

Abstract
Sporadic inclusion body myositis (IBM) is a muscle disease with two separate pathogenic components, degeneration and inflammation. Typically, nonnecrotic myofibers are focally surrounded and invaded by CD8+ T cells and macrophages. Both attacked and nonattacked myofibers express high levels of human leukocyte antigen class I (HLA-I) molecules, a prerequisite for antigen presentation to CD8+ T cells. However, only a subgroup of HLA-I+ myofibers is attacked by immune cells. By using IHC, we classified myofibers from five patients with sporadic IBM as attacked (AIBM) or nonattacked (NIBM) and isolated the intracellular contents of myofibers separately by laser microdissection. For comparison, we isolated myofibers from control persons (HCTRL). The samples were analyzed by microarray hybridization and quantitative PCR. HLA-I up-regulation was observed in AIBM and NIBM, whereas HCTRL were negative for HLA-I. In contrast, the inducible chain of the interferon (IFN) γ receptor (IFNGR2) and several IFN-γ–induced genes were up-regulated in AIBM compared with NIBM and HCTRL fibers. Confocal microscopy confirmed segmental IFNGR2 up-regulation on the membranes of AIBM, which positively correlated with the number of adjacent CD8+ T cells. Thus, the differential up-regulation of the IFN-γ signaling cascade observed in the attacked fibers is related to local inflammation, whereas the ubiquitous HLA-I expression on IBM muscle fibers does not require IFNGR expression.
Sporadic inclusion body myositis (sIBM) is the most common inflammatory myopathy in adults >50 years presenting with progressive weakness and atrophy of both proximal and distal muscle groups and leading to disability within 5 to 10 years after diagnosis.1, 2 Because sIBM, in contrast to polymyositis (PM) or dermatomyositis (DM), is notoriously refractive to immunosuppressive therapies,3, 4 it is assumed that sIBM is not a primary autoimmune myopathy but that the inflammatory changes are secondary to as yet unknown viral or degenerative triggers.5 Therefore, sIBM is regarded as the paradigm of a myopathy with distinct degenerative and inflammatory pathogenetic components.1, 6
The mechanism of inflammatory myofiber injury is unique in that CD8+ T cells and macrophages focally surround and invade initially nonnecrotic myofibers.7 Several articles6, 8, 9 provide a review of this information. The myoinvasive CD8+ cytotoxic T cells likely recognize (still unknown) antigens presented by HLA class I (HLA-I) molecules on the myofiber surface.10 Antigen-driven recruitment of the myocytotoxic CD8+ T cells is supported by evidence that the T cells are clonally expanded, using a restricted repertoire of T-cell receptors11, 12, 13, 14, 15 and persisting over time in individual patients.12, 15, 16 Moreover, the CD8+ T cells form specific immunological synapses with vectorial excretion of perforin toward the attacked myofibers.17 This cytotoxicity cannot be assessed in in vitro co-culturing experiments because only alloreactive CD8+ T cells were strongly cytotoxic to allogenic myotubes in co-cultures.18, 19
HLA-I is not detectable on myofibers of healthy subjects.20 In sIBM, HLA-I is uniformly up-regulated on all myofibers21, 22, 23, 24; however, CD8+ T cells attack myofibers in a strictly focal pattern.25 Obviously, HLA-I expression is only a necessary, but not sufficient, precondition for a myofiber to be attacked. There seem to be additional factors rendering muscle fibers susceptible to inflammatory attack. The aim of the present study was to identify molecular patterns that are differentially regulated in invaded versus noninvaded muscle fibers, focusing on the pathways of antigen presentation and processing.
To address the question of why certain myofibers are attacked, whereas others are spared, we used the unique properties of the distinct morphological characteristics of myofibers that remain clearly distinguishable, even under conditions required for laser microdissection (ie, nonembedded tissue and short incubation times, as described in Materials and Methods). We separately laser microdissected attacked and nonattacked muscle fibers while excluding inflammatory cells. We compared attacked myofibers from IBM muscle (AIBM), nonattacked sIBM myofibers (NIBM), and healthy control myofibers (HCTRL) using microarray transcriptome analysis, quantitative PCR (qPCR), and confocal microscopy. PM or DM could not serve as controls because PM is heterogeneous26 and DM lacks CD8+-mediated immunopathological features. We found that the attacked muscle fibers in sIBM differentially overexpressed members of the interferon (IFN) γ signaling cascade. The signature of these molecular changes suggests that they are induced by locally secreted pro-inflammatory cytokines and, therefore, are a consequence, rather than the cause, of the focal inflammatory attack. In contrast, the ubiquitous up-regulation of HLA-I on all IBM myofibers seems to be independent of IFN-γ–mediated signaling and, therefore, triggered before inflammation by as yet unknown mechanisms.
Materials and Methods
Patients and Healthy Controls
Muscle biopsy specimens were obtained from five patients with sIBM (IBM-1 to IBM-5) and three healthy controls (C-1 to C-3) (Table 1). IBM biopsy samples were obtained at the Neuromuscular Laboratory, Mayo Clinic, Rochester, MN (A.G.E.). The study was approved by the local Institutional Review Board (approval no. 1278–03). Patients' consent was obtained according to the Declaration of Helsinki.27 None of the five patients with IBM had received prior anti-inflammatory treatment. The diagnosis of definitive IBM was made according to published criteria.28 Congophilic deposits were visualized in Congo red–stained sections viewed under rhodamine optics.
Table 1.
Overview of the Basic Characteristics of Patients and Healthy Controls
| Patient or control | Diagnosis | Age (years) | Sex | Biopsied muscle | No. of isolated myofibers to reach >100,000 μm2⁎ |
|---|---|---|---|---|---|
| Patient no. | |||||
| IBM-1 | sIBM | 75 | Male | Biceps brachii | 43/37 |
| IBM-2 | sIBM | 70 | Male | Vastus lateralis | 42/35 |
| IBM-3 | sIBM | 79 | Female | Biceps brachii | 42/38 |
| IBM-4 | sIBM | 69 | Male | Triceps brachii | 50/41 |
| IBM-5 | sIBM | 72 | Male | Triceps brachii | 50/47 |
| Control no. | |||||
| C-1 | Nonspecific myalgia, depression | 57 | Male | Biceps brachii | 55 |
| C-2 | Nonspecific myalgia | 61 | Male | Biceps brachii | 60 |
| C-3 | Nonspecific myalgia | 52 | Male | Rectus femoris | 57 |
Five patients with sIBM and three controls were included in the study. Their clinical and histopathological characteristics are listed. The congophilic deposits were visualized in Congo red–stained sections viewed under rhodamine optics. All biopsy specimens of patients with sIBM showed rimmed vacuoles, autoaggressive inflammatory exudates, and congophilic inclusions. IBM-2 presented additional mitochondrial dysfunction. IBM-4 and IBM-5 additionally displayed atrophic myofibers. C-1 had a clinically silent leukocytosis with C-reactive protein elevation at biopsy. C-2 showed no abnormalities on muscle biopsy or laboratory results. C-3 presented with status post statin therapy with transient slight creatine kinase elevation; however, the muscle histology was negative and there were no other abnormalities. The biopsied muscle is included, along with the number of laser-microdissected myofibers that, in each case, compose a total of 100,000 μm2 of myofiber area. The number of myofibers varies in the patients with sIBM given the high variability in myofiber diameter. Control fibers, in general, were smaller on average and, thus, more fibers had to be sampled from controls.
For patients, data are given as AIBM/NIBM.
Three individuals who had clinically presented with nonspecific myalgia, but had normal serum creatine kinase values and normal diagnostic biopsy findings (H.L., University of Munich, Munich, Germany), served as controls. C-1 had clinically silent leukocytosis with slight C-reactive protein elevation at biopsy. All muscle blocks were stored at −80°C. We did not include healthy individuals. For the experiments performed herein, it is absolutely essential that the samples are shock frozen immediately after surgery because otherwise the RNA quality will not be sufficient anymore. RNA, in contrast to DNA or proteins, is degraded rapidly. Therefore, we were limited to diagnostic biopsy material. Samples from autopsy cases or from accidents would not fulfill the listed requirements.
Antibodies
The following antibodies were used in this study. Mouse anti-human CD8α antibody clone LT8 (Serotec, Oxford, UK), which we had previously labeled with the Cy3-mAb Labeling Kit (GE/Amersham, Freiburg, Germany), was used at 2 μg/mL. Mouse anti-human HLA–avidin-biotin complex (ABC) antibody clone W6/32 (directly labeled with Alexa 488 by the manufacturer; AbD Serotec, Düsseldorf, Germany) was used at 0.5 μg/mL. Rabbit anti-human IFNGR2 antibody, polyclonal (order no. HPA001535; Sigma, Deisenhofen, Germany), was used at 1.6 μg/mL with a goat anti-rabbit Alexa 594–labeled secondary antibody (order no. A-11037; Invitrogen, Darmstadt, Germany) at 4 μg/mL. When unlabeled mouse anti-human CD8α antibody clone LT8 was used (dilution, 10 μg/mL; Serotec), a secondary goat anti-mouse Alexa 488–labeled antibody (order no. A-11029; Invitrogen) was used at 2 μg/mL. For negative controls, we used isotype-matched IgGs [mouse IgG1, order no. 555746 (BD Pharmingen, Heidelberg, Germany); mouse IgG2a, order no. MCA929XZ (AbD Serotec)] for monoclonal antibodies and purified IgG (IgG from rabbit serum, order no. I5006; Sigma) for polyclonal antibodies.
Laser Microdissection
Before the experiments, the RNA quality of each biopsy was assessed by Agilent Bioanalyzer 2100 following the recommendations of the manufacturer (Agilent Technologies, Palo Alto, CA). RNA was heat denatured at 70°C and diluted to concentrations of 5 to 15 ng/μL. RNA, 1 μL, was then used for the Agilent Bioanalyzer chip (Agilent Technologies, Böblingen, Germany). We used a staining and laser microdissection protocol similar to our previously published method.29 Briefly, 10-μm cryostat sections from muscle biopsy specimens of patients 1 to 5 and healthy controls 1 to 3 were double stained for CD8α and HLA-ABC with the antibodies previously described. To inhibit RNase activity, all aqueous solutions were pretreated with diethylpyrocarbonate and contained 3 U/μL Protector RNase inhibitor (Roche, Mannheim, Germany) and 2% purified bovine serum albumin (B4287; Sigma). Sections were mounted on positron emission tomographic films (P.A.L.M. Microlaser, Bernried, Germany) that had previously been baked at 180°C for 4 hours, UV irradiated, and coated with poly-l-lysine. Mounted sections were stored at −80°C. After drying in a desiccator, rehydrating in PBS for 10 seconds, and blocking by PBS with 2% bovine serum albumin for 3 minutes, the sections were co-incubated for 5 minutes with the Cy-3–labeled anti-CD8α antibody and the Alexa 488–labeled anti-HLA-ABC antibody. This protocol preserves RNA to a large extent (see Supplemental Figure S1 at http://ajp.amjpathol.org). The tissue was then rinsed with PBS and immediately imaged using a P.A.L.M. Microbeam-Z microscope (P.A.L.M. Microlaser, Bernried, Germany). We defined a myofiber as being attacked when at least three CD8+ T cells were clearly adhering to the myofiber or at least one CD8+ T cell was invading the fiber. We focused on fibers that were surrounded and superficially invaded by inflammatory cells, avoiding fibers that were deeply invaded. A nonattacked myofiber was defined as such if no CD8+ T cells could be seen in the proximity (Figure 1, A and B). Intermediate myofibers not corresponding to either category were not sampled. The fibers were dissected from transversally cut biopsy sections. When dissecting the myofiber content, we left a safety margin, avoiding contamination with surrounding and superficially invading immune cells (Figure 1, C and D). The specimens were examined within 10 minutes after staining for attacked and nonattacked myofibers, which were marked electronically and subsequently microdissected and laser pressure catapulted into respective reaction tubes (either attacked or nonattacked) containing 20-μL TRIzol (Invitrogen), which were then immediately stored on dry ice. Because of a strong variability in myofiber diameter typically seen in sIBM, we chose to sample equal areas rather than equal numbers of myofibers. The area of each collected myofiber (automatically calculated by the P.A.L.M. software, P.A.L.M. Microlaser) was recorded, and a total of 100,000-μm2 myofiber area was collected from each patient with sIBM (attacked and nonattacked) and each control subject. To minimize contamination with inflammatory cells, we assessed the dried sections for artifacts by comparing them with embedded tissue (stained under the same conditions and embedded with Fluorescent Mounting Medium; Dako, Glostrup, Denmark). Moreover, we checked for possible contamination by examining our microarray data for the expression of lymphocyte-, monocyte-, and macrophage-specific transcripts (see Supplemental Table S1 at http://ajp.amjpathol.org).
Figure 1.
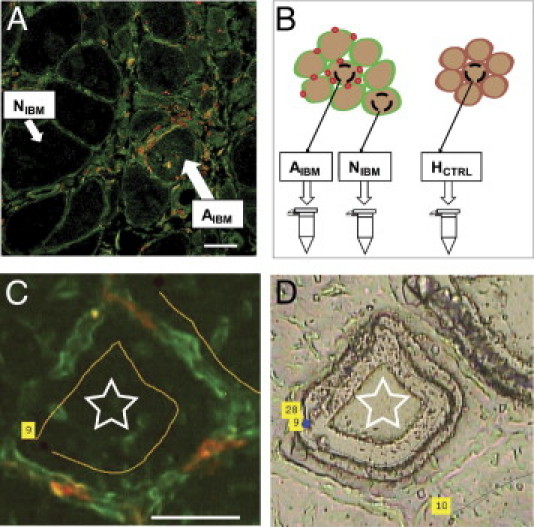
Experimental approach for separate analysis of attacked and nonattacked myofiber subsets in sIBM. A: The focal nature of inflammatory infiltrates in IBM. Muscle tissue was stained with anti-CD8 (red) and anti-HLA-ABC (green) antibodies, as described in Materials and Methods. A shows a confocal image, C shows unembedded dry tissue. All myofibers are HLA-ABC positive, but only some are attacked by CD8+ T cells (AIBM), whereas others are spared (NIBM). B: Myofibers in direct contact with at least three CD8+ T cells or invaded by at least one CD8+ T cell were defined as AIBM. Myofibers not in contact with any CD8+ T cells were defined as NIBM. Ambiguous myofibers were not sampled. Laser-capture microdissection was used to pressure catapult the different types of myofibers into collecting tubes. In total, 100,000 μm2 of AIBM and NIBM myofibers was sampled from each patient with sIBM, as was the same amount of HCTRL myofibers from each control subject. C: Double immunostaining for HLA-ABC (green) and CD8 (red), according to the protocol described in Materials and Methods. Only myofiber tissue (star) was isolated, avoiding surrounding lymphocytes (red). D: Corresponding bright-light image after the myofiber was dissected and catapulted out of the tissue. C and D: The numbers in the yellow fields refer to apparatus parameters. Scale bars = 50 μm (A and C).
RNA Isolation and Linear Transcriptome Amplification
After obtaining 100,000 μm2 of each group of myofibers, the samples were pooled according to their classification. RNA was then extracted using the TRIzol method, with some modifications as recommended by the linear amplification protocol. Three rounds of linear T7-based transcriptome amplification were performed using the ExpressArt-mRNA Amplification Kit, Pico Version (AmpTec, Hamburg, Germany), according to the instructions of the supplier, with the following exceptions: for synthesis of the first cDNA strand, the master mix was prewarmed at 45°C, and 1 μL of RNase R was added to each reaction for the RNA removal step. The resulting yields of amplified RNA (aRNA) were between 30 and 40 μg, derived from <10 ng of input total RNA.
Microarray Hybridization and Analysis
Amplified cDNA, 15 μg, was biotinylated and concomitantly transcribed into aRNA using the BioArray High Yield RNA labeling kit (Enzo Life Sciences, Plymouth Meeting, PA). Biotinylated aRNA was hybridized to HG U133 GeneChip arrays (Affymetrix, High Wycombe, UK). All chips were normalized using the robust multiarray average or the GeneChip robust multiarray average procedure in R packages from Bioconductor (Fred Hutchinson Center, Seattle, WA) (http://www.bioconductor.org, last accessed December 2008). For each probe set, the q values were calculated.30 The q values mirror the false discovery rate and illustrate the degree of heterogeneity between different samples (ie, herein, between different patients). Thus, a low q value reflects a high significance of a particular expression change across all samples. To achieve a high concordance across all patients, we considered only transcripts with a q value of ≤20%.
TaqMan Real-Time qPCR
Expression levels of the following genes were measured in all 13 samples (attacked and nonattacked myofibers from each of the five patients and C-1 to C-3) by real-time qPCR using the 5700 Real-Time PCR System (Applied Biosystems, Foster City, CA): HLA-A, HLA-B, HLA-C, HLA-E, HLA-F, HLA-G, IFN-γ receptor α chain (IFNGR1), IFN-γ receptor β chain (IFNGR2), STAT1, class II transactivator (CIITA), proteasome subunit β-type 8 (PSMB8), HLA-DRA, HLA-DRB, HLA-DPA, HLA-DPB, HLA-DQA, HLA-DQB, CCL5, and STAT3. aRNA was reverse transcribed into cDNA using primer D from the ExpressArt-mRNA Amplification Kit. Three replicates per sample were assayed for each gene in a 96-well format plate. For data normalization across samples, cyclophilin was used as a housekeeping gene. Normalization of the CT values of each gene and determination of fold changes in gene expression were calculated according to the comparative CT method, also known as the 2−ΔCT method.31, 32 For some genes, TaqMan gene expression assays (Applied Biosystems) were used; others were analyzed using custom-made primers and probes (Metabion, Martinsried, Germany). Table 2 lists the primers and probes used in this study.
Table 2.
Genes Analyzed with TaqMan qPCR
| Gene | TaqMan qPCR primers/probes |
|---|---|
| HLA-A | Fwd: 5′-CTGAGATGGGAGCTGTCTTC-3′ |
| Rev: 5′-CTATCTGAGCTCTTCCTCCT-3′ | |
| P: 5′-FAM-GTAAAGTGTGAGACAGCTGCCTTG-TAMRA-3′ | |
| HLA-B | Fwd: 5′-CTGAGATGGGAGCCGTCTT-3′ |
| Rev: 5′-CTCCTTTTCCACCTGAACTC-3′ | |
| P: 5′-FAM-GAGCTTGAAAAGCCTGAGAGAGC-TAMRA-3′ | |
| HLA-C | Fwd: 5′-GAGCTGGGAGCCATCTTCC-3′ |
| Rev: 5′-CTGTTGCTGCACGCAGCCT-3′ | |
| P: 5′-FAM-CCATCATGGGCATCGTTGCTGG-TAMRA-3′ | |
| HLA-E | Fwd: 5′-GTCACCCTGAGATGGAAGC-3′ |
| Rev: 5′-CTTGGATCTGTGGTCTCTGG-3′ | |
| P: 5′-FAM-CCATCGTGGGCATCATTGCTGG-TAMRA-3′ | |
| HLA-F | Fwd: 5′-CCTCCAAAGGCACACGTTG-3′ |
| Rev: 5′-GATAGAAACAGAGGGAGCTAC-3′ | |
| P: 5′-FAM-CAAGACACACGTGACCCACCAC-TAMRA-3′ | |
| HLA-G | Fwd: 5′-CCACAGATACCTGGAGAACG-3′ |
| Rev: 5′-GATCATACTGACCTGGCAGC-3′ | |
| P: 5′-FAM-CAAGACACACGTGACCCACCAC-TAMRA-3′ | |
| IFNGR1 | Fwd: 5′-CATCACGTCATACCAGCCATTT-3′ |
| Rev: 5′-CTGGATTGTCTTCGGTATGCAT-3′ | |
| P: 5′-FAM-GGTCTGTGAAGAGCCGTTGTCTC-TAMRA-3′ | |
| IFNGR2 | Fwd: 5′-CCACCAAGCATCCCATTACA-3′ |
| Rev: 5′-CCTTGGACAAGGACAGCTC-3′ | |
| P: 5′-GACCCAACTCAGCCCATCTTAGA-3′ | |
| STAT1 | Fwd: 5′-GAGCAGGTTCACCAGCTTTATG-3′ |
| Rev: 5′-GAAAACGGATGGTGGCAAATG-3′ | |
| P: 5′-FAM-CAAGACTGGGAGCACGCTGCCAA-TAMRA-3′ | |
| CIITA | Fwd: 5′-ACGCCCTGCTGGGTCC-3′ |
| Rev: 5′-AACTCCATGGTGGCACACTG-3′ | |
| P: 5′-FAM-ACCTGTCAGAGCCCCAAGGCAGC-TAMRA-3′ | |
| PSMB8 | Fwd: 5′-GTCCTACATTAGTGCCTTACG-3′ |
| Rev: 5′-GATAGTACAGCCTGCATTCC-3′ | |
| P: 5′-FAM-GCTGTGCAGACTGTCAGTAC-TAMRA-3′ | |
| HLA-DRA | Fwd: 5′-GGCTTGGATGAGCCTCTTC-3′ |
| Rev: 5′-GGACCATCTTCATCATCAAGG-3′ | |
| P: 5′-FAM-CAAGCACTGGGAGTTTGATGCTC-TAMRA-3′ | |
| HLA-DRB | Fwd: 5′-GGAGAGGTTTACACCTGCC-3′ |
| Rev: 5′-GCAAGATGCTGAGTGGAGTC-3′ | |
| P: 5′-FAM-GAATGGAGAGCACGGTCTGAATC-TAMRA-3′ | |
| HLA-DPA | Fwd: 5′-CACAAGTTCCATTACCTGACC-3′ |
| Rev: 5′-GAGCAAGAAAGTTCAACGAGG-3′ | |
| P: 5′-CTTCTATGACTGCAGGGTGGAGC-3′ | |
| HLA-DPB | Fwd: 5′-GGAGTGGAAGGCACAGTCT-3′ |
| Rev: 5′-GAGCAAGAAAGTTCAACGAGG-3′ | |
| P: 5′-CGGAGTAAGACATTGACGGGAGC-3′ | |
| HLA-DQA | Fwd: 5′-CACCAAGGGCCATTGTGAAT-3′ |
| Rev: 5′-CCAGAGAATAGTGCTAGGTC-3′ | |
| P: 5′-FAM-CCATCTACAGGAGCAGAAGAATGG-TAMRA-3′ | |
| HLA-DQB | Fwd: 5′-CCAGAGCAAGATGCTGAGTG-3′ |
| Rev: 5′-GTGCAGAAGCCCTTTCTGAC-3′ | |
| P: 5′-FAM-GGCTGGGCCTTATCATCCGTCAA-TAMRA-3′ | |
| CCL5 | Applied Biosystems assay ID Hs00174575_m1 |
| STAT3 | Applied Biosystems assay ID Hs01047580_m1 |
| PPIA | Applied Biosystems no. 4333763 |
| GAPDH | Applied Biosystems no. 4333764 |
Classic and nonclassic HLA-I genes were analyzed, as were the major HLA-II genes DR, DP, and DQ. Moreover, we examined the expression of genes downstream of IFN-γ. Forward and reverse primers, and FAM/TAMRA-labeled probes, are listed. All primer/probe sets were designed by us, with the exceptions of IFNGR133 and CIITA.34
Fwd, forward; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; P, probe; PPIA, cyclophilin; Rev, reverse.
Histological Features and Confocal Microscopy
To demonstrate CD8+ T-cell interaction with HLA-I–positive myofibers, we used 10-μm cryosections fixed in ice-cold acetone. To block nonspecific binding, sections were incubated with 2% bovine serum albumin and 2% mouse serum in PBS. Then, we stained for CD8α and HLA-ABC using the antibodies previously described. To confirm IFNGR2 expression on the protein level, we fixed the tissue in 4% paraformaldehyde at room temperature and washed with PBS, then blocked unspecific binding with 2% bovine serum albumin–2% goat serum in PBS, and subsequently stained for CD8α (same as previously described, unlabeled) and rabbit anti-human IFNGR2 antibody (Sigma). After washing in PBS, sections were incubated with goat anti-mouse Alexa 488–labeled antibody and goat anti-rabbit Alexa 594–labeled antibody (both from Invitrogen). For negative controls, we used isotype-matched IgGs (mouse IgG1 for anti-CD8 and mouse IgG2a for anti-HLA-ABC) for monoclonal antibodies and purified IgG (rabbit IgG) for polyclonal antibodies. The staining was visualized using a Zeiss Axiovert 200M inverted microscope (Zeiss, Göttingen, Germany). For confocal microscopy, a Leica TCS SP2 system was used (Leica Microsystems, Wetzlar, Germany). For image analysis, we used the ImageJ software written by Wayne Rasband at the US NIH, version 1.42m (http://rsbweb.nih.gov/ij/index.html, last accessed September 2010).
Quantitative Analysis of IFNGR2 Signal Distribution
Cryosections were stained for IFNGR2 and CD8 and imaged as previously described. Myofibers were photographically documented and classified into four groups according to their extent of membrane circumference positivity for IFNGR2: group 1, >80%; group 2, 50% to 80%; group 3, 20% to 50%; and group 4, <20%. For each myofiber, we counted the number of adjacent CD8+ T cells and calculated the medians. Two investigators (J.I. and Ingrid Eiglmeier) independently performed this analysis for each patient. A Mann-Whitney U-test was performed to determine whether the medians of adjacent CD8+ T cells of group 1 (>80% IFNGR2 positivity) and group 4 (<20% IFNGR2 positivity) differed significantly. The two-tailed P value was calculated, as was the Mann-Whitney U-test value. This analysis was performed for four of five patients with sIBM because of lack of biopsy material from patient 4.
Results
Hints from Microarray Analysis of Attacked and Nonattacked Myofibers
We isolated AIBM, NIBM, and HCTRL myofibers from five patients and three control individuals, as described in Materials and Methods and illustrated in Figure 1. At least 100,000 μm2 of total myofiber area was isolated from each fiber subset of each patient and from each control. We amplified RNA from these laser-microdissected samples and hybridized the aRNA to HG U133 GeneChip microarrays. The microarray data were cross-checked for inadvertent sampling of inflammatory cells (see Supplemental Table S1 at http://ajp.amjpathol.org). We focused on transcripts that were regulated with q values of ≤20% (Table 3) to ensure that interpatient variation is minimized. Of the 34 transcripts with a q value of ≤20%, 19 belonged to HLA-I, HLA-II, or inflammatory signaling genes. Next, we explored whether these molecules are differentially expressed in AIBM versus NIBM versus HCTRL myofibers. Of these 34 transcripts, six were polymorphic HLA-I (ie, HLA-A, HLA-B, or HLA-C). Both AIBM and NIBM myofibers showed a strong up-regulation of HLA-ABC expression compared with HCTRL myofibers (average fold change for AIBM/HCTRL = 16.78, NIBM/HCTRL = 12.95, and AIBM/NIBM = 1.29). Moreover, seven nonpolymorphic HLA-I (ie, HLA-E, HLA-F, or HLA-G) transcripts were up-regulated, with low q values (average fold change for AIBM/HCTRL = 23.63, NIBM/HCTRL = 13.74, and AIBM/NIBM = 1.62).
Table 3.
Microarray Data: Transcripts Most Significantly Regulated across All Patients and Controls
| Probe set ID | Gene title | Raw expression values |
Fold change |
|||||
|---|---|---|---|---|---|---|---|---|
| AIBM | NIBM | HCTRL | q value (%) | AIBM/HCTRL | AIBM/NIBM | NIBM/HCTRL | ||
| 205132_at | Actin, α, cardiac muscle 1 | 9926.55 | 5275.13 | 338.44 | 0.00 | 29.33 | 1.88 | 15.59 |
| 201891_s_at⁎ | β-2-Microglobulin | 12,436.09 | 11,093.17 | 1570.33 | 0.00 | 7.92 | 1.12 | 7.06 |
| 216526_x_at⁎ | MHC, class I, B; MHC, class I, C; MHC class I polypeptide-related sequence A; MHC class I polypeptide-related sequence B | 8959.71 | 7976.71 | 705.27 | 0.00 | 12.70 | 1.12 | 11.31 |
| 209140_x_at⁎ | MHC, class I, B; MHC, class I, C; MHC class I polypeptide-related sequence A; MHC class I polypeptide-related sequence B | 9521.62 | 7469.66 | 297.92 | 0.00 | 31.96 | 1.27 | 25.07 |
| 217456_x_at⁎ | MHC, class I, E | 1503.90 | 827.43 | 75.69 | 0.00 | 19.87 | 1.82 | 10.93 |
| 200905_x_at⁎ | MHC, class I, E | 3383.98 | 1638.89 | 57.90 | 0.00 | 58.44 | 2.06 | 28.30 |
| 221875_x_at⁎ | MHC, class I, F | 1959.54 | 1293.23 | 101.36 | 0.00 | 19.33 | 1.52 | 12.76 |
| 211529_x_at⁎ | MHC, class I, G | 250.88 | 163.19 | 12.87 | 0.00 | 19.49 | 1.54 | 12.68 |
| 209040_s_at⁎ | Proteasome (prosome, macropain) subunit, β type, 8 (large multifunctional peptidase 7) | 1751.19 | 613.20 | 4.05 | 0.00 | 432.37 | 2.86 | 151.40 |
| 202296_s_at | RER1 retention in endoplasmic reticulum 1 homolog (Saccharomyces cerevisiae) | 16.24 | 23.27 | 706.64 | 0.00 | 0.02 | 0.70 | 0.03 |
| 200887_s_at⁎ | STAT 1, 91 kDa | 7963.49 | 6296.56 | 94.89 | 0.00 | 83.92 | 1.26 | 66.35 |
| AFFX-HUMISGF3A/M97935_3_at⁎ | STAT 1, 91 kDa | 1928.89 | 1828.85 | 34.57 | 0.00 | 55.80 | 1.05 | 52.91 |
| 221087_s_at | Apolipoprotein L, 3 | 605.96 | 373.82 | 25.70 | 5.12 | 23.58 | 1.62 | 14.55 |
| 208812_x_at⁎ | MHC, class I, C | 9337.37 | 6540.77 | 430.46 | 5.12 | 21.69 | 1.43 | 15.19 |
| 214459_x_at⁎ | MHC, class I, C | 6556.01 | 4679.25 | 371.30 | 5.12 | 17.66 | 1.40 | 12.60 |
| 204806_x_at⁎ | MHC, class I, F | 753.67 | 512.69 | 59.87 | 5.12 | 12.59 | 1.47 | 8.56 |
| 211071_s_at | Myeloid/lymphoid or mixed-lineage leukemia (trithorax homologue, Drosophila), translocated to 11 | 4511.40 | 2944.13 | 205.20 | 5.12 | 21.99 | 1.53 | 14.35 |
| 202237_at | Nicotinamide N-methyltransferase | 1944.88 | 1365.55 | 21.27 | 5.12 | 91.44 | 1.42 | 64.20 |
| 212845_at | Sterile α motif domain containing 4A | 22.95 | 44.53 | 554.77 | 5.12 | 0.04 | 0.52 | 0.08 |
| 215076_s_at | Collagen, type III, α 1 (Ehlers-Danlos syndrome type IV, autosomal dominant) | 6542.53 | 3846.48 | 12.69 | 8.84 | 515.45 | 1.70 | 303.05 |
| 213932_x_at⁎ | MHC, class I, A | 9857.13 | 7352.34 | 2127.78 | 8.84 | 4.63 | 1.34 | 3.46 |
| 201137_s_at⁎ | MHC, class II, DP β 1 | 1817.92 | 684.36 | 37.08 | 8.84 | 49.02 | 2.66 | 18.46 |
| 225061_at | DnaJ (Hsp40) homologue, subfamily A, member 4 | 1906.51 | 1525.02 | 29.68 | 11.67 | 64.24 | 1.25 | 51.39 |
| 211528_x_at⁎ | MHC, class I, G | 195.97 | 149.47 | 16.23 | 11.67 | 12.08 | 1.31 | 9.21 |
| 200814_at⁎ | Proteasome (prosome, macropain) activator subunit 1 (PA28 α) | 4007.01 | 3496.38 | 405.11 | 11.67 | 9.89 | 1.15 | 8.63 |
| 232500_at | Chromosome 20 open reading frame 74 | 1047.84 | 210.09 | 12.61 | 14.41 | 83.07 | 4.99 | 16.66 |
| 217436_x_at⁎ | MHC, class I, J (pseudogene) | 141.93 | 114.16 | 19.59 | 14.41 | 7.24 | 1.24 | 5.83 |
| 226470_at | γ-Glutamyltransferase 7 | 51.17 | 51.41 | 1893.54 | 16.21 | 0.03 | 1.00 | 0.03 |
| 228098_s_at | Myosin regulatory light chain interacting protein | 794.08 | 85.92 | 27.30 | 16.21 | 29.09 | 9.24 | 3.15 |
| 200743_s_at | Tripeptidyl peptidase I | 1741.52 | 1749.31 | 337.65 | 16.21 | 5.16 | 1.00 | 5.18 |
| 238431_at | Transcribed locus | 1251.44 | 347.59 | 22.14 | 20.02 | 56.52 | 3.60 | 15.70 |
| 215313_x_at⁎ | MHC, class I, A | 11,623.07 | 9764.78 | 968.10 | 20.02 | 12.01 | 1.19 | 10.09 |
| 204070_at | Retinoic acid receptor responder (tazarotene induced) 3 | 3065.52 | 1516.95 | 88.87 | 20.02 | 34.49 | 2.02 | 17.07 |
| 209118_s_at | Tubulin, α 1a | 1608.79 | 1037.65 | 48.98 | 20.02 | 32.85 | 1.55 | 21.19 |
Raw expression values are provided along with fold expression changes showing regulation of transcripts in AIBM vs HCTRL, AIBM vs NIBM, and NIBM vs HCTRL. The q value mirrors the false discovery rate. The lower the q value, the higher the significance of the results across all samples (see Materials and Methods). Herein, we show transcripts that were expressed with a q value of ≤20%.
MHC, major histocompatibility complex; RER, retention in endoplasmic reticulum.
Among these 34 transcripts, 19 can be attributed to components of the classic and nonclassic HLA-I, HLA-II, IFN-γ–inducible genes and immunoproteasome components.
Two probe sets of STAT1, a mediator of IFN signaling, were regulated, with a q value of 0%, thus being highly significant across all patients. STAT1 was strongly expressed in AIBM and NIBM and undetectable in HCTRL. HLA-DPB1 was the only HLA-II transcript with a q value <20%. In contrast to HLA-I, HLA-DPB1 was up-regulated 2.66-fold in AIBM versus NIBM myofibers. PSMB8, a major constituent of the immunoproteasome, had a q value of 0% as well and was differentially regulated in AIBM versus NIBM (fold change = 2.86).
Several transcripts important for inflammatory signaling and antigen presentation were overexpressed in global microarrays from sIBM biopsy specimens by other researchers (see Supplemental Table S2 at http://ajp.amjpathol.org). Although only some of the transcripts had significantly low q values, our results allow us to attribute these regulation patterns to specific myofiber subsets. On the other hand, Ig genes were absent in our myofiber-specific microarray studies (see Supplemental Table S1 at http://ajp.amjpathol.org). This ensures that cells of the B lineage, and not myofibers, are the source of Igs in sIBM tissue.35, 36
TaqMan qPCR Confirms Global HLA-I Up-Regulation in sIBM Muscle Fibers
We analyzed aRNA from laser-microdissected AIBM, NIBM, and HCTRL myofibers using TaqMan qPCR. Relative expression values were calculated for all TaqMan results using the 2−ΔCT method.31, 32
We found that all members of the HLA-I family were up-regulated in both AIBM and NIBM compared with HCTRL myofibers (Figure 2). This agrees with our microarray data and with previous immunohistological studies21 and supports the overall validity of our experimental approach.
Figure 2.
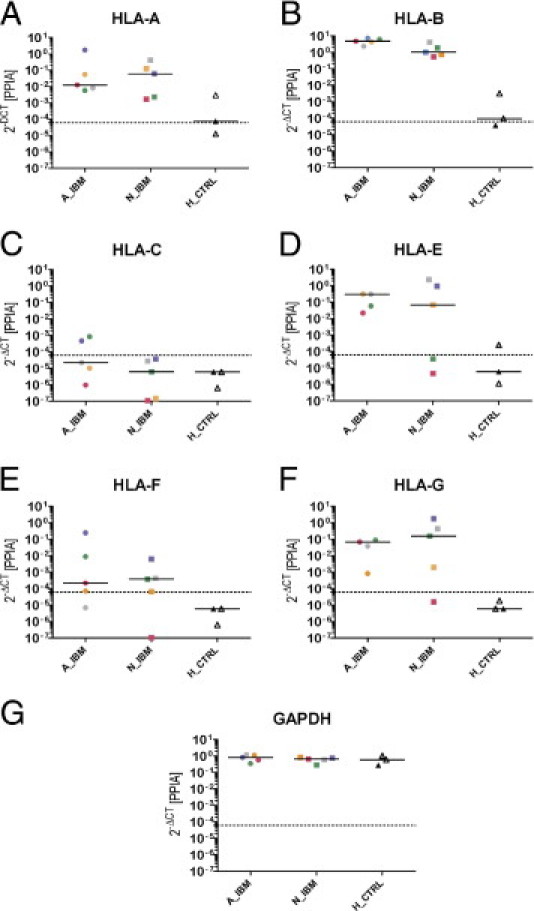
Comparison of transcript levels in attacked, nonattacked, and healthy control myofibers using TaqMan qPCR. Relative expression values were calculated using the 2−ΔCT method with cyclophilin as the endogenous control. Each symbol represents the mean of triplicate experiments performed on one sample. Five AIBM samples (circles), five NIBM samples (squares), and three HCTRL samples (triangles) were included in the study. Colors correspond to individual patients (IBM-1, pink; IBM-2, green; IBM-3, gray; IBM-4, blue; IBM-5, yellow) and controls (C-1, white; C-2, gray; C-3, black), and medians are shown for each subset. Dashed line, detection limit. The following are shown: HLA-A (A), HLA-B (B), HLA-C (C), HLA-E (D), HLA-F (E), HLA-G (F), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (G). Data for HLA-E and HLA-G from AIBM of patient 4 were out of range (>100) and are omitted. HLA-A and HLA-B were strongly up-regulated in both attacked and nonattacked sIBM myofibers. The nonclassic HLA (ie, HLA-E, HLA-F, and HLA-G) displayed a pattern similar to that of HLA-A and HLA-B. GAPDH was used as an additional endogenous control. As expected, its expression pattern largely mirrors that of cyclophilin in that it is unchanged in all samples.
Differential Regulation of IFN-γ Downstream Signaling Pathways
We found several IFN-γ–inducible molecules to be differentially up-regulated in AIBM versus NIBM and HCTRL myofibers. To see whether myofibers are susceptible to IFN-γ, we analyzed the expression of both chains of IFNGR and its major downstream mediator STAT1 in AIBM versus NIBM and HCTRL myofibers with TaqMan qPCR. Both IFNGR1 and IFNGR2 were overexpressed in AIBM. In NIBM and HCTRL myofibers, the expression of both IFNGR chains ranged below the detection limit in most cases (Figure 3, A and B). STAT1 was up-regulated in AIBM and below the detection limit in NIBM and HCTRL (Figure 3C). Two other major downstream effector proteins of IFN-γ, the immunoproteasome component PSMB837 and CIITA,38 were up-regulated in AIBM compared with NIBM and HCTRL (Figure 3, D and E). In one patient, the levels of all three downstream transcripts (ie, STAT1, PSMB8, and CIITA) were particularly high compared with the other patients. This reflects a certain interindividual variability that is to be expected in such a cohort.
Figure 3.
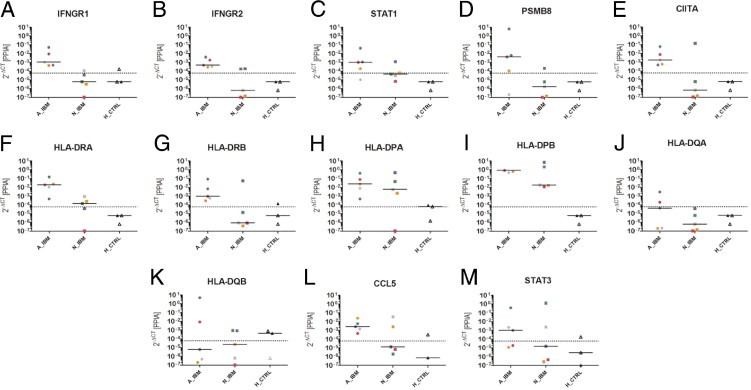
Comparison of transcript levels in attacked, nonattacked, and healthy control myofibers using TaqMan qPCR. Relative expression values were calculated using the 2−ΔCT method with cyclophilin as the endogenous control. The following are shown: IFNGR1 (A), IFNGR2 (B), STAT1 (C), PSMB8 (D), CIITA (E), HLA-DRA (F), HLA-DRB (G), HLA-DPA (H), HLA-DPB (I), HLA-DQA (J), HLA-DQB (K), CCL5 (L), and STAT3 (M). The legend to Figure 2 provides a detailed explanation of all symbols. IFNGR1, IFNGR2, STAT1, CIITA, and PSMB8, as well as HLA-DRA and HLA-DRB, were overexpressed in attacked myofibers compared with nonattacked myofibers and healthy controls. For HLA-DP, the difference between attacked and nonattacked myofibers was less pronounced. HLA-DQ was only detectable in two of five patients with sIBM.
Next, we investigated whether IFN-γ cascade up-regulation results in effective HLA-II overexpression in AIBM. We looked at the expression of the respective α and β chains of HLA-DR, HLA-DP, and HLA-DQ. HLA-DRA and HLA-DRB were up-regulated in AIBM compared with NIBM and HCTRL myofibers (Figure 3, F and G). In the case of HLA-DPA and HLA-DPB, the difference in overexpression between AIBM and NIBM was less striking (Figure 3, H and I). HLA-DQA and HLA-DQB showed the same tendency, albeit with lower expression levels (Figure 3, J and K).
Because tumor necrosis factor (TNF)-α induces chemokine ligand (CCL) 5 [regulated on activation normal T-cell expressed and secreted (RANTES)] via STAT3 in vascular smooth muscle cells and many other nonlymphoid cell types,39 we investigated the transcriptional activity of the CCL5-STAT3 system in our laser-microdissected myofiber subsets from patients with sIBM. Although RANTES and STAT3 levels were generally low, and the expression patterns were heterogeneous across all patients and controls, median values indicate a tendency for higher CCL5 expression in attacked myofibers compared with nonattacked myofibers and healthy controls (Figure 3, L and M).
Confocal Microscopy Confirms Differential IFNGR2 Expression in AIBM versus NIBM Myofibers
On binding of IFN-γ, IFNGR1 is internalized in a complex with STAT1, whereas IFNGR2, also known as the inducible chain of the IFN-γ receptor, remains on the cell surface.40 To evaluate IFNGR2 expression on the protein level, we double stained cryosections from five patients with sIBM and three controls for IFNGR2 and CD8. We found that a proportion of myofibers of patients with sIBM stained positive for IFNGR2. The IFNGR2 was not uniformly distributed in patients with sIBM. Some myofibers were positive for IFNGR2 along their entire surface. Other myofibers showed only partial segmental membrane staining, and yet others were entirely negative. This variability of IFNGR2 membrane staining was similar in all five patients with sIBM who were included in our study. By contrast, myofibers from healthy controls did not stain positive for IFNGR2 (Figure 4).
Figure 4.
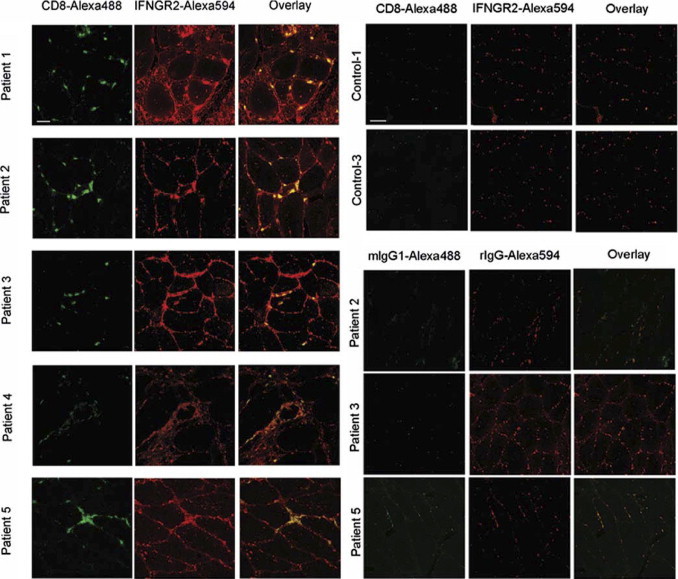
IFNGR2 expression on the protein level in patients with sIBM and healthy controls. IFNGR2 (Alexa 594 in red) and CD8 (Alexa 488 in green) were stained as described in Materials and Methods and visualized with confocal microscopy. Technical controls were performed with rat IgG (rIgG) for anti-IFNGR2 antibody and mouse Ig isotype 1 (mIgG1) for anti-CD8 antibody under identical experimental conditions. Left to right: Images are organized as follows: green channel (CD8 or respective control), red channel (IFNGR2 or respective control), and overlay.
We then asked whether the extent of IFNGR2 expression might be related to the extent of inflammatory changes around individual fibers. To this end, we classified myofibers into four groups according to the extent of IFNGR2 surface expression as follows: group 1, >80%; group 2, 50% to 80%; group 3, 20% to 50%; and group 4, <20% surface staining (Figure 5). Control sections stained in the same experiment were uniformly negative for IFNGR2 (Figure 4). Two independent observers (J.I. and Ingrid Eiglmeier) evaluated the numbers of attacking CD8+ T cells that were in direct contact with each myofiber for each classified myofiber (Figure 6). A Mann-Whitney U-test was performed to determine whether the distributions of adjacent CD8+ T-cell numbers per myofiber in groups 1 and 4 differed significantly in all analyzed patients (Figure 6). In patient IBM-1, the median CD8+ T-cell numbers were 5 and 1 (observer Ingrid Eiglmeier: Mann-Whitney U-test = 9.5, P < 0.0001) and 6 and 1 (observer J.I.: Mann-Whitney U-test = 5.5, P < 0.0001), respectively. In patient IBM-2, the median CD8+ T-cell numbers were 4 and 0 (observer I.E.: Mann-Whitney U-test = 0, P < 0.0278) and 5 and 0.5 (observer J.I.: Mann-Whitney U-test = 0, P < 0.0223), respectively. In patient IBM-3, the median CD8+ T-cell numbers were 5 and 1 (observer Ingrid Eiglmeier: Mann-Whitney U-test = 3, P < 0.0072) and 5.5 and 1 (observer J.I.: Mann-Whitney U-test = 0.5, P < 0.0011), respectively. In patient IBM-5, the median CD8+ T-cell numbers were 6 and 0 (observer IE: Mann-Whitney U-test = 13, P < 0.0001) and 7 and 0 (observer J.I.: Mann-Whitney U-test = 10.5, P < 0.0001), respectively. (All P values were two tailed.) The results reveal a positive correlation between the density of the focal inflammatory infiltrate and the extent of IFNGR2 surface expression, indicating that IFNGR2 is up-regulated on myofibers by locally produced pro-inflammatory cytokines.
Figure 5.
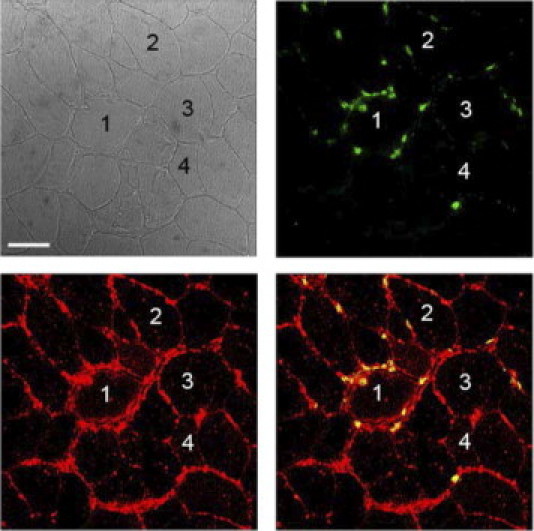
IHC demonstrates differential IFNGR2 expression in patients with sIBM. IFNGR2 (Alexa 594 in red) and CD8 (Alexa 488 in green) were stained as described in Materials and Methods and visualized with confocal microscopy. Although some myofibers were strongly positive for IFNGR2 along their entire surface, others were only segmentally positive or negative. The strongly positive myofibers were often heavily attacked by CD8+ T cells. Herein, representative examples from one patient are shown. Four groups were defined according to IFNGR2 membrane positivity: group 1, >80% of the myofiber membrane positive for INFGR2; group 2, 50% to 80% of the myofiber membrane positive for INFGR2; group 3, 20% to 50% of the myofiber membrane positive for INFGR2; and group 4, <20% of the myofiber membrane positive for INFGR2. Images are organized clockwise from top left as follows: transmission, green channel (CD8 or respective control), red channel (IFNGR2 or respective control), and overlay. Scale bar = 50 μm.
Figure 6.
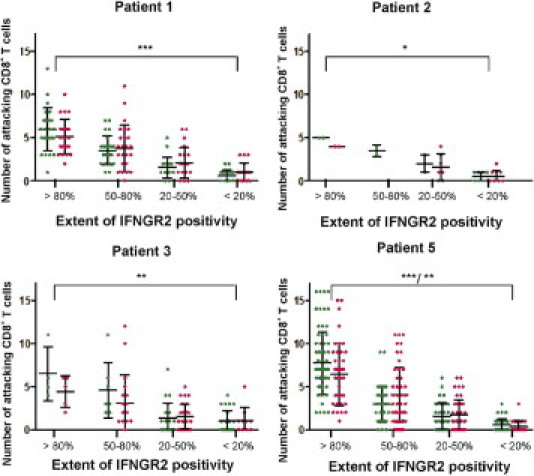
Relationship between extent of membrane positivity for IFNGR2 and number of adjacent CD8+ T cells. Each dot represents an individual myofiber. Each myofiber was assigned to a group according to the criteria discussed in Results and in Figure 5, and the number of adjacent CD8+ T cells was counted. In the previously described graphs, the number of adjacent CD8+ T cells was plotted against the respective group for each myofiber. Results from two independent observers (green and pink) are shown side by side. Mann-Whitney U-tests were performed to determine statistical significance (*P < 0.01, **P < 0.001, and ***P < 0.0001). Patient IBM-4 was not included because of lack of material for a systematic analysis.
Discussion
In sIBM, essentially all muscle fibers express HLA-I.6 By contrast, the attacking CD8+ T cells follow a strictly focal pattern. In the present study, we sought to identify differences between the following: AIBM, NIBM, and HCTRL. We focused on the molecular pathways of antigen presentation. We did not investigate PM or DM because PM is heterogeneous26 and DM lacks CD8+-mediated muscle fiber injury. We found high levels of HLA-I in both AIBM and NIBM myofibers. In contrast, the receptor for IFN-γ was overexpressed in AIBM compared with NIBM myofibers, indicating increased susceptibility of AIBM to IFN-γ downstream signaling.
Ubiquitous Up-Regulation of HLA-I
Most tissues constitutively express HLA-I molecules. Muscle is an exception in that HLA-I is normally undetectable but up-regulated in inflammatory myopathies.21, 24, 41 In sIBM, HLA-I expression is essentially ubiquitous on all myofibers. Consistent with this well-established histological feature, we observed up-regulation of classic and nonclassic HLA-I transcripts in both AIBM and NIBM (Figure 7). This confirms and extends previous observations that nonclassic HLA-G is up-regulated on muscle fibers in inflammatory myopathies.42 Because up-regulation of HLA-I on sIBM myofibers is independent of the presence of local inflammatory cells,6 and because it may occur in the absence of an IFN-γ signature (as shown by our present results from NIBM), it appears that HLA-I is induced by unknown triggers upstream of the IFN-γ–related changes. Other inflammatory mediators (eg, TNF-α) may precede and subsequently potentiate the IFN-γ cascade, as suggested by our findings on the up-regulation of CCL5-STAT3 in attacked myofibers, which is a TNF-α–inducible chemokine system.39 IFN-α is a further important inflammatory mediator implicated in the pathogenesis of inflammatory myopathies. In juvenile DM, IFN-α up-regulated HLA-I and several IFN-γ–inducible genes, possibly through up-regulation of IL1-α/β.43, 44 The interplay between IFN-α and IFN-γ demonstrated in juvenile DM may play a role in sIBM as well.
Figure 7.
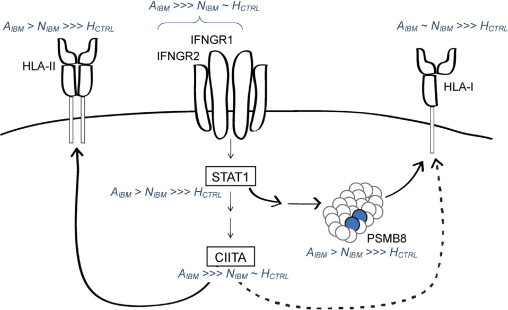
Scheme for changes in the molecular pattern of inflammatory signaling, antigen processing, and presentation in attacked myofibers. HLA-I molecules are up-regulated in all IBM myofibers. We show the pronounced effect of IFN-γ–induced CIITA on HLA-II by a continuous arrow and the relatively small effect of CIITA on HLA-I expression (Gobin et al, 1998) as a dotted arrow. However, the effect of CIITA cannot be held responsible for the initial IFN-γ–independent and probably ubiquitous HLA-I up-regulation. In AIBM, we found up-regulation of IFN-γ pathway components IFNGR1 and IFNGR2. The response to IFN-γ explains the up-regulation of IFN-γ–inducible genes, such as STAT1, CIITA, PSMB8, and HLA-II, in AIBM.
The nonhistone nuclear protein alarmin high-mobility group box 1 protein may be a further important upstream inducer of HLA-I in sIBM. High-mobility group box 1 protein up-regulation has been demonstrated in myofibers of patients with inflammatory myopathies early in the disease course. Moreover, high-mobility group box 1 protein up-regulated HLA-I on muscle cells in vitro.45
Another possible explanation for IFN-γ–independent HLA-I induction may be a viral infection, given the known association of inflammatory myopathies with human T-lymphotropic virus I and HIV.46, 47, 48 Moreover, viral infection induced HLA-I on cultured human myoblasts independently of IFN-γ.49 Although there is no current evidence that HLA-I can be induced by degenerative factors, the HLA-I up-regulation in sIBM might be related to the observed degenerative changes.50, 51, 52 Amyloid-β precursor protein and embryonic myosin variants showed reasonable q values between 20% and 40% (data not shown). These findings, which need to be extended and confirmed in future studies, would be consistent with the concept that inflammatory and degenerative changes in sIBM are mutually related.53, 54 An additional aspect not addressed in our study is the expression of αB-crystallin in so-called X fibers.55 Previous observations that X fibers are otherwise morphologically normal, and are not (yet?) attacked by inflammatory cells, suggested that αB-crystallin expression is induced in these fibers by an unknown proximal triggering event, perhaps infection with an unknown virus. It will be interesting to apply our microdissection approach to the investigation of X fibers in future studies.
The precise pathogenetic role of HLA-I overexpression remains uncertain. In animal models, overexpression of major histocompatibility complex I was associated with inflammatory and degenerative myopathic changes, possibly related to an endoplasmic reticulum stress response.56, 57, 58 Furthermore, HLA-I expression is definitively a prerequisite for CD8+ T-cell–mediated muscle fiber injury because CD8+ T cells recognize HLA-I–bound antigenic peptides. Previous studies59 have demonstrated HLA-I up-regulation in human myoblasts incubated by IFN-γ. The difference might be explained by the fact that mature myofibers differ from less differentiated myotubes and myoblasts. However, as supported by our findings, HLA-I is only a necessary, but by no means sufficient, precondition for a CD8+ T-cell–mediated attack, and HLA-I expression alone cannot explain the focal character of inflammation in sIBM.
Up-Regulation of IFN-γ Receptor in Attacked Myofibers
The crucial upstream protein required for initiation of downstream IFN-γ signaling is the IFN-γ receptor, which was expressed by AIBM but not NIBM or HCTRL myofibers by qPCR (Figure 7). When IFN-γ binds to the IFN-γ receptor, the complex of IFN-γ, IFNGR1, and STAT1 translocates into the nucleus while IFNGR2 remains on the cell surface.40, 60 In sIBM muscle, IFNGR2 protein expression, as demonstrated by immunofluorescence staining, was heterogeneous and related to the presence of inflammatory cells. Quantitative analysis of the relationship between inflammatory cells and segmental IFNGR2 expression revealed a positive correlation between the extent of IFNGR2 surface expression on myofibers and the number of adjacent CD8+ T cells in all patients, confirming our qPCR findings.
Up-Regulation of IFN-γ–Induced Transcripts
An important downstream component of the IFN-γ signaling cascade is STAT1 (Figure 7). A role for STAT1 overexpression has been demonstrated for DM and PM.61 Our qPCR results revealed that STAT1 is differentially overexpressed in AIBM myofibers, which is consistent with our observation that IFNGR2 is preferentially expressed in AIBM myofibers.
CIITA (Figure 7) is the most important transcription factor for the induction of HLA-II.62 To some extent, CIITA can induce HLA-I as well.63 CIITA is strongly regulated by IFN-γ, which can induce CIITA in nonlymphoid cells.38 We show, for the first time to our knowledge, an up-regulation of CIITA in CD8+ T-cell–attacked myofibers of patients with sIBM. Literature on the expression of HLA-II in sIBM is controversial. Global tissue profiling has demonstrated an increase of HLA-DP, HLA-DQ, and HLA-DR in sIBM; however, the cellular source remained uncertain.35 Immunohistochemical studies addressing the expression of HLA-II on myofibers in inflammatory myopathies are sparse. One study64 analyzed the expression of HLA-II in DM and PM and provided evidence that the HLA-DR expression is independent of inflammatory infiltrates. However, in sIBM, we found differential HLA-DR overexpression in CD8+ T-cell–attacked myofibers (Figure 7). This discrepancy might be related to differences between the different myopathies, with IBM being dominated by CD8+ T cells and DM by CD4+ T cells; PM is a heterogeneous disease. The pathogenic relevance of IFN-γ–induced HLA-II expression is unknown, probably initiated by TNF-α.65 However, (auto-) antigen presentation to CD4+ T cells is unlikely because the endomysial inflammatory infiltrate in sIBM consists mainly of CD8+ T cells.7
The IFN-γ–inducible catalytic immunoproteasome subunit PSMB8 (LMP-2) generates peptides with an increased affinity for HLA-I.35 Previous global microarray profiling studies35, 36 showed a strong overexpression of PSMB8 in inflammatory myopathies, without revealing the cellular source. Our microarray and qPCR data attribute up-regulation of PSMB8 to AIBM myofibers. This indicates an up-regulated IFN-γ signature in AIBM, consistent with our other findings.
It seems likely that additional IFN-γ–inducible transcripts are up-regulated in AIBM myofibers. qPCR showed up-regulation of the CCL5-STAT3 system in AIBM myofibers (Figure 3, L and M). CCL5, also known as RANTES, is synergistically up-regulated by TNF-α and IFN-γ66, 67 and would be expected to attract activated T cells,39, 68 thereby contributing to the local pro-inflammatory loop. Previous work69 has shown that TNF-α up-regulates the TSP1-CD36-CD47 complex in cultured myoblasts and that the TSP1-CD36-CD47 was up-regulated in sIBM. The multifold synergies between TNF-α and IFN-γ likely play an important role in sIBM pathophysiological features. As demonstrated herein, multiple transcripts that are known to be synergistically up-regulated by TNF-α and IFN-γ, such as the CCL5-STAT3 system and IFNGR1/2, are induced on attacked myofibers. Indeed, previous studies70, 71 have suggested that the myofibers themselves are capable of producing IFN-γ–inducible chemokines that could potentially direct activated T cells. Clearly, the role of chemokines needs to be further explored in future studies.
Comparison with Other Microarray Studies in Human Inflammatory Myopathies
Previous microarray studies35, 36 in IBM, PM, DM, and degenerative muscle diseases were global and did not distinguish between muscle-derived transcripts and genes overexpressed by leukocytes present in the biopsy specimen. Determining the source of transcripts overexpressed in inflammatory myopathies is all the more important given that the ability of muscle fibers to produce certain chemokines and other proteins under inflammatory conditions has repeatedly been demonstrated.70, 71, 72
Regarding IBM, previously reported global microarray studies have demonstrated a strong up-regulation of HLA-I, HLA-II, PSMB8, several IFN-induced genes, and certain cytokines, such as RANTES (CCL5) and monocyte induced gamma-interferon (MIG, CXCL9) (see Supplemental Table S2 at http://ajp.amjpathol.org). By comparison, our study allows changes of expression patterns, in particular the overexpression of IFN-γ–inducible transcripts, to be attributed, for the first time to our knowledge, to particular subgroups of fibers. On the other hand, our data attribute the previously described Ig overexpression to tissue-invading B cells (see Supplemental Table S1 at http://ajp.amjpathol.org).36, 73
Conclusion and Model
In conclusion, we found that the receptor for IFN-γ is selectively up-regulated in attacked IBM myofibers (Figure 7). This raises the crucial question of what comes first: focal inflammation or focal induction of antigen presentation and processing pathways in the attacked muscle fiber? Although we have not yet identified the initial trigger that leads to the up-regulation of HLA-I expression, we may delineate the sequence of later events.
Our findings suggest a scenario that helps explain the focal nature of the inflammatory attack in IBM muscle. First, ubiquitous HLA-I expression is induced on essentially all myofibers by unknown, possibly infectious or inflammatory, proximal triggers, thereby providing a necessary precondition for antigen recognition by CD8+ T cells. Second, a CD8+ T cell recognizes a surface-exposed HLA-I–bound antigen on a myofiber. This initial encounter with antigen occurs as a random event. However, once such a contact is established, a specific immunological synapse is formed, with vectorial orientation of perforin toward the area of contact.17 On antigen recognition and activation, the T cell starts to secrete pro-inflammatory cytokines and chemokines, thereby attracting and activating additional inflammatory cells, including macrophages, dendritic cells, and B cells. The cytokines produced by the inflammatory cells induce IFN-γ receptor expression on the attacked and perhaps adjacent muscle fibers, which thereby become susceptible to IFN-γ–mediated stimulation. IFN-γ–mediated signaling induces a cascade of secondary changes in the attacked fibers, increasing their susceptibility to inflammatory attack. In this way, an initial antigen recognition event by a CD8+ T cell on an HLA-I–positive myofiber would be amplified, leading to strong focal inflammatory changes.
Acknowledgments
We thank Joachim Malotka and Ingrid Eiglmeier for excellent technical assistance and Drs. Dieter Jenne and Naoto Kawakami for helpful discussion of the manuscript.
Footnotes
Supported by a scholarship from the Gerhard C. Starck Foundation, Düsseldorf, Germany (J.I.). H.L. is an investigator at the MRC Centre for Neuromuscular Diseases and at TREAT-NMD (EC, 6th FP, proposal 036825).
R.Hoh. and K.D. contributed equally to this work.
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi:10.1016/j.ajpath.2011.05.055.
Contributor Information
Reinhard Hohlfeld, Email: reinhard.hohlfeld@med.uni-muenchen.de.
Klaus Dornmair, Email: dornmair@neuro.mpg.de.
Supplementary data
Analysis of RNA quality under immunofluorescent staining conditions. Agilent Bioanalyzer gel electrophoresis was run according to the manufacturer's recommendations. RNA quality was assessed by evaluation of the delineation of the bands. As a positive control, we used unstained tissue (directly from −80°C) that shows clearly delineated 18S and 28S RNA bands (unstained). Under standard conditions (ie, after a traditional immunofluorescent staining and laser microdissection protocol that takes at least 1.5 hours at room temperature), RNA is rapidly degraded, severely compromising transcriptome analysis. In this study, a rapid staining protocol that largely preserves RNA was used (optimized). nt, nucleotide.
References
- 1.Dalakas M.C. Sporadic inclusion body myositis: diagnosis, pathogenesis and therapeutic strategies. Nat Clin Pract Neurol. 2006;2:437–447. doi: 10.1038/ncpneuro0261. [DOI] [PubMed] [Google Scholar]
- 2.Dalakas M.C. Immunotherapy of myositis: issues, concerns and future prospects. Nat Rev Rheumatol. 2010;6:129–137. doi: 10.1038/nrrheum.2010.2. [DOI] [PubMed] [Google Scholar]
- 3.Griggs R.C. The current status of treatment for inclusion-body myositis. Neurology. 2006;66:S30–S32. doi: 10.1212/01.wnl.0000192262.29924.9e. [DOI] [PubMed] [Google Scholar]
- 4.Wiendl H. Idiopathic inflammatory myopathies: current and future therapeutic options. Neurotherapeutics. 2008;5:548–557. doi: 10.1016/j.nurt.2008.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hilton-Jones D., Miller A., Parton M., Holton J., Sewry C., Hanna M.G. Inclusion body myositis: MRC Centre for Neuromuscular Diseases, IBM workshop, London, 13 June 2008. Neuromuscul Disord. 2010;20:142–147. doi: 10.1016/j.nmd.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 6.Dalakas M.C. Inflammatory muscle diseases: a critical review on pathogenesis and therapies. Curr Opin Pharmacol. 2010;10:346–352. doi: 10.1016/j.coph.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 7.Engel A.G., Arahata K. Monoclonal antibody analysis of mononuclear cells in myopathies, II: phenotypes of autoinvasive cells in polymyositis and inclusion body myositis. Ann Neurol. 1984;16:209–215. doi: 10.1002/ana.410160207. [DOI] [PubMed] [Google Scholar]
- 8.Stangel M., Mix E., Zettl U.K., Gold R. Oxides and apoptosis in inflammatory myopathies. Microsc Res Tech. 2001;55:249–258. doi: 10.1002/jemt.1174. [DOI] [PubMed] [Google Scholar]
- 9.Dalakas M.C. Inflammatory, immune, and viral aspects of inclusion-body myositis. Neurology. 2006;66:S33–S38. doi: 10.1212/01.wnl.0000192129.65677.87. [DOI] [PubMed] [Google Scholar]
- 10.Dustin M.L., Long E.O. Cytotoxic immunological synapses. Immunol Rev. 2010;235:24–34. doi: 10.1111/j.0105-2896.2010.00904.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lindberg C., Oldfors A., Tarkowski A. Restricted use of T cell receptor V genes in endomysial infiltrates of patients with inflammatory myopathies. Eur J Immunol. 1994;24:2659–2663. doi: 10.1002/eji.1830241114. [DOI] [PubMed] [Google Scholar]
- 12.Amemiya K., Granger R.P., Dalakas M.C. Clonal restriction of T-cell receptor expression by infiltrating lymphocytes in inclusion body myositis persists over time: studies in repeated muscle biopsies. Brain. 2000;123(Pt 10):2030–2039. doi: 10.1093/brain/123.10.2030. [DOI] [PubMed] [Google Scholar]
- 13.Bender A., Behrens L., Engel A.G., Hohlfeld R. T-cell heterogeneity in muscle lesions of inclusion body myositis. J Neuroimmunol. 1998;84:86–91. doi: 10.1016/s0165-5728(97)00246-4. [DOI] [PubMed] [Google Scholar]
- 14.Seitz S., Schneider C.K., Malotka J., Nong X., Engel A.G., Wekerle H., Hohlfeld R., Dornmair K. Reconstitution of paired T cell receptor alpha- and beta-chains from microdissected single cells of human inflammatory tissues. Proc Natl Acad Sci U S A. 2006;103:12057–12062. doi: 10.1073/pnas.0604247103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hofbauer M., Wiesener S., Babbe H., Roers A., Wekerle H., Dornmair K., Hohlfeld R., Goebels N. Clonal tracking of autoaggressive T cells in polymyositis by combining laser microdissection, single-cell PCR, and CDR3-spectratype analysis. Proc Natl Acad Sci U S A. 2003;100:4090–4095. doi: 10.1073/pnas.0236183100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dimitri D., Benveniste O., Dubourg O., Maisonobe T., Eymard B., Amoura Z., Jean L., Tiev K., Piette J.C., Klatzmann D., Herson S., Boyer O. Shared blood and muscle CD8+ T-cell expansions in inclusion body myositis. Brain. 2006;129:986–995. doi: 10.1093/brain/awl020. [DOI] [PubMed] [Google Scholar]
- 17.Goebels N., Michaelis D., Engelhardt M., Huber S., Bender A., Pongratz D., Johnson M.A., Wekerle H., Tschopp J., Jenne D., Hohlfeld R. Differential expression of perforin in muscle-infiltrating T cells in polymyositis and dermatomyositis. J Clin Invest. 1996;97:2905–2910. doi: 10.1172/JCI118749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hohlfeld R., Engel A.G. Lysis of myotubes by alloreactive cytotoxic T cells and natural killer cells: relevance to myoblast transplantation. J Clin Invest. 1990;86:370–374. doi: 10.1172/JCI114711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hohlfeld R., Engel A.G. Coculture with autologous myotubes of cytotoxic T cells isolated from muscle in inflammatory myopathies. Ann Neurol. 1991;29:498–507. doi: 10.1002/ana.410290509. [DOI] [PubMed] [Google Scholar]
- 20.McDouall R.M., Dunn M.J., Dubowitz V. Expression of class I and class II MHC antigens in neuromuscular diseases. J Neurol Sci. 1989;89:213–226. doi: 10.1016/0022-510x(89)90023-3. [DOI] [PubMed] [Google Scholar]
- 21.Karpati G., Pouliot Y., Carpenter S. Expression of immunoreactive major histocompatibility complex products in human skeletal muscles. Ann Neurol. 1988;23:64–72. doi: 10.1002/ana.410230111. [DOI] [PubMed] [Google Scholar]
- 22.Bartoccioni E., Gallucci S., Scuderi F., Ricci E., Servidei S., Broccolini A., Tonali P. MHC class I, MHC class II and intercellular adhesion molecule-1 (ICAM-1) expression in inflammatory myopathies. Clin Exp Immunol. 1994;95:166–172. doi: 10.1111/j.1365-2249.1994.tb06032.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jain A., Sharma M.C., Sarkar C., Bhatia R., Singh S., Handa R. Major histocompatibility complex class I and II detection as a diagnostic tool in idiopathic inflammatory myopathies. Arch Pathol Lab Med. 2007;131:1070–1076. doi: 10.5858/2007-131-1070-MHCCIA. [DOI] [PubMed] [Google Scholar]
- 24.Emslie-Smith A.M., Arahata K., Engel A.G. Major histocompatibility complex class I antigen expression, immunolocalization of interferon subtypes, and T cell-mediated cytotoxicity in myopathies. Hum Pathol. 1989;20:224–231. doi: 10.1016/0046-8177(89)90128-7. [DOI] [PubMed] [Google Scholar]
- 25.Arahata K., Engel A.G. Monoclonal antibody analysis of mononuclear cells in myopathies, I: quantitation of subsets according to diagnosis and sites of accumulation and demonstration and counts of muscle fibers invaded by T cells. Ann Neurol. 1984;16:193–208. doi: 10.1002/ana.410160206. [DOI] [PubMed] [Google Scholar]
- 26.Chahin N., Engel A.G. Correlation of muscle biopsy, clinical course, and outcome in PM and sporadic IBM. Neurology. 2008;70:418–424. doi: 10.1212/01.wnl.0000277527.69388.fe. [DOI] [PubMed] [Google Scholar]
- 27.Lynoe N., Sandlund M., Dahlqvist G., Jacobsson L. Informed consent: study of quality of information given to participants in a clinical trial. BMJ. 1991;303:610–613. doi: 10.1136/bmj.303.6803.610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dalakas M.C. Clinical, immunopathologic, and therapeutic considerations of inflammatory myopathies. Clin Neuropharmacol. 1992;15:327–351. doi: 10.1097/00002826-199210000-00001. [DOI] [PubMed] [Google Scholar]
- 29.Junker A., Ivanidze J., Malotka J., Eiglmeier I., Lassmann H., Wekerle H., Meinl E., Hohlfeld R., Dornmair K. Multiple sclerosis: T-cell receptor expression in distinct brain regions. Brain. 2007;130:2789–2799. doi: 10.1093/brain/awm214. [DOI] [PubMed] [Google Scholar]
- 30.Storey J.D., Tibshirani R. Statistical methods for identifying differentially expressed genes in DNA microarrays. Methods Mol Biol. 2003;224:149–157. doi: 10.1385/1-59259-364-X:149. [DOI] [PubMed] [Google Scholar]
- 31.Schmittgen T.D., Livak K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 32.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 33.Wang Y., Liu D., Chen P., Koeffler H.P., Tong X., Xie D. Negative feedback regulation of IFN-gamma pathway by IFN regulatory factor 2 in esophageal cancers. Cancer Res. 2008;68:1136–1143. doi: 10.1158/0008-5472.CAN-07-5021. [DOI] [PubMed] [Google Scholar]
- 34.Butticè G., Miller J., Wang L., Smith B.D. Interferon-gamma induces major histocompatibility class II transactivator (CIITA), which mediates collagen repression and major histocompatibility class II activation by human aortic smooth muscle cells. Circ Res. 2006;98:472–479. doi: 10.1161/01.RES.0000204725.46332.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Greenberg S.A., Sanoudou D., Haslett J.N., Kohane I.S., Kunkel L.M., Beggs A.H., Amato A.A. Molecular profiles of inflammatory myopathies. Neurology. 2002;59:1170–1182. doi: 10.1212/wnl.59.8.1170. [DOI] [PubMed] [Google Scholar]
- 36.Greenberg S.A., Bradshaw E.M., Pinkus J.L., Pinkus G.S., Burleson T., Due B., Bregoli L., O'Connor K.C., Amato A.A. Plasma cells in muscle in inclusion body myositis and polymyositis. Neurology. 2005;65:1782–1787. doi: 10.1212/01.wnl.0000187124.92826.20. [DOI] [PubMed] [Google Scholar]
- 37.Kloetzel P.M. Antigen processing by the proteasome. Nat Rev Mol Cell Biol. 2001;2:179–187. doi: 10.1038/35056572. [DOI] [PubMed] [Google Scholar]
- 38.van den Elsen P.J., Holling T.M., Kuipers H.F., Van der Stoep N. Transcriptional regulation of antigen presentation. Curr Opin Immunol. 2004;16:67–75. doi: 10.1016/j.coi.2003.11.015. [DOI] [PubMed] [Google Scholar]
- 39.Kovacic J.C., Gupta R., Lee A.C., Ma M., Fang F., Tolbert C.N., Walts A.D., Beltran L.E., San H., Chen G., St Hilaire C., Boehm M. Stat3-dependent acute Rantes production in vascular smooth muscle cells modulates inflammation following arterial injury in mice. J Clin Invest. 2010;120:303–314. doi: 10.1172/JCI40364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Larkin J., III, Johnson H.M., Subramaniam P.S. Differential nuclear localization of the IFNGR-1 and IFNGR-2 subunits of the IFN-gamma receptor complex following activation by IFN-gamma. J Interferon Cytokine Res. 2000;20:565–576. doi: 10.1089/10799900050044769. [DOI] [PubMed] [Google Scholar]
- 41.van der Pas J., Hengstman G.J., ter Laak H.J., Borm G.F., van Engelen B.G. Diagnostic value of MHC class I staining in idiopathic inflammatory myopathies. J Neurol Neurosurg Psychiatry. 2004;75:136–139. [PMC free article] [PubMed] [Google Scholar]
- 42.Wiendl H., Behrens L., Maier S., Johnson M.A., Weiss E.H., Hohlfeld R. Muscle fibers in inflammatory myopathies and cultured myoblasts express the nonclassical major histocompatibility antigen HLA-G. Ann Neurol. 2000;48:679–684. [PubMed] [Google Scholar]
- 43.Tezak Z., Hoffman E.P., Lutz J.L., Fedczyna T.O., Stephan D., Bremer E.G., Krasnoselska-Riz I., Kumar A., Pachman L.M. Gene expression profiling in DQA1*0501+ children with untreated dermatomyositis: a novel model of pathogenesis. J Immunol. 2002;168:4154–4163. doi: 10.4049/jimmunol.168.8.4154. [DOI] [PubMed] [Google Scholar]
- 44.Feldman B.M., Rider L.G., Reed A.M., Pachman L.M. Juvenile dermatomyositis and other idiopathic inflammatory myopathies of childhood. Lancet. 2008;371:2201–2212. doi: 10.1016/S0140-6736(08)60955-1. [DOI] [PubMed] [Google Scholar]
- 45.Grundtman C., Bruton J., Yamada T., Ostberg T., Pisetsky D.S., Harris H.E., Andersson U., Lundberg I.E., Westerblad H. Effects of HMGB1 on in vitro responses of isolated muscle fibers and functional aspects in skeletal muscles of idiopathic inflammatory myopathies. FASEB J. 2010;24:570–578. doi: 10.1096/fj.09-144782. [DOI] [PubMed] [Google Scholar]
- 46.Illa I., Nath A., Dalakas M. Immunocytochemical and virological characteristics of HIV-associated inflammatory myopathies: similarities with seronegative polymyositis. Ann Neurol. 1991;29:474–481. doi: 10.1002/ana.410290505. [DOI] [PubMed] [Google Scholar]
- 47.Leon-Monzon M., Illa I., Dalakas M.C. Polymyositis in patients infected with human T-cell leukemia virus type I: the role of the virus in the cause of the disease. Ann Neurol. 1994;36:643–649. doi: 10.1002/ana.410360414. [DOI] [PubMed] [Google Scholar]
- 48.Cupler E.J., Leon-Monzon M., Miller J., Semino-Mora C., Anderson T.L., Dalakas M.C. Inclusion body myositis in HIV-1 and HTLV-1 infected patients. Brain. 1996;119(Pt 6):1887–1893. doi: 10.1093/brain/119.6.1887. [DOI] [PubMed] [Google Scholar]
- 49.Bao S., King N.J., Dos Remedios C.G. Flavivirus induces MHC antigen on human myoblasts: a model of autoimmune myositis? Muscle Nerve. 1992;15:1271–1277. doi: 10.1002/mus.880151109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Askanas V., Engel W.K. Sporadic inclusion-body myositis: a proposed key pathogenetic role of the abnormalities of the ubiquitin-proteasome system, and protein misfolding and aggregation. Acta Myol. 2005;24:17–24. [PubMed] [Google Scholar]
- 51.Askanas V., Engel W.K. Inclusion-body myositis: muscle-fiber molecular pathology and possible pathogenic significance of its similarity to Alzheimer's and Parkinson's disease brains. Acta Neuropathol. 2008;116:583–595. doi: 10.1007/s00401-008-0449-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vattemi G., Nogalska A., King E.W., D'Agostino C., Checler F., Askanas V. Amyloid-beta42 is preferentially accumulated in muscle fibers of patients with sporadic inclusion-body myositis. Acta Neuropathol. 2009;117:569–574. doi: 10.1007/s00401-009-0511-6. [DOI] [PubMed] [Google Scholar]
- 53.Karpati G., O'Ferrall E.K. Sporadic inclusion body myositis: pathogenic considerations. Ann Neurol. 2009;65:7–11. doi: 10.1002/ana.21622. [DOI] [PubMed] [Google Scholar]
- 54.Schmidt J., Barthel K., Wrede A., Salajegheh M., Bahr M., Dalakas M.C. Interrelation of inflammation and APP in sIBM: iL-1 beta induces accumulation of beta-amyloid in skeletal muscle. Brain. 2008;131:1228–1240. doi: 10.1093/brain/awn053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Banwell B.L., Engel A.G. AlphaB-crystallin immunolocalization yields new insights into inclusion body myositis. Neurology. 2000;54:1033–1041. doi: 10.1212/wnl.54.5.1033. [DOI] [PubMed] [Google Scholar]
- 56.Nagaraju K., Raben N., Loeffler L., Parker T., Rochon P.J., Lee E., Danning C., Wada R., Thompson C., Bahtiyar G., Craft J., Hooft Van H.R., Plotz P. Conditional up-regulation of MHC class I in skeletal muscle leads to self-sustaining autoimmune myositis and myositis-specific autoantibodies. Proc Natl Acad Sci U S A. 2000;97:9209–9214. doi: 10.1073/pnas.97.16.9209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nagaraju K., Casciola-Rosen L., Lundberg I., Rawat R., Cutting S., Thapliyal R., Chang J., Dwivedi S., Mitsak M., Chen Y.W., Plotz P., Rosen A., Hoffman E., Raben N. Activation of the endoplasmic reticulum stress response in autoimmune myositis: potential role in muscle fiber damage and dysfunction. Arthritis Rheum. 2005;52:1824–1835. doi: 10.1002/art.21103. [DOI] [PubMed] [Google Scholar]
- 58.Li C.K., Knopp P., Moncrieffe H., Singh B., Shah S., Nagaraju K., Varsani H., Gao B., Wedderburn L.R. Overexpression of MHC class I heavy chain protein in young skeletal muscle leads to severe myositis: implications for juvenile myositis. Am J Pathol. 2009;175:1030–1040. doi: 10.2353/ajpath.2009.090196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hohlfeld R., Engel A.G. HLA expression in myoblasts. Neurology. 1991;41:2015. doi: 10.1212/wnl.41.12.2015-c. [DOI] [PubMed] [Google Scholar]
- 60.Ahmed C.M., Johnson H.M. IFN-gamma and its receptor subunit IFNGR1 are recruited to the IFN-gamma-activated sequence element at the promoter site of IFN-gamma-activated genes: evidence of transactivational activity in IFNGR1. J Immunol. 2006;177:315–321. doi: 10.4049/jimmunol.177.1.315. [DOI] [PubMed] [Google Scholar]
- 61.Illa I., Gallardo E., Gimeno R., Serrano C., Ferrer I., Juarez C. Signal transducer and activator of transcription 1 in human muscle: implications in inflammatory myopathies. Am J Pathol. 1997;151:81–88. [PMC free article] [PubMed] [Google Scholar]
- 62.Leibundgut-Landmann S., Waldburger J.M., Krawczyk M., Otten L.A., Suter T., Fontana A., Acha-Orbea H., Reith W. Mini-review: specificity and expression of CIITA, the master regulator of MHC class II genes. Eur J Immunol. 2004;34:1513–1525. doi: 10.1002/eji.200424964. [DOI] [PubMed] [Google Scholar]
- 63.Gobin S.J., Peijnenburg A., van Eggermond M., van Zutphen M., van den Berg R., van den Elsen P.J. The RFX complex is crucial for the constitutive and CIITA-mediated transactivation of MHC class I and beta2-microglobulin genes. Immunity. 1998;9:531–541. doi: 10.1016/s1074-7613(00)80636-6. [DOI] [PubMed] [Google Scholar]
- 64.Englund P., Lindroos E., Nennesmo I., Klareskog L., Lundberg I.E. Skeletal muscle fibers express major histocompatibility complex class II antigens independently of inflammatory infiltrates in inflammatory myopathies. Am J Pathol. 2001;159:1263–1273. doi: 10.1016/S0002-9440(10)62513-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Keller C.W., Fokken C., Turville S.G., Lünemann A., Schmidt J., Münz C., Lunemann J.D. TNF-alpha induces macroautophagy and regulates MHC class II expression in human skeletal muscle cells. J Biol Chem. 2011;286:3970–3980. doi: 10.1074/jbc.M110.159392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lee A.H., Hong J.H., Seo Y.S. Tumour necrosis factor-alpha and interferon-gamma synergistically activate the RANTES promoter through nuclear factor kappaB and interferon regulatory factor 1 (IRF-1) transcription factors. Biochem J. 2000;350(Pt 1):131–138. [PMC free article] [PubMed] [Google Scholar]
- 67.Hiroi M., Ohmori Y. The transcriptional coactivator CREB-binding protein cooperates with STAT1 and NF-kappa B for synergistic transcriptional activation of the CXC ligand 9/monokine induced by interferon-gamma gene. J Biol Chem. 2003;278:651–660. doi: 10.1074/jbc.M204544200. [DOI] [PubMed] [Google Scholar]
- 68.Flier J., Boorsma D.M., van Beek P.J., Nieboer C., Stoof T.J., Willemze R., Tensen C.P. Differential expression of CXCR3 targeting chemokines CXCL10, CXCL9, and CXCL11 in different types of skin inflammation. J Pathol. 2001;194:398–405. doi: 10.1002/1096-9896(200108)194:4<397::aid-path899>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 69.Salajegheh M., Raju R., Schmidt J., Dalakas M.C. Upregulation of thrombospondin-1(TSP-1) and its binding partners, CD36 and CD47, in sporadic inclusion body myositis. J Neuroimmunol. 2007;187:166–174. doi: 10.1016/j.jneuroim.2007.04.022. [DOI] [PubMed] [Google Scholar]
- 70.Raju R., Vasconcelos O., Granger R., Dalakas M.C. Expression of IFN-gamma-inducible chemokines in inclusion body myositis. J Neuroimmunol. 2003;141:125–131. doi: 10.1016/s0165-5728(03)00218-2. [DOI] [PubMed] [Google Scholar]
- 71.Tateyama M., Fujihara K., Misu T., Itoyama Y. CCR7+ myeloid dendritic cells together with CCR7+ T cells and CCR7+ macrophages invade CCL19+ nonnecrotic muscle fibers in inclusion body myositis. J Neurol Sci. 2009;279:47–52. doi: 10.1016/j.jns.2008.12.024. [DOI] [PubMed] [Google Scholar]
- 72.Hohlfeld R., Goebels N., Engel A.G. Cellular mechanisms in inflammatory myopathies. Baillieres Clin Neurol. 1993;2:617–635. [PubMed] [Google Scholar]
- 73.Bradshaw E.M., Orihuela A., McArdel S.L., Salajegheh M., Amato A.A., Hafler D.A., Greenberg S.A., O'Connor K.C. A local antigen-driven humoral response is present in the inflammatory myopathies. J Immunol. 2007;178:547–556. doi: 10.4049/jimmunol.178.1.547. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Analysis of RNA quality under immunofluorescent staining conditions. Agilent Bioanalyzer gel electrophoresis was run according to the manufacturer's recommendations. RNA quality was assessed by evaluation of the delineation of the bands. As a positive control, we used unstained tissue (directly from −80°C) that shows clearly delineated 18S and 28S RNA bands (unstained). Under standard conditions (ie, after a traditional immunofluorescent staining and laser microdissection protocol that takes at least 1.5 hours at room temperature), RNA is rapidly degraded, severely compromising transcriptome analysis. In this study, a rapid staining protocol that largely preserves RNA was used (optimized). nt, nucleotide.