

Abstract
The cellular DNA damage response (DDR) ensures genomic stability and protects against genotoxic stresses. Conversely, defects in the DDR contribute to genome instability, with the resulting accumulated genetic changes capable of inducing neoplastic transformation. Thus, DDR is central to both the mechanism of oncogenesis and cancer therapy. Specifically, DDR is accomplished via a complicated meshwork of evolutionary conserved proteins, including ATM, ATR, and phospho-H2AX (γH2AX). GLTSCR2 is a nucleolar protein believed to function as a tumor suppressor, although its exact molecular mechanisms have yet to be fully elucidated. As a result of our research pertaining to the role of GLTSCR2 in tumor suppression, we have determined that GLTSCR2 is involved in DDR. Under genotoxic conditions, such as cellular exposure to UV radiation or radiomimetic drugs, GLTSCR2 expression increased and later mobilized to the nucleoplasm. Moreover, GLTSCR2 knockdown attenuated both the presence of phospho-H2AX at the nuclear foci and the phosphorylation of multiple DDR proteins, including ATM, ATR, Chk2, Chk1, and H2AX. In addition, the decreased expression of GLTSCR2 sensitized cells to DNA damage, delayed DNA repair, and abolished G2/M checkpoint activation. Our observations indicate that GLTSCR2 is a key component of DDR and GLTSCR2 seems to act as a tumor suppressor by participating in optimal DDR because DNA damage is a frequent and crucial event in oncogenesis.
The cellular DNA damage response (DDR), which includes the processes of cell cycle checkpoint activation and DNA damage repair, ensures genomic stability and protects against genotoxic stresses, such as DNA double-strand break (DSB). Specifically, this response occurs via a complicated network of evolutionary conserved pathways,1,2 which sequentially sense areas of damage, recruit DDR proteins into macromolecular foci, and activate checkpoint proteins to halt cell cycle progression.3 For instance, DSBs activate ATM, ATR, and DNA-dependent protein kinase, resulting in the phosphorylation of H2AX at sites of DNA damage. Phospho-H2AX (γH2AX) then serves as a nidus for the accumulation of additional DNA repair proteins, including MDC1, BRCA1, and 53BP1.4–6 In this way, defects in DDR can contribute to genome instability and ultimately the accumulation of genetic changes that lead to neoplastic transformation. Thus, DDR is central to the mechanisms of both oncogenesis and cancer treatment outcomes.
Previously, the nucleolar protein GLTSCR2 has been shown to be a putative tumor suppressor gene because it is capable of inducing PTEN-dependent apoptotic cell death and inhibiting tumor growth.7–9 Conversely, both the suppression of GLTSCR2 transcripts and translates and the overall allelic loss were identified in several brain tumors.10 However, much remains to be elucidated about the biological function and molecular mechanisms of GLTSCR2 associated with tumor suppression. Through our research pertaining to the role of GLTSCR2 in the process of neoplastic transformation, we discovered that GLTSCR2 is involved in DDR via the ATM-Chk2 and ATR-Chk1 pathways. Because the genetic damage is a frequent and key event that occurs in tumor development and growth, our findings imply that GLTSCR2 may act as a tumor suppressor by participating in DDR.
Materials and Methods
Cell Culture and Treatment
SK-Hep-1 cells obtained from the American Type Culture Collection (Manassas, VA) were cultured to subconfluence in a humidified 5% CO2 incubator maintained at 37°C in a Dulbecco's modified Eagle's medium containing 10% fetal calf serum, 2 mmol/L l-glutamine, 100 U/mL of penicillin, 100 μg/mL of streptomycin, and 10 mmol/L HEPES. The radiomimetic drug neocarzinostatin (NCS; Sigma-Aldrich, St. Louis, MO) was then added to fresh cell media, with a final concentration of 50 ng/mL. For UV treatment, cells were first washed in PBS, after which a minimal volume of serum-free culture medium was exposed to light from a 254-nm UV-C lamp (model XL-1000/F; Spectronics Corporation, Westbury, NY) at the indicated dose. After UV irradiation, cells were recultured in serum-containing medium at 37°C for the appropriate times.
Antibodies and Reagents
The rabbit polyclonal anti-GLTSCR2 antibody used here has been previously described and characterized,8 whereas the anti-phospho-H2AX (S139), anti-nucleolin, anti-phospho-Chk1 (S345), anti-Chk1, anti-phospho-Chk2 (T98), anti-Chk2, anti-phospho-Histone H3 (S10), anti-phospho-ATM (S1981), anti-ATM, anti-phospho-ATR (S428), and anti-ATR antibodies were purchased from Cell Signaling Technology (Danvers, MA). In addition, anti-tubulin was obtained from Santa Cruz Biotechnology Inc. (Santa Cruz, CA). All other remaining reagents were obtained from Sigma-Aldrich Inc. (St. Louis, MO), unless otherwise specified.
Construction of Tet-Off Adenoviral-Mediated System
The assembly and production of the recombinant adenovirus were performed per manufacturer instruction (Adeno-X Tet-Off Expression System 1; Clontech, Mountain View, CA). Initially, a GFP-GLTSCR2 construct [consisting of a full-length GLTSCR2 cDNA fused to a C-terminal Green fluorescent protein (GFP) tag] was cloned into pTRE-Shuttle2 vector (Clontech, Mountain View, CA) containing a tetracycline-responsive element (TRE) upstream of the cytomegalovirus (CMV) minimal promoter. Next, the resulting TRE-GFP-GLTSCR2 expression unit was excised from pTRE-Shuttle2 vector using the I-CeuI and PI-SceI restriction enzymes before ligation to the Adeno-X System 1 Viral DNA (Clontech).11 This resulting recombinant Adeno-X-GFP-GLTSCR2 vector (Ad-GFP/GLT) was then packaged into infectious adenoviral particles by the transfection of HEK293 cells. Recombinant adenoviruses were subsequently harvested through the lysis of transfected cells. To transiently express GLTSCR2, cells were coinfected with a recombinant adenovirus (Ad-GFP/GLT) and a regulation virus (Adeno-X Tet-Off Virus) in serum-free media for 12 hours, after which the fresh complete medium was changed.
Generation of GLTSCR2 Knockdown Stable Cell Lines
For GLTSCR2 expression knockdown, feline leukemia virus–based lentiviral GLTSCR2 short hairpin RNA (shRNA) vectors were purchased from Genecopoeia Inc. (Rockville, MD), with a GLTSCR2-targeted sequence of 5′-GAGACCGGTTCAAGAGCTT-3′ and scrambled sequence of 5′-CGATACTGAACGAATC-3′. Pseudovirus containing lentiviral GLTSCR2 shRNA vectors were then prepared by co-transfecting the Lenti-PacFIV packaging mix (Genecopoeia, Rockville, MD) into the 293Ta producer cell line, mediated by the EndoFectin Lenti transfection reagent (per manufacturer instruction). Either the lentiviral stocks of GLTSCR2 or the control shRNA vector as then incubated with separate sets of cells. After 48 hours, clones of GLTSCR2 knockdown cells were selected by puromycin (1 μg/mL) treatment so that protein expression could be analyzed using Western blot and immunocytochemistry.
Clonogenic Cell Survival Assay
To determine clonogenic survival after UV or NCS exposure, SK-shGLT and SK-shScr cells were trypsinized, after which various dilutions of the cells were resuspended in 2 mL of the growth media (12% serum and 0.3% agarose) and then plated in 6-well plates containing 2 mL of a second growth media (12% serum and 0.6% agarose). Next, the plates were incubated at 37°C in an atmosphere of 5% CO2 for 2 weeks, at which time the resulting colonies were stained with 0.005% Crystal Violet for scoring purposes.
Immunocytochemistry
After culture on coverslips coated with poly-l-lysine, the cells were fixed with 4% paraformaldehyde in PBS and permeabilized with 0.1% Triton X-100. The cells were then incubated with the primary antibody at 4°C overnight, after blocking with 1% bovine serum albumin in PBS. Next, the cells were stained with a secondary antibody that was conjugated with fluorescein isothiocyanate (FITC) or Texas Red and subsequently stained with DAPI to allow nuclear visualization by confocal microscopy (META 510, Zeiss, Jena, Germany).
Immunoblot Analysis
Proteins extracted from cells were separated by SDS-PAGE on polyacrylamide gels and subsequently transferred by electrophoresis onto nitrocellulose membranes. The membranes were then probed first with the primary antibody and then a horseradish peroxidase–coupled secondary antibody. Protein staining was detected with a chemiluminescence system (Amersham-Pharmacia Biotech, Piscataway, NJ) and quantified by densitometric analysis (Perkin-Elmer, Waltham, MA).
G2/M Checkpoint Analysis
After the cells were harvested, washed with PBS, and fixed with 1% formaldehyde for 10 minutes at 37°C, they were permeabilized with 0.1% Triton X-100 in PBS at room temperature for 15 minutes. Next, the cells were washed with PBS, blocked with the incubation buffer (0.5% bovine serum albumin in PBS) for 10 minutes, and stained with anti-phospho-histone H3 (S10) antibody overnight at 4°C. After a second staining with the FITC-conjugated secondary antibody, the cells were washed and resuspended in PBS containing 50 μg/mL of propidium iodide (PI). At least 10,000 cells were analyzed by Cytomics FC500 using CXP software (Beckman Coulter, Brea, CA).
Comet Assay
All comet assays were performed per the Trevigen Comet Assay kit protocol. Briefly, the cells were first treated with UV or NCS, then trypsinized at 37°C for 5 minutes. On average, the resulting final cell density was roughly 1 × 105 cells/mL. A total of 50 μL of the cell suspension was then mixed with 500 μL of molten low-melt agarose at 37°C. Next, 75 μL of the cell/agarose mixture was transferred onto glass slides, which were then immersed in a prechilled lysis buffer (2.5M NaCl, 100 mmol/L EDTA, 10 mmol/L Tris, pH 10.0, 1% Triton X-100, and 10% dimethyl sulfoxide) for 1 hour, before being electrophoresed in a 1× Tris-borate-EDTA buffer at 1 V/cm for 20 minutes at room temperature. After electrophoresis, slides were dehydrated in 70% alcohol for 5 minutes and air-dried overnight. Cells were then stained with diluted SYBR Green for 1 hour. At this time, fluorescence microscopy was used for visualization and all images were captured with a cooled CCD camera. For all colonies of at least 100 cells, the mean ± SE was calculated for each treatment group.
Statistical Analysis
In all cases, results are presented as mean ± SD. Statistical analysis was performed using SPSS software, version 12.0 (SPSS Inc., Chicago, IL). A two-tailed unpaired Student's t-test or analysis of variance test was used for data analysis. Differences were defined as statistically significant if P < 0.05.
Results
Up-Regulation and Nucleoplasmic Mobilization of GLTSCR2 in Response to DNA Damage
To determine whether GLTSCR2 is involved in the DDR, we initially assessed the changes in GLTSCR2 expression and localization within SK-Hep-1 cells after exposure to UV or ionizing radiation (IR) or treatment with the IR-mimetic agent NCS. As shown in Figure 1A, GLTSCR2 expression began to increase 10 minutes after UV exposure, with the peak levels between 30 minutes and 2 hours. Increases in phospho-ATM and γH2AX, both key proteins associated with cellular response to DNA damage, paralleled the increases in GLTSCR2 (Figure 1A). GLTSCR2 up-regulation was also observed in NCS-treated) or IR-exposed cells (Figure 1A). We next evaluated the intracellular redistribution of GLTSCR2 after treatment with UV or NCS because most, if not all, nucleolar proteins shuttle between the nucleolus and nucleoplasm during periods of genotoxic stress to perform their specific functions.12 Both control and UV-exposed cells were immunostained with anti-GLTSCR2 and anti-nucleolin antibodies before visualization with the Nomarski differential interference contrast–equipped inverted confocal microscope to identify DAPI-stained nuclei. Although among control cells most of the GLTSCR2 signal was located in the nucleolus,in UV-exposed cells GLTSCR2 was dispersed diffusely throughout the nucleoplasm, implying mobilization from the nucleolus (Figure 1B). This release of GLTSCR2 is not the result of nucleolar disassembly secondary to UV exposure because the nucleolar protein nucleolin remained localized in the nucleolus and the nucleolar architecture was observed to be well preserved (Figure 1B). Like in the UV-exposed cells, a nucleoplasmic mobilization of GLTSCR2 was also observed in NCS-treated or IR-exposed cells (Figure 1B). In combination, these findings suggest that GLTSCR2 is up-regulated and mobilized to the nucleoplasm in response to DNA damage.
Figure 1.
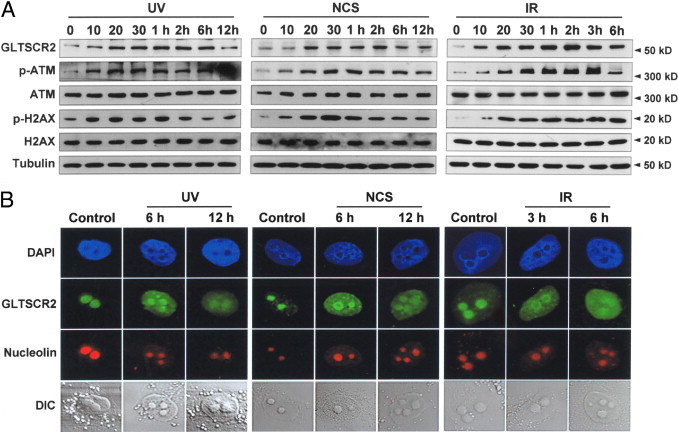
GLTSCR2 is up-regulated and mobilized to the nucleoplasm in response to genotoxic stresses. A: SK-Hep-1 cells were exposed to UV (10 J/m2) (left) or irradiated with gamma rays (2 Gy) (right) and cultured under normal conditions for the indicated durations or treated with NCS (50 ng/mL) for varying durations as indicated (middle). Protein lysates were subjected to Western blots for the relative quantitative evaluation of GLTSCR2, phospho-ATM, ATM, phospho-H2AX, and H2AX. Tubulin was used as the loading control. B: Cells were untreated or treated as in panel A and fixed after the indicated times. Then, cells were co-immunostained with anti-GLTSCR2 and antinucleolin antibodies and viewed after nuclear staining with DAPI under the differential interference contrast–equipped inverted confocal microscope. Representative images are shown.
Down-Regulation of GLTSCR2 Disrupts Nuclear Foci Formation of γH2AX
In cases of UV-associated DSBs, histone H2AX is quickly phosphorylated to γH2AX (p-S139) to generate nuclear foci around the DSB site, a process that is critical for efficient DDR and genome stability maintenance.5 To further characterize the role of GLTSCR2 in DDR, we quantified the phosphorylation levels and γH2AX nuclear foci formation efficiency in cells exposed to UV. Initially, SK-Hep-1 cells were infected with either a doxycycline-inducible (Tet-Off system) GFP-tagged GLTSCR2-expressing adenovirus (Ad-GLT/GFP) or a control virus (Ad-GFP) and placed in media containing differing concentrations of doxycycline for 24 hours. H2AX phosphorylation status was then determined using Western blot. As depicted in Figure 2A, H2AX phosphorylation occurred in proportion to GLTSCR2 expression levels. To further demonstrate the effect of GLTSCR2 in H2AX phosphorylation, SK-Hep-1 cells were stably infected with either a GLTSCR2-targeting shRNA expression lentivirus (shGLT) or a scrambled shRNA lentivirus (shScr). Subsequent immunoblotting confirmed that the expression of GLTSCR2 was significantly reduced by more than 80% in stable cells (Figure 2B). After exposure to UV, the phosphorylation status of H2AX was determined again using Western blot. As shown in Figure 2C, GLTSCR2 knockdown both reduced and delayed H2AX phosphorylation. Next, we evaluated whether GLTSCR2 knockdown affected γH2AX nuclear foci formation. SK-shGLT and SK-shScr cells were either exposed to UV or treated with NCS for a total of 6 hours of genotoxic stress and then immediately immunostained with an anti-γH2AX antibody. As shown in Figure 2, D and E, γH2AX nuclear foci formation was significantly reduced in GLTSCR2 knockdown cells when compared with SK-shScr cells. These results imply that GLTSCR2 plays an important role in both H2AX phosphorylation and nuclear foci formation.
Figure 2.
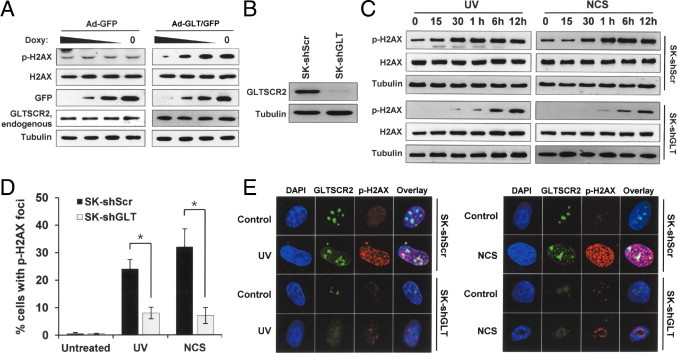
GLTSCR2 is involved in the phosphorylation of H2AX. A: SK-Hep-1 cells were infected by Ad-GFP or Ad-GLT/GFP and incubated in varying concentrations of doxycycline (50, 20, 10 ng/mL) for 24 hours. The resulting protein lysates were then analyzed using Western blot, where anti-H2AX, anti-H2AX, anti-GFP, and anti-GLTSCR2 antibodies were used to detect each target protein. Tubulin was used as the loading control. B: SK-Hep-1 cells were stably infected by either shRNA targeted to GLTSCR2 (SK-shGLT) or scrambled shRNA (SK-shScr), and GLTSCR2 knockdown was assessed using Western blot using an anti-GLTSCR2 antibody. C: SK-shScr and SK-shGLT cells were either exposed to UV (10 J/m2) and harvested at the indicated time points (left) or treated with NCS (50 ng/mL) for the indicated times (right). The resulting cell lysates were then analyzed using Western blot to detect phosphorylated H2AX or the total form of H2AX. Tubulin was used as the loading control. D: SK-shScr or SK-shGLT cells were either untreated or exposed to UV (10 J/m2) or NCS (50 ng/mL). After 12 hours of UV or NCS exposure, the cells were co-immunostained with anti-GLTSCR2 and anti-H2AX antibodies. Next, more than 200 cells were identified so that the number of cells with H2AX nuclear foci formation could be counted by confocal microscopy after nuclear staining with DAPI. For all three independent experiments, data are presented as mean ± SD (*P < 0.01). E: Representative picture of D.
Suppression of GLTSCR2 Sensitizes Cells to DNA Damage
To determine whether GLTSCR2 knockdown alters cell susceptibility to DNA damage, we performed a clonogenic survival assay after exposing two clones (1 and 2) of SK-shGLT and SK-shScr cells to either UV or NCS. Although GLTSCR2 overexpression is known to be cytotoxic,8 SK-shGLT cells were more sensitive to UV and NCS exposure than the control SK-shScr cells (Figure 3, A and B). These findings indicate that GLTSCR2 contributes to cell survival during the DDR. Differences in colony survival rate may be due to either mitotic catastrophe caused by entry into mitosis in the presence of DNA damage or incomplete DNA repair. To determine whether SK-shGLT cells undergo mitotic catastrophe after DNA damage, we performed co-immunofluorescence staining for phosphorylated histone H3 and activated caspase-3. However, significant differences between SK-shScr and SK-shGLT cells in mitotic catastrophe were not observed (Figure 3C). Next, Comet assays of individual cell nuclei were performed to determine whether DNA repair is delayed by defective damage sensing due to GLTSCR2 knockdown. SK-shGLT cells had significantly longer and denser comet movement (ie, evidence of excessive, unrepaired DNA damage in their respective genomes) when compared with SK-shScr cells (Figure 3, D and E). These results demonstrate that DNA damage persists longer after UV and NCS exposure in GLTSCR2-knockdowned SK-shGLT cells.
Figure 3.
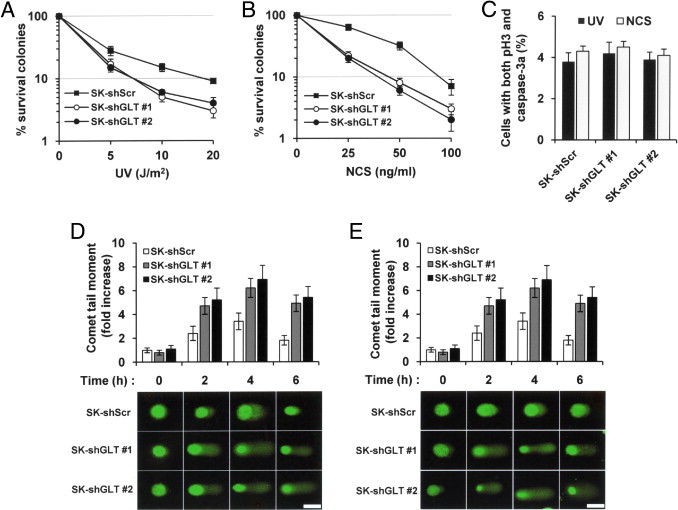
Down-regulation of GLTSCR2 sensitizes cells to DNA damage. A–B: Control (SK-shScr) and GLTSCR2 knockdown (SK-shGLT, 1 and 2) cells were exposed to the indicated doses of UV (A) or treated with the indicated concentrations of NCS (B). Clonogenic assays were then performed, as described in Materials and Methods, with the number of surviving colonies shown here. All data are from the three independent experiments are presented as mean ± SD (P < 0.01). C: Indicated cells were treated as in panel A and co-immunostainined with anti-phospho-histone H3 (pH3) and anti-active-caspase-3 (caspase-3a) antibodies. Data represent the mean numbers of cells that were positive for both pH3 and caspase-3a as determined by three independent experiments. One hundred cells were counted per sample. D–E: Excessive, persistent DNA damage in GLTSCR2-knockdown SK-shGLT cells. Repair of DSBs was detected in SK-shScr and SK-shGLT cells at the indicated time points both before and after exposure to UV (D) or NCS (E). The histograms display relative comet tail length quantification, as measured by comet tail movement and normalized to control cells (SK-shScr). Assays were performed in quadruplicate, and all data are presented as mean ± SD. The lower panels denote representative examples of single cells that were gel electrophoresed under alkaline conditions and subsequently stained with SYBR Green to demonstrate fast-migrating damaged DNA in the comet tails. Scale bar = 10 μm.
GLTSCR2 Regulates Chk1 Activation and G2/M Checkpoint Activation after DNA Damage
Checkpoint activation is crucial to the cellular responses to DNA damage because this process either inhibits further cell cycle progression until the damaged DNA is repaired or induces apoptosis when repair is not possible.13 Consequently, we assessed whether GLTSCR2 is also involved in G2/M checkpoint regulation. SK-shScr and SK-shGLT cells were exposed to either UV or NCS, after which the resulting cell lysates were evaluated using Western blot to quantify the levels of several specific proteins related to checkpoint activation, including ATM, ATR, Chk1, and Chk2. As shown in Figure 4A, treatment with UV or NCS resulted in a time-dependent increase of activated ATM (phosphorylated at Ser1981) in both SK-shScr and SK-shGLT cells. However, not only was the phosphorylation of ATM markedly delayed, but also the levels of phosphorylated ATM were significantly lower in SK-shGLT cells at each time point. Similarly, phosphorylation of the ATM-downstream substrate Chk2 was also reduced in SK-shGLT cells when compared with SK-shScr cells. Like the ATM-Chk2 pathway, both the phosphorylation of ATR (another apical kinase in the DDR) and its target protein Chk1 were markedly reduced in SK-shGLT cells. Such results indicate that the activity of both the ATM-Chk2 and ATR-Chk1 pathway are reduced by the down-regulation of GLTSCR2. Next, we further evaluated the role of GLTSCR2 in G2/M checkpoint activation via G2/M checkpoint assay. Although an arrest of the cell cycle at the G2/M transition allows DNA lesion repair under normal circumstances, such an arrest at the G2/M transition cannot occur if the G2/M checkpoint is abolished. Down-regulation of GLTSCR2 with transient transfection of shGLT did not affect cell cycle profile of Sk-Hep-1 cells in time course experiment (Figure 4B). However, as shown in Figure 4, C and D, DNA damage secondary to either UV or NCS exposure, respectively, reduced the phospho-histone H3 antibody-marked mitotic population to 0.6% and 0.4% in SK-shScr cells, whereas these same mitotic populations were 1.6% in untreated control cells. Conversely, in SK-shGLT cells the corresponding mitotic populations were 1.2% and 0.9% after UV and NCS exposure and 1.8% in untreated cells. These findings indicate that G2/M checkpoint could not be activated in cells with decreased levels of GLTSCR2 expression.
Figure 4.
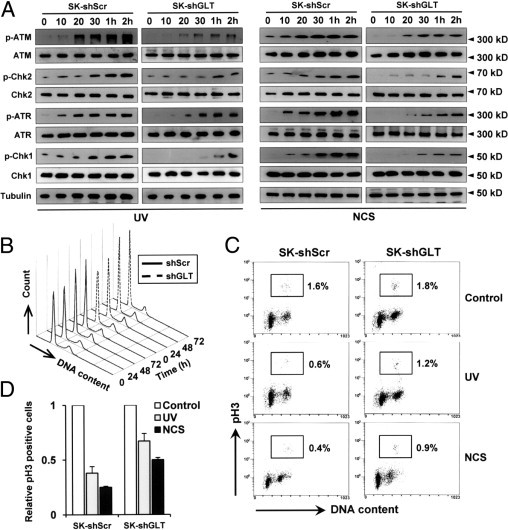
GLTSCR2 is involved in G2/M checkpoint activation. A: Control (SK-shScr) and GLTSCR2 knockdown (SK-shGLT) cells were exposed to 10 J/m2 UV (left) or 50 ng/mL of NCS (right). Cells were harvested at the indicated time points and the resulting lysates were analyzed using Western blot analysis to detect the presence of phospho-ATM, ATM, phospho-Chk2, Chk2, phospho-ATR, ATR, phospho-Chk1, and Chk1. All loadings were normalized to tubulin. B: SK-Hep-1 cells were transfected with shRNA targeted to GLTSCR2 (shGLT) or scrambled shRNA (shScr). Cell cycle profiles were analyzed at the indicated time points after PI staining. Representative plots of three independent experiments are shown. C: SK-shScr and SK-shGLT cells were untreated (control) or treated as in panel A. After 12 hours of UV or NCS exposure, the number of cells in mitotic phase was determined by flow cytometric analysis and stained with FITC-conjugated anti-phospho-histone H3 antibody and PI. The total percentages of cells in mitotic phase are shown here. D: Data represent relative phospho-histone H3-positive cells compared with untreated control cells; data are presented as mean ± SD.
Discussion
Efficient repair of damaged DNA is mediated by a complicated network of sensor and effector molecules and is essential for both cell survival and neoplastic transformation suppression. Although several reports suggest that GLTSCR2 primarily functions as a putative tumor suppressive gene,7–10 the molecular mechanisms for this inhibition of tumor development and/or growth are poorly understood. Here, we report that GLTSCR2 is a member of a class of nucleolar proteins, which are all involved in the DDR via the modulation of the ATM-Chk2 and ATR-Chk1 pathways.
Our preliminary data indicate that exogenous genotoxic stresses, such as cellular exposure to UV or NCS, mediate the up-regulation of GLTSCR2 and subsequent translocation to the nucleoplasm. Previously, several nucleolar proteins have been demonstrated to shuttle between the nucleolus and nucleoplasm in response to ribotoxic stresses, thereby directly inhibiting cell cycle progression and/or participating in DDR.14 Specifically, nucleophosmin mobilizes to the nucleoplasm, where it acts to stabilize p53 by inactivating MDM2, ultimately inhibiting cell cycle progression.15 In addition, DNA damage facilitates the redistribution of the nucleolar tumor suppressor protein ARF throughout the nucleoplasm.16 Our data, which show that cellular DNA damage up-regulates and redistributes GLTSCR2 to the nucleoplasm, provide further evidence that GLTSCR2 actively participates in the DDR. However, unlike the nuclear foci patterns associated with the ATM or γH2AX response, GLTSCR2 was diffusely dispersed throughout the nucleus without any indication of foci formation. Furthermore, GLTSCR2 was not detected in either the ATM or γH2AX immunoprecipitates (data not shown). Together, these findings suggest that GLTSCR2 likely modulates and facilitates the damage response proteins in the DDR process, rather than directly repairing DNA damage.
When genomic damage occurs, H2AX is phosphorylated at serine 139 to generate γH2AX, a molecule that serves to aggregate nuclear foci at the sites of DNA damage, which act as platforms for DDR proteins.17 Such processes are critical not only for recruiting downstream DNA damage and repair proteins but also for the amplification of DDR signaling.18 Our data show that the shRNA-targeted down-regulation of GLTSCR2 reduces both the phosphorylation of H2AX and the resulting γH2AX formation of nuclear foci. Under normal circumstances, H2AX is phosphorylated by the apical kinases ATM or ATR19,20 and dephosphorylated by one of several protein phosphatases, including PP2A, PP4, and Wip1.21–23 Accordingly, two possibilities exist for the role of GLTSCR2 in the DDR process: either GLTSCR2 up-regulates H2AX activation via the ATM-Chk2 and/or ATR-Chk1 pathway (thereby mediating the phosphorylation of these apical kinases) or negatively modulates the activity of the γH2AX phosphatases. Although the latter scenario cannot be completely discarded, the attenuation of ATM and ATR phosphorylation in SK-shGLT cells under genotoxic conditions strongly suggests the involvement of GLTSCR2 in both the ATM-Chk2-γH2AX and ATR-Chk1-γH2AX pathways.
In cases where DNA is incompletely repaired, certain checkpoint proteins are activated, which halt the cell cycle via a Chk-1– or Chk-2–dependent degradation of Cdc25A, ultimately preventing cells with unrepaired DNA from entering mitosis.24,25 Here, we demonstrate that not only does GLTSCR2 mediate the activation of ATM-Chk2 and ATR-Chk1 activation in response to DNA damage but also the G2/M checkpoint is unable to inhibit cell cycle progression in GLTSCR2-knockdown SK-shGLT cells. In combination with the aforementioned comet and clonogenic assays, these results demonstrate that GLTSCR2 down-regulation results in DNA repair delay and G2/M checkpoint inactivation.
The molecular mechanism for DDR modulation by GLTSCR2 is unclear and currently under investigation. However, it may be explained in part by histone modification in DDR. Acetylation/deacetylation of histones plays a pivotal role in DDR by allowing the DDR protein to access sites of DNA damage and to interact with chromatin.26 The interaction of ATM with chromatin is crucial for optimal ATM activation and subsequent recruitment of DDR proteins.27 Knockdown of GLTSCR2 may induce improper or inadequate histone modification in DDR, thereby resulting in suppression of ATM-H2AX activation and DDR. Thus, histone modification, including acetylation/deacetylation or methylation, is a putative mechanism for the involvement of GLTSCR2 in DDR.
In summary, GLTSCR2 is up-regulated and redistributed throughout the nucleoplasm in response to cellular DNA damage. Down-regulation of GLTSCR2 not only decreases γH2AX foci formation and thereby sensitizes cells to DNA damage but also prevents G2/M checkpoint activation. Although the exact molecular mechanisms for this checkpoint regulation have not been elucidated, our results support the categorization of GLTSCR2 as a DNA damage responsive protein.
Footnotes
Supported by a grant from the Korea Science and Engineering Foundation funded by the Korea government (R13-2002-020-02002-0).
References
- 1.Rouse J., Jackson S.P. Interfaces between the detection, signaling, and repair of DNA damage. Science. 2002;297:547–551. doi: 10.1126/science.1074740. [DOI] [PubMed] [Google Scholar]
- 2.Harper J.W., Elledge S.J. The DNA damage response: ten years after. Mol Cell. 2007;28:739–745. doi: 10.1016/j.molcel.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 3.Chen Y., Chen P.L., Chen C.F., Jiang X., Riley D.J. Never-in-mitosis related kinase 1 functions in DNA damage response and checkpoint control. Cell Cycle. 2008;7:3194–3201. doi: 10.4161/cc.7.20.6815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stucki M., Jackson S.P. GammaH2AX and MDC1: anchoring the DNA-damage-response machinery to broken chromosomes. DNA Repair. 2006;5:534–543. doi: 10.1016/j.dnarep.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 5.Paull T.T., Rogakou E.P., Yamazaki V., Kirchgessner C.U., Gellert M., Bonner W.M.A. Critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol. 2000;10:886–895. doi: 10.1016/s0960-9822(00)00610-2. [DOI] [PubMed] [Google Scholar]
- 6.Mah L.J., El-Osta A., Karagiannis T.C. GammaH2AX: a sensitive molecular marker of DNA damage and repair. Leukemia. 2010;24:679–686. doi: 10.1038/leu.2010.6. [DOI] [PubMed] [Google Scholar]
- 7.Okahara F., Itoh K., Nakagawara A., Murakami M., Kanaho Y., Maehama T. Critical role of PICT-1, a tumor suppressor candidate, in phosphatidylinositol 3,4,5-trisphosphate signals and tumorigenic transformation. Mol Biol Cell. 2006;17:4888–4895. doi: 10.1091/mbc.E06-04-0301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yim J.H., Kim Y.J., Ko J.H., Cho Y.E., Kim S.M., Kim J.Y., Lee S., Park J.H. The putative tumor suppressor gene GLTSCR2 induces PTEN-modulated cell death. Cell Death Differ. 2007;14:1872–1879. doi: 10.1038/sj.cdd.4402204. [DOI] [PubMed] [Google Scholar]
- 9.Okahara F., Ikawa H., Kanaho Y., Maehama T. Regulation of PTEN phosphorylation and stability by a tumor suppressor candidate protein. J Biol Chem. 2004;279:45300–45303. doi: 10.1074/jbc.C400377200. [DOI] [PubMed] [Google Scholar]
- 10.Kim Y.J., Cho Y.E., Kim Y.W., Kim J.Y., Lee S., Park J.H. Suppression of putative tumour suppressor gene GLTSCR2 expression in human glioblastomas. J Pathol. 2008;216:218–224. doi: 10.1002/path.2401. [DOI] [PubMed] [Google Scholar]
- 11.Hemmati P.G., Gillissen B., von Haefen C., Wendt J., Stärck L., Güner D., Dörken B., Daniel P.T. Adenovirus-mediated overexpression of p14ARF induces p53 and Bax-independent apoptosis. Oncogene. 2002;21:3149–3161. doi: 10.1038/sj.onc.1205458. [DOI] [PubMed] [Google Scholar]
- 12.Sherr C.J. The INK4a/ARF network in tumour suppression. Nat Rev Mol Cell Biol. 2001;2:731–737. doi: 10.1038/35096061. [DOI] [PubMed] [Google Scholar]
- 13.Smith J., Tho L.M., Xu N., Gillespie D.A. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv Cancer Res. 2010;108:73–112. doi: 10.1016/B978-0-12-380888-2.00003-0. [DOI] [PubMed] [Google Scholar]
- 14.Tembe V., Henderson B.R. Protein trafficking in response to DNA damage. Cell Signal. 2007;19:1113–1120. doi: 10.1016/j.cellsig.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 15.Gjerset R.A. DNA damage, p14ARF, nucleophosmin (NPM/B23), and cancer. J Mol Histol. 2006;37:239–251. doi: 10.1007/s10735-006-9040-y. [DOI] [PubMed] [Google Scholar]
- 16.Lee C., Smith B.A., Bandyopadhyay K., Gjerset R.A. DNA damage disrupts the p14ARF-B23(nucleophosmin) interaction and triggers a transient subnuclear redistribution of p14ARF. Cancer Res. 2005;65:9834–9842. doi: 10.1158/0008-5472.CAN-05-1759. [DOI] [PubMed] [Google Scholar]
- 17.Yuan J., Adamski R., Chen J. Focus on histone variant H2AX: to be or not to be. FEBS Lett. 2010;584 doi: 10.1016/j.febslet.2010.05.021. 3717-3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bassing C.H., Suh H., Ferguson D.O., Chua K.F., Manis J., Eckersdorff M., Gleason M., Bronson R., Lee C., Alt F.W. Histone H2AX: a dosage-dependent suppressor of oncogenic translocations and tumors. Cell. 2003;114:359–370. doi: 10.1016/s0092-8674(03)00566-x. [DOI] [PubMed] [Google Scholar]
- 19.Burma S., Chen B.P., Murphy M., Kurimasa A., Chen D.J. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem. 2001;276:42462–42467. doi: 10.1074/jbc.C100466200. [DOI] [PubMed] [Google Scholar]
- 20.Ward I.M., Chen J. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J Biol Chem. 2001;276:47759–47762. doi: 10.1074/jbc.C100569200. [DOI] [PubMed] [Google Scholar]
- 21.Chowdhury D., Keogh M.C., Ishii H., Peterson C.L., Buratowski S., Lieberman J. Gamma-H2AX dephosphorylation by protein phosphatase 2A facilitates DNA double-strand break repair. Mol Cell. 2005;20:801–809. doi: 10.1016/j.molcel.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 22.Chowdhury D., Xu X., Zhong X., Ahmed F., Zhong J., Liao J., Dykxhoorn D.M., Weinstock D.M., Pfeifer G.P., Lieberman J. A PP4-phosphatase complex dephosphorylates gamma-H2AX generated during DNA replication. Mol Cell. 2008;31:33–46. doi: 10.1016/j.molcel.2008.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Macurek L., Lindqvist A., Voets O., Kool J., Vos H.R., Medema R.H. Wip1 phosphatase is associated with chromatin and dephosphorylates gammaH2AX to promote checkpoint inhibition. Oncogene. 2010;29:2281–2291. doi: 10.1038/onc.2009.501. [DOI] [PubMed] [Google Scholar]
- 24.Mailand N., Podtelejnikov A.V., Groth A., Mann M., Bartek J., Lukas J. Regulation of G(2)/M events by Cdc25A through phosphorylation-dependent modulation of its stability. EMBO J. 2002;21:5911–5920. doi: 10.1093/emboj/cdf567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiao Z., Chen Z., Gunasekera A.H., Sowin T.J., Rosenberg S.H., Fesik S., Zhang H. Chk1 mediates S and G2 arrests through Cdc25A degradation in response to DNA-damaging agents. J Biol Chem. 2003;278:21767–21773. doi: 10.1074/jbc.M300229200. [DOI] [PubMed] [Google Scholar]
- 26.Pena A.N., Pereira-Smith O.M. The role of the MORF/MRG family of genes in cell growth, differentiation: DNA repair, and thereby aging. Ann N Y Acad Sci. 2007;1100:299–305. doi: 10.1196/annals.1395.031. [DOI] [PubMed] [Google Scholar]
- 27.Kim Y.C., Gerlitz G., Furusawa T., Catez F., Nussenzweig A., Oh K.S., Kraemer K.H., Siloh Y., Bustin M. Activation of ATM depends on chromatin interactions occurring before induction of DNA damage. Nat Cell Biol. 2009;11:92–96. doi: 10.1038/ncb1817. [DOI] [PMC free article] [PubMed] [Google Scholar]