

Abstract
Cytoglobin (Cygb) is a recently discovered vertebrate globin with molecular characteristics that are similar to myoglobin. To study the biological function of Cygb in vivo, we generated Cygb knockout mice and investigated their susceptibility to N,N-diethylnitrosamine (DEN)–induced tumorigenesis. Four-week-old male mice were administered DEN in drinking water at a dose of 25 ppm for 25 weeks or 0.05 ppm for 36 weeks. Cygb deficiency promoted the DEN-induced development of liver and lung tumors. All Cygb+/− and Cygb−/− mice treated with 25-ppm DEN exhibited liver tumors, compared with 44.4% of their wild-type counterparts. Lung tumors were present only in Cygb-deficient mice. More than 40% of Cygb−/− mice developed liver and lung tumors at the nontoxic dose of DEN (0.05 ppm), which did not induce tumors in wild-type mice. Cygb loss was associated with increased cancer cell proliferation, elevated extracellular signal–regulated kinase and Akt activation, overexpression of IL-1β, IL-6, Tnfα, and Tgfβ3 mRNAs, and hepatic collagen accumulation. Cygb-deficient mice also exhibited increased nitrotyrosine formation and dysregulated expression of cancer-related genes (cyclin D2, p53, Pak1, Src, Cdkn2a, and Cebpa). These results suggest that Cygb deficiency induces susceptibility to cancer development in the liver and lungs of mice exposed to DEN. Thus, globins such as Cygb will shed new light on the biological features of organ carcinogenesis.
Cytoglobin (Cygb) was originally identified in 2001 as a protein up-regulated in rat hepatic stellate cells under profibrotic conditions. Accordingly, Cygb was originally termed a stellate cell activation-associated protein1 until it was identified as the fourth globin in mammals.2,3 Human Cygb displays approximately 25% amino acid identity with vertebrate myoglobin and hemoglobin and 16% identity with human neuroglobin. The Cygb gene is localized to chromosome 17q25.3 in humans and chromosome 11E2 in mice.
Unlike myoglobin, which is tissue restricted to cardiomyocytes and skeletal myofibers, hemoglobin in erythrocytes, and neuroglobin in the nervous system, Cygb is ubiquitously expressed in the cytoplasm of mesenchymal fibroblastic cells in many organs, including the heart, lung, liver, kidneys, small intestine, and spleen. The presence of Cygb in the nucleus of these cells has also been reported.4,5 In particular, Cygb was present in stellate cells and myofibroblasts in the liver and pancreas, reticulocytes in the spleen, mesenchymal cells in the submucosal layer of the gut, and the mesangium and stromal cells of the kidney.4 An interesting aspect of Cygb expression is its presence in visceral cells, with a strong storage ability for vitamin A. Thus, Cygb may facilitate the diffusion of oxygen through tissues, scavenge nitric oxide (NO) or other reactive oxygen species, or serve a protective function during oxidative stress.6 However, the precise physiological role of Cygb in vivo remains unresolved. Cygb is considered a hypoxia-responsive molecule because its mRNA expression is augmented under hypoxia in fibroblastic cell lineages and rat brain.7 Hypoxia-inducible factor 1 is assumed to be an important transcription factor for Cygb because hypoxia-responsive elements at positions -141, -144, and -448 are essential for the activation of CYGB gene expression, and the binding of hypoxia-inducible factor 1 to this area has been confirmed.8,9 In contrast, CYGB overexpression rescues the human neuronal cell line TE671 from prooxidant Ro19-8022–induced DNA damage.10 CYGB overexpression also protected human neuroblastoma SH-SY5Y cells from H2O2-induced cell death.11,12 Furthermore, the in vitro and in vivo overexpression of Cygb in rat hepatic stellate cells protected these cells against oxidative stress and inhibited their differentiation into an active phenotype.13 Together, these reports suggest that Cygb may act as a cytoprotective and radical-scavenging molecule in addition to its function as a gas carrier.
Although the function of Cygb in vivo remains largely unknown, down-regulation of CYGB has been reported in several human cancerous tissues and human cancer cell lines. Decreased expression of CYGB and the hypermethylation of the CYGB promoter has been reported in patients with tylosis, non–small-cell lung carcinoma tissues, head and neck cancers, ovarian cancers, and breast cancers.14–18 McRonald et al14 reported that CYGB gene expression in tylosis with esophagus cancer was reduced to approximately 70% compared with the normal esophagus and was accompanied by hypermethylation of the promoter. Xinarianos et al15 reported a significant reduction of CYGB mRNA expression in non-small-cell lung carcinoma tissues and hypermethylation of CYGB, compared with healthy samples. Similar results were reported in head and neck, ovarian, and breast cancer tissues.16–18 In addition, Shivapurkar et al19 reported high levels of CYGB promoter methylation in lung, breast, bladder, and colon cancers and in leukemia in humans. The augmented growth of NCI-H661 lung cancer cells that were CYGB silenced by RNA interference and the suppression of NCI-H228 cell proliferation in cells stably transfected with plasmids containing CYGB cDNA have also been reported.19 These reports indicate a tumor suppressor function of Cygb.
To study the biological function of Cygb at the tissue level, we first generated Cygb-deficient (Cygb−/−) mice and observed that, after treatment with N,N-diethylnitrosamine (DEN), Cygb−/− mice showed a high incidence of tumor development in the liver and lungs. These results indicate the tumor suppressor role of Cygb in vivo.
Materials and Methods
Materials
All experimental reagents were obtained from Sigma Chemical Co (St. Louis, MO) or Wako Pure Chemical Co (Osaka, Japan), unless otherwise stated.
Construction of the Targeting Vector
Mice lacking exon 1 of the Cygb gene were generated using the lox-P system, as previously described.20 The targeting vector (pTVneo/Cygb) was constructed from PCR DNA fragments from 129Sv mouse genomic DNA. Two DNA fragments were used as the 3′ and 5′ arms. One fragment contained Cygb exons 2, 3, and 4 (6.5 kb); and the other contained the transcription initiation site in exon 1 and the 5′ upstream sequence of the Cygb gene (6.0 kb). The neomycin-resistance gene, driven by the phosphoglycerate kinase 1 promoter, flanking the lox-P sequences was inserted into the arms (Figure 1A).
Figure 1.
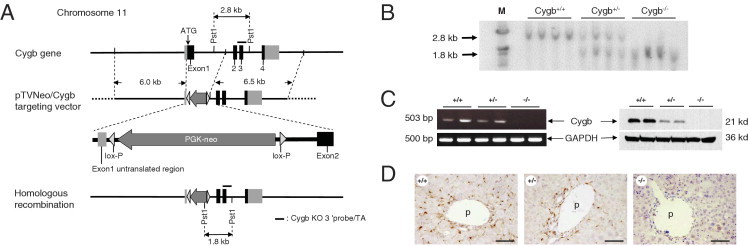
Generation of Cygb-deficient mice. A: Strategy for inactivation of the Cygb gene by homologous recombination in embryonic stem cells. A partial genomic map of the Cygb gene with coding exons (black boxes), noncoding regions (light gray boxes), and flanking introns (solid lines) is shown (top). Targeting vector pTVneo/Cygb with homology to the Cygb gene locus is shown (middle). The translation initiation site in exon 1 was replaced with a neomycin-resistance cassette. The predicted Cygb gene locus after homologous recombination is shown (bottom). B: Southern blot analysis shows the sizes of the wild-type (2.8-kb) and disrupted (1.8-kb) Cygb fragments after PstI cleavage. M, molecular weight marker. C: RT-PCR and immunoblot analysis of Cygb expression in the liver from Cygb+/+, Cygb+/−, and Cygb−/− mice. GAPDH was used as a loading control. D: Representative IHC images of Cygb in the liver of Cygb+/+, Cygb+/−, and Cygb−/− mice. Cygb is present along the sinusoids of Cygb+/+ mice, whereas it is undetectable in Cygb−/− mice. p, portal vein. Scale bar = 50 μm.
Embryonic stem cells (1 × 107 cells/mL) were transfected with a linearized targeting vector (20 μg) by electroporation and cultured in selection medium containing 150 μg/mL geneticine (G418). Of 480 neomycin-resistant clones, 6 (1.2%) were homologous recombinants by Southern blotting using the 5′ probe (data not shown).
Production of Cygb-Deficient Mice
Two clones were aggregated with C57BL/6-DBA2 F1 mouse morulae, and one produced chimeric mice that transmitted the knockout (KO) construct. Chimeric males were mated with C57BL/6J females to obtain Cygb heterozygous mice that were backcrossed to the C57BL/6J background for more than nine generations. To assess the role of the Cygb gene in development, we intercrossed Cygb heterozygous mice. The litter sizes were normal, and analysis of the tail biopsy specimens at the age of 4 weeks, from 102 offspring from heterozygote crosses, revealed the presence of homozygous mutant mice at a frequency of 24%. The homozygotes appeared normal morphologically and histopathologically 1 month after birth. Four-week-old Cygb+/+ (wild-type), Cygb+/− (heterozygote), and Cygb−/− (homozygote) male mice were used in this study.
All mice were cared for according to the guidelines approved by the Institutional Animal Care and Use Committee of Osaka City University, Osaka.
Genotyping of Mice
PCR genotyping of mouse tail DNA produced the expected 338-bp product from wild-type mice using the following primer pairs: forward, 5′-CTCCCAGCCGGGACCGCGGTGGCCTT-3′; and reverse, 5′-GGAGCCGAGGCCGGTGCGTGCGAGGC-3′. A 529-bp product was diagnostic for the Cygb KO allele using the described forward primer and the following reverse primer: 5′-GTGGGGTGGGATTAGATAAATGCCTGCTCT-3′. PCR was performed in a 15-μL reaction mixture containing 1 μL of extract from mouse tail DNA, which was digestion extracted using LYPPO (Gene Modification R&D Co Ltd, Osaka), according to the manufacturer's protocol; 1 μmol/L of each primer; 2.5% to 5% dimethyl sulfoxide; and 0.5 U GO Taq polymerase (Promega, San Luis Obispo, CA). PCRs were performed for 40 cycles, each cycle being 1 minute at 94°C, 30 seconds at 70°C, and 1 minute at 72°C.
Southern blot analysis confirmed the Cygb-null allele. Genomic tail DNA (5 μg) was cleaved with PstI, subjected to agarose gel electrophoresis, blotted onto nylon membranes, and hybridized to the 32P-labeled Cygb KO 3′ probe/TA (Figure 1A). DNA fragments of 2.8 and 1.8 kb represented the Cygb wild-type and null alleles, respectively.
RT-PCR was performed to confirm the absence of Cygb mRNA in tissues. Total RNA was extracted from the homogenates of liver tissues using the RNeasy Mini Kit (Qiagen, Valencia, CA). cDNA was synthesized using 1 μg of total RNA, ReverTra Ace (Toyobo, Osaka), and oligo (dT)12–18 primers, according to the manufacturer's instructions. Thirty-five PCR cycles (30 seconds at 94°C, 30 seconds at 68°C, and 1 minute at 72°C) were run using the following mouse Cygb primers: forward, 5′-GCGACATGGAGATAGAGCGT-3′; and reverse, 5′-CTGTACCCAGCCCACTTCCT-3′. This generated a 503-bp product and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) primers: forward, 5′-CGCCTGGTCACCAGG-3′; and reverse, 5′-CAGTTGGTGGTGCAGGA-3′). A 500-bp product was generated.
Administration of DEN
DEN (0.95 g/mL) was obtained from Sigma Chemical Co. A stock solution of DEN was prepared by dissolving 1 g (1.06 mL) of DEN in 400 mL of water. The stock solution was diluted 100-fold before use to obtain a final concentration of 25-mg DEN per 1000-mL water (25 ppm) for the high-dose experiment. The 25-ppm solution was further diluted 500-fold to obtain a final concentration of 0.05-mg DEN per 1000-mL water (0.05 ppm) for the low-dose experiment. The diluted solution (25 or 0.05 ppm) was placed in a shaded serving bottle and administered to the animals instead of water. The diluted solution was prepared weekly. The administration of DEN to male mice began at the age of 4 weeks for 25 weeks in the high-dose experiment (25 ppm) and for 36 weeks in the low-dose experiment (0.05 ppm). Each experiment contained three groups (Cygb+/+, Cygb+/−, and Cygb−/−) with a minimum of seven mice per group. Mice were sacrificed after the completion of DEN treatment.
Necropsy
At necropsy, mice were weighed, anesthetized, and examined for grossly visible lesions in whole organs. Livers and lungs were excised, weighed (in the case of liver), and examined for macroscopic lesions. The number of macroscopic abnormal masses ≥1 mm was determined in addition to the size of the mass by taking the average of the largest and smallest length of the mass. For histological examination, 2- to 3-mm-thick sections from nontumor or tumor tissues were fixed in 10% formalin for 24 hours and embedded in paraffin. The samples were then sectioned at 5 μm and stained with H&E.
IHC and TUNEL Assays
For immunohistochemistry (IHC), paraffin sections were dewaxed in xylene and rehydrated in decreasing concentrations of ethanol. The primary antibodies and conditions used for IHC are listed in Table 1. Negative controls with no primary antibody were used to assess nonspecific staining. The secondary antibodies used were horseradish peroxidase–conjugated goat anti-rabbit IgG (1:200; Dako, Glostrup, Denmark), rabbit anti-goat IgG (1:200; Dako), or rabbit anti-mouse IgG (1:200; Dako). 3,3′-Diaminobenzidine (Dako) was used as the chromogen. TdT-mediated dUTP-biotin nick-end labeling (TUNEL) staining was performed using the In situ Apoptosis Detection Kit (MK500; TaKaRa Bio Inc., Shiga, Japan), according to the manufacturer's protocol. All sections were counterstained with Meyer's hematoxylin.
Table 1.
Primary Antibodies Used for IHC Analyses
| Antigen⁎ | Source | Name/clone; catalog no. | Incubation |
|---|---|---|---|
| AFP | US Biological, Swampscott, MA | F4100-16A (Go) | Overnight 4°C, 1:20 |
| CK19 | Santa Cruz Biotechnology, Santa Cruz, CA | Polyclonal (Rb); sc-33111 | Overnight 4°C, 1:100 |
| CRBP-1 | Santa Cruz Biotechnology | Polyclonal (Rb); sc-30106 | Overnight 4°C, 1:100 |
| Cygb | Our laboratory† | Polyclonal (Rb) | Overnight 4°C, 1:100 |
| Erk | Cell Signaling, Danvers, MA | Monoclonal (Rb); 4695 | Overnight 4°C, 1:300 |
| PCNA | Dako, Glostrup, Denmark | Monoclonal (Mo)‡; clone: PC-10 | Overnight 4°C, 1:200 |
| Phosphorylated Erk | Cell Signaling | Monoclonal (Rb); 4370 | Overnight 4°C, 1:200 |
| α-SMA | Dako | Monoclonal (Mo)‡; clone: 1A4 | 30 minutes at room temperature, 1:100 |
| Desmin | Santa Cruz Biotechnology | Polyclonal (Go); sc-7559 | Overnight 4°C, 1:100 |
AFP, alpha-fetoprotein.
All antigens were retrieved by autoclaving for 15 minutes in 0.01 mol/L citrate buffer containing 0.05% Tween 20 (pH 6.0), except for desmin, which was used in Tris-EDTA buffer (pH 9.0).
Data taken from Kawada et al.1
For mouse primary antibodies, after antigen retrieval, sections were incubated with goat anti-mouse IgG Fab fragments (Jackson ImmunoResearch Laboratories) for 1 hour at room temperature (1:100) to block nonspecific background staining.
Quantification of Liver Fibrosis
Morphometric image analysis was performed in liver tissue specimens with a computerized system, consisting of a photomicroscope, a digital camera, and LuminaVision 2.4 bioimaging software (Mitani Corporation, Tokyo, Japan) to quantitatively assess fibrosis. The proportion of the area stained with Sirius red in the liver sections was calculated as the sum of the pixelwise-bound stain measurements divided by the number of summed pixels.
Quantitative Real-Time PCR
Total RNA was extracted from liver and liver tumor tissues using the RNeasy Mini Kit (Qiagen, Valencia, CA). cDNAs were synthesized as previously described. Gene expression was measured by quantitative real-time PCR using cDNA, real-time PCR Master Mix Reagents (Toyobo, Osaka), or TaqMan Fast Universal PCR Master Mix (Applied Biosystems, Foster City, CA), and a set of gene-specific oligonucleotide primers and probes (Table 2) using an Applied Biosystems Prism 7500 (Applied Biosystems). GAPDH levels were measured and used to normalize the relative abundance of mRNA.
Table 2.
Primers Used for RT-qPCR
| Gene | Sequence |
|---|---|
| AFP | |
| Forward | 5′-CACACCCGCTTCCCTCAT-3′ |
| Reverse | 5′-TTTTCGTGCAATGCTTTGGA-3′ |
| Bcl2 | |
| Forward | 5′-AAGGGCTTCACACCCAAATCT-3′ |
| Reverse | 5′-CTTCTACGTCTGCTTGGCTTTGA-3′ |
| Cdkn2a | |
| Forward | 5′-GCTCTGGCTTTCGTGAACATG-3′ |
| Reverse | 5′-GTGCGGCCCTCTTCTCAA-3′ |
| Cebpα | |
| Forward | 5′-CGCAAGAGCCGAGATAAAGC-3′ |
| Reverse | 5′-CGGTCATTGTCACTGGTCAACT-3′ |
| Col1α1 | |
| Forward | 5′-GACATCCCTGAAGTCAGCTGC-3′ |
| Reverse | 5′-TCCCTTGGGTCCCTCGAC-3′ |
| Cyclin D1 | |
| Forward | 5′-GCCCGGAGGGATTTGC-3′ |
| Reverse | 5′-AGACGGAACACTAGAACCTAACAGATT-3′ |
| Cyclin D2 | |
| Forward | 5′-AAGGCAGATACTCATCAAACACAGA-3′ |
| Reverse | 5′-CTGGTGCACGCATGCAA-3′ |
| Fos | |
| Forward | 5′-CCCCAAACTTCGACCATGAT-3′ |
| Reverse | 5′-GGAGGATGACGCCTCGTAGTC-3′ |
| GAPDH | |
| Forward | 5′-TGCACCACCAACTGCTTAG-3′ |
| Reverse | 5′-GGATGCAGGGATGATGTTC-3′ |
| IL-6 | |
| Forward | 5′-CGCTATGAAGTTCCTCTCTGCAA-3′ |
| Reverse | 5′-CACCAGCATCAGTCCCAAGA-3′ |
| IL-1β | |
| Forward | 5′-CCATGGCACATTCTGTTCAAA-3′ |
| Reverse | 5′-GCCCATCAGAGGCAAGGA-3′ |
| Jun | |
| Forward | 5′-CCGCCCCTGTCCCCTAT-3′ |
| Reverse | 5′-TCCTCATGCGCTTCCTCTCT-3′ |
| Pak1 | |
| Forward | 5′-CGTATTGCGGGTGTTTGCTA-3′ |
| Reverse | 5′-CACAGCAGGAGAACCAAAACC-3′ |
| p53 | |
| Forward | 5′-GCATGAACCGCCGACCTAT-3′ |
| Reverse | 5′-CAGAAGGTTCCCACTGGAGTCT-3′ |
| Src | |
| Forward | 5′-CCTCCCGCACCCAGTTC-3′ |
| Reverse | 5′-CATCAGCATGTTTGGAGTAGTAAGC-3′ |
| TGFβ3 | |
| Forward | 5′-AGGGCCCTGGACACCAATTAC-3′ |
| Reverse | 5′-CCTTAGGTTCTGGGACCCATTTC-3′ |
| TNFα | |
| Forward | 5′-CCTCACACTCAGATCTTCTCA-3′ |
| Reverse | 5′-GCTGCTCCTCCACTTGGTG-3′ |
| Probe | 5′-GCAAGCCTGTAGCCCACGTCGTAGCAAA-3′ |
Immunoblotting
Protein samples (10 to 20 μg) were subjected to SDS-PAGE and transferred to Immobilon P membranes (Millipore Corp, Bedford, MA). After blocking, membranes were probed with primary antibodies against Cygb (1:500) from our laboratory (Table 1), Akt (1:1000; Cell Signaling, Danvers, MA), phosphorylated Akt (1:500; Cell Signaling), extracellular signal–regulated kinase (Erk; 1:500; Cell Signaling), phosphorylated Erk (1:1000; Cell Signaling), cyclin D1 (1:5000; Cell Signaling), nitrotyrosine (1:500; Cell Signaling), or GAPDH (1:2000; Santa Cruz Biotechnology, Santa Cruz, CA). Membranes were then labeled with horseradish peroxidase–conjugated secondary antibodies. Immunoreactive bands were visualized using the ECL detecting reagent (GE Healthcare UK Ltd, Buckinghamshire, UK) and documented with an LAS 1000 (Fuji Photo Film, Kanagawa, Japan).
Data Analyses
The data presented as bar graphs are the means ± SDs in all experiments. Statistical analyses were performed using the Student's t-test, and P < 0.05 indicated statistical significance.
Results
Characterization of the Cygb−/− Mice
Cygb-deficient mice were generated by deleting exon 1 of the mouse Cygb gene (Figure 1A) and backcrossed on the C57BL/6J background. Southern blotting (Figure 1B) and PCR genotyping using mouse tail DNA (data not shown) confirmed the deletion of the Cygb gene. Both Cygb mRNA and protein expression were absent in Cygb−/− mouse livers, compared with wild-type livers (Figure 1C). Furthermore, IHC indicated that the Cygb protein was present in sinusoidal lining cells in addition to perivascular cells around the portal and central veins, as previously reported,1 in Cygb+/+ mice, whereas it was undetected in Cygb−/− mice (Figure 1D). Together, these data demonstrate the successful production of the Cygb−/− mice.
The mice homozygous for the disrupted allele appeared normal both morphologically and histopathologically 1 month after birth. Next, we examined whether Cygb influenced the toxicity and carcinogenesis of DEN in mice.
Cygb Deficiency Strongly Promotes DEN-Induced Tumorigenesis
DEN is a commonly used chemical carcinogen for the liver because it is activated by cytochrome P-450 enzymes in hepatocytes.21 C57BL/6J mice were reported to be relatively resistant to liver tumor development under DEN treatment.22,23 In this study, we examined whether the loss of Cygb could influence the toxicity and carcinogenesis of DEN in the tumor-resistant C57BL/6J strain. We administered 25-ppm DEN to the drinking water of Cygb-deficient male mice at the age of 4 weeks. Unexpectedly, as early as 25 weeks after DEN treatment, 100% of Cygb−/− and Cygb+/− mice developed tumors in the liver and lungs, whereas the tumor incidence in wild-type mice was 44.4% (Figure 2, A and B). The liver/body weight ratio was approximately 50% more than in wild-type animals (Figure 2C) because of these tumor masses. Cygb−/− mice displayed significantly larger lesions and produced tumors more frequently than wild-type mice (Figure 2C).
Figure 2.
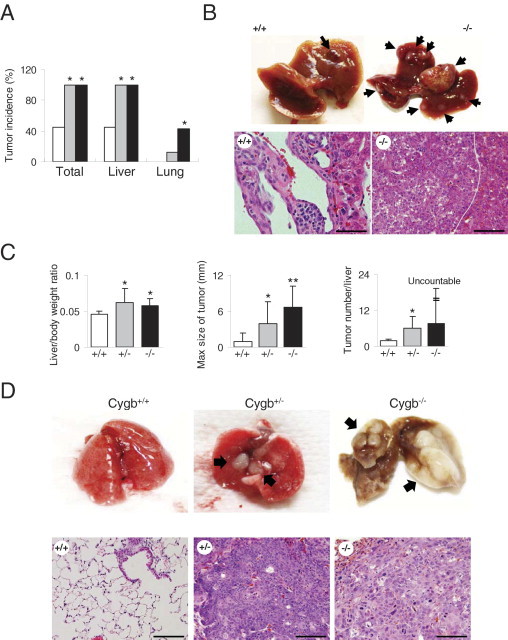
Tumor development in Cygb-deficient mice treated with 25-ppm DEN for 25 weeks. A: Tumor incidence in total, livers, and lungs from DEN-treated Cygb+/+ (white bars), Cygb+/− (gray bars), and Cygb−/− (black bars) mice (n = 7 to 12). *P < 0.05 compared with wild type. B: Representative gross photographs of livers (top) from wild-type (+/+) and Cygb-KO (−/−) mice treated with DEN. There was a marked increase in tumor multiplicity in Cygb-deficient mice compared with wild-type mice (arrows). Representative H&E-stained paraffin sections (bottom) of hemangioma from wild-type and poorly differentiated hepatocellular carcinoma composed of small immature neoplastic cells with mitotic figures (the line indicates the boundary of tumor and nontumor areas), from Cygb−/− mice. Scale bar = 100 μm. C: Determination of liver/body weight ratios, maximum (Max) tumor sizes, and liver tumor numbers for Cygb+/+ (white bars), Cygb+/− (gray bars), and Cygb−/− (black bars) mice (n = 7 to 12). Values are given as the mean ± SD. *P < 0.05, **P < 0.01. D: Representative gross photographs (top) and microphotographs (bottom) of lungs from Cygb+/+, Cygb+/−, and Cygb−/− DEN-treated mice. Lung tumors were only found in Cygb+/− and Cygb−/− mice. Arrows indicate lung tumors that were classified as squamous cell carcinomas. Scale bar = 100 µm.
DEN has induced tumors in the liver and in the lungs of the B6C3F1 mouse strain, although with a lower frequency.24 Consistent with these studies, lung tumors were found in Cygb+/− and Cygb−/− mice, with a frequency of 11.7% and 57.1%, respectively, whereas Cygb+/+ mice showed complete resistance (Figure 2, A and D).
Next, using a low dose of DEN, at which wild-type mice do not develop liver tumors, we examined whether Cygb deficiency functioned as a tumor promoter. We took advantage of earlier observations that, when DEN was administered to mice at a nontoxic dose, it failed to induce liver cancer, unless combined with a tumor promoter, such as phenobarbital.25,26 We maintained wild-type and Cygb-deficient male mice during DEN administration at the nontoxic dose of 0.05 ppm for 36 weeks. As a result, >40% of Cygb−/− mice developed tumors in the liver or lungs, whereas, as expected, wild-type mice exhibited no tumor formation (Figure 3). Cygb+/− mice showed an intermediate sensitivity. These findings confirm the tumor-promoting effects of Cygb depletion.
Figure 3.
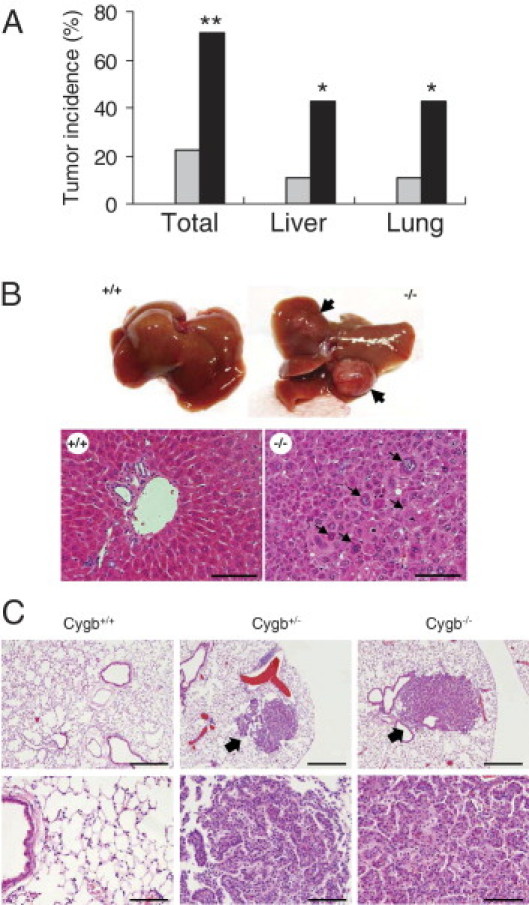
Tumor development in Cygb-deficient mice treated with 0.05-ppm DEN for 36 weeks. A: Tumor incidence in total, livers, and lungs from 0.05-ppm DEN-treated Cygb+/+ (white bars), Cygb+/− (gray bars), and Cygb−/− (black bars) mice (n = 7 to 15). *P < 0.05, **P < 0.01 compared with the wild type. B: Gross photographs of livers (top) from Cygb+/+ and Cygb−/− mice treated with 0.05-ppm DEN, as previously mentioned; arrows indicate lung tumors. Representative photomicrographs of H&E-stained paraffin sections of liver parenchyma (bottom) from wild-type and moderately differentiated hepatocellular carcinoma composed of large cells that vary in size and shape (arrows), from Cygb−/− mice. Scale bar = 100 μm. C: Representative photomicrographs of H&E-stained paraffin sections of lungs from Cygb+/+, Cygb+/−, and Cygb−/− mice treated with 0.05-ppm DEN for 36 weeks. Scale bars: 400 μm (top); 100 μm (bottom). Arrows indicate lung tumors that were classified as adenocarcinomas. No lung tumor was observed in Cygb+/+ mice.
To define the histological features of the liver tumors, liver samples were evaluated IHC. The liver tumors in C57BL/6 mice (wild type) administered DEN in drinking water were dominantly composed of cholangiomas, hemangiomas, hemangiosarcomas, and, to a lesser degree, hepatocellular carcinomas.27–29 In this study, Cygb-deficient and wild-type mice developed similar histological alterations in the liver, including hemangioma (Figure 2B), hepatocellular carcinoma (Figures 2B and 3B), which stained positive for alpha-fetoprotein (AFP) (Figure 4A), and cholangioma, which stained positive for CK19 (Figure 4A). Quantitative real-time PCR (RT-qPCR) analysis showed increased mRNA expression of AFP in Cygb+/− and Cygb−/− mice compared with their wild-type counterparts (Figure 4B).
Figure 4.
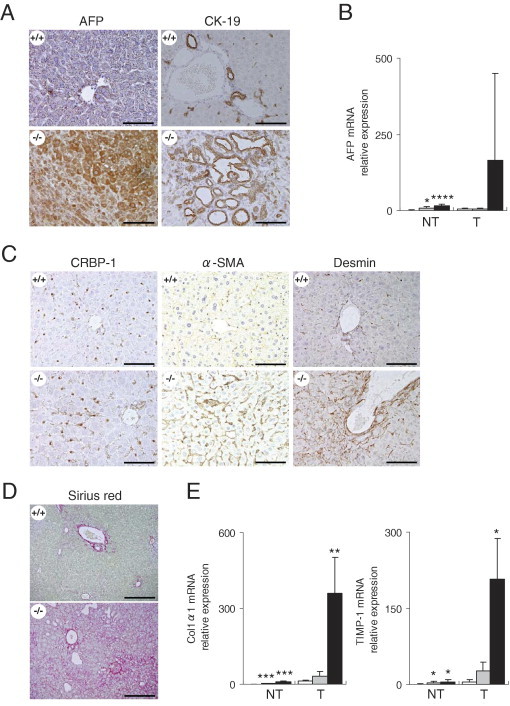
Expression of tumor markers and liver fibrosis development in DEN-treated Cygb-deficient mice. Cygb-deficient mice from Figure 2 were subjected to histological and biochemical analyses. A: Paraffin-embedded liver sections from Cygb+/+ and Cygb−/− mice were stained with AFP and cytokeratin 19 (CK-19). Scale bar = 100 μm. B: Expression of AFP mRNA in Cygb+/+ (white bars), Cygb+/− (gray bars), and Cygb−/− (black bars) mice livers was determined by RT-qPCR (n = 7 to 12). Levels were normalized to GAPDH. Values are given as the mean ± SD of all experiments. NT, nontumor area; T, liver tumor. *P < 0.05, ****P < 0.0001. C: Paraffin-embedded liver sections were IHC stained for the detection of CRBP-1, α-SMA, and desmin. Scale bar = 100 μm. D and E: Development of liver fibrosis. D: Sirius red staining for collagen deposition in paraffin-embedded liver sections. Scale bar = 100 μm. There was marked deposition of collagen (red) around the hepatocytes (pericellular fibrosis) in Cygb−/− mice. E: Relative levels of collagen (Col) 1α1 and tissue inhibitor of matrix metalloproteinase-1 (TIMP-1) mRNA in the nontumor area (NT; n = 7 to 12) and in liver tumors (T; n = 3 to 5) of Cygb+/+ (white bars), Cygb+/− (gray bars), and Cygb−/− (black bars) mice were determined by RT-qPCR and normalized to GAPDH mRNA. Values are given as the mean ± SD of all experiments. *P < 0.05, **P < 0.01, and ***P < 0.001.
In the liver, Cygb was present in stellate cells.1,4 We used cellular retinol-binding protein-1 and α-smooth muscle actin antibodies to detect stellate cell expression. The presence of cellular retinol-binding protein-1– and α-smooth muscle actin–expressing cells in the liver parenchyma from DEN-treated homozygote mice indicated that stellate cells were present along sinusoids, even in the absence of Cygb (Figure 4C). The expression of α-smooth muscle actin and desmin, another marker of stellate cells, was markedly augmented in Cygb−/− mice (Figure 4C). Next, we assessed whether liver fibrosis developed in these mice. Sirius red staining for collagen deposition in paraffin-embedded sections of liver samples collected from Cygb+/+ and Cygb−/− mice treated with 25-ppm DEN for 25 weeks showed marked deposition of collagen (red) around the hepatocytes (pericellular fibrosis) in Cygb−/− mice (Figure 4D). Morphometric image analysis was performed with a computerized system, consisting of a photomicroscope, a digital camera, and LuminaVision 2.4 bioimaging software, to quantitatively assess fibrosis. The Sirius red–positive area of Cygb−/− mice (7.67% ± 7.82%, n = 3) was significantly greater than that in Cygb+/+ mice (mean ± SD, 1.36% ± 0.36%, n = 3; P < 0.05, Kruskal-Wallis test). Pericellular fibrosis with collagen deposition was accompanied by significantly augmented mRNA expression of collagen 1α1 and tissue inhibitor of matrix mettaloproteinase-1 in the livers of Cygb−/− mice, compared with that of Cygb+/+ mice treated with 25-ppm DEN for 25 weeks (Figure 4E). Thus, the augmented occurrence of pericellular fibrosis and fibrotic reactions in Cygb deficiency may be involved in the development of liver cancer.
Cygb Loss Is Associated with Increased Cancer Cell Proliferation
Two important cellular processes in tumorigenesis are cell apoptosis and proliferation. To examine how Cygb loss affected these two processes in the livers of C57BL/6J mice, we determined the labeling indexes of TUNEL, as a marker of apoptotic cells, and proliferating cell nuclear antigen (PCNA), as a marker of proliferating cells in the liver, both in the tumor and adjacent nontumor areas of Cygb+/+ and Cygb−/− mice treated with 25-ppm DEN in drinking water for 25 weeks. Liver tumors in Cygb−/− mice exhibited reduced apoptotic cell death relative to liver tumors in Cygb+/+ mice (Figure 5, A and C) and showed elevated mRNA expression of the antiapoptotic protein Bcl-2 (Figure 5E). Liver tumors in the Cygb−/− mice exhibited more proliferating cells than the tumors of the wild-type mice, as shown by PCNA labeling (Figure 5, B and D), in addition to elevated cyclin D1 mRNA expression (Figure 5F). These results suggest that stimulation of proliferating neoplastic cells is the primary cellular mechanism for increased liver tumorigenesis in Cygb-deficient mice.
Figure 5.
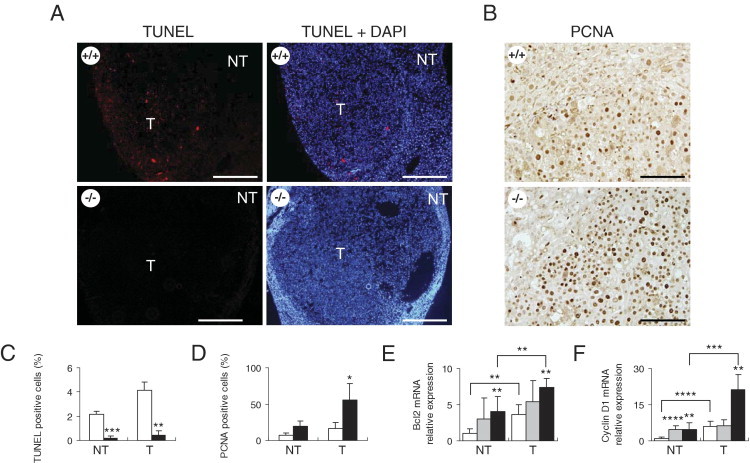
Tumor-bearing Cygb-deficient mice are associated with increased cancer cell proliferation and reduced apoptosis. A: Paraffin-embedded liver sections from wild-type (+/+) and homozygous (−/−) mice treated with 25-ppm DEN for 25 weeks were TUNEL labeled (left) and counterstained with DAPI (right). Apoptotic cells were present in the tumor (T) area in wild-type mice. NT, nontumor area. Scale bar = 400 μm. B: Paraffin-embedded liver sections were stained with PCNA. Scale bar = 100 μm. The frequency of apoptotic (C) or proliferate (D) cells was determined by counting TUNEL- or PCNA-positive cells, respectively (cells with the nucleus stained dark brown in the case of PCNA), in at least 1000 cells from each liver (n = 3). Expression levels of Bcl-2 (E) and cyclin D1 (F) mRNA in the NT (n = 7 to 12) and T (n = 3 to 5) areas from 25-ppm DEN-treated mice were determined by RT-qPCR and normalized to GAPDH. Cygb+/+ (white bars), Cygb+/− (gray bars), and Cygb−/− (black bars) mice. Values are given as the mean ± SD of all experiments. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Cygb-Deficient Mice Exhibit Elevated Phosphorylated Akt and Erk and Liver Inflammation
To identify the signaling pathways responsible for enhanced hepatocyte survival and proliferation, we examined the effects of Cygb deficiency on the major pathways implicated in liver cancer.30 As expected, Cygb loss was associated with an increase in both Akt phosphorylation and abundance in the livers (Figure 6A). This observation was also evident for Erk signaling (Figure 6, A and B). Consistent with increased Erk phosphorylation, Cygb-deficient mice exhibited increased expression of cyclin D1 (Figure 6A), and Jun and Fos mRNA in nontumor and tumor areas, relative to wild type (Figure 6C). These results suggested that the Akt and Erk pathways are activated in response to Cygb deficiency. The increased levels of Akt and Erk activation correlated with a marked elevation of IL-6 mRNA in both nontumor and tumor areas in Cygb-deficient mice (Figure 6D). IL-6 is a tumor-promoting cytokine that is required for Erk activation and contributes to alterations in Akt signaling.31 Knowing that IL-6 functions as a downstream mediator for both IL-1 and tumor necrosis factor-α,32 we examined the expression of these two cytokines. Remarkably, IL-1β and Tnfα levels (Figure 6D) increased 10- and 30-fold, respectively, at the mRNA level in the nontumor area of the liver and increased further in the tumors, relative to wild type. Cygb-deficient mice also had increased expression of Tgfβ3 mRNA (Figure 6D). These data suggest that Cygb loss can trigger inflammation and lead to the long-term elevation of tumor-promoting cytokines, resulting in the development of tumors.
Figure 6.
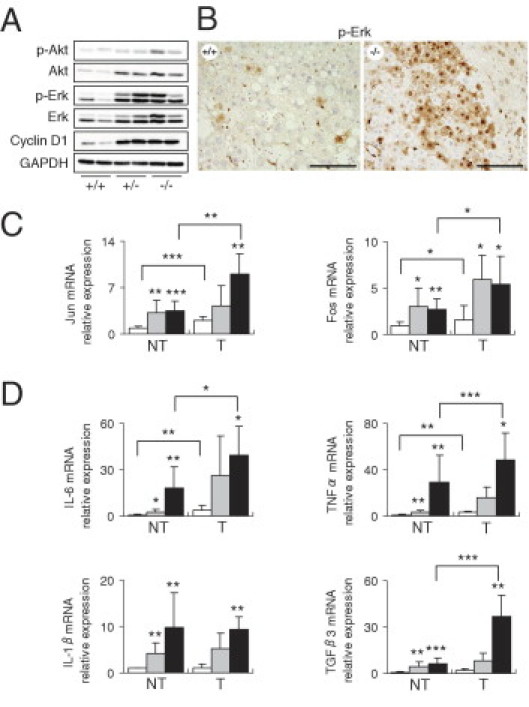
Tumor-bearing Cygb-deficient mice exhibited elevated phosphorylated Akt (p-Akt), phosphorylated Erk (p-Erk), and inflammation. Cygb-deficient mice from Figure 2 were subjected to additional biochemical and gene expression analyses. A: The liver was lysed and gel separated, and the levels of p-Akt, p-Erk, total Akt, total Erk, and cyclin D1 were examined by immunoblot analyses. All blots were reprobed with anti-GAPDH as a loading control. B: Paraffin-embedded liver sections were stained for phosphorylated Erk. Scale bar = 100 μm. C and D: Relative mRNA levels of Jun and Fos (C) and IL-6, Tnfα, IL-1β, and Tgfβ3 (D) in the nontumor (NT; n = 7 to 12) and tumor (T; n = 3 to 5) areas of the liver were determined by RT-qPCR and normalized to GAPDH. Cygb+/+ (white bars), Cygb+/− (gray bars), and Cygb−/− (black bars) mice. Values are given as the mean ± SD of all experiments. *P < 0.05, **P < 0.01, and ***P < 0.001.
Nitrotyrosine Accumulation in the Livers of DEN-Treated Cygb-Deficient Mice
Long-term administration of DEN has induced the expression of the inducible isoform of NO synthase and 3-nitrotyrosine, a marker of peroxynitrite formation, in preneoplastic and neoplastic rat liver tissues.33 In this study, we detected the overproduction of nitrotyrosine in tumor and nontumor liver tissues of Cygb-deficient mice, compared with wild-type mice, as shown by IHC (Figure 7A) and immunoblot analyses (Figure 7B). These results indicate the high production of NO, together with superoxide, in Cygb-deficient mice.
Figure 7.
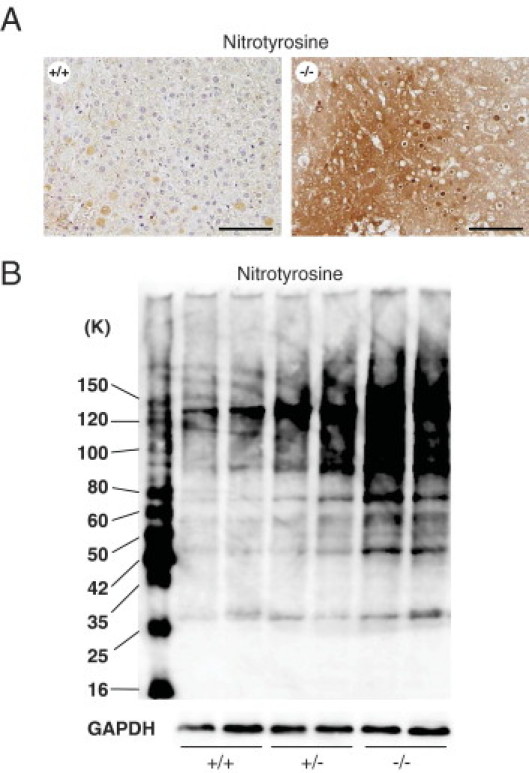
Peroxynitrite formation in the livers of Cygb-deficient mice. A: Paraffin-embedded liver sections from wild-type (+/+) and homozygous (−/−) mice treated with 25-ppm DEN for 25 weeks were stained with antinitrotyrosine. Nitrotyrosine-containing proteins were strongly expressed in the cytoplasm and nuclei of cancer cells in the tumor area, particularly in the inclusions of cancer cells. Scale bar = 100 μm. B: Protein homogenates of liver tissues from Cygb+/+, Cygb+/−, and Cygb−/− mice treated with 25-ppm DEN for 25 weeks were subjected to immunoblot detection for nitrotyrosine. GAPDH was used as a loading control. K, kDa (molecular mass).
Dysregulation of Genes Associated with Cell Proliferation and Differentiation in Cygb-Deficient Mice
To further screen for cellular alterations caused by Cygb gene disruption, we compared gene expression profiles between Cygb-deficient mice and their wild-type counterparts after 25-ppm DEN treatment for 25 weeks. We observed the altered expression of cancer genes, including p53, cyclin D2, Pak1 (p21-activated kinase), Src, Cdkn2a, and Cebpa (CCAAT/enhancer-binding protein α) (data not shown). We examined in detail the mRNA levels of these genes by RT-qPCR. Consistent with the high rate of cellular proliferation in the liver of Cygb-deficient mice (Figure 5, B and D), we observed overexpression of cyclin D2 (Figure 8A) and p53 (Figure 8B), which have displayed high expression in astrocytomas, a type of brain tumor.34 Pak1 promotes malignant tumor progression, and the Src proto-oncogene has shown increased expression in human skin tumors and leukemia.35–38 In this study, we found that the mRNA expression of Pak1, in addition to Src, increased fivefold in the livers of Cygb−/− mice, relative to the wild type (Figure 8, C and D).
Figure 8.
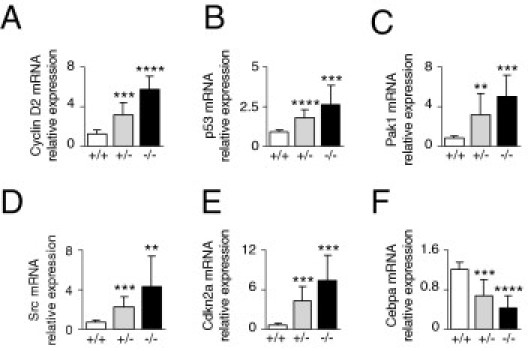
Altered regulation of cancer genes in Cygb-deficient mice. RT-qPCR analysis of cell growth–related gene transcripts cyclin D2 (A), p53 (B), and Pak1 (C) and cell differentiation and apoptosis-related gene transcripts Src (D), Cdkn2a (E), and Cebpa (F) in liver from mice treated with 25-ppm DEN for 25 weeks (n = 7 to 12). Levels are normalized to GAPDH. Cygb+/+ (white bars), Cygb+/− (gray bars), and Cygb−/− (black bars) mice. Values are given as the mean ± SD of all experiments. **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Cdkn2a, a tumor suppressor that negatively regulates the cell cycle, displayed increased expression at the mRNA level in the livers of Cygb-deficient mice (Figure 8E), consistent with other studies39,40 on sarcomas and lung tumors.
Cebpa has the ability to inhibit proliferation, particularly in hepatocytes.41,42 Down-regulation of CEBPA has been reported in human myeloid leukemia.43 Consistent with these studies, our results showed decreased expression of Cebpa mRNA in the liver of Cygb-deficient mice, relative to the wild type (Figure 8F).
Discussion
In the present study, we showed that loss of Cygb in C57BL/6J mice markedly increased their susceptibility to DEN-induced tumorigenesis. In the absence of Cygb, liver tumors developed earlier, were larger, and were more numerous compared with wild-type mice. By administering low-dose DEN to adult mice (0.05 ppm), which failed to induce liver cancer in wild-type mice, we observed the tumor-promoting effects of Cygb deficiency.
We observed high levels of Cygb expression in hepatic stellate cells, a liver-specific pericyte, from which Cygb was originally discovered by proteomic analysis of primary cultured rat cells and other stromal cells in the visceral organs, including the pancreas, gut, spleen, lung, and kidney.1,4 These stromal cells are pericytes localized around the capillaries of the organs that are capable of vitamin A storage. Thus, we propose that Cygb may be an indicator of a vitamin A–storing phenotype of myofibroblasts of endoderm origin. However, as previously reported,1,4,5 Cygb is ubiquitously expressed in all body organs in human, as in mice and rats. At the mRNA level, high expression is evident in the adult human heart and liver; modest expression is evident in the brain, kidney, trachea, and placenta; and low expression is evident in the adult skeletal muscle.44 Several cancer cell lines, including HepG2 cells,44 the NCI-H2228 lung cancer cell line, and HCC 1569 breast cancer cells,19 also display CYGB mRNA expression. These observations indicate the role of Cygb in the regulation of cellular function originating from the epithelia. In this context, cancer development in the liver and lungs in Cygb−/− mice is anticipated. However, the role of mesenchymal cells that express Cygb highly in tumor development should be further evaluated because these myofibroblasts represent important environmental factors during tumor formation.
Cygb expression is augmented under hypoxia in the liver, heart, brain, and skeletal muscle and in HN33 cells (an immortalized mouse hippocampal cell line), BEAS-2B cells (a transformed human bronchial epithelial cell line), and HeLa cells (a human cervix carcinoma cell line).16 Overexpression of Cygb protects mouse neuroblastoma N2a cells and human neuroblastoma SH-SY5Y cells under H2O2 exposure and the human neuronal cell line TE671 under prooxidant Ro19-8022 stimulation.11,12,44 Overexpression of Cygb in the liver, induced by adeno-associated–virus-induced transfection, protects the liver from oxidative injury.13 Conversely, the role of Cygb as an NO scavenger in rat hepatocytes and NIH3T3 fibroblasts may protect cells from NO-induced toxicity.8,33 These reports and our findings regarding the accumulation of nitrotyrosine protein adducts in Cygb-deficient mice indicate the cytoprotective and antioxidative properties of Cygb.
Several tumor suppressor genes are located in both arms of chromosome 17. TP53, a known tumor suppressor gene at 17p13.1, is one of the most frequently mutated genes in cancers, including hepatocellular carcinoma.45 BRCA1 (breast cancer 1, early onset) at 17q12 is a human tumor suppressor gene encoding the breast cancer type 1 susceptibility protein,46 which is present in the breast and other tissues, aiding the repair of damaged DNA and the destruction of the cell when DNA cannot be repaired. If BRCA1 is damaged, cells duplicate uncontrollably, leading to cancer. Other known breast cancer–associated genes include septin, DMC1, and HER2/ErbB2.47,48 Because CYGB exists on chromosome 17q25, genes on this chromosome appear commonly in the tumorigenesis of epithelial cells and mutation or epigenetic modification of these genes appears to trigger malignant transformation.
Clinically, liver cancer develops from a fibrotic liver, with chronic trauma induced by alcohol abuse and hepatitis virus B/C infection.49 Chronic inflammation offers an appropriate environment for cancer development, by producing multiple growth factors, extracellular matrices, and neovascularization involving local hypoxia.50 In this context, the augmented occurrence of pericellular fibrosis and fibrotic reactions (Figure 4, C–E) in Cygb deficiency may be involved in the development of liver cancer.
In conclusion, to our knowledge, this is the first report that Cygb deficiency induces susceptibility to cancer development in the liver and lungs of mice receiving DEN treatment. Thus, Cygb-deficient mice may provide a useful animal model to study cancer development in the liver and lungs; globins, such as Cygb, may shed new light on the biological features of organ carcinogenesis.
Acknowledgment
We thank Drs. Masaru Enomoto, Hideki Fujii, and Thoru Komiya for their valuable comments during this study.
Footnotes
Supported by a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science [grant 21390232 (2009) to N.K.]; grants from the Ministry of Health, Labour, and Welfare of Japan (2008); and the Thrust Area Research grant from Osaka City University (2008).
References
- 1.Kawada N., Kristensen D.B., Asahina K., Nakatani K., Minamiyama Y., Seki S., Yoshizato K. Characterization of a stellate cell activation-associated protein (STAP) with peroxidase activity found in rat hepatic stellate cells. J Biol Chem. 2001;276:25318–25323. doi: 10.1074/jbc.M102630200. [DOI] [PubMed] [Google Scholar]
- 2.Burmester T., Ebner B., Weich B., Hankeln T. Cytoglobin: a novel globin type ubiquitously expressed in vertebrate tissues. Mol Biol Evol. 2002;19:416–421. doi: 10.1093/oxfordjournals.molbev.a004096. [DOI] [PubMed] [Google Scholar]
- 3.Sawai H., Kawada N., Yoshizato K., Nakajima H., Aono S., Shiro Y. Characterization of the heme environmental structure of cytoglobin, a fourth globin in humans. Biochemistry. 2003;42:5133–5142. doi: 10.1021/bi027067e. [DOI] [PubMed] [Google Scholar]
- 4.Nakatani K., Okuyama H., Shimahara Y., Saeki S., Kim D.H., Nakajima Y., Seki S., Kawada N., Yoshizato K. Cytoglobin/STAP, its unique localization in splanchnic fibroblast-like cells and function in organ fibrogenesis. Lab Invest. 2004;84:91–101. doi: 10.1038/labinvest.3700013. [DOI] [PubMed] [Google Scholar]
- 5.Shigematsu A., Adachi Y., Matsubara J., Mukaide H., Koike-Kiriyama N., Minamino K., Shi M., Yanai S., Imamura M., Taketani S., Ikehara S. Analyses of expression of cytoglobin by immunohistochemical studies in human tissues. Hemoglobin. 2008;32:287–296. doi: 10.1080/03630260802017261. [DOI] [PubMed] [Google Scholar]
- 6.Sugimoto H., Makino M., Sawai H., Kawada N., Yoshizato K., Shiro Y. Structural basis of human cytoglobin for ligand binding. J Mol Biol. 2004;339:873–885. doi: 10.1016/j.jmb.2004.04.024. [DOI] [PubMed] [Google Scholar]
- 7.Li R.C., Lee S.K., Pouranfar F., Brittian K.R., Clair H.B., Row B.W., Wang Y., Gozal D. Hypoxia differentially regulates the expression of neuroglobin and cytoglobin in rat brain. Brain Res. 2006;1096:173–179. doi: 10.1016/j.brainres.2006.04.063. [DOI] [PubMed] [Google Scholar]
- 8.Fordel E., Geuens E., Dewilde S., Rottiers P., Carmeliet P., Grooten J., Moens L. Cytoglobin expression is upregulated in all tissues upon hypoxia: an in vitro and in vivo study by quantitative real-time PCR. Biochem Biophys Res Commun. 2004;319:342–348. doi: 10.1016/j.bbrc.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 9.Guo X., Philipsen S., Tan-Un K.C. Study of the hypoxia-dependent regulation of human CYGB gene. Biochem Biophys Res Commun. 2007;364:145–150. doi: 10.1016/j.bbrc.2007.09.108. [DOI] [PubMed] [Google Scholar]
- 10.Hodges N.J., Innocent N., Dhanda S., Graham M. Cellular protection from oxidative DNA damage by over-expression of the novel globin cytoglobin in vitro. Mutagenesis. 2008;23:293–298. doi: 10.1093/mutage/gen013. [DOI] [PubMed] [Google Scholar]
- 11.Fordel E., Thijs L., Martinet W., Lenjou M., Laufs T., Van Bockstaele D., Moens L., Dewilde S. Neuroglobin and cytoglobin overexpression protects human SH-SY5Y neuroblastoma cells against oxidative stress-induced cell death. Neurosci Lett. 2006;410:146–151. doi: 10.1016/j.neulet.2006.09.027. [DOI] [PubMed] [Google Scholar]
- 12.Fordel E., Thijs L., Martinet W., Schrijvers D., Moens L., Dewilde S. Anoxia or oxygen and glucose deprivation in SH-SY5Y cells: a step closer to the unraveling of neuroglobin and cytoglobin functions. Gene. 2007;398:114–122. doi: 10.1016/j.gene.2007.03.022. [DOI] [PubMed] [Google Scholar]
- 13.Xu R., Harrison P.M., Chen M., Li L., Tsui T.Y., Fung P.C., Cheung P.T., Wang G., Li H., Diao Y., Krissansen G.W., Xu S., Farzaneh F. Cytoglobin overexpression protects against damage-induced fibrosis. Mol Ther. 2006;13:1093–1100. doi: 10.1016/j.ymthe.2005.11.027. [DOI] [PubMed] [Google Scholar]
- 14.McRonald F.E., Liloglou T., Xinarianos G., Hill L., Rowbottom L., Langan J.E., Ellis A., Shaw J.M., Field J.K., Risk J.M. Down-regulation of the cytoglobin gene, located on 17q25, in tylosis with oesophageal cancer (TOC): evidence for trans-allele repression. Hum Mol Genet. 2006;15:1271–1277. doi: 10.1093/hmg/ddl042. [DOI] [PubMed] [Google Scholar]
- 15.Xinarianos G., McRonald F.E., Risk J.M., Bowers N.L., Nikolaidis G., Field J.K., Liloglou T. Frequent genetic and epigenetic abnormalities contribute to the deregulation of cytoglobin in non-small cell lung cancer. Hum Mol Genet. 2006;15:2038–2044. doi: 10.1093/hmg/ddl128. [DOI] [PubMed] [Google Scholar]
- 16.Shaw R.J., Omar M.M., Rokadiya S., Kogera F.A., Lowe D., Hall G.L., Woolgar J.A., Homer J., Liloglou T., Field J.K., Risk J.M. Cytoglobin is upregulated by tumour hypoxia and silenced by promoter hypermethylation in head and neck cancer. Br J Cancer. 2009;101:139–144. doi: 10.1038/sj.bjc.6605121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Presneau N., Dewar K., Forgetta V., Provencher D., Mes-Masson A.M., Tonin P.N. Loss of heterozygosity and transcriptome analyses of a 1.2 Mb candidate ovarian cancer tumor suppressor locus region at 17q25.1-q25.2. Mol Carcinog. 2005;43:141–154. doi: 10.1002/mc.20096. [DOI] [PubMed] [Google Scholar]
- 18.Chua P.J., Yip G.W., Bay B.H. Cell cycle arrest induced by hydrogen peroxide is associated with modulation of oxidative stress related genes in breast cancer cells. Exp Biol Med (Maywood) 2009;234:1086–1094. doi: 10.3181/0903-RM-98. [DOI] [PubMed] [Google Scholar]
- 19.Shivapurkar N., Stastny V., Okumura N., Girard L., Xie Y., Prinsen C., Thunnissen F.B., Wistuba I.I., Czerniak B., Frenkel E., Roth J.A., Liloglou T., Xinarianos G., Field J.K., Minna J.D., Gazdar A.F. Cytoglobin, the newest member of the globin family, functions as a tumor suppressor gene. Cancer Res. 2008;68:7448–7456. doi: 10.1158/0008-5472.CAN-08-0565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sinal C.J., Tohkin M., Miyata M., Ward J.M., Lambert G., Gonzalez F.J. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell. 2000;102:731–744. doi: 10.1016/s0092-8674(00)00062-3. [DOI] [PubMed] [Google Scholar]
- 21.Verna L., Whysner J., Williams G.M. N-nitrosodiethylamine mechanistic data and risk assessment: bioactivation, DNA-adduct formation, mutagenicity, and tumor initiation. Pharmacol Ther. 1996;71:57–81. doi: 10.1016/0163-7258(96)00062-9. [DOI] [PubMed] [Google Scholar]
- 22.Diwan B.A., Rice J.M., Ohshima M., Ward J.M. Interstrain differences in susceptibility to liver carcinogenesis initiated by N-nitrosodiethylamine and its promotion by phenobarbital in C57BL/6NCr, C3H/HeNCrMTV- and DBA/2NCr mice. Carcinogenesis. 1986;7:215–220. doi: 10.1093/carcin/7.2.215. [DOI] [PubMed] [Google Scholar]
- 23.Drinkwater N.R., Ginsler J.J. Genetic control of hepatocarcinogenesis in C57BL/6J and C3H/HeJ inbred mice. Carcinogenesis. 1986;7:1701–1707. doi: 10.1093/carcin/7.10.1701. [DOI] [PubMed] [Google Scholar]
- 24.Vesselinovitch S.D., Koka M., Mihailovich N., Rao K.V. Carcinogenicity of diethylnitrosamine in newborn, infant, and adult mice. J Cancer Res Clin Oncol. 1984;108:60–65. doi: 10.1007/BF00390974. [DOI] [PubMed] [Google Scholar]
- 25.Gray R., Peto R., Brantom P., Grasso P. Chronic nitrosamine ingestion in 1040 rodents: the effect of the choice of nitrosamine, the species studied, and the age of starting exposure. Cancer Res. 1991;51:6470–6491. [PubMed] [Google Scholar]
- 26.Ward J.M., Diwan B.A., Ohshima M., Hu H., Schuller H.M., Rice J.M. Tumor-initiating and promoting activities of di(2-ethylhexyl) phthalate in vivo and in vitro. Environ Health Perspect. 1986;65:279–291. doi: 10.1289/ehp.8665279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Iwai S., Murai T., Makino S., Min W., Morimura K., Mori S., Hagihara A., Seki S., Fukushima S. High sensitivity of fatty liver Shionogi (FLS) mice to diethylnitrosamine hepatocarcinogenesis: comparison to C3H and C57 mice. Cancer Lett. 2007;246:115–121. doi: 10.1016/j.canlet.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 28.Koen H., Pugh T.D., Goldfarb S. Hepatocarcinogenesis in the mouse: combined morphologic-stereologic studies. Am J Pathol. 1983;112:89–100. [PMC free article] [PubMed] [Google Scholar]
- 29.Koen H., Pugh T.D., Nychka D., Goldfarb S. Presence of alpha-fetoprotein-positive cells in hepatocellular foci and microcarcinomas induced by single injections of diethylnitrosamine in infant mice. Cancer Res. 1983;43:702–708. [PubMed] [Google Scholar]
- 30.Whittaker S., Marais R., Zhu A.X. The role of signaling pathways in the development and treatment of hepatocellular carcinoma. Oncogene. 2010;29:4989–5005. doi: 10.1038/onc.2010.236. [DOI] [PubMed] [Google Scholar]
- 31.Grivennikov S.I., Greten F.R., Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kamimura D., Ishihara K., Hirano T. IL-6 signal transduction and its physiological roles: the signal orchestration model. Rev Physiol Biochem Pharmacol. 2003;149:1–38. doi: 10.1007/s10254-003-0012-2. [DOI] [PubMed] [Google Scholar]
- 33.Ahn B., Han B.S., Kim D.J., Ohshima H. Immunohistochemical localization of inducible nitric oxide synthase and 3-nitrotyrosine in rat liver tumors induced by N-nitrosodiethylamine. Carcinogenesis. 1999;20:1337–1344. doi: 10.1093/carcin/20.7.1337. [DOI] [PubMed] [Google Scholar]
- 34.Kheirollahi M., Mehr-Azin M., Kamalian N., Mehdipour P. Expression of cyclin D2, P53, Rb and ATM cell cycle genes in brain tumors. Med Oncol. 2011;28:7–14. doi: 10.1007/s12032-009-9412-8. [DOI] [PubMed] [Google Scholar]
- 35.Kumar R., Gururaj A.E., Barnes C.J. p21-activated kinases in cancer. Nat Rev Cancer. 2006;6:459–471. doi: 10.1038/nrc1892. [DOI] [PubMed] [Google Scholar]
- 36.Siu M.K., Wong E.S., Chan H.Y., Kong D.S., Woo N.W., Tam K.F., Ngan H.Y., Chan Q.K., Chan D.C., Chan K.Y., Cheung A.N. Differential expression and phosphorylation of Pak1 and Pak2 in ovarian cancer: effects on prognosis and cell invasion. Int J Cancer. 2010;127:21–31. doi: 10.1002/ijc.25005. [DOI] [PubMed] [Google Scholar]
- 37.Barnekow A., Paul E., Schartl M. Expression of the c-src proto-oncogene in human skin tumors. Cancer Res. 1987;47:235–240. [PubMed] [Google Scholar]
- 38.McClain K.L. Expression of oncogenes in human leukemias. Cancer Res. 1984;44:5382–5389. [PubMed] [Google Scholar]
- 39.Maelandsmo G.M., Berner J.M., Florenes V.A., Forus A., Hovig E., Fodstad O., Myklebost O. Homozygous deletion frequency and expression levels of the CDKN2 gene in human sarcomas: relationship to amplification and mRNA levels of CDK4 and CCND1. Br J Cancer. 1995;72:393–398. doi: 10.1038/bjc.1995.344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Belinsky S.A., Swafford D.S., Middleton S.K., Kennedy C.H., Tesfaigzi J. Deletion and differential expression of p16INK4a in mouse lung tumors. Carcinogenesis. 1997;18:115–120. doi: 10.1093/carcin/18.1.115. [DOI] [PubMed] [Google Scholar]
- 41.Timchenko N.A., Wilde M., Nakanishi M., Smith J.R., Darlington G.J. CCAAT/enhancer-binding protein alpha (C/EBP alpha) inhibits cell proliferation through the p21 (WAF-1/CIP-1/SDI-1) protein. Genes Dev. 1996;10:804–815. doi: 10.1101/gad.10.7.804. [DOI] [PubMed] [Google Scholar]
- 42.Diehl A.M., Johns D.C., Yang S., Lin H., Yin M., Matelis L.A., Lawrence J.H. Adenovirus-mediated transfer of CCAAT/enhancer-binding protein-alpha identifies a dominant anti-proliferative role for this isoform in hepatocytes. J Biol Chem. 1996;271:7343–7350. doi: 10.1074/jbc.271.13.7343. [DOI] [PubMed] [Google Scholar]
- 43.Pabst T., Mueller B.U., Harakawa N., Schoch C., Haferlach T., Behre G., Hiddemann W., Zhang D.E., Tenen D.G. AML1-ETO downregulates the granulocytic differentiation factor C/EBPalpha in t(8;21) myeloid leukemia. Nat Med. 2001;7:444–451. doi: 10.1038/86515. [DOI] [PubMed] [Google Scholar]
- 44.Asahina K., Kawada N., Kristensen D.B., Nakatani K., Seki S., Shiokawa M., Tateno C., Obara M., Yoshizato K. Characterization of human stellate cell activation-associated protein and its expression in human liver. Biochem Biophys Acta. 2002;1577:471–475. doi: 10.1016/s0167-4781(02)00477-3. [DOI] [PubMed] [Google Scholar]
- 45.Caron de Fromentel C., Soussi T. TP53 tumor suppressor gene: a model for investigating human mutagenesis. Genes Chromosomes Cancer. 1992;4:1–15. doi: 10.1002/gcc.2870040102. [DOI] [PubMed] [Google Scholar]
- 46.Deng C.X. BRCA1: cell cycle checkpoint, genetic instability, DNA damage response and cancer evolution. Nucleic Acids Res. 2006;34:1416–1426. doi: 10.1093/nar/gkl010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Harada H., Nagai H., Tsuneizumi M., Mikami I., Sugano S., Emi M. Identification of DMC1, a novel gene in the TOC region on 17q25.1 that shows loss of expression in multiple human cancers. J Hum Genet. 2001;46:90–95. doi: 10.1007/s100380170115. [DOI] [PubMed] [Google Scholar]
- 48.Coussens L., Yang-Feng T.L., Liao Y.C., Chen E., Gray A., McGrath J., Seeburg P.H., Libermann T.A., Schlessinger J., Francke U., Levinson A., Ullrich A. Tyrosine kinase receptor with extensive homology to EGF receptor shares chromosomal location with neu oncogene. Science. 1985;230:1132–1139. doi: 10.1126/science.2999974. [DOI] [PubMed] [Google Scholar]
- 49.Llovet J.M., Burroughs A., Bruix J. Hepatocellular carcinoma. Lancet. 2003;362:1907–1917. doi: 10.1016/S0140-6736(03)14964-1. [DOI] [PubMed] [Google Scholar]
- 50.Brunt E.M. Pathology of nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol. 2010;7:195–203. doi: 10.1038/nrgastro.2010.21. [DOI] [PubMed] [Google Scholar]