

Abstract
Fatty acyl-coenzyme A oxidase 1 (ACOX1) knockout (ACOX1−/−) mice manifest hepatic metabolic derangements that lead to the development of steatohepatitis, hepatocellular regeneration, spontaneous peroxisome proliferation, and hepatocellular carcinomas. Deficiency of ACOX1 results in unmetabolized substrates of this enzyme that function as biological ligands for peroxisome proliferator-activated receptor–α (PPARα) in liver. Here we demonstrate that sustained activation of PPARα in ACOX1−/− mouse liver by these ACOX1 substrates results in endoplasmic reticulum (ER) stress. Overexpression of transcriptional regulator p8 and its ER stress–related effectors such as the pseudokinase tribbles homolog 3, activating transcription factor 4, and transcription factor CCAAT/-enhancer-binding protein homologous protein as well as phosphorylation of eukaryotic translation initiation factor 2α, indicate the induction of unfolded protein response signaling in the ACOX1−/− mouse liver. We also show here that, in the liver, p8 is a target for all three PPAR isoforms (-α, -β, and -γ), which interact with peroxisome proliferator response elements in p8 promoter. Sustained activation of p8 and unfolded protein response–associated ER stress in ACOX1−/− mouse liver contributes to hepatocyte apoptosis and liver cell proliferation culminating in the development of hepatocarcinogenesis. We also demonstrate that human ACOX1 transgene is functional in ACOX1−/− mice and effectively prevents metabolic dysfunctions that lead to ER stress and carcinogenic effects. Taken together, our data indicate that progressive PPARα- and p8-mediated ER stress contribute to the hepatocarcinogenesis in ACOX1−/− mice.
Fatty liver disease is a burgeoning chronic liver disorder, commencing with hepatic steatosis and steatohepatitis, that has a propensity to progress toward cirrhosis and liver cancer.1–3 The pathological spectrum of fatty liver disease, caused by either alcoholic or nonalcoholic conditions, suggests a possible convergence, at some critical juncture, of pathogenetic mechanisms that enable the progression of steatosis toward steatohepatitis.3–5 This transition could contribute to events responsible for liver cell death, hepatocellular proliferation, and stellate cell activation, all of which are critical in the progression to cirrhosis and liver cancer.3,6 Increasingly, mice with genetic ablations of selected pathways in hepatic lipid metabolism and of other signal transduction networks are serving as important models to investigate the pathogenetic mechanism of fatty liver disease.7–13 For example, disruption of peroxisomal fatty acyl-CoA oxidase 1 (ACOX1) gene in the mouse has been shown to result in the development of steatohepatitis and increased expression of genes regulated by peroxisome proliferator-activated receptor α (PPARα).8,10 ACOX1 is the first and a rate-limiting enzyme of the PPARα-regulated and peroxisome proliferator–inducible fatty acid β–oxidation system.11–14 Mice deficient in ACOX1 exhibit growth retardation, high levels of very long chain fatty acids, microvesicular steatohepatitis, apoptosis, liver regeneration, and oxidative stress, and these mice eventually develop hepatocellular carcinomas.8,10,15,16
We hypothesized that the unmetabolized substrates of ACOX1, mainly fatty acids and their derivatives, function as endogenous ligands for PPARα to induce sustained peroxisome proliferation and elevation of PPARα target gene expression, contributing to the development of liver cancer.8,10,14–17 Steatohepatitis, increased levels of hepatic hydrogen peroxide content, and increased hepatocellular apoptosis and proliferation resulting from disturbances in fatty acid oxidation in the ACOX1−/− mouse,10 suggested a potential role for endoplasmic reticulum (ER) stress in this model, as excess lipid accumulation in tissues is linked to the development of ER stress.9,18–22 In this study, we first demonstrate that ACOX1-deficient livers exhibit progressive ER stress with activation of unfolded protein response signaling, with increases in the levels of ER stress response markers such as phosphorylated eukaryotic translation initiation factor 2α (p-eIF2α), activating transcription factor (ATF) 4, glucose regulated protein 78 (GRP78), C/EBP homologous protein (CHOP/CHOP10), GADD45α, pseudokinase tribbles homolog 3 (TRB3), Bax, BclII, and Nrf2.18–29 Furthermore, we provide evidence to suggest that ER stress is induced by hyperactivation of PPARα, which in turn upregulates the expression of stress-modulated transcription factor p8 [also known as nuclear protein 1 (NUPR1)]26–29 and its ER stress–related downstream targets ATF4, CHOP, and TRB3. Thus, sustained PPARα activation and its induction of p8 expression in the liver by the unmetabolized substrates of ACOX1 in mice nullizygous for ACOX1, contributes to ER stress and may contribute to the development of hepatocellular carcinoma. The data presented here also indicate that transgenic expression of human ACOX1 in ACOX1−/− mice prevents the development of metabolic dysfunctions including steatohepatitis spontaneous activation of PPARα, steatohepatitis, liver cancer, and unfolded protein response signaling. These observations indicate that human ACOX1 gene functionally replaces mouse ACOX1 gene and that this enzyme keeps the PPARα endogenous ligands in check and prevents sustained activation of this transcription factor.
Materials and Methods
ACOX1−/− Mice and Generation of ACOX1−/− with Human ACOX1 Transgene (ACOX1−/−h+ Humanized Mice)
The human bacterial artificial chromosome (BAC) clone CTD2336I6, containing the human ACOX1 genomic DNA and its promoter, was purchased from Invitrogen Corporation (Carlsbad, CA). This BAC DNA was trimmed to ∼90 kb using homologous recombination in bacteria to excise flanking genes and then replaced with an ampicilin resistance gene using primers shown in Table 1. BAC transgenic mice were generated by zygotic pronuclear microinjection in the Transgenic and Targeted Mutagenesis Laboratory, Northwestern University (Chicago, IL). Human BAC transgenic founders were then backcrossed with ACOX1+/− mice8,10 to generate ACOX1+/− mice containing human ACOX1 transgene (ACOX1+/−h+). The ACOX1+/−h+ mice were then mated with ACOX1+/− mice to generate ACOX1−/− humanized mice (ACOX1−/−h+), and ACOX1−/− and wild-type littermates. Mice used in this study were housed using 12-hour light, 12-hour dark cycle, in a pathogen-free animal facility and maintained on standard rodent chow (Harlan-Teklad, Indianapolis, IN) and water ad libitum. The PPARα synthetic ligand Wy-14,643 [4-chloro-6-(2,3-xylidino)−2-pyrimidinylthio]acetic acid] (0.125% w/w) was administered in chow.10,30,31 For cell proliferation analysis, mice were administered bromodeoxyuridine (BrdUrd) in drinking water (0.5 mg/mL) for 4 days and their livers processed for immunohistochemical localization of BrdUrd.32,33 All animal procedures used in this study were reviewed and preapproved by the Northwestern University Institutional Animal Care and Use Committee.
Table 1.
Oligonucleotide Sequences of Trimming BAC Clone, Genotyping, Real-Time PCR Primers, and Northern Probes
| Trimming BAC clone | 5′-GGGTGCAAATTGCCCGGTGCCTTCTGTTTCCCAGGCAGCTCTGTG-3′ |
| 5′-CAGCTTACCTCTCAGGAATGCTACGTTTTGAACATCAAGAATGGAAA-3′ | |
| ACOX-amp | |
| Sense | 5′-GCCATGGATATGTTCCAGAAGGTAGCTTGGTCTGACAGTTACCAATGC-3′ |
| Antisense | 5′-GCTGCCATTGAGGCTTTTAACAAAGGTGGCACTTTTC-3′ |
| Genotyping | |
| Neo | |
| Sense | 5′-TATTCGGCTATGACTGGGCACA-3′ |
| Antisense | 5′-GATGGATACTTTCTCGGCAGGA-3′ |
| mACOX1 exon | |
| Sense | 5′-CCGCAAGCCATCCGACATTC-3′ |
| Antisense | 5′-ATTCAGTGGGTCAGGCGACTGC-3′ |
| hACOX1BAC | |
| Sense | 5′-ATTGCCCGGTGCCTTCTGTTTC-3′ |
| Antisense | 5′-AGCCGGTGAGCGTGGGTCTC-3′ |
| qPCR/Probe | |
| hACOX1 | |
| Forward | 5′-TCTGTCTGGGCCGCTGTCACTC-3′ |
| Reverse | 5′-CCTAGGAGGCAGCCTCAGGACG-3′ |
| mACOX1 | |
| Forward | 5′-GCCAAGGCGACCTGAGTGAGC-3′ |
| Reverse | 5′-ACCGCAAGCCATCCGACATTC-3′ |
| ATF6 | |
| Forward | 5′-CAGTTGCTCCATCTCCTCTCC-3′ |
| Reverse | 5′-TGGGACACTGGCATTGGTTTG-3′ |
| XBP1 | |
| Forward | 5′-CCTGAGCCCGGAGGAGAA-3′ |
| Reverse | 5′-CTCGAGCAGTCTGCGCTG-3′ |
| XBP1s | |
| Forward | 5′-ACACGCTTGGGAATGGACAC-3′ |
| Reverse | 5′-CCATGGGAAGATGTTCTGGG-3′ |
| ATF4 | |
| Forward | 5′-ACTATCTGGAGGTGGCCAAG-3′ |
| Reverse | 5′-CATCCAACGTGGTCAAGAGC-3′ |
| GRP78 | |
| Forward | 5′-CGTGGAGATCATAGCCAACG-3′ |
| Reverse | 5′-ATACGCCTCAGCAGTCTCCT-3′ |
| Trb3 | |
| Forward | 5′-CCCACAGGCACAGAGTACAC-3′ |
| Reverse | 5′-CGTCCTCTCACAGTTGCTGA-3′ |
| CHOP | |
| Forward | 5′-AGCCTGGTATGAGGATCTGC-3′ |
| Reverse | 5′-CTCCTGCTCCTTCTCCTTCA-3′ |
| Gadd45α | |
| Forward | 5′-CCAAGCTGCTCAACGTAGA-3′ |
| Reverse | 5′-CCACTGATCCATGTAGCGAC-3′ |
| P8 | |
| Forward | 5′-ACCAAGAGAGAAGCTGCTGC-3′ |
| Reverse | 5′-CTCCCTCTCCAGAACCTCACT-3′ |
| PPARγ | |
| Forward | 5′-CCACAGTTGATTTCTCCAGCATTTC-3′ |
| Reverse | 5′-CAGGTTCTACTTTGATCGCACTTTG-3′ |
| aP2 | |
| Forward | 5′-GAAGTGGGAGTGGGCTTTGC-3′ |
| Reverse | 5′-TGTGGTCGACTTTCCATCCC-3′ |
| Bax | |
| Forward | 5′-ACCAAGAAGCTGAGCGAGTG-3′ |
| Reverse | 5′-CTCACGGAGGAAGTCCAGTG-3′ |
| Bcl2 | |
| Forward | 5′-TCTTCTCCTTCCAGCCTGAG-3′ |
| Reverse | 5′-CCCACCGAACTCAAAGAAGG-3′ |
| 18S | |
| Forward | 5′-AAACGGCTACCACATCCAAG-3′ |
| Reverse | 5′-CCTCCAATGGATCCTCGTTA-3′ |
BAC = bacterial artificial chromosome.
Morphology
Livers were fixed in 4% paraformaldehyde and embedded in paraffin. Sections 5 μm in thickness were stained with hematoxylin and eosin (H&E) or Sirius Red, or processed for immunohistochemical localization of BrdUrd.31,32 Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) assay (Roche Molecular Biochemicals, Mannheim, Germany) was performed with fluorescein-dUTP following the manufacturer's instructions. Electron microscopy and histochemical localization of peroxisomal catalase were performed as described elsewhere.10,31
Serum and Hepatic Lipid
Sera were analyzed for very long chain fatty acids C26:0/C22:0 and C24:0/C22:0 as described previously.8 For hepatic triglyceride and cholesterol levels, 100 mg of liver was homogenized for extraction of lipid by chloroform/methanol extraction assay. Total lipid was resuspended in 5% Triton X-100 in phosphate-buffered saline, and levels of triglyceride and total cholesterol were then quantified according to the manufacturer's procedures.34
Cell Culture
Mouse primary hepatocytes from 1-month-old C57BL6/J mice were isolated by collagenase II perfusion and cultured as reported.33 Wy-14,643 (25 μmol/L or 50 μmol/L) was added to the culture medium, and cells were harvested 48 hours later. Mouse embryonic fibroblasts were prepared from ACOX1−/− fetuses that were harvested at day 13.5 of gestation.35 Fetuses were dissected and incubated with 20 mL of 0.05% trypsin (Gibco, Carlsbad, CA) at 37°C for 25 minutes. Cells were cultured in Dulbecco's modified Eagle's medium with 10% fetal bovine serum and 1% L-glutamine. For siRNA transfection, mouse p8 siRNA oligos (catalog no. sc-40793) and its negative control (catalog no. sc-37007) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The ACOX1−/− mouse embryonic fibroblasts were cultured in 60-mm plates overnight. Cells were transfected with 1 μg or 3 μg of p8 siRNA or with 3 μg negative control siRNA using Lipofectamine 2000 (Invitrogen). RNA was extracted from the cells 2 days after transfection and used for quantitative PCR (qPCR) analysis.
Gene Expression Analysis
For qPCR, total RNA was isolated with TRIzol reagent (Invitrogen) and reverse transcribed using 2 μg of total RNA. The primer sequences used are listed in Table 1. Northern and Western blotting procedures used were as described elsewhere.31,32
Chromatin Immunoprecipitation and Electrophoretic Mobility Shift Assay
Chromatin was prepared from liver nuclei and used for chromatin immunoprecipitation (ChIP) assays as described elsewhere.34 For the gel shift assay, the in vitro translated PPARα, PPARγ, and PPARβ/δ protein was incubated with γ32P-labeled wild-type P1, P2, or P3 duplex oligonucleotides, and DNA–protein complexes were resolved using 4% acrylamide nondenatured gels.36 Primers and oligonucleotides used for these assays are listed in Table 2.
Table 2.
Oligonucleotide Sequences of ChIP Assay and EMSA Primers
| ChIP assay | |
| P1 | |
| Forward | 5′-CTGACGGAAGGAAGGTTGTT-3′ |
| Reverse | 5′-TGGCTCATTTAGAGACACGC-3′ |
| P2 | |
| Forward | 5′-CAGCCAGACCAGAAATTGTC-3′ |
| Reverse | 5′-GAGCCCATTGCACTTAGATG-3′ |
| P3 | |
| Forward | 5′-CTCAGACCTCAGGCCACTCT-3′ |
| Reverse | 5′-CTTGGTGAGCTGGTGAGAAA-3′ |
| EMSA | |
| P1 | |
| Sense | 5′-GGCACACCATCAAAGGGGAAAAGGTCAGAGGTTGGAAAACCTGAGCTCA-3′ |
| Antisense | 5′-GAGACTCTGAGCTCAGGTTTTCCAACCTCTGACCTTTTCCCCTTTGAT-3′ |
| P2 | |
| Sense | 5′-GGATATGTCCCTGAGAGTTGCAGCTGACCCACAGCCTGCTGTGAGGCCACT-3′ |
| Antisense | 5′-GGAATCAGTGGCCTCACAGCAGGCTGTGGGTCAGCTGCAACTCTCAGGGA-3′ |
| P3 | |
| Sense | 5′-GAGTAAAATTGCAACATTCCAAACTCAGACCTCAGGCCACTCTACACTTGGT-3′ |
| Antisense | 5′-GGTTGAACCAAGTGTAGAGTGGCCTGAGGTCTGAGTTTGGAATGTTGCAATT-3′ |
ChIP = chromatin immunoprecipitation; EMSA = electrophoretic mobility shift assay.
Statistical Analysis
Data were analyzed using one-way analysis of variance, with values of P < 0.05 considered to be significant.
Results
Steatohepatitis, Lipoapoptosis, Hepatocellular Regeneration, and Hepatocarcinogenesis in ACOX1−/− Mice
Young ACOX1−/− mice exhibit severe hepatic steatosis and evidence of hepatocellular regeneration (Figure 1, A–I, and Figure 2, A–I). The hepatic triglyceride, but not total cholesterol, was increased in ACOX1−/− mice at the age of 5 months (see Supplemental Figure S1 at http://ajp.amjpathol.org). A reduction in the extent of hepatic fatty change occurs in older ACOX1−/− mice, which by then develop grossly visible liver tumors (Figure 1D). Clusters of foamy macrophages loaded with Oil Red O–positive material are seen scattered in the liver parenchyma in ACOX1−/− mice 8 months or older (Figure 1G). These livers also displayed increased amounts of fibrosis as evidenced by portal-to-portal and portal-to–central vein bridging strands of Sirius red–positive collagen (Figure 1, H and I). Regenerated hepatocytes are resistant to fatty change (Figure 2, B and C) and display abundant granular cytoplasm indicative of peroxisome proliferation (Figure 2B). These cells repopulate liver and gradually replace steatotic hepatocytes. BrdUrd immunohistochemical staining confirmed increase in liver cell proliferation in ACOX1−/− mice (Figure 2, D–F). Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling staining of liver sections revealed age-related increased level of hepatocyte apoptotic activity in ACOX1−/− mice, reflecting death of some steatotic liver cells (Figure 2, G–I).
Figure 1.
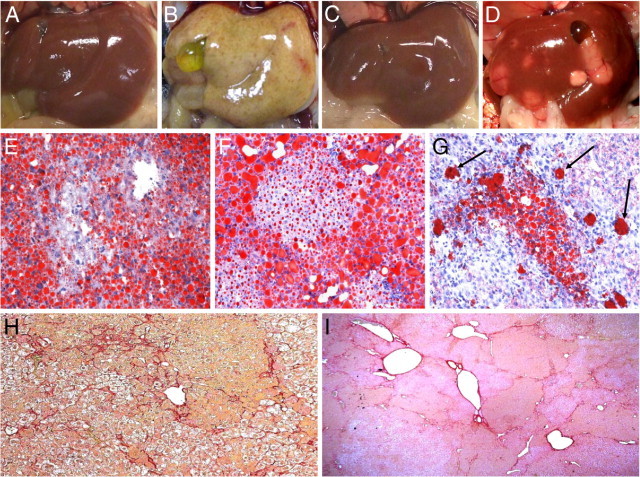
Progression from steatohepatitis to hepatocellular carcinoma in ACOX1−/− mice. Liver of 2-month-old wild-type (A) and ACOX1−/− mouse (B); 1-year-old wild-type (C) and ACOX1−/− mouse liver with liver tumors (D). Oil Red O staining of liver from 2-month-old (E), 5-month-old (F), and 8-month-old (G) ACOX1−/− mice. Proliferating hepatocytes reveal lessening of steatosis. By 8 months of age, only a few clusters of hepatocytes and a few macrophages (arrows) remain steatotic (G). Sirius red staining of liver from an 8-month-old (H) and a 1-year-old (I) ACOX1−/− mouse.
Figure 2.
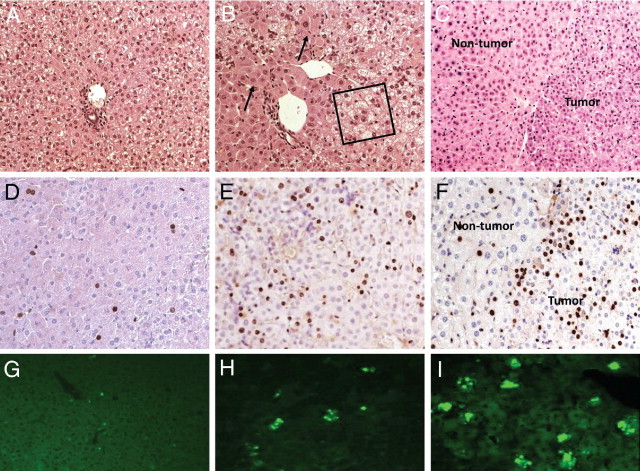
Age-related hepatocellular proliferation with abatement of hepatic steatosis in ACOX1−/− mice. H&E (A–C), BrdUrd (D–F), and terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (G–I) staining of liver from 2-week-old (A, D, and G), 5-month-old (B, E, and H), and 1-year-old (C, F, and I) ACOX1−/− mice. Boxed area in B indicates microvesicular steatosis; arrows point to regenerated hepatocytes. In C and F, tumor and nontumor areas are shown.
Aggravated ER Stress in ACOX1−/− Mouse Liver
ACOX1−/− mice exhibit steatohepatitis, increased hepatic levels of H2O2, and hepatocellular regeneration.8,10 Progression of hepatic manifestations in ACOX1−/− mice prompted us to examine whether these mice would manifest ER stress in view of the sustained activation of PPARα by ACOX1 substrates that remain unmetabolized in the absence of ACOX1 and function as endogenous activators of this nuclear receptor.10,14,15 In agreement with this, serum C26:0/C22:0 and C24:0/C22:0 ratios increase approximately threefold in ACOX1−/− mice compared with wild-type mice, whereas these ACOX1−/− humanized mice (discussed below) showed levels similar to those in wild-type mice (see Supplemental Figure S2 at http://ajp.amjpathol.org).
The expression levels of selected ER stress–related genes revealed increases in ATF4, CHOP, GADD45α, p53, and p8 in a time-dependent manner in ACOX1−/− mouse livers (Figure 3, A and B). qPCR results further confirmed that ER stress response genes CHOP, TRB3, Bax, BclII, and p8 are also increased in ACOX1−/− mouse liver (Figure 3C). Transcription factor p8 mRNA expression in the wild-type mouse liver was low, but in the ACOX1−/− mouse, p8 mRNA expression increased with age (Figure 3, A and C). Reduction of GRP78 protein content in ACOX1−/− mouse livers was noted on Western blotting (Figure 3B). In ACOX1−/− mouse liver, the p-eIF2α protein level increased significantly beginning at 1 month of age, whereas the total amount of eIF2α protein did not change (Figure 3B). An increase in p-eIF2α was associated with upregulation of ATF4 expression and its target gene, transcription factor CHOP, in ACOX1−/− mice (Figure 3A).
Figure 3.
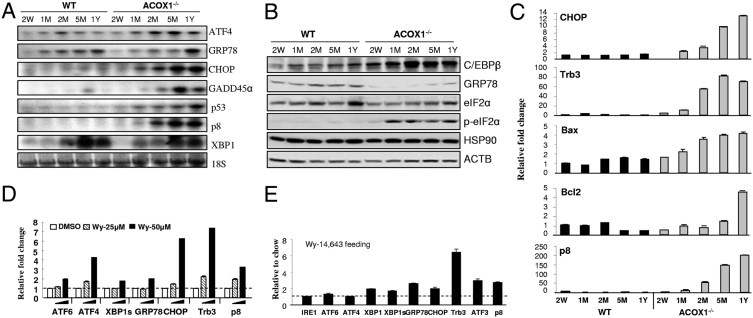
Spontaneous PPARα activation in ACOX1−/− mouse liver aggravates ER stress. A: Northern blot analysis of wild-type (WT) and ACOX1−/− mouse liver RNA. Mice 2 weeks old (W), 1, 2, and 5 months old (M), and 1 year old (Y). B: Western blot analysis of liver protein (20 μg protein). ACTB served as loading control. C: qPCR analysis of liver RNA from wild-type (WT) and ACOX1−/− mice. D: qPCR analysis of ER stress gene expression in primary hepatocytes treated with Wy-14,643. E: ER stress response gene expression in the liver of wild-type mice fed Wy-14,643 for 4 days was analyzed by qPCR. Fold change presented represents normalization of Wy-14,643 group against chow control. 18S was used as internal control.
Induction of ER Stress in Liver by Upregulation of Transcription Factor p8 by PPAR Isoforms
Fatty acids and their metabolic derivatives serve as potent endogenous ligands in ACOX1−/− mice to activate PPARα, which may further induce ER stress in ACOX1−/− mice.8,10,14,15 Primary hepatocytes treated with synthetic PPARα ligand Wy-14,643 (50 μmol/L) in the culture medium showed increases in the mRNA levels of ER stress response genes, in particular, ATF4, CHOP, TRB3, and p8 (Figure 3D). Wild-type mice fed Wy-14,643 (0.125% w/w) in powdered chow for 4 days revealed increases in mRNA expression for ER stress response genes in liver as assessed by qPCR. The relative mRNA expression of GRP78, Trb3, ATF3, and p8 were increased approximately threefold compared with the chow controls (Figure 3E). These data indicate the development of significant ER stress in liver with activation of PPARα. Furthermore, p8 mRNA levels increased in livers of wild-type mice but not in PPARα−/− mice fed Wy-14,643 (Figure 4, A and B). Likewise, spontaneous activation of PPARα by endogenous ligands in ACOX1−/− mouse liver caused a marked increase in hepatic p8 mRNA content (Figure 4B). Expression of GRP78 in untreated and Wy-14, 643–treated PPARα−/− mouse livers was reduced as compared with that in wild-type mice, suggesting PPARα-activated dependency of ER stress (see Supplemental Figure S3 at http://ajp.amjpathol.org). Overexpression of PPARγ in the liver by adenovirus transduction also resulted in an increase in p8 mRNA expression (Figure 4C). Levels of p8 mRNA also increased in the liver of a mouse treated with PPARβ/δ ligand L-165,041 (Figure 4D). Based on these observations, we consider p8 as a possible target gene for all three PPAR isoforms.
Figure 4.
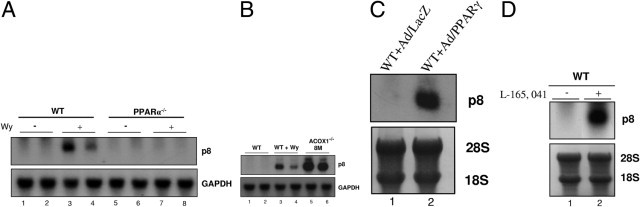
Induction of p8 mRNA in liver by PPAR isoforms. Northern blot analyses: (A) p8 mRNA in wild-type (PPARα+/+) (WT; lanes 3 and 4) and PPARα−/− (lanes 7 and 8) mouse livers treated for 4 days with PPARα ligand Wy-14,643 (0.125% w/w). Untreated controls (lanes 1 and 2; lanes 5 and 6). B: Endogenous ligand-mediated activation of PPARα in ACOX1−/− mouse liver increases hepatic p8 mRNA content. Wild-type mice without (lanes 1 and 2) or with Wy-14,643 treatment (lanes 3 and 4) and untreated 8-month-old ACOX1−/− mice (lanes 5 and 6). GAPDH is shown as loading control (A and B). C: Expression of p8 in mouse liver in response to PPARγ overexpression using adenovirus PPARγ (Ad/PPARγ) (lane 2). Ad/LacZ (lane 1) served as control. D: PPARβ/δ activation increases p8 mRNA content in liver. WT mouse injected i.p with PPARβ/δ ligand, L-165,041 (1 mg/day for 5 days). 28S and 18S are shown as loading controls.
We found three putative peroxisome proliferator-response elements (PPREs)17 in the p8 promoter and designated these as P1, P2, and P3 (Figure 5A). These consist of an imperfect hexamer separated by 1 bp (DR1-like). The nucleotide sequences and positions of PPREs in p8 are as follows: p8-P1 (5′-AGGTCAGAGGTTG-3′; −2714/−2702); p8-P2 (5′-TGACCCACAGCCT-3′; −1339/−1327), and p8-P3 (5′-AGACCTCAGGCCA-3′; −583/−571). Gel mobility shift assays revealed that all three in vitro translated PPAR isoforms (-α, -β/δ, and -γ) and RXRα bind variably as PPAR/RXR heterodimers to P1 and P3 sites. PPARα and PPARγ bind more prominently to P1 and P3, respectively (Figure 5B). Of the three isoforms, only PPARα showed a minimally detectable binding to P2 (Figure 5B). Competitive electrophoretic mobility shift assay (EMSA) confirmed binding specificity of P1 and P3 to PPARα and PPARγ, respectively (Figure 5C). ChIP assays revealed that PPARα binds all three PPREs on p8 promoter in Wy-14,643–fed wild-type mouse liver, but not in PPARα−/− mouse liver (Figure 5D). ChIP assay for PPARγ occupancy on p8 promoter showed that PPARγ bound to P1 and P3 PPRE in both wild-type and PPARα−/− mouse liver (Figure 5E).
Figure 5.
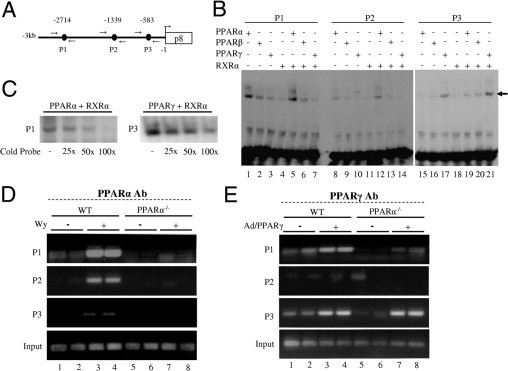
Regulation of p8 promoter by PPAR isoforms. A: Schematic representation of p8 promoter upstream of the first ATG with three potential PPREs (P1 at −2714, P2 at −1339, and P3 at −583). Arrows represent primers used for ChIP analysis. B: Binding analyses of PPAR isoforms using EMSA. Arrow represents product in EMSA binding assay. C: Specific binding for P1 with PPARα/RXRα and P3 with PPARγ/RXRα were confirmed by competitive EMSA using 25-, 50-, or 100-fold molar excess of cold oligonucleotide. D and E: ChIP assays to examine recruitment of PPARα (D) and PPARγ (E) on p8 promoter. Chromatin extracts, prepared from liver of wild-type and PPARα−/− mice fed normal or Wy-14-643 (0.125% w/w)–containing chow for 4 days, were used for immuonoprecipitation with PPARα antibody (PPARα Ab). Promoter-occupancy was observed on all three PPREs (P1, P2, and P3) with differential affinity on treatment with PPARα agonist (lanes 3 and 4), whereas no binding was observed in PPARα−/− mice (lanes 5–8). For in vivo occupancy by PPARγ, chromatin extracts prepared from wild-type and PPARα−/− mice injected with Ad/PPARγ were immmunoprecipitated using PPARγ antibody (PPARγ Ab). Input represents control in D and E.
Our data suggest that PPARα exerts a direct action on the mouse p8 promoter to upregulate its expression resulting in progressively increased expression of p8 in ACOX1−/− mice. When p8 was knocked down using siRNAs in PPARα−/− mouse embryonic fibroblasts, there was an alleviation of ER stress in response to reduced expression of p8. The expression of ER stress response genes, such as ATF4, CHOP, XBP1s, GRP78, and Trb3 was inhibited (see Supplemental Figure S4 at http://ajp.amjpathol.org). These results are consistent with an earlier report that p8 may regulate ATF4, CHOP, and TRB3.37 These results further extend our knowledge that p8, a novel PPARα target gene, can induce ER stress.
Generation of ACOX1−/− Mice with Human ACOX1 Transgene
We generated human ACOX1 transgenic mice using the BAC transgenic technique (Figure 6A) and crossed these with ACOX1−/− mice to generate ACOX1−/−h+ mice. We measured the expression levels of mouse ACOX1 (mACOX1) (see Supplemental Figure S5A at http://ajp.amjpathol.org) and human ACOX1 (hACOX1) (see Supplemental Figure S5B at http://ajp.amjpathol.org) mRNA in different tissues, and show that hACOX1 mRNA is present in many tissues in ACOX1−/−h+ mice. As anticipated, the age- and sex-matched ACOX1−/−h+ humanized mice appeared similar in body size to wild-type mice, suggesting that the human ACOX1 is functionally capable of preventing deficiency of mouse ACOX1. ACOX1−/−h+ humanized mice did not develop fatty liver, and hepatic triglyceride content appeared similar to that of wild-type control mice (see Supplemental Figure S1 at http://ajp.amjpathol.org). The spontaneous and sustained activation of PPARα target genes noted in ACOX1−/− mouse liver was not observed in ACOX1−/−h+ mouse liver (Figure 6, B and C). Serum C26:0/C22:0 and C24:0/C22:0 ratios in humanized mice were similar to that noted in wild-type mice (see Supplemental Figure S2 at http://ajp.amjpathol.org).
Figure 6.
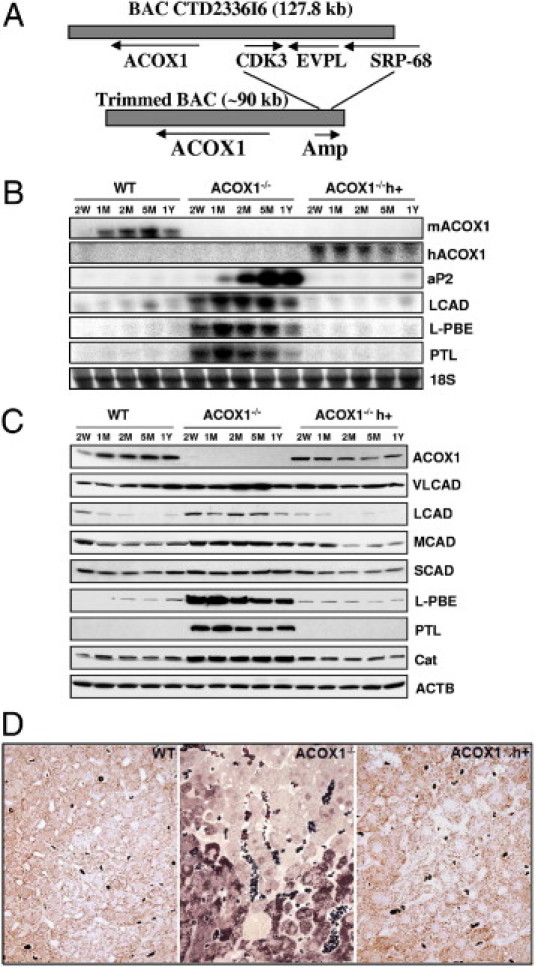
Generation of ACOX1−/−h+ mice. A: Human ACOX1 BAC clone CTD2336I6. AMP (ampicillin-resistant gene); CDK3 (cyclin-dependent kinase 3); EVPL (envoplakin); and SRP68 (signal recognition particle 68 kDa). B: Northern blot analysis of PPARα target genes involved in fatty acid oxidation in liver. C: Western blot analysis. Protein levels of PPARα-regulated genes involved in fatty acid oxidation. D: Diaminobenzidine histochemical localization of peroxisomal catalase to assess peroxisome population in liver. Dark brown dots in the cytoplasm represent peroxisomes in these semithin sections (0.5 μmol/L). In ACOX1−/− livers regenerated hepatocytes show peroxisome proliferation (intense brown dots) which differ from hepatocytes with microvesicular steatosis (foamy cytoplasm).
As expected, in both ACOX1−/− and ACOX1−/−h+ mouse livers, no mouse ACOX1 (mACOX1) mRNA was detected (Figure 6B). Human ACOX1 mRNA was detected in ACOX1−/−h+ mouse liver (Figure 6B). ACOX1 protein was undetected in ACOX1−/− but was present in ACOX1−/−h+ mouse liver (Figure 6C). As expected, the mRNA and protein concentrations of PPARα target genes such as LCAD, L-PBE, and PTL14 increased in ACOX1−/− mouse livers because of activation of PPARα by unmetabolized biological ligands (Figure 6B). Of interest is that a PPARγ target gene aP2 mRNA level is also increased in ACOX1−/− mouse liver, implying that other PPARγ target genes may also be upregulated in ACOX1 deficiency (Figure 6B). Expression of human ACOX1 transgene in ACOX1−/−h+ mouse liver abrogated the spontaneous activation of PPARα, resulting in the failure of induction of the target genes (Figure 6, B and C). Likewise, PPARγ target gene aP2 induction was also abrogated in ACOX1−/−h+ mouse liver (Figure 6B). Regenerated hepatocytes in ACOX1−/− mouse liver showed profound increase in the number of peroxisomes, indicating endogenous activation of PPARα, whereas hepatocytes with microvesicular steatosis in these livers revealed no peroxisome proliferation (Figure 6D). Livers of ACOX1−/−h+ mice showed no evidence of steatosis and steatohepatitis and no peroxisome proliferation (Figure 6D), indicating that the introduction of human ACOX1 gene prevents the development of ACOX1 null phenotypic characteristics.
Prevention of Liver Carcinogenesis in ACOX1−/−h+ Mice by ER Stress Relief
ACOX1 null mice with introduced human ACOX1 gene (ACOX1−/−h+) did not develop steatosis and steatohepatitis. There was no evidence of spontaneous PPARα activation, as there was no increase in the mRNA levels of PPARα target genes in these ACOX1−/−h+ mouse livers (Figure 6, B–D). ACOX1−/−h+ mice fed Wy-14,643, a synthetic PPARα ligand, showed induction of human ACOX1 mRNA in liver, and this up-regulation of human ACOX1 mRNA appeared essentially similar to the induction of mouse ACOX1 mRNA in wild-type mice fed Wy-14643 (see Supplemental Figure S5, C and D at http://ajp.amjpathol.org). Human ACOX1 gene in these BAC transgenic mice contains a PPRE consisting of 5′-AGGTCA G CTGTCA-3′, and this gene responds to the transcriptional activation by PPARα ligand Wy-14,643.38–40 These findings negate the mistaken notion that human ACOX1 promoter is nonfunctional or noninducible and minimizes the potential risk to human beings of chronic exposure to PPARα ligands.41 If human ACOX1 gene is uninducible as proposed,41 one would expect severe metabolic problems and hepatic dysfunction in human beings, since these individuals would not be able to properly metabolize endogenous PPARα ligands.10,17,40 Furthermore, none of the ACOX1−/−h+ mice developed hepatocellular carcinomas, whereas, as expected, all ACOX1−/− mice developed liver tumors by 8 to 14 months of age. To ascertain whether the ER stress noted in ACOX1−/− mouse liver (Figure 3, A–C) is relieved in ACOX1−/−h+ mouse liver, we studied the expression of these ER stress response genes CHOP, TRB3, Bax, BclII, and p8 by qPCR in mice 2 weeks to 1 year old (data not shown). The ER stress response increased progressively with age in ACOX1−/− mouse liver, whereas the expression of these genes in ACOX1−/−h+ mouse liver appeared similar to that seen in wild-type controls, implying prevention of ER stress by human ACOX1 transgene expression. Furthermore, in ACOX1−/−h+ mouse liver, increases in other ER stress–related genes such as GRP78, p-elF2a, C/EBP were not noted (data not shown), indicating the prevention of ER stress in ACOX1−/− mice with the expression of human ACOX1 gene.
Discussion
ACOX1 deficiency, which affects the metabolism of very long chain fatty acyl-CoAs and certain other fatty acid derivatives, presents an ideal murine model system for studying the role of steatohepatitis as well as sustained hyperactivation of PPARα in the development of liver tumors.8,10 Furthermore, because disturbances of fatty acid metabolism in liver constitute a common thread linking steatosis and steatohepatitis caused by alcoholic and nonalcoholic conditions, as well as by consumption of high-fat or high-carbohydrate diets, the insights gained from the current work might be applicable for further understanding of the pathogenesis of fatty liver diseases.5,18–22,37,42–44 In the present study, we show that ACOX1 deletion is associated with augmented levels of ER stress in the liver, most likely caused by a cascade of events initiated by fatty acid intermediates that remain as unmetabolized, putatively toxic substrates of this key lipid-metabolizing enzyme.10,17,44 Sustained enhancement of ER stress in mice nullizygous for ACOX1 gene is likely to initiate fatty change in liver and contribute to a plethora of events leading to liver cancer.10 Our experimental results are consistent with the following scenario. Initially, the disruption of ACOX1 gene in the mouse leads to disturbances in the peroxisomal fatty acid β-oxidation pathway, resulting in increased intracellular levels of unmetabolized very long chain saturated fatty acids and their fatty acyl-CoAs.10,44 These unoxidized toxic substrates of ACOX1, as well as lipid derivatives such as triglycerides, compromise liver function, contributing to the development of fat-laden liver cells and promoting their apoptosis by inducing ER stress. Death of hepatocytes overloaded with fat results in the release of toxic fatty acids/derivatives that incite the onset of steatohepatitis and exaggerated ER stress.5,18 Apoptotic cell death of some steatotic hepatocytes in ACOX1−/− liver then imparts a signal for neighboring hepatocytes to proliferate. These newly formed hepatocytes become resistant to steatosis, but manifest pleiotropic changes consistent with sustained PPARα activation by endogenous ligands of this receptor that remain unmetabolized in the absence of ACOX1.10,17 PPARα activation increases fatty acid oxidation systems that increased activity of mitochondrial β-oxidation system can metabolize a bulk of short- and medium-chain fatty acid to prevent steatotic alteration in hepatocytes that exhibit enormous increases in peroxisome populations.10 Accordingly, a functional ACOX1 under normal physiological conditions plays a pivotal role in minimizing ER stress in liver by degrading endogenous PPARα ligands.
In accordance with the foregoing, we show evidence for a defective unfolded protein response signaling in the ACOX1−/− mouse liver, as exemplified by increases in ER stress effectors such as phosphorylated eIF-2α (p-eIF2α), ATF4, CHOP (CHOP10), GADD45α, TRB3, and the transcriptional regulator p8 (Figures 3 and 4). These age-progressive alterations are indicative of unresolved, ER stress in the ACOX1−/− mouse liver. TRB3, a novel target for ATF4/CHOP pathway, is known to participate in CHOP-dependent cell death.45,46 CHOP is known to interfere with C/EBPα, a transcription factor involved in hepatic gluconeogenesis and lipid homeostasis.19,42,43 Increased levels of CHOP expression with antecedent decrease in C/EBPα and increased phosphorylation of eIF2α (together with decreased fatty acid oxidation) result in disturbances in hepatic lipid metabolism, contributing to the development of fatty liver in ACOX1−/− mice. Increased phosphorylation of eIF2α leads to upregulation of transcription factor ATF4, which further activates CHOP and GADD34.20–22,24 Induction of proapoptotic transcription factor CHOP expression in ACOX1−/− mouse liver indicates prolonged ER stress, which is closely associated with the progression of apoptosis (Figures 2 and 3). In ACOX1−/− mouse liver, GRP78 is generally decreased at both mRNA and protein levels. Several studies have shown that GRP78 expression is either increased or decreased under ER stress.22 A decrease in GRP78 expression has been shown to enhance mitochondrial permeability and augmented apoptosis in H460 cells during ER stress.47 Knockdown of GRP78 by siRNA increased CHOP expression.48 Reduction in GRP78 signaling in ACOX1−/− mouse liver, beginning at a very early age (ie, 2 weeks of age) further confirms that ER stress begins early in ACOX1 null livers and is possibly linked to the steatohepatitis phase. It is worth noting that GRP78 protein increased in older ACOX1−/− mouse livers (Figure 3B). Furthermore, in the ACOX1−/− mouse liver, age-related increases in proapoptotic Bax expression were noted when compared with the expression of the antiapoptotic factor Bcl-2 (Figure 3C). The intracellular ratio of Bax/Bcl-2 proteins can influence the ability of a cell to respond to an apoptotic signal,49 and, in large part, we show that this ratio is higher in ACOX1−/− mouse liver than in wild-type mice. It is also important to note that TRB3, a novel target for ATF4/CHOP pathway, is also known to participate in CHOP-dependent cell death.49 TRB3 has been shown to be regulated by PPARα,50 and because PPARα is activated in a sustained manner in ACOX1−/− mouse liver, the high levels of TRB3 expression appear to be consistent with the proposed regulatory pathway (Figure 7). TRB3 is also upregulated by reactive oxygen species as well as free fatty acids.51,52 These factors also appear to be operative in ACOX1−/− mouse liver, in addition to CHOP and PPARα, in inducing TRB3.
Figure 7.
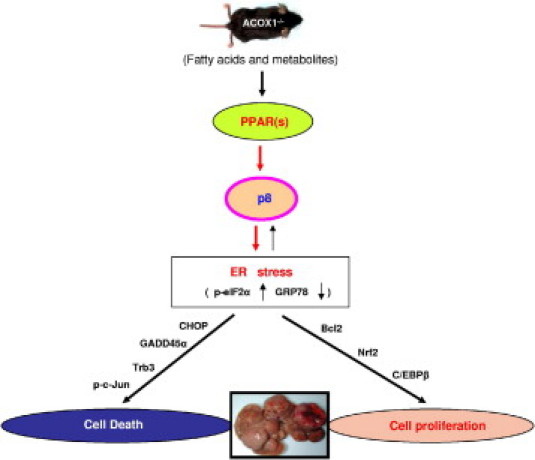
Model to illustrate the role of PPARs and p8 in inducing ER stress in ACOX1-deficient mouse liver phenotype. In ACOX1−/− mouse liver, there is evidence for the activation of both PPARα and PPARγ (target gene induction described in Figure 6B) and thus contribute to p8 induction and p8-regulated ER stress genes. In addition, p8 is induced by PPARβ/δ synthetic ligand, but whether p8 is activated spontaneously in ACOX1−/− liver is unclear.
Of interest is that this study presents evidence for the regulatory role of PPARα in the upregulation of transcription regulator p8 in ACOX1−/− mouse liver (Figures 4 and 5). Generally, p8 is considered a survival and cell proliferation signal with an impact on tumor progression and metastasis.37,42 It also mediates apoptosis by upregulating the ER stress response genes ATF4, CHOP, and TRB3.37 In ACOX1−/− mouse liver, p8, ATF4, CHOP, GADD45α, and TRB3 show a similar pattern of increased expression. It should be noted that p8 is induced by ATF4, and evidence suggests that p8 also acts as a positive regulator of the transcriptional activator ATF4.29,53 The data presented here demonstrate that spontaneous induction of p8 in ACOX1−/− mouse liver is due to, at least in part, to sustained activation of PPARα and also to PPARγ, in view of the increase in the mRNA content of its target gene aP2 (Figure 6B). The induction of p8 mRNA by the synthetic ligands of PPARα and PPARβ/δ in wild-type mice (Figure 4) further supports the proposed regulatory function of PPARs. We show here that the p8 gene promoter contains functional PPRE that appear to respond to all three PPAR isoforms (Figure 5). Thus, it appears that p8 is regulated by ATF4 and PPARs and possibly by other factors. Further studies are required to assess the role of transcription coactivators such as mediator subunit Med1 in p8-mediated ER stress.54
In conclusion, these studies present evidence for the role of ER stress in ACOX1−/− mouse liver in the age-progressive phenomenon of steatosis, steatohepatitis, fibrosis, and the development of hepatocelular carcinoma. The progressive ER stress, which may be initiated by the fatty acid derivatives that remain unmetabolized and serve to activate PPARα, may be critical for the development of hepatocellular carcinoma in ACOX1−/− mice. Furthermore, all three PPAR isoforms appear to regulate the transcription of nuclear protein p8, which is a key player in ER stress. Finally, our data with ACOX1−/−mice expressing human ACOX1 transgene (ACOX1−/−h+ humanized mice) clearly establish that functional expression of the human ACOX1 gene prevents the development of phenotypic features of ACOX1 deficiency and abrogates ER stress in liver.
Footnotes
Supported in part by National Institutes of Health grants GM23750 and DK083163 (J.K.R) and by the China Scholarship Council (L.B.).
Supplemental material for this article can be found at http://ajp.amjapthol.org or at doi: 10.1016/j.ajpath.2011.04.030.
Supplementary data
Hepatic triglyceride and cholesterol content. Mouse liver samples (100 mg) from 5 month old mice (wild-type; ACOX1−/− and ACOX1−/−h+) were used for lipid extraction and the levels of triglyceride and total cholesterol were quantified using the commercial kits. *P<0.05 compared to wild type or ACOX1−/−h+ mice.
Changes in the level of C26:0/C22:0 (A) and C24:0/C22:0 (B) fatty acids in plasma of wild type, ACOX1−/− and ACOX1−/−h+ humanized mice.
Northern blot analysis of GRP78 mRNA level in liver of wild type and PPARα−/− mice untreated or treated with Wy-14,643. Mice were fed control chow or chow with 0.125% Wy-14,643 for 3 days. A reduction in GRP78 mRNA expression in untreated PPARα-/- mice compared to wild type control. There is slight increase of GRP78 mRNA in wild type mice after Wy-14,643 treatment but not changed in PPARα−/− mice. 18S and 28S were used as loading controls.
Reduction of p8 in ACOX1−/− MEFs downregulates ER stress response gene expression. siRNA knockdown with 0 μg , 1 μg or 3 μg of p8 siRNA in ACOX1−/− MEF (P2) for 2 days, 3 μg of negative control siRNA was used as control. The fold presented is a normalization of p8 groups against negative control.
Expression of human ACOX1 mRNA in ACOX1−/−h+ humanized mice. (A) Mouse ACOX1 (mACOX1) mRNA tissue distribution in wild-type mice. (B) Human ACOX1 (hACOX1) mRNA tissue distribution in ACOX1−/−h+ humanized mice quantified by Q-PCR. ACOX1 mRNA was quantified by Q-PCR and normalized against 18S rRNA. In A and B the tissues used are indicated: brain, heart, liver, lung, skeletal muscle (SKM), small intestine (S. Intestine), white adipose tissue (WAT), and brown adipose tissue (BAT). (C) Induction of mouse ACOX1 mRNA (mACOX1) in response to Wy-14, 643 (Wy) treatment in wild-type mouse and in ACOX1−/−h+ humanized mouse liver. (D) Marked induction of human ACOX1 mRNA (hACOX1) in the liver of ACOX1−/−h+ humanized mice treated with Wy-14,643. No hACOX1 mRNA is detected in ACOX1−/− and WT mouse livers. Wy-14,643 (0.125%) was fed for 1 week. ACOX1−/− mouse liver is used as control.
References
- 1.Grundy S.M. Obesity, metabolic syndrome, and cardiovascular disease. J Clin Endocrinol Metab. 2004;89:2595–2600. doi: 10.1210/jc.2004-0372. [DOI] [PubMed] [Google Scholar]
- 2.Angulo P., Lindor K.D. Non-alcoholic fatty liver disease. J Gastroenterol Hepatol. 2002;17:S186–S190. doi: 10.1046/j.1440-1746.17.s1.10.x. [DOI] [PubMed] [Google Scholar]
- 3.Reddy J.K., Rao M.S. Lipid metabolism and liver inflammation: II. Fatty liver disease and fatty acid oxidation. Am J Physiol Gastrointest Liver Physiol. 2006;290:G852–G858. doi: 10.1152/ajpgi.00521.2005. [DOI] [PubMed] [Google Scholar]
- 4.Anderson N., Borlak J. Molecular mechanisms and therapeutic targets in steatosis and steatohepatitis. Pharmacol Rev. 2008;60:311–357. doi: 10.1124/pr.108.00001. [DOI] [PubMed] [Google Scholar]
- 5.Malhi H., Gores G.J. Molecular mechanisms of lipotoxicity in nonalcoholic fatty liver disease. Semin Liver Dis. 2008;28:360–369. doi: 10.1055/s-0028-1091980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bugianesi E., Leone N., Vanni E., Marchesini G., Brunello F., Carucci P., Musso A., De Paolis P., Capussotti L., Salizzoni M., Rizzetto M. Expanding the natural history of nonalcoholic steatohepatitis: from cryptogenic cirrhosis to hepatocellular carcinoma. Gastroenterology. 2002;123:134–140. doi: 10.1053/gast.2002.34168. [DOI] [PubMed] [Google Scholar]
- 7.Anstee Q.M., Goldin R.D. Mouse models in non-alcoholic fatty liver disease and steatohepatitis research. Int J Exp Pathol. 2006;87:1–16. doi: 10.1111/j.0959-9673.2006.00465.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fan C.Y., Pan J., Chu R., Lee D., Kluckman K.D., Usuda N., Singh I., Yeldandi A.V., Rao M.S., Maeda N., Reddy J.K. Hepatocellular and hepatic peroxisomal alterations in mice with a disrupted peroxisomal fatty acyl-coenzyme A oxidase gene. J Biol Chem. 1996;271:24698–24710. doi: 10.1074/jbc.271.40.24698. [DOI] [PubMed] [Google Scholar]
- 9.Purushotham A., Schug T.T., Xu Q., Surapureddi S., Guo X., Li X. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab. 2009;9:327–338. doi: 10.1016/j.cmet.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fan C.Y., Pan J., Usuda N., Yeldandi A.V., Rao M.S., Reddy J.K. Steatohepatitis, spontaneous peroxisome proliferation and liver tumors in mice lacking peroxisomal fatty acyl-CoA oxidase: Implications for peroxisome proliferator-activated receptor alpha natural ligand metabolism. J Biol Chem. 1998;273:15639–15645. doi: 10.1074/jbc.273.25.15639. [DOI] [PubMed] [Google Scholar]
- 11.Reddy J.K. Nonalcoholic steatosis and steatohepatitis: III. Peroxisomal β-oxdiation, PPARα and steatohepatitis. Am J Physiol Gastrointest Liver Physiol. 2001;281:G1333–G1339. doi: 10.1152/ajpgi.2001.281.6.G1333. [DOI] [PubMed] [Google Scholar]
- 12.Horie Y., Suzuki A., Kataoka E., Sasaki T., Hamada K., Sasaki J., Mizuno K., Hasegawa G., Kishimoto H., Iizuka M., Naito M., Enomoto K., Watanabe S., Mak T.W., Nakano T. Hepatocyter-specific Pten deficiency results in steatohepatitis and hepatocellular carcinomas. J Clin Invest. 2004;113:1774–1783. doi: 10.1172/JCI20513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee S.S., Pineau T., Drago J., Lee E.J., Owens J.W., Kroetz D.L., Fernandez-Salguero P.M., Westphal H., Gonzalez F.J. Targeted disruption of the alpha isoform of the peroxisome proliferator-activated receptor gene in mice results in abolishment of the pleiotropic effects of peroxisome proliferators. Mol Cell Biol. 1995;15:3012–3022. doi: 10.1128/mcb.15.6.3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reddy J.K., Hashimoto T. Peroxisomal beta-oxidation and peroxisome proliferator-activated receptor alpha: an adaptive metabolic system. Annu Rev Nutr. 2001;21:193–230. doi: 10.1146/annurev.nutr.21.1.193. [DOI] [PubMed] [Google Scholar]
- 15.Yeldandi A.V., Rao M.S., Reddy J.K. Hydrogen peroxide generation in peroxisome proliferator-induced oncogenesis. Mutat Res. 2000;448:159–177. doi: 10.1016/s0027-5107(99)00234-1. [DOI] [PubMed] [Google Scholar]
- 16.Rusyn I., Asakura S., Pachkowski B., Bradford B.U., Denissenko M.F., Peters J.M., Holland S.M., Reddy J.K., Cunningham M.L., Swenberg J.A. Expression of base excision DNA repair genes is a sensitive biomarker for in vivo detection of chemical-induced chronic oxidative stress: identification of the molecular source of radicals responsible for DNA damage by peroxisome proliferators. Cancer Res. 2004;64:1050–1057. doi: 10.1158/0008-5472.can-03-3027. [DOI] [PubMed] [Google Scholar]
- 17.Pyper S.R., Viswakarma N., Yu S., Reddy J.K. PPARalpha: energy combustion, hypolipidemia, inflammation and cancer. Nucl Recept Signal. 2010;8:e002. doi: 10.1621/nrs.08002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaplowitz N., Than T.A., Shinohara M., Ji C. Endoplasmic reticulum stress and liver injury. Semin Liver Dis. 2007;27:367–377. doi: 10.1055/s-2007-991513. [DOI] [PubMed] [Google Scholar]
- 19.Basseri S., Austin R.C. ER stress and lipogenesis: a slippery slope toward hepatic steatosis. Dev Cell. 2008;15:795–796. doi: 10.1016/j.devcel.2008.11.013. [DOI] [PubMed] [Google Scholar]
- 20.Rutkowski D.T., Wu J., Back S.H., Callaghan M.U., Ferris S.P., Iqbal J., Clark R., Miao H., Hassler J.R., Fornek J., Katze M.G., Hussain M.M., Song B., Swathirajan J., Wang J., Yau G.D., Kaufman R.J. UPR pathways combine to prevent hepatic steatosis caused by ER stress-mediated suppression of transcriptional master regulators. Dev Cell. 2008;15:829–840. doi: 10.1016/j.devcel.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Flamment M., Kammoun H.L., Hainault I., Ferre P., Foufelle F. Endoplasmic reticulum stress: a new actor in the development of hepatic steatosis. Curr Opin Lipidol. 2010;21:239–246. doi: 10.1097/MOL.0b013e3283395e5c. [DOI] [PubMed] [Google Scholar]
- 22.Kammoun H.L., Chabanon H., Hainault I., Luquet S., Magnan C., Koike T., Ferré P., Foufelle F. GRP78 expression inhibits insulin and ER stress-induced SREBP-1c activation and reduces hepatic steatosis in mice. J Clin Invest. 2009;119:1201–1215. doi: 10.1172/JCI37007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Armstrong J.L., Flockhart R., Veal G.J., Lovat P.E., Redfern C.P. Regulation of endoplasmic reticulum stress-induced cell death by ATF4 in neuroectodermal tumor cells. J Biol Chem. 2010;285:6091–6100. doi: 10.1074/jbc.M109.014092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oyadomari S., Harding H.P., Zhang Y., Oyadomari M., Ron D. Dephosphorylation of translation initiation factor 2alpha enhances glucose tolerance and attenuates hepatosteatosis in mice. Cell Metab. 2008;7:520–532. doi: 10.1016/j.cmet.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Naidoo N. The endoplasmic reticulum stress response and aging. Rev Neurosci. 2009;20:23–37. doi: 10.1515/revneuro.2009.20.1.23. [DOI] [PubMed] [Google Scholar]
- 26.Lin J.H., Walter P., Benedict Yen T.S. Endoplasmic reticulum stress in disease pathogenesis. Annu Rev Pathol Mech Dis. 2008;3:399–425. doi: 10.1146/annurev.pathmechdis.3.121806.151434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tabas I. The role of endoplasmic reticulum stress in the progression of atherosclerosis. Circ Res. 2010;107:839–850. doi: 10.1161/CIRCRESAHA.110.224766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chowdhury U.R., Samant R.S., Fodstad O., Shevde L.A. Emerging role of nuclear protein 1 (NUPR1) in cancer biology. Cancer Metastasis Rev. 2009;28:225–232. doi: 10.1007/s10555-009-9183-x. [DOI] [PubMed] [Google Scholar]
- 29.Goruppi S., Iovanna J.L. Stress-inducible protein p8 is involved in several physiological and pathological processes. J Biol Chem. 2010;285:1577–1581. doi: 10.1074/jbc.R109.080887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reddy J.K., Rao M.S., Azarnoff D.L., Sell S. Mitogenic and carcinogenic effects of a hypolipidemic peroxsiome proliferator, [4-chloro-6-(2,3-xylidino)-2-pyrimidinylthio]acetic acid] (Wy-14,643), in rat and mouse liver. Cancer Res. 1979;39:152–161. [PubMed] [Google Scholar]
- 31.Jia Y., Qi C., Kashireddi P., Surapureddi S., Zhu Y.J., Rao M.S., Le Roith D., Chambon P., Gonzalez F.J., Reddy J.K. Transcription coactivator PBP, the peroxisome proliferator-activated receptor (PPAR)-binding protein, is required for PPARalpha-regulated gene expression in liver. J Biol Chem. 2004;279:24427–24434. doi: 10.1074/jbc.M402391200. [DOI] [PubMed] [Google Scholar]
- 32.Qi C., Zhu Y., Pan J., Yeldandi A.V., Rao M.S., Maeda N., Subbarao V., Pulikuri S., Hashimoto T., Reddy J.K. Mouse steroid receptor coactivator-1 is not essential for peroxisome proliferator-activated receptor alpha-regulated gene expression. Proc Natl Acad Sci USA. 1999;96:1585–1590. doi: 10.1073/pnas.96.4.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vluggens A., Androletti P., Viswakarma N., Jia Y., Matsumoto K., Kulik W., Khan M., Huang J., Guo D., Yu S., Sarkar J., Singh I., Rao M.S., Wanders R.J., Reddy J.K., Cherkaoui-Malki M. Functional significance of the two ACOX1 isoforms and their crosstalks with PPARα and RXRα. Lab Invest. 2010;90:696–708. doi: 10.1038/labinvest.2010.46. [DOI] [PubMed] [Google Scholar]
- 34.Huang J., Iqbal J., Saha P.K., Liu J., Chan L., Hussain M.M., Hussain M.M., Moore D.D., Wang L. Molecular characterization of the role of orphan receptor small heterodimer partner in development of fatty liver. Hepatology. 2007;46:147–157. doi: 10.1002/hep.21632. [DOI] [PubMed] [Google Scholar]
- 35.Qi C., Surapureddi S., Zhu Y.J., Yu S., Kashireddy P., Rao M.S., Reddy J.K. Transcriptional coactivator PRIP, the peroxisome proliferator-activated receptor gamma (PPARgamma)-interacting protein, is required for PPARgamma-mediated adipogenesis. J Biol Chem. 2003;278:25281–25284. doi: 10.1074/jbc.C300175200. [DOI] [PubMed] [Google Scholar]
- 36.Viswakarma N., Yu S., Naik S., Kashireddy P., Matsumoto K., Sarkar J., Sarkar J., Surapureddi S., Jia Y., Rao M.S., Reddy J.K. Transcriptional regulation of Cidea, mitochondrial cell death-inducing DNA fragmentation factor alpha-like effector A, in mouse liver by peroxisome proliferator-activated receptor alpha and gamma. J Biol Chem. 2007;282:18613–18624. doi: 10.1074/jbc.M701983200. [DOI] [PubMed] [Google Scholar]
- 37.Carracedo A., Lorente M., Egia A., Blázquez C., García S., Giroux V., Malicet C., Villuendas R., Gironella M., González-Feria L., Piris M.A., Iovanna J.L., Guzmán M., Velasco G. The stress-regulated protein p8 mediates cannabinoid-induced apoptosis of tumor cells. Cancer Cell. 2006;9:301–312. doi: 10.1016/j.ccr.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 38.Reddy J.K., Goel S.K., Nemali M.R., Carrino J.J., Laffler T.G., Reddy M.K., Sperbeck S.J., Osumi T., Hashimoto T., ND Lalwani N.D., Rao M.S. Transcription regulation of peroxisomal fatty acyl-CoA oxidase and enoyl-CoA hydratase/3-hydroxyacyl-CoA dehydrogenase in rat liver by peroxisome proliferators. Proc Natl Acad Sci USA. 1996;83:1747–1751. doi: 10.1073/pnas.83.6.1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Varanasi U., Chu R., Huang Q., Castellon R., Yeldandi A.V., Reddy J.K. Identification of a peroxisome proliferator-responsive element upstream of the human peroxisomal fatty acyl coenzyme A oxidase gene. J Biol Chem. 1996;271:2147–2155. doi: 10.1074/jbc.271.4.2147. [DOI] [PubMed] [Google Scholar]
- 40.Reddy J.K. Peroxisome proliferators and peroxisome proliferator-activated receptor alpha: biotic and xenobiotic sensing. Am J Pathol. 2004;164:2305–2321. doi: 10.1016/s0002-9440(10)63787-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Woodyatt N.J., Lambe K.G., Myers K.A., Tugwood J.D., Roberts R.A. The peroxisome proliferator (PP) response element upstream of the human acyl-CoA oxidase gene is inactive among a sample human population: significance for species differences in response to PPs. Carcinogenesis. 1999;20:369–372. doi: 10.1093/carcin/20.3.369. [DOI] [PubMed] [Google Scholar]
- 42.Salazar M., Carracedo A., Salanueva I.J., Hernández-Tiedra S., Lorente M., Egia A., Vázquez P., Blázquez C., Torres S., García S., Nowak J., Fimia G.M., Piacentini M., Cecconi F., Pandolfi P.P., González-Feria L., Iovanna J.L., Guzmán M., Boya P., Velasco G. Cannabinoid action induces autophagy-mediated cell death through stimulation of ER stress in human glioma cells. J Clin Invest. 2009;118:1359–1372. doi: 10.1172/JCI37948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jaeschke H., Gores G.J., Cederbaum A.I., Hinson J.A., Pessayre D., Lemasters J.J. Mechanisms of hepatotoxicity. Toxicol Sci. 2002;65:166–176. doi: 10.1093/toxsci/65.2.166. [DOI] [PubMed] [Google Scholar]
- 44.Hashimoto T., Fujita T., Usuda N., Cook W., Qi C., Peters J.M., Gonzalez F.J., Yeldandi A.V., Rao M.S., Reddy J.K. Peroxisomal and mitochondrial fatty acid beta-oxidation in mice nullizygous for both peroxisome proliferator-activated receptor alpha and peroxisomal fatty acyl-CoA oxidase: Genotype correlation with fatty liver phenotype. J Biol Chem. 1999;274:19228–19236. doi: 10.1074/jbc.274.27.19228. [DOI] [PubMed] [Google Scholar]
- 45.Ohoka N., Yoshi S., Hattori T., Onozaki K., Hayashi H. TRB3, a novel ER stresss-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. EMBO J. 2005;24:1243–1255. doi: 10.1038/sj.emboj.7600596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim I., Xu W., Reed J.C. Cell death an dendoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2008;7:1013–1030. doi: 10.1038/nrd2755. [DOI] [PubMed] [Google Scholar]
- 47.Shu C.W., Sun F.C., Cho J.H., Lin C.C., Liu P.F., Chen P.Y., Chang M.D., Fu H.W., Lai Y.K. GRP78 and Raf-1 cooperatively confer resistance to endoplasmic reticulum-induced apoptosis. J Cell Physiol. 2008;215:627–635. doi: 10.1002/jcp.21340. [DOI] [PubMed] [Google Scholar]
- 48.Zu K., Bihani T Lin A., Park Y.M., Mori K., Ip C. Enhanced selenium effect on growth arrest by BiP/GRP78 knockdown in p53-null human prostate cancer cells. Oncogene. 2006;25:546–554. doi: 10.1038/sj.onc.1209071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Perlman H., Zhang X., Chen M.W., Walsh K., Buttyan R. An elevated bax/bcl-2 ratio corresponds with the onset of prostate epithelial cell apoptosis. Cell Death Differentiation. 1999;6:48–54. doi: 10.1038/sj.cdd.4400453. 1. [DOI] [PubMed] [Google Scholar]
- 50.Koo S.H., Satoh H., Herzig S., Lee C.H., Hedrick S., Kulkarni R., Evans R.M., Olefsky J., Montminy M. PGC-1 promotes insulin resistance in liver through PPAR-α-dependent induction of TRB-3. Nat Med. 2004;10:530–534. doi: 10.1038/nm1044. [DOI] [PubMed] [Google Scholar]
- 51.Morse E., Schroth J., You Y.H., Pizzo D.P., Okada S., Ramachandrarao S., Vallon V., Sharma K., Cunard R. TRB3 is stimulated in diabetic kidneys, regulated by the ER stress marker CHOP and is a suppressor of podocyte MCP-1. Am J Physiol Renal Physiol. 2010;299:F965–F972. doi: 10.1152/ajprenal.00236.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Morgan N.G. Fatty acids and β-cell toxicity. Curr Opin Clin Nutr Metab Care. 2009;12:117–122. doi: 10.1097/MCO.0b013e328321e423. [DOI] [PubMed] [Google Scholar]
- 53.Million Passe C.M., Cooper G., Quirk C.C. The murine p8 gene promoter is activated by activating factor 4 (ATF4) in the gonadotrope-derived LβT2 cell line. Endocrine. 2006;30:81–91. doi: 10.1385/endo:30:1:81. [DOI] [PubMed] [Google Scholar]
- 54.Bai L., Jia Y., Viswakarma N., Huang J., Vluggens A., Wolins N., Jafari N., Rao M.S., Borensztajn J., Yang G., Reddy J.K. Transcription coactivator Mediator subunit Med1 is required for the development of fatty liver in the mouse. Hepatology. 2011;53:1164–1174. doi: 10.1002/hep.24155. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Hepatic triglyceride and cholesterol content. Mouse liver samples (100 mg) from 5 month old mice (wild-type; ACOX1−/− and ACOX1−/−h+) were used for lipid extraction and the levels of triglyceride and total cholesterol were quantified using the commercial kits. *P<0.05 compared to wild type or ACOX1−/−h+ mice.
Changes in the level of C26:0/C22:0 (A) and C24:0/C22:0 (B) fatty acids in plasma of wild type, ACOX1−/− and ACOX1−/−h+ humanized mice.
Northern blot analysis of GRP78 mRNA level in liver of wild type and PPARα−/− mice untreated or treated with Wy-14,643. Mice were fed control chow or chow with 0.125% Wy-14,643 for 3 days. A reduction in GRP78 mRNA expression in untreated PPARα-/- mice compared to wild type control. There is slight increase of GRP78 mRNA in wild type mice after Wy-14,643 treatment but not changed in PPARα−/− mice. 18S and 28S were used as loading controls.
Reduction of p8 in ACOX1−/− MEFs downregulates ER stress response gene expression. siRNA knockdown with 0 μg , 1 μg or 3 μg of p8 siRNA in ACOX1−/− MEF (P2) for 2 days, 3 μg of negative control siRNA was used as control. The fold presented is a normalization of p8 groups against negative control.
Expression of human ACOX1 mRNA in ACOX1−/−h+ humanized mice. (A) Mouse ACOX1 (mACOX1) mRNA tissue distribution in wild-type mice. (B) Human ACOX1 (hACOX1) mRNA tissue distribution in ACOX1−/−h+ humanized mice quantified by Q-PCR. ACOX1 mRNA was quantified by Q-PCR and normalized against 18S rRNA. In A and B the tissues used are indicated: brain, heart, liver, lung, skeletal muscle (SKM), small intestine (S. Intestine), white adipose tissue (WAT), and brown adipose tissue (BAT). (C) Induction of mouse ACOX1 mRNA (mACOX1) in response to Wy-14, 643 (Wy) treatment in wild-type mouse and in ACOX1−/−h+ humanized mouse liver. (D) Marked induction of human ACOX1 mRNA (hACOX1) in the liver of ACOX1−/−h+ humanized mice treated with Wy-14,643. No hACOX1 mRNA is detected in ACOX1−/− and WT mouse livers. Wy-14,643 (0.125%) was fed for 1 week. ACOX1−/− mouse liver is used as control.