicated for 48 hours. The results are mean ± SE of three experiments. *P < 0.05 versus untreated cells; **P < 0.05 versus leptin.
We then performed three-dimensional MCF-7 cell cultures, which closely mimic some in vivo biologic features of tumors.27 We showed that BRL treatment (10 μmol/L) inhibited the enhanced cell-cell adhesion induced by leptin (1000 ng/mL) exposure as evidenced by the extent of aggregation scored by measuring the spheroid diameters (Figure 1C). In three-dimensional cultures, BRL decreased cell growth compared with untreated cells and reversed the enhanced leptin cell numbers. Moreover, the effects of leptin and/or BRL on cell proliferation were assessed by [3H]-thymidine DNA incorporation assay. Leptin stimulated the growth of MCF-7 cells; this effect was completely inhibited by BRL treatment (Figure 1D). Similar results were also obtained using the natural PPARγ ligand PGJ2 (see Supplemental Figure S2 at http://ajp.amjpathol.org). The inhibitory effects exerted by PPARγ ligands were no longer noticeable in the presence of the specific PPARγ antagonist GW9662 as well as PPARγ RNAi, demonstrating direct involvement of this nuclear receptor in antagonizing leptin-induced tumor growth (Figure 1D).
In ERα-negative breast cancer cells BT-20, BRL also reversed leptin-induced cell aggregation and cell proliferation (see Supplemental Figure S3 at http://ajp.amjpathol.org), suggesting that BRL effects are estrogen independent.
BRL Represses Activation of Leptin Signal Transduction Pathways in MCF-7 Cells
Leptin exerts its biologic function through binding to its receptors, which mediate a downstream signal by activating multiple signaling pathways.28,29 We examined the effects of BRL on ObRs and its transduction pathways. Stimulation of MCF-7 cells with leptin resulted in an increase in ObRL and ObRS, which was reversed by treatment with BRL (Figure 2A). Similar results were also obtained in MCF-7 xenografts (Figure 2B). In addition, as expected, leptin significantly induced phosphorylation of MAPK/STAT3/Akt in in vivo and in vitro models, whereas treatment with BRL completely abrogated the leptin activation of these signaling pathways (Figure 2, C and D). All these data suggest that the inhibitory action of BRL on leptin-induced tumor growth, cell adhesion, and proliferation involves, at least in part, the ability of PPARγ ligand to modulate ObR expression and antagonize its signaling pathways.
Figure 2.
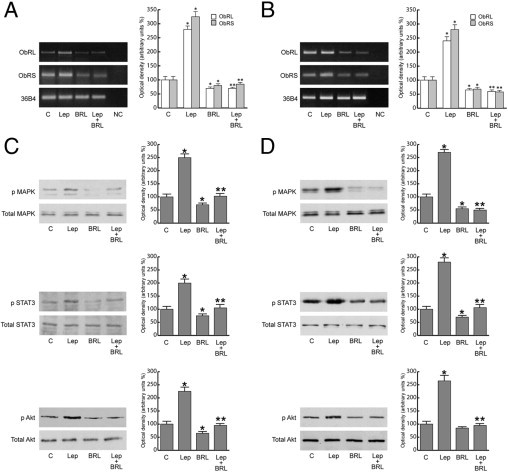
BRL represses leptin (Lep) signaling in MCF-7 cells and xenografts. RT-PCR of ObRL and ObRS mRNA from MCF-7 cells stimulated for 48 hours (A) and from xenografts (B). 36B4 mRNA levels were determined as control. NC indicates RNA sample without the addition of reverse transcriptase (negative control). Representative Western blot analysis on protein extracts from MCF-7 cells stimulated for 15 minutes (C) and xenografts excised from control and treated mice (D) showing MAPK/STAT3/Akt activation. The immunoblots were stripped and reprobed with total MAPK/STAT3/Akt as loading control. The results are mean ± SE of three separate experiments in which the band intensities were evaluated in terms of optical density arbitrary units and expressed as the percentage of the control assumed to be 100%. *P < 0.05 versus untreated cells; **P < 0.05 versus leptin.
Modulation of Ob Expression and Its Transcriptional Activity by BRL
To assess whether BRL can also affect Ob, we performed RT-PCR in MCF-7 cells. We showed in in vitro and in vivo models that leptin increased Ob mRNA, whereas BRL reversed this effect (Figure 3A). This latter result led us to ascertain whether PPARγ activation can modulate leptin transcriptional activity. To this aim, we transiently transfected MCF-7 cells with a plasmid containing leptin regulatory sequences p1774 (−2922/+30) and found that leptin induced luciferase activity, which was inhibited by treatment with BRL (Figure 3B). The Ob promoter contains multiple transcription factor binding motifs, including CRE and Sp1 sites and one GRE site (Figure 3B). To determine which cis-acting elements in the Ob promoter can mediate the previously mentioned effects, Ob promoter–deleted constructs −1975/+30 (p1775), −1546/+30 (p1776), and −805/+30 (p1778) were tested. In transfection experiments performed using p1775 and p1776 constructs, the responsiveness to leptin was still observed, and BRL still inhibited the increase induced by leptin. In contrast, in the presence of the construct p1778, no up-regulatory effects were noticeable on leptin exposure (Figure 3B). Thus, we performed site-directed mutagenesis assays to evaluate which site was responsible for leptin-induced Ob promoter activation. We found that only the mutation of the GRE site was involved in mediating the stimulatory effect of leptin (Figure 3C), addressing that the up-regulatory effect of leptin requires the GRE motif. To explore whether leptin affects the activity of GR, we tested its nuclear translocation and phosphorylation at S211. Leptin induced GR nuclear localization with a concomitant increase in S211 phosphorylated GR levels, which were significantly reduced by pretreatment with the MAPK inhibitor PD98059 and the JAK/STAT inhibitor AG490. In contrast, the PI3K inhibitor LY294002 did not affect GR phosphorylation status (Figure 4).
Figure 3.
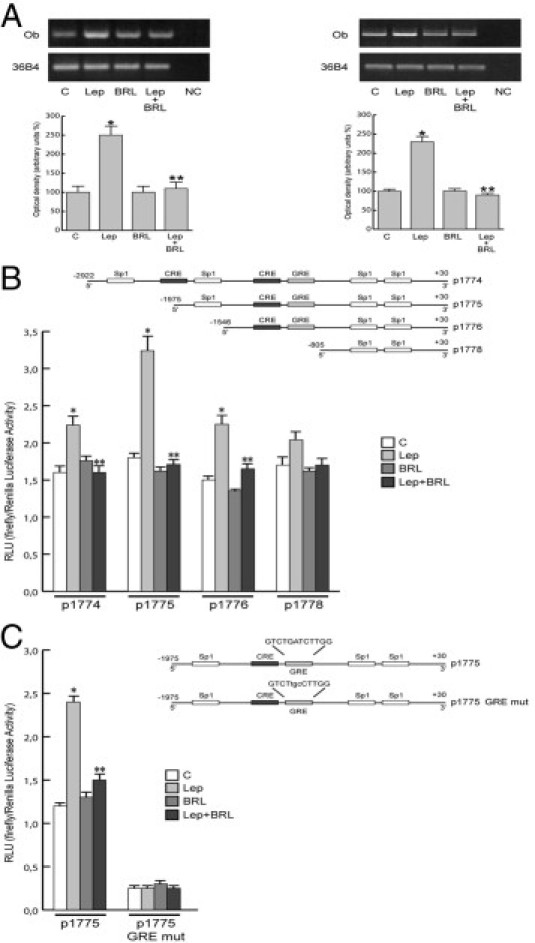
BRL negatively regulates leptin (Lep)-induced Ob expression and its transcriptional activity. A: RT-PCR of Ob was performed in MCF-7 cells stimulated for 48 hours (left panel) and in xenografts (right panel). 36B4 mRNA levels were determined as control. NC, negative control. MCF-7 cells were transiently transfected with luciferase plasmids containing the human leptin promoter (p1774) or its deletions (p1775, p1776, and p1778) (B) or p1775 mutated in the GRE site (p1775 GRE mut) (C). Schematic maps of the human leptin promoter constructs are included. Cells were untreated or treated with leptin (1000 ng/mL) and/or 10 μmol/L BRL for 24 hours. RLU indicates relative light units. The results are mean ± SE of three separate experiments. *P < 0.05 versus untreated cells; **P < 0.05 versus leptin.
Figure 4.
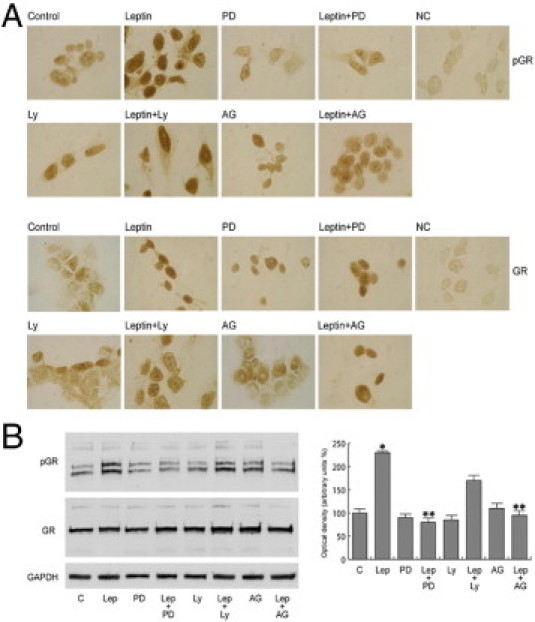
Leptin (Lep) induces nuclear translocation of GR and its phosphorylation (pGR). MCF-7 cells were untreated or treated with 1000 ng/mL of leptin for 15 minutes or pretreated with 10 μmol/L PD98059 (PD), AG490 (AG), or LY294002 (LY). A: Immunostaining of pGR and GR. No immunodetection was observed replacing the antibodies with horse serum [negative control (NC)]. B: Representative Western blotting for pGR on S211 and GR. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as loading control. The results are mean ± SE of three separate experiments for pGR in which the band intensities were evaluated in terms of optical density arbitrary units and expressed as the percentage of the control assumed to be 100%. *P < 0.05 versus untreated cells; **P < 0.05 versus leptin.
PPARγ Reverses Leptin-Induced Effects on Ob Promoter at the GRE Site through Corepressor Recruitment
To provide insight into the molecular mechanism by which the GRE motif modulates Ob promoter activity, we performed electrophoretic mobility shift assay experiments using as a probe the GRE sequence present in the Ob regulatory region. We observed the formation of a complex in nuclear extracts from MCF-7 cells (Figure 5A, lane 1), which was abrogated by 100-fold molar excess of unlabeled probe (Figure 5A, lane 2), demonstrating the specificity of the DNA binding complex. This inhibition was no longer observed using a mutated oligodeoxyribonucleotide as competitor (Figure 5A, lane 3). Leptin and BRL induced a slight increase in the specific band (Figure 5A, lanes 4 and 5), whereas an enhanced DNA binding complex was observed on combined treatments (Figure 5A, lane 6). The inclusion of anti-GR and anti-PPARγ antibodies in the reactions attenuated the specific bands, suggesting the presence of both proteins in the complex (Figure 5A, lanes 7 and 8). Note that the leptin-induced increase in the DNA binding complex was no longer noticeable when a synthetic oligodeoxyribonucleotide corresponding to the CRE or Sp1 motif was used as probe (data not shown).
Figure 5.
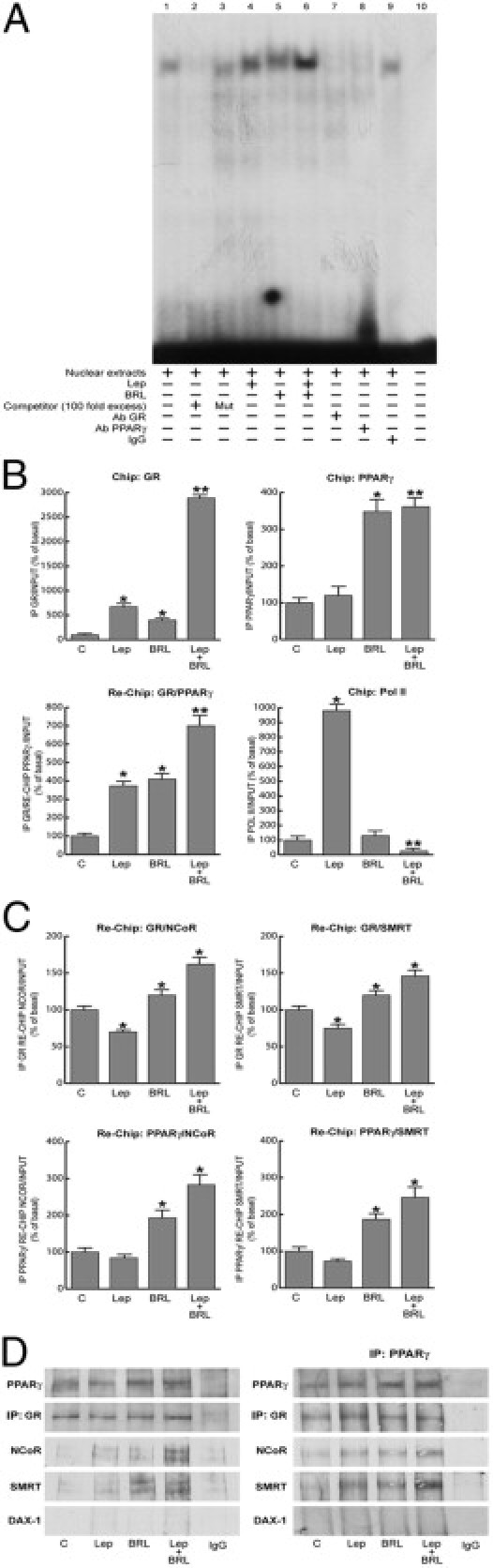
PPARγ binds and recruits corepressors to the GRE site in the Ob promoter. A: Nuclear extracts from MCF-7 cells were incubated with GRE probe (lane 1). A 100-fold molar excess of unlabeled (lane 2) and mutated (Mut) (lane 3) probe was added. MCF-7 nuclear extracts treated with 1000 ng/mL of leptin and/or 10 μmol/L BRL for 6 hours (lanes 4, 5, and 6). Anti-GR (lane 7), anti-PPARγ (lane 8) antibodies (Abs) or IgG (lane 9) were added to the mixture. Lane 10 shows probe alone. B: ChIP with the anti-GR, anti-PPARγ, and anti-Pol II antibodies. ChIP with the anti-GR antibody was re-ChIP with anti-PPARγ antibody. C: ChIP with the anti-GR or anti-PPARγ antibodies was re-ChIP with either anti-NCoR or anti-SMRT antibodies. The leptin (Lep) promoter sequence including the GRE site was detected by real-time-PCR with specific primers (see Materials and Methods). The results are mean ± SE of three separate experiments expressed as the percentage of control. *P < 0.05 versus untreated cells; **P < 0.05 versus leptin. D: MCF-7 cells were treated with 1000 ng/mL of leptin and/or 10 μmol/L BRL for 48 hours. Immunoprecipitation was performed using anti-GR (left panel) or anti-PPARγ (right panel) antibodies and then blotted with anti-PPARγ, GR, NCoR, SMRT, or DAX-1 antibodies.
The interaction of GR and PPARγ receptors with the Ob promoter was further elucidated by chromatin immunoprecipitation (ChIP) assays. Using anti-GR or anti-PPARγ antibodies, protein-chromatin complexes were immunoprecipitated from MCF-7 cells treated for 1 hour with leptin and/or BRL. Real-time PCR was used to determine the recruitment of GR and PPARγ to the Ob region containing the GRE site. The results indicate that GR was constitutively bound to the Ob promoter in untreated cells and that this recruitment was increased on leptin or BRL exposure and to a higher extent after combined treatments. Immunoprecipitation with anti-PPARγ antibody showed enhanced recruitment of this nuclear receptor to the Ob promoter in the presence of BRL and BRL plus leptin treatments. Similar results were also obtained by GR/PPARγ Re-ChIP assay. We revealed after leptin treatment an enhanced association of RNA Pol II that was drastically reduced by leptin plus BRL exposure (Figure 5B).
To assess whether the decrease in Ob promoter transcriptional activity might be caused by the cooperative interaction between GR/PPARγ and negative transcriptional regulators, we investigated the involvement of NCoR and SMRT, which interact with and function as negative coregulators of GR and PPARγ.30–33 A coimmunoprecipitation assay was performed on nuclear protein fractions from MCF-7 cells treated with leptin and/or BRL. The formation of GR and PPARγ complex was clearly detected in all the conditions tested. Moreover, GR/NCoR and GR/SMRT complexes were slightly revealed in untreated cells, but this association was enhanced by combined treatments. No interaction of the orphan nuclear receptor DAX-1, a corepressor for GR,34 was observed under the same experimental conditions (Figure 5D). Similar results were obtained in MCF-7 cells immmunoprecipitated with anti-PPARγ antibody (Figure 5D).
Re-ChIP assays demonstrated increased NCoR and SMRT occupancy of the GRE-containing region of the Ob promoter after BRL exposure and particularly on combined treatments (Figure 5C). In the presence of a MAPK inhibitor able to interfere with GR phosphorylation, the recruitment of corepressors to the Ob promoter was markedly reduced (data not shown).
BRL Abrogates Leptin-Activated Estrogen Signaling in MCF-7 Cells
We previously demonstrated that leptin can transactivate ERα and enhance aromatase gene expression in breast cancer cells.6,8 Thus, because PPARγ ligands interfere with leptin signaling, we investigated the ability of BRL to reverse the leptin effects on estrogen signaling.
In MCF-7 cells transiently transfected with XETL construct, we observed that BRL significantly inhibited ERE-dependent transactivation induced by leptin (Figure 6A), and this effect was reversed by pretreatment with GW9662 (data not shown). Similar results were reproduced in ER-negative CHO cells in which ERα was ectopically expressed (Figure 6A).
Figure 6.
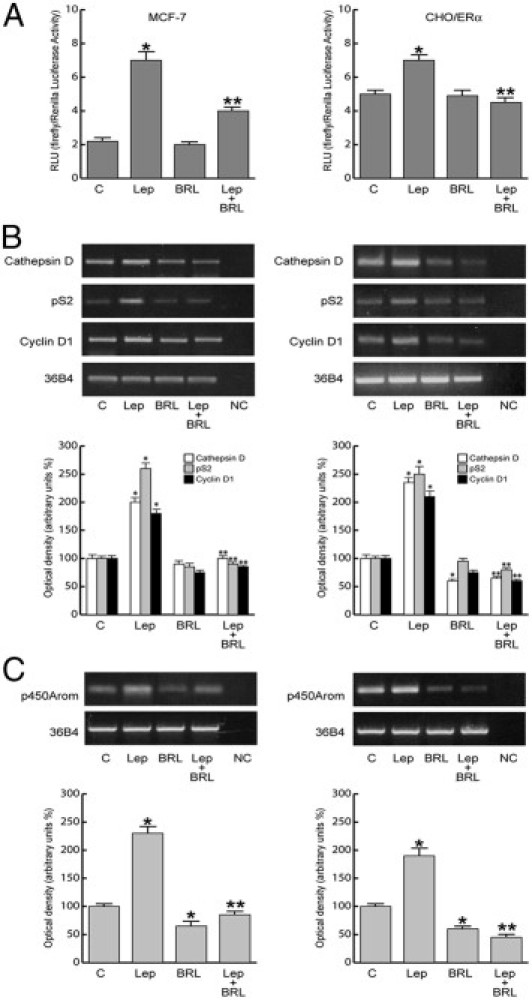
BRL antagonizes estrogen signaling induced by leptin (Lep). A: MCF-7 cells were transfected with XETL plasmid. CHO cells were cotransfected with XETL and HEGO plasmids (CHO/ERα). RLU, relative light unit. The results are mean ± SE of three independent experiments performed in triplicate. *P < 0.05 versus untreated cells; *P < 0.05 versus leptin. Cathepsin D, pS2, and cyclin D1 (B) and aromatase (C) mRNA expression in MCF-7 cells treated for 48 hours as indicated (left panel) and in xenografts (right panel). 36B4 mRNA levels were determined as control. NC indicates negative control. The results are mean ± SE of three separate experiments in which the band intensities were evaluated in terms of optical density arbitrary units and expressed as the percentage of the control assumed to be 100%. *P < 0.05 versus untreated cells; **P < 0.05 versus leptin.
These data correlated well with the ability of BRL to abrogate in in vivo and in vitro models the up-regulatory effects of leptin on mRNA expression levels of the classic estrogen genes cathepsin, pS2, and cyclin D1 (Figure 6B). Besides, in MCF-7 cell cultures and in xenografts, BRL reversed the increase in aromatase expression induced by leptin (Figure 6C). These results indicate that treatment with BRL counteracting leptin action on estrogen signaling may contribute to reduce breast cancer cell growth and progression.
Discussion
For the first time, we demonstrate that activation of PPARγ reverses leptin-mediated promotion of breast tumor growth either in vivo in MCF-7 xenografts implanted in female nude mice or in vitro in MCF-7 three-dimensional and monolayer cultured breast cancer cells.
Elevated leptin levels strongly correlate with obesity, hyperinsulinemia, and insulin resistance, conditions found in most patients with diabetes mellitus.35 PPARγ ligands, thiazolidinediones, able to reduce hyperglycemia and hyperinsulinemia in insulin-resistant states, also down-regulate gene expression of pluripotent hormone leptin in vivo and in vitro.11,12 Besides, previous observations have reported that PPARγ agonists inhibited leptin-induced proliferation in hepatic stellate cells by suppression of MAPK activation14 and abolished leptin-directed migration of endothelial cells through Akt.13 Thus, regulation and function of leptin and PPARγ are possibly interrelated and relevant in obese patients in whom metabolic changes are major contributors to the development of breast carcinoma.
The present results show that the PPARγ ligand BRL prevents the development of leptin-induced MCF-7 tumor xenografts in the presence of significantly lower plasma leptin levels and inhibits the increased cell-cell aggregation and proliferation observed on leptin exposure. The in vitro results were also reproduced in ERα-negative breast cancer cells BT20, addressing how the mechanism by which PPARγ activation affects breast tumor cell growth is not tightly related to estrogen dependency. Moreover BRL down-regulates the enhanced expression of ObRs induced by leptin and inhibits MAPK/Akt/STAT3 leptin downstream signaling pathways. These results are in agreement with data obtained in other cell models showing that PPARγ ligands suppress ObR mRNA and its promoter activity and block leptin signaling.13,14,36
It emerges from recent studies that leptin and estrogen systems are involved in a functional cross talk. For example, leptin has been shown to directly transactivate ERα8 and positively modulate aromatase activity.6
We demonstrated in MCF-7 cells that BRL, interfering with leptin signaling, could reverse the effects of leptin on estrogen signal specifically, counteracting ERα activation and its classic target genes in either in vitro or in vivo models.
Furthermore, the leptin-enhanced aromatase expression was completely abrogated by BRL treatment as we observed in xenografts and in monolayer cultures of MCF-7 cells, underlying the ability of this new class of oral antidiabetic drugs to inhibit local estrogen production, which represents an important factor of tumor microenvironment able to maintain tumor growth and progression.
Activated PPARγ is also known to inhibit the expression of Ob in adipose tissue.12,37 Herein, we demonstrated in MCF-7 cells the ability of BRL to reverse the leptin-induced Ob mRNA expression and its transcriptional activity, showing how PPARγ negatively interferes in the short autocrine loop maintained by leptin on Ob gene in breast cancer cells. The human Ob promoter contains multiple transcription regulatory elements, including several CRE, Sp1, and NFkB sites and one GRE site.38 Functional experiments using Ob promoter–deleted constructs and site-directed mutagenesis studies have shown that the up-regulatory effects induced by leptin on Ob promoter activity completely reversed by BRL occurred through the GRE site. These latter results fit well with the evidence that leptin induced GR translocation and phosphorylation at S211, which was inhibited by MAPK and JAK/STAT inhibitors. It was reported that the S211 phosphorylated GR strongly correlates with GR transcriptional activation; indeed, inhibition of this phosphorylation is associated with decreased nuclear retention of GR and inhibited gene transcription.39
The important role of GRE in regulating Ob promoter activity was also shown by electrophoretic mobility shift assays. We found in nuclear extracts from MCF-7 cells treated with leptin plus BRL a strong increase in the GRE-DNA binding that was immunodepleted by anti-GR and anti-PPARγ antibodies, suggesting the presence of the two proteins in the complex.
The physiologic relevance of GRE in the Ob promoter in vivo is pointed out by ChIP analysis showing that the GR/PPARγ occupancy of the GRE-containing promoter region, induced by leptin plus BRL treatment, is concomitant with a decrease in RNA Pol II recruitment and a reduction in Ob transcriptional activity.
It is known that members of the nuclear hormone receptor superfamily, including GR and PPARγ, once activated, can interact physically and modulate target gene transcription.40,41 We have shown a strong association of PPARγ with GR in the nuclear fraction of untreated MCF-7 cells that was further potentiated by leptin plus BRL treatments.
PPARγ and GR can regulate transcription by several distinct mechanisms, and their functions, as shown for other corticosteroid receptors, seem to depend not only on ligand binding, which is known to regulate receptor conformation, but also on the context of the gene and associated promoter factors that contribute to create a gene-specific topography, achieving specific profiles of gene expression.42,43 Our proposed model for PPARγ-mediated repression of the Ob gene involves its interaction with GR and recruitment of the corepressors NCoR and SMRT, which share the same molecular architecture, interact with many of the same transcription factors, and assemble into similar corepressor complexes.44 NCoR and SMRT are recruited by PPARγ and GR to regulate the transcription of different genes.45,46 Overexpression of NCoR and SMRT represses PPARγ-mediated gene transcription in certain cell types,47 and recently, increasing evidence suggests that these two corepressors modulate adipogenesis most likely via their ability to repress PPARγ action.48,49 The present results suggest that in MCF-7 cells on BRL and to a higher extent leptin plus BRL stimulation, NCoR and SMRT are recruited on the GRE site of the Ob promoter together with GR and PPARγ.
A hypothetical model of the possible mechanism through which PPARγ activation may modulate leptin expression and function in reducing breast cancer growth is shown in Figure 7. Leptin, through JAK/STAT/MAPK activation, may phosphorylate GR and induce its transactivation, resulting in an increase in leptin promoter activity. This up-regulatory effect on Ob is counteracted by PPARγ through the recruitment of corepressors NCoR and SMRT on the GRE site of the Ob regulatory region in the presence of GR and PPARγ.
Figure 7.
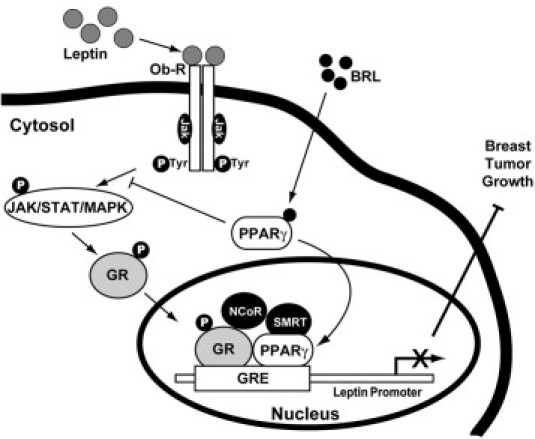
Molecular mechanism through which PPARγ counteracts leptin expression and function in breast cancer. Leptin, through JAK/STAT/MAPK activation, increases GR phosphorylation (pGR) and its nuclear translocation. pGR transactivates leptin promoter by binding to GRE motif. In the presence of BRL, PPARγ binds to GRE and, through the formation of GR/PPARγ complex, allows the recruitment of NCoR and SMRT corepressors, thus inhibiting Ob transcription and reducing breast tumor growth.
Moreover, activation of PPARγ also decreases ObRs, inhibits its transductional pathways, and negatively interferes with estrogen signaling through the down-regulation of aromatase gene expression and the inhibition of ERα transactivation.
In conclusion, these data suggest that PPARγ ligands rather than ameliorate metabolic parameters may represent pharmacologic tools to be exploited in the novel therapeutic adjuvant strategies for breast cancer treatment.
Acknowledgments
We thank Dr. Vittoria Rago for her support in immunostaining assays and Dr. Pasquale Cicirelli for his technical assistance.
Footnotes
Supported by Associazione Italiana Ricerca Cancro grants IG 1482, IG 8804, and MFAG 6180, ex 60% Ministero Istruzione Università Ricerca.
S.C., L.M., and D.B. contributed equally to this work.
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.04.026.
Supplementary data
Staining of MCF-7 xenograft tumors. Representative tumor sections from mice at week 12 were formalin fixed, paraffin embedded, sectioned, and stained with H&E Y or incubated with a mouse monoclonal antibody directed against human cytokeratin 18 as described in Materials and Methods. Cytokeratin 18 expression appears as brown cytoplasmic staining.
PGJ2 reverses leptin (Lep)-induced growth in MCF-7 cells. A: MCF-7 three-dimensional cultures treated with leptin (1000 ng/mL) and/or PGJ2 (10 μmol/L) for 48 hours (top panel). The extent of aggregation was scored by measuring the spheroid diameters using an ocular micrometer in 10 optical fields under ×10 magnification (bottom panel). B: Cell numbers obtained from three-dimensional spheroids (left panel) and proliferation of MCF-7 cells determined by [3H]thymidine incorporation (right panel). The results are mean ± SE from at least three experiments. *P < 0.05 versus control; **P < 0.05 versus leptin.
BRL reverses leptin (Lep)-induced growth in ERα-negative BT-20 cells. A: BT-20 three-dimensional cultures treated with leptin (1000 ng/mL) and/or BRL (10 μmol/L) for 48 hours (top panel). The extent of aggregation was scored by measuring the spheroid diameters using an ocular micrometer in 10 optical fields under ×10 magnification (bottom panel). B: Cell numbers obtained from three-dimensional spheroids (left panel) and proliferation of BT-20 cells determined by [3H]thymidine incorporation (right panel). The results are mean ± SE from at least three experiments. *P < 0.05 versus control; **P < 0.05 versus leptin.
References
- 1.Calle E.E., Kaaks R. Overweight, obesity and cancer: epidemiological evidence and proposed mechanisms. Nat Rev Cancer. 2004;4:579–591. doi: 10.1038/nrc1408. [DOI] [PubMed] [Google Scholar]
- 2.Chlebowski R.T., Aiello E., McTiernan A. Weight loss in breast cancer patient management. J Clin Oncol. 2002;20:1128–1143. doi: 10.1200/JCO.2002.20.4.1128. [DOI] [PubMed] [Google Scholar]
- 3.Ahima RS, Flier JS: Leptin. Annu Rev Physiol 20006, 2:413–437 [DOI] [PubMed]
- 4.Garofalo C., Surmacz E. Leptin and cancer. J Cell Physiol. 2006;207:12–22. doi: 10.1002/jcp.20472. [DOI] [PubMed] [Google Scholar]
- 5.Vona-Davis L., Rose D.P. Adipokines as endocrine, paracrine and autocrine factors in breast cancer risk and progression. Endocr Relat Cancer. 2007;14:189–206. doi: 10.1677/ERC-06-0068. [DOI] [PubMed] [Google Scholar]
- 6.Catalano S., Marsico S., Giordano C., Mauro L., Rizza P., Panno M.L., ò S. Leptin enhances, via AP-1, expression of aromatase in the MCF-7 cell line. J Biol Chem. 2003;278:28668–28676. doi: 10.1074/jbc.M301695200. [DOI] [PubMed] [Google Scholar]
- 7.Mauro L., Catalano S., Bossi G., Pellegrino M., Barone I., Morales S., Giordano C., Bartella V., Casaburi I., ò S. Evidences that leptin up-regulates E-cadherin expression in breast cancer: effects on tumor growth and progression. Cancer Res. 2007;67:3412–3421. doi: 10.1158/0008-5472.CAN-06-2890. [DOI] [PubMed] [Google Scholar]
- 8.Catalano S., Mauro L., Marsico S., Giordano C., Rizza P., Rago V., Montanaro D., Maggiolini M., Panno M.L., ó S. Leptin induces, via ERK1/ERK2 signal, functional activation of estrogen receptor α in MCF-7 cells. J Biol Chem. 2004;279:19908–19915. doi: 10.1074/jbc.M313191200. [DOI] [PubMed] [Google Scholar]
- 9.Desvergne B., Wahli W. Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr Rev. 1999;20:649–688. doi: 10.1210/edrv.20.5.0380. [DOI] [PubMed] [Google Scholar]
- 10.Quinn C.E., Hamilton P.K., Lockhart C.J., McVeigh G.E. Thiazolidinediones: effects on insulin resistance and the cardiovascular system. Br J Pharmacol. 2008;153:636–645. doi: 10.1038/sj.bjp.0707452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De Vos P., Lefebvre A.M., Miller S.G., Guerre-Millo M., Wong K., Saladin R., Hamann L.G., Staels B., Briggs M.R., Auwerx J. Thiazolidinediones repress ob gene expression in rodents via activation of peroxisome proliferator-activated receptor γ. J Clin Invest. 1996;98:1004–1009. doi: 10.1172/JCI118860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rieusset J., Auwerx J., Vidal H. Regulation of gene expression by activation of the peroxisome proliferator-activated receptor γ with rosiglitazone (BRL 49653) in human adipocytes. Biochem Biophys Res Commun. 1999;265:265–271. doi: 10.1006/bbrc.1999.1657. [DOI] [PubMed] [Google Scholar]
- 13.Goetze S., Bungenstock A., Czupalla C., Eilers F., Stawowy P., Kintscher U., Spencer-Hänsch C., Graf K., Nürnberg B., Law R.E., Fleck E., Gräfe M. Leptin induces endothelial cell migration through Akt, which is inhibited by PPARγ-ligands. Hypertension. 2002;40:748–754. doi: 10.1161/01.hyp.0000035522.63647.d3. [DOI] [PubMed] [Google Scholar]
- 14.Lee J.I., Paik Y.H., Lee K.S., Lee J.W., Kim Y.S., Jeong S., Kwon K.S., Lee D.H., Kim H.G., Shin Y.W., Kim M.A. A peroxisome-proliferator activated receptor-γ ligand could regulate the expression of leptin receptor on human hepatic stellate cells. Histochem Cell Biol. 2007;127:495–502. doi: 10.1007/s00418-007-0282-x. [DOI] [PubMed] [Google Scholar]
- 15.Rocchi S., Auwerx J. Peroxisome proliferator-activated receptor-γ: a versatile metabolic regulator. Ann Med. 1999;31:342–351. doi: 10.3109/07853899908995901. [DOI] [PubMed] [Google Scholar]
- 16.Lefebvre A.M., Chen I., Desreumaux P., Najib J., Fruchart J.C., Geboes K., Briggs M., Heyman R., Auwerx J. Activation of the peroxisome proliferators-activated receptor γ promotes the development of colon tumors in C57BL/6J-APCMin/+ mice. Nat Med. 1998;4:1053–1057. doi: 10.1038/2036. [DOI] [PubMed] [Google Scholar]
- 17.Saez E., Tontonoz P., Nelson M.C., Alvarez J.G., Ming U.T., Baird S.M., Thomazy V.A., Evans R.M. Activators of the nuclear receptors PPARγ enhance colon polyp formation. Nat Med. 1998;4:1058–1061. doi: 10.1038/2042. [DOI] [PubMed] [Google Scholar]
- 18.Chinery R., Coffey R.J., Graves-Deal R., Kirkland S.C., Sanchez S.C., Zackert W.E., Oates J.A., Morrow J.D. Prostaglandin J2 and 15-deoxy-Δ 12,14-prostaglandin J2 induce proliferation of cyclooxygenase-depleted colorectal cancer cells. Cancer Res. 1999;59:2739–2746. [PubMed] [Google Scholar]
- 19.Elstner E., Müller C., Koshizuka K., Williamson E.A., Park D., Asou H., Shintaku P., Said J.W., Heber D., Koeffler H.P. Ligands for peroxisome proliferator-activated receptorγ and retinoic acid receptor inhibit growth and induce apoptosis of human breast cancer cells in vitro and in BNX mice. Proc Natl Acad Sci U S A. 1998;95:8806–8811. doi: 10.1073/pnas.95.15.8806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grommes C., Landreth G.E., Heneka M.T. Antineoplastic effects of peroxisome proliferator-activated receptor γ agonists. Lancet Oncol. 2004;5:419–429. doi: 10.1016/S1470-2045(04)01509-8. [DOI] [PubMed] [Google Scholar]
- 21.Bonofiglio D., Gabriele S., Aquila S., Catalano S., Gentile M., Middea E., Giordano F., ò S. Estrogen receptor α binds to peroxisome proliferator-activated receptor (PPAR) response element and negatively interferes with PPAR γ signalling in breast cancer cells. Clin Cancer Res. 2005;11:6139–6147. doi: 10.1158/1078-0432.CCR-04-2453. [DOI] [PubMed] [Google Scholar]
- 22.Bonofiglio D., Aquila S., Catalano S., Gabriele S., Belmonte M., Middea E., Qi H., Morelli C., Gentile M., Maggiolini M., ò S. Peroxisome proliferator-activated receptor γ activates p53 gene promoter binding to the nuclear factor-κB sequence in human MCF7 breast cancer cells. Mol Endocrinol. 2006;20:3083–3092. doi: 10.1210/me.2006-0192. [DOI] [PubMed] [Google Scholar]
- 23.Bonofiglio D., Gabriele S., Aquila S., Qi H., Belmonte M., Catalano S., ò S. Peroxisome proliferator-activated receptor γ activates fas ligand gene promoter inducing apoptosis in human breast cancer cells. Breast Cancer Res Treat. 2009;113:423–434. doi: 10.1007/s10549-008-9944-1. [DOI] [PubMed] [Google Scholar]
- 24.Andrews N.C., Faller D.V. A rapid micropreparation technique for extraction of DNA-binding proteins from limiting numbers of mammalian cells. Nucleic Acids Res. 1991;19:2499. doi: 10.1093/nar/19.9.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morelli C., Garofalo C., Sisci D., del Rincon S., Cascio S., Tu X., Vecchione A., Sauter E.R., Miller W.H., Jr, Surmacz E. Nuclear insulin receptor substrate 1 interacts with estrogen receptor α at ERE promoters. Oncogene. 2004;23:7517–7526. doi: 10.1038/sj.onc.1208014. [DOI] [PubMed] [Google Scholar]
- 26.Sirianni R., Chimento A., Malivindi R., Mazzitelli I., ò S., Pezzi V. Insulin-like growth factor-I, regulating aromatase expression through steroidogenic factor 1, supports estrogen-dependent tumor Leydig cell proliferation. Cancer Res. 2007;67:8368–8377. doi: 10.1158/0008-5472.CAN-06-4064. [DOI] [PubMed] [Google Scholar]
- 27.Mauro L., Surmacz E. IGF-I receptor, cell-cell adhesion, tumor development and progression. J Mol Histol. 2004;35:247–253. doi: 10.1023/b:hijo.0000032356.98363.a2. [DOI] [PubMed] [Google Scholar]
- 28.Sweeney G. Leptin signalling. Cell Signal. 2002;14:655–663. doi: 10.1016/s0898-6568(02)00006-2. [DOI] [PubMed] [Google Scholar]
- 29.Ahima R.S., Osei S.Y. Leptin signaling. Physiol Behav. 2004;81:223–241. doi: 10.1016/j.physbeh.2004.02.014. [DOI] [PubMed] [Google Scholar]
- 30.Cohen R.N. Nuclear receptor corepressors and PPARγ. Nucl Recept Signal. 2006;4:e003. doi: 10.1621/nrs.04003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ricote M., Glass C.K. PPARs and molecular mechanisms of transrepression. Biochim Biophys Acta. 2007;1771:926–935. doi: 10.1016/j.bbalip.2007.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van der Laan S., Meijer O.C. Pharmacology of glucocorticoids: beyond receptors. Eur J Pharmacol. 2008;585:483–491. doi: 10.1016/j.ejphar.2008.01.060. [DOI] [PubMed] [Google Scholar]
- 33.Ronacher K., Hadley K., Avenant C., Stubsrud E., Simons S.S., Jr, Louw A., Hapgood J.P. Ligand-selective transactivation and transrepression via the glucocorticoid receptor: role of cofactor interaction. Mol Cell Endocrinol. 2009;299:219–231. doi: 10.1016/j.mce.2008.10.008. [DOI] [PubMed] [Google Scholar]
- 34.Zhou J., Oakley R.H., Cidlowski J.A. DAX-1 (dosage-sensitive sex reversal-adrenal hypoplasia congenita critical region on the X-chromosome, gene 1) selectively inhibits transactivation but not transrepression mediated by the glucocorticoid receptor in a LXXLL-dependent manner. Mol Endocrinol. 2008;22:1521–1534. doi: 10.1210/me.2007-0273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zimmet P., Boyko E.J., Collier G.R., de Courten M. Etiology of the metabolic syndrome: potential role of insulin resistance, leptin resistance, and other players. Ann N Y Acad Sci. 1999;892:25–44. doi: 10.1111/j.1749-6632.1999.tb07783.x. [DOI] [PubMed] [Google Scholar]
- 36.Tang Y., Zheng S., Chen A. Curcumin eliminates leptin's effects on hepatic stellate cell activation via interrupting leptin signaling. Endocrinology. 2009;150:3011–3020. doi: 10.1210/en.2008-1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kallen C.B., Lazar M.A. Antidiabetic thiazolidinediones inhibit leptin (ob) gene expression in 3T3-L1 adipocytes. Proc Natl Acad Sci U S A. 1996;93:5793–5796. doi: 10.1073/pnas.93.12.5793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gong D.W., Bi S., Pratley R.E., Weintraub B.D. Genomic structure and promoter analysis of the human obese gene. J Biol Chem. 1996;271:3971–3974. doi: 10.1074/jbc.271.8.3971. [DOI] [PubMed] [Google Scholar]
- 39.Chen W., Dang T., Blind R.D., Wang Z., Cavasotto C.N., Hittelman A.B., Rogatsky I., Logan S.K., Garabedian M.J. Glucocorticoid receptor phosphorylation differentially affects target gene expression. Mol Endocrinol. 2008;22:1754–1766. doi: 10.1210/me.2007-0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ialenti A., Grassia G., Di Meglio P., Maffia P., Di Rosa M., Ianaro A. Mechanism of the anti-inflammatory effect of thiazolidinediones: relationship with the glucocorticoid pathway. Mol Pharmacol. 2005;67:1620–1628. doi: 10.1124/mol.104.004895. [DOI] [PubMed] [Google Scholar]
- 41.Nie M., Corbett L., Knox A.J., Pang L. Differential regulation of chemokine expression by peroxisome proliferator-activated receptor γ agonists: interactions with glucocorticoids and β2-agonists. J Biol Chem. 2005;280:2550–2561. doi: 10.1074/jbc.M410616200. [DOI] [PubMed] [Google Scholar]
- 42.Hall J.M., Couse J.F., Korach K.S. The multifaceted mechanisms of estradiol and estrogen receptor signaling. J Biol Chem. 2001;276:36869–36872. doi: 10.1074/jbc.R100029200. [DOI] [PubMed] [Google Scholar]
- 43.Katzenellenbogen B.S., Katzenellenbogen J.A. Biomedicine: defining the “S” in SERMs. Science. 2002;295:2380–2381. doi: 10.1126/science.1070442. [DOI] [PubMed] [Google Scholar]
- 44.Ghisletti S., Huang W., Jepsen K., Benner C., Hardiman G., Rosenfeld M.G., Glass C.K. Cooperative NCoR/SMRT interactions establish a corepressor-based strategy for integration of inflammatory and anti-inflammatory signaling pathways. Genes Dev. 2009;23:681–693. doi: 10.1101/gad.1773109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gurnell M., Wentworth J.M., Agostini M., Adams M., Collingwood T.N., Provenzano C., Browne P.O., Rajanayagam O., Burris T.P., Schwabe J.W., Lazar M.A., Chatterjee V.K. A dominant-negative peroxisome proliferator-activated receptor γ (PPARγ) mutant is a constitutive repressor and inhibits PPARγ-mediated adipogenesis. J Biol Chem. 2000;275:5754–5759. doi: 10.1074/jbc.275.8.5754. [DOI] [PubMed] [Google Scholar]
- 46.Wang Q., Blackford J.A., Jr, Song L.N., Huang Y., Cho S., Simons S.S., Jr Equilibrium interactions of corepressors and coactivators with agonist and antagonist complexes of glucocorticoid receptors. Mol Endocrinol. 2004;18:1376–1395. doi: 10.1210/me.2003-0421. [DOI] [PubMed] [Google Scholar]
- 47.Krogsdam A.M., Nielsen C.A., Neve S., Holst D., Helledie T., Thomsen B., Bendixen C., Mandrup S., Kristiansen K. Nuclear receptor corepressor-dependent repression of peroxisome-proliferator-activated receptor delta-mediated transactivation. Biochem J. 2002;363:157–165. doi: 10.1042/0264-6021:3630157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Powell E., Kuhn P., Xu W. Nuclear receptor cofactors in PPARγ-mediated adipogenesis and adipocyte energy metabolism. PPAR Res. 2007;2007:53843. doi: 10.1155/2007/53843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Samarasinghe S.P., Sutanto M.M., Danos A.M., Johnson D.N., Brady M.J., Cohen R.N. Altering PPARγ ligand selectivity impairs adipogenesis by thiazolidinediones but not hormonal inducers. Obesity (Silver Spring) 2009;17:965–972. doi: 10.1038/oby.2008.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Staining of MCF-7 xenograft tumors. Representative tumor sections from mice at week 12 were formalin fixed, paraffin embedded, sectioned, and stained with H&E Y or incubated with a mouse monoclonal antibody directed against human cytokeratin 18 as described in Materials and Methods. Cytokeratin 18 expression appears as brown cytoplasmic staining.
PGJ2 reverses leptin (Lep)-induced growth in MCF-7 cells. A: MCF-7 three-dimensional cultures treated with leptin (1000 ng/mL) and/or PGJ2 (10 μmol/L) for 48 hours (top panel). The extent of aggregation was scored by measuring the spheroid diameters using an ocular micrometer in 10 optical fields under ×10 magnification (bottom panel). B: Cell numbers obtained from three-dimensional spheroids (left panel) and proliferation of MCF-7 cells determined by [3H]thymidine incorporation (right panel). The results are mean ± SE from at least three experiments. *P < 0.05 versus control; **P < 0.05 versus leptin.
BRL reverses leptin (Lep)-induced growth in ERα-negative BT-20 cells. A: BT-20 three-dimensional cultures treated with leptin (1000 ng/mL) and/or BRL (10 μmol/L) for 48 hours (top panel). The extent of aggregation was scored by measuring the spheroid diameters using an ocular micrometer in 10 optical fields under ×10 magnification (bottom panel). B: Cell numbers obtained from three-dimensional spheroids (left panel) and proliferation of BT-20 cells determined by [3H]thymidine incorporation (right panel). The results are mean ± SE from at least three experiments. *P < 0.05 versus control; **P < 0.05 versus leptin.