

Abstract
Obliterative bronchiolitis after lung transplantation is a chronic inflammatory and fibrotic condition of small airways. The fibrosis associated with obliterative bronchiolitis might be reversible. Matrix metalloproteinases (MMPs) participate in inflammation and tissue remodeling. MMP-2 localized to myofibroblasts in post-transplant human obliterative bronchiolitis lesions and to allograft fibrosis in a rat intrapulmonary tracheal transplant model. Small numbers of infiltrating T cells were also observed within the fibrosis. To modulate inflammation and tissue remodeling, the broad-spectrum MMP inhibitor SC080 was administered after the allograft was obliterated, starting at post-transplant day 21. The allograft lumen remained obliterated after treatment. Only low-dose (2.5 mg/kg per day) SC080 significantly reduced collagen deposition, reduced the number of myofibroblasts and the infiltration of T cells in association with increased collagenolytic activity, increased MMP-2 gene expression, and decreased MMP-8, MMP-9, and MMP-13 gene expression. In in vitro experiments using cultured myofibroblasts, a relatively low concentration of SC080 increased MMP-2 activity and degradation of type I collagen. Moreover, coculture with T cells facilitated persistence of myofibroblasts, suggesting a role for T-cell infiltration in myofibroblast persistence in fibrosis. By combining low-dose SC080 with cyclosporine in vivo at post-transplant day 28, partial reversal of obliterative fibrosis was observed at day 42. Thus, modulating MMP activity might reverse established allograft airway fibrosis by regulating inflammation and tissue remodeling.
Chronic allograft dysfunction after lung transplantation is manifested by obliterative bronchiolitis (OB), a fibroproliferative obstructive lesion in small airways, and its clinical correlate, bronchiolitis obliterans syndrome (BOS).1,2 Once the fibrotic process of OB is initiated, conventional immunosuppression is usually ineffective.3 The traditional pathological perspective is that fibrosis is the end result of damage: scar tissue, with no possibility of return to the pre-existing structure.4 However, increasing evidence suggests that fibrosis still undergoes dynamic remodeling and is potentially a reversible process. For example, the resolution of liver fibrosis is well documented both clinically and experimentally. In animal experiments, up-regulation or overexpression of matrix metalloproteinases (MMPs) capable of degrading interstitial type I and type III collagen (including MMP-1,5 MMP-8,6 MMP-13,7and MMP-2 and MMP-148,9) is associated with the regression of liver fibrosis. Pulmonary fibrosis has also been shown to be conditionally reversible.10
One possible mechanism rendering fibrosis unlikely to resolve is the aberrant persistence of myofibroblasts, an active form of fibroblasts positive for α-smooth muscle actin (α-SMA), which leads to production of extracellular matrix (ECM) in excess of MMP-dependent ECM degradation.11 Unresolved inflammation can be an important contributor to this mechanism.10 Accumulating evidence suggests that chronic fibrotic conditions are mediated by complex interactions between immune and nonimmune cells, in which the persistence of a relatively low grade of inflammation continuously stimulates resident stromal cells12,13 and provides survival signals to myofibroblasts.14 For instance, the resolution of liver fibrosis encountered in alcohol-induced and virus-related fibrosis occurs only after remedy of the underlying cause.15,16 Moreover, in experimental models of fibrosis, reversal of fibrosis has occurred in one-hit injury models such as bleomycin-induced pulmonary fibrosis,17 in which the initial tissue injury leads to fibrosis but the tissue injury or inflammation is not continuous.8,9
Along those lines, OB after lung transplantation is a fibrotic and chronic inflammatory condition18 in which myofibroblasts persist.19 The intrapulmonary tracheal transplant model of OB is a unique animal model in which persistent alloantigen from the donor trachea within the pulmonary milieu causes continuous alloantigen-induced inflammation and results in robust fibrosis in the allograft lumen.20 We have previously demonstrated that myofibroblasts expressing high levels of collagen and MMP-2 and MMP-14 play a central role in the remodeling of established allograft airway fibrosis.20 Given that MMPs also play important but complex roles in the trafficking of immune responsive cells,20 MMPs involved in both tissue remodeling and inflammation may play key roles in the reversal of fibrosis.
We therefore hypothesized that allograft airway fibrosis is a potentially reversible process involving MMPs. Here, we demonstrate expression patterns of MMPs in established human OB lesions and describe the roles of MMPs in the remodeling of collagen matrix, myofibroblasts, and immune responsive cells using in vivo and in vitro models with SC080, a general MMP inhibitor. Finally, we demonstrate for the first time reversibility of allograft airway fibrosis by combining immunosuppression with a low dose of SC080.
Materials and Methods
Tissue Samples of BOS Lungs
Human lung tissue samples were obtained from patients with an established diagnosis of BOS at the time of retransplantation. The patients were four men and seven women, with a mean age of 36.9 ± 4.2 years, a mean transplantation period of 47.9 ± 8.4 months, and original diagnoses of pulmonary fibrosis,3 cystic fibrosis,3 bronchiectasis,2 bronchiolitis obliterans secondary to bone marrow transplantation,2 and chronic obstructive pulmonary disease.1 Use of these tissues was approved by the University Health Network Research Ethics Board.
Animal Model
Male Brown-Norway and Lewis rats (250 to 300 g) were purchased from Charles River Laboratories International (Wilmington, MA). Intrapulmonary tracheal transplantation was performed as described previously.21 In brief, a trachea retrieved from a donor rat was placed into the lumen of a plastic catheter 2.1 mm in diameter. The catheter was inserted into the parenchyma of the recipient's left lung, and a blunt needle was used to push the donor trachea out into the lung tissue. After the catheter was removed, bleeding and air leakage were controlled by a small surgical clip placed at the site of insertion. All animals received care in compliance with the Guide to the Care and Use of Experimental Animals formulated by the Canadian Council on Animal Care. The experimental protocol was approved by the Animal Care Committee of the Toronto General Research Institute.
MMP Inhibitor Treatment
To assess the effect of SC080 (a gift from Dr. Teresa Sunyer, Pfizer, St. Louis, MO), recipients of intrapulmonary allograft tracheal transplant (Brown-Norway to Lewis) were randomized to the following groups: sacrifice on day 21 (control, n = 3); SC080 from day 21 to day 35 after tracheal transplant at 2.5, 5.0, or 10 mg/kg per day, q.d., or 5.0 or 10 mg/kg per day, b.i.d. (n = 5 per group); or methylcellulose-based vehicle-treated controls (n = 5 per group). In addition, we performed intrapulmonary isograft tracheal transplant (Lewis to Lewis) to act as a further control (n = 3). SC080 was administered through an orogastric tube, as described previously.20 All of the treated animals were sacrificed on day 35, at 12 hours after the final dose of drug administration. Grafts were explanted and divided into three sections. The middle section was fixed in 10% formalin; the other sections were snap-frozen for histological and gene-expression analysis.
For treatment with SC080 in combination with cyclosporine, intrapulmonary allograft recipients did not receive any additional treatment for the first 28 days. We waited an extra 7 days, compared with the timing in the experiment described above, to allow for more robust fibrosis. The animals were then randomly allocated at day 28 into five groups (n = 6/group): including i) sacrifice on day 28 (control); ii) SC080 (2.5 mg/kg per day) in combination with cyclosporine (10 mg/kg per day; Novartis Pharmaceuticals Canada, Mississauga, ON, Canada); iii) vehicle in combination with cyclosporine; iv) SC080 in combination with saline (subcutaneous injection); v) vehicle in combination with saline. Treated animals were sacrificed on day 42.
Primary Lung Fibroblast Culture
Lung tissue from a healthy Lewis rat was minced and placed onto a 100-mm culture dish and incubated for 1 hour without medium. After the cells attached to the dish, the primary culture was grown in high-glucose Dulbecco's modified Eagle's medium with 10% fetal bovine serum and 1% penicillin/streptomycin at 37°C and 5% CO2. Once the cells reached confluence, the cells were passaged. Cells at passage 5 to passage 7 were used in the following experiments.
SC080 Treatment for Myofibroblasts
The rat pulmonary fibroblasts were trypsinized to remove them from their culture substrate, and 5 × 104 cells were transferred onto glass coverslips in a 12-well plate (BD Biosciences). The myofibroblast phenotype was induced by stimulating cells with 5 ng/mL of human recombinant transforming growth factor β-1 (TGF-β1; R&D Systems, Minneapolis, MN) for 24 to 48 hours. In one experiment, myofibroblasts stimulated with TGF-β1 for 48 hours were cultured for a further 48 hours in culture medium containing 2% fetal bovine serum with different concentrations of SC080 dissolved in 30% 2-hydroxypropyl-β-cyclodextrin (Sigma-Aldrich, St. Louis, MO). The culture medium containing SC080 was replaced every 12 hours.
In another experiment, the pulmonary fibroblasts were seeded onto a six-well plate at 1.5 × 105cells/well and treated with SC080 (250 nmol/L) or 30% 2-hydroxypropyl-β-cyclodextrin plus 5 ng/mL recombinant human TGF-β1 for 3 days in culture medium containing 2% fetal bovine serum. The same amount of SC080 or vehicle was added every 24 hours without changing the medium. At the fourth day, cells were washed twice with PBS and fasted in 800 μL of serum-free Dulbecco's modified Eagle's medium containing TGF-β1 (5 ng/mL) and either SC080 (250 nmol/L) or vehicle for 24 hours. After confirming that the cell number and morphology did not differ between groups (<10% in average cell number), the cell supernatant was centrifuged at 8000 × g for 1 minute to remove floating cells. The volume of the supernatant was adjusted with serum-free medium and then was used for SDS-PAGE zymography and collagen degradation assays. These experiments were repeated three times, to confirm reproducibility.
Myofibroblast Coculture with T Lymphocytes
Pulmonary fibroblasts (5 × 104 cells) were plated onto a 12-well plate and the myofibroblast phenotype was induced as described above. Cells were observed under a light microscope, to confirm that the numbers of cells were similar among wells. Peripheral blood mononuclear cells were isolated from the peripheral blood of a normal Lewis rat using Lympholyte density gradient separation medium (Cedarlane Laboratories, Burlington, ON, Canada; Burlington, NC), and T cells were isolated using magnetic activated cell sorting (MACS cell separation with anti-T-cell OX52 MicroBeads; Miltenyi Biotech, Auburn, CA). Myofibroblasts were continuously cultured in RPMI 1640 with 1% fetal bovine serum. The insert of a Transwell permeable support (0.4-μm pore; BD Biosciences, Mississauga, Canada; San Jose, CA) was placed in each well of the myofibroblast culture. In one group, isolated T cells (20 × 105 cells) were added to the insert; in a second group, T cells were added to the myofibroblast culture under the insert, leaving the insert free of cells; in a third group, only the insert was placed, without any T cells. Cells were then cultured for 48 hours.
Cyclosporine Treatment for Myofibroblasts
Pulmonary fibroblasts (5 × 104 cells) were plated onto a 12-well plate and the myofibroblast phenotype was induced as described above. Myofibroblasts were incubated with different concentrations of cyclosporine (0, 50, 500, and 5000 ng/mL) for 96 hours without medium exchange.
Histology and Immunofluorescence Labeling
Formalin-fixed, paraffin-embedded tissue was sectioned (5 μm in thickness) onto a glass slide and was used for all of the histological and immunofluorescence staining for tissue. H&E, Masson's trichrome, and Picrosirius Red (PSR) collagen staining were performed according to standard protocols. Graft obliteration after intrapulmonary tracheal transplantation was morphometrically quantified on H&E-stained sections. A low magnification image (×20) was acquired, covering the whole trachea, and the border outlining the area inside the tracheal cartilage and the border outlining the area inside the luminal opening were traced manually using Photoshop version 6.0 software (Adobe, San Jose, CA). The area inside each trace was calculated using ImageJ software (version 1.30; NIH, Bethesda, MD) to yield the whole luminal area (the total area of the lumen inside of the tracheal cartilage) and the open luminal area (the area that is not occupied by fibrous tissue). The obliterated area was calculated by subtracting the open luminal area from the whole luminal area, and the luminal occlusion ratio was calculated by dividing the obliterated area by the whole luminal area. PSR collagen staining was observed under both regular and polarized light. Observation of PSR staining under polarized light is a special technique to visualize intact interstitial collagen fibers, taking advantage of their birefringence characteristics to exclude denatured collagen.22 Five images of PSR-stained slides were acquired at medium magnification (×200) from the inside of the graft lumen under polarized light. The image was then binarized by a preoptimized threshold, using ImageJ software, and the positive staining area was calculated (n = 5 or 6 per group).
Anti-human MMP-9, α-SMA, CD3 (Dako Canada, Mississauga, Canada), MMP-2 (Biomol, Plymouth Meeting, PA), MMP-14, CD79a (Abcam, Cambridge, MA) and anti-rat CD68 (Serotec, Raleigh, NC) antibodies were used for immunofluorescence labeling, as described previously.20 All of the anti-human antibodies have been reported to cross-react with rat tissues. TUNEL in combination with α-SMA was performed using a fluorescein in situ cell death detection kit (Roche Applied Science, Indianapolis, IN), as described previously.23 For semiquantification of T cells, B cells, and macrophages, slides were stained for CD3, CD79a, and CD68, respectively. Ten high-power-field images (×400) were acquired from two sections per animal for all animals (n = 5 or 6 per group), and the number of positive cells was counted in images; counting was done blinded to treatment.
Cultured cells on coverslips were fixed with 4% paraformaldehyde for 60 minutes, permeabilized with 0.1% Triton-X, blocked with 6% goat serum in PBS, and then incubated with primary anti-α-SMA antibodies with or without anti-CD3 antibodies, followed by secondary goat-anti-mouse antibody conjugated with Alexa-555 and Alexa-488 (Invitrogen, Carlsbad, CA), respectively. Cells were washed three times with PBS between steps. In some experiments, incubation for 1 hour at 37°C using the Roche fluorescein in situ cell death detection kit was followed by immunofluorescence staining for α-SMA.
For semiquantification of cells cultured on coverslips, images at medium magnification (×200) were systematically acquired from five different parts of a coverslip (top, left, bottom, right, and center). To quantify the number of myofibroblasts, an image for red fluorescence (α-SMA) was first binarized with a preoptimized threshold using ImageJ software. After merging the picture with the blue channel (nucleus) with or without the green channel (CD3), the nuclei surrounded by red color were counted as the number of myofibroblasts. In an experiment in which T cells were cocultured with myofibroblasts, nuclei associated with CD3+ cells were excluded.
Fluorescence in Situ Gelatin Zymography
To localize gelatinolytic activity, in situ zymography was performed using fluorescein isothiocyanate-conjugated DQ-gelatin (Invitrogen). Fresh-frozen sections (8 μm thick) were first stained with Cy3-conjugated α-SMA (Sigma-Aldrich) and Hoechst dye for 15 minutes without fixation, and then washed with PBS. The tissue was incubated with or without MMP inhibitor [SC080, 20 μmol/L; EDTA 25 mmol/L; or 1,10-phenanthroline (pan MMP inhibitor; Sigma-Aldrich), 1 mmol/L] at room temperature for 1 hour. After solution was removed, DQ-gelatin mixed with reaction buffer (0.05 mol/L Tris-HCl, 0.15 mol/L NaCl, 5 mmol/L CaCl2, 200 μmol/L sodium azide, pH 7.6) was applied to the sections. Images were visualized in 3 hours.
Tissue Collagenolytic Activity Assay
EnzChek fluorescein isothiocyanate-collagen (Invitrogen Canada, Burlington, ON, Canada) was used according to the manufacturer's instructions. Frozen tracheal samples were homogenized in 50 mmol/L Tris-HCl buffer, pH 7.6, containing 1.5 mmol/L NaCl, 0.5 mmol/L CaCl2, 1 μmol/L ZnCl2, 0.01% Brij 35, and 0.25% (v/v) Triton X-100. After incubation at 37°C for 1 hour and centrifugation at 2500 gat 4°C for 10 minutes, the supernatant of the tissue lysate was recovered and further analyzed. After the tissue lysate was mixed with DQ-collagen and then incubated for 6 hours with or without 1 mmol/L of 1,10-phenanthroline using a black half-volume 96-well plate (Nalge Nunc International, Rochester, NY) at 37°C, fluorescence was read using a fluorescent plate reader (Gemini XPS; Molecular Devices, Sunnyvale, CA) at the emission wavelength of 413/488 nm.
SDS-PAGE Zymography
Cell culture supernatant (7 μL from each well) was loaded for electrophoresis on Novex precast 10% Tris-glycine gels containing 0.1% gelatin (Invitrogen) for 2 hours at 125 V. After renaturation, the gels were incubated at 37°C for 5 hours, to avoid overdevelopment and consequent saturation. Gels were stained with 0.5% Coomassie Blue G250 (Thermo Scientific, Worcester, MA) for 30 minutes and then were destained in 30% methanol-10% acetic acid. The gels were scanned using a GS-800 densitometer (Bio-Rad, Hercules, CA).
Collagen Degradation Assay for Cell Culture Supernatant
The remaining cell supernatant was concentrated using a protein concentration kit, Amicon Ultra-0.5 (Millipore, Billerica, MA), with centrifugation at 14,000 × g, 4°C, for 30 minutes, which yielded 20 μL of concentrated medium. MMPs were activated with 4-aminophenylmercuric acetate (Sigma-Aldrich) for 30 minutes at 37°C with or without a high concentration of SC080 (250 μmol/L) and then were incubated with 25 μg of rat-tail-derived type I collagen (Sigma-Aldrich) dissolved in 50 μL of reaction buffer (50 nmol/L Tris-HCI, 150 mmol/L NaCl, 5 mmol/L CaCl2) with 1 μL of MMP-2-specific inhibitor I (EMD Chemicals, Gibbstown, NJ) dissolved in ethanol (13 μmol/L) or ethanol alone. After 10 hours of incubation, the digestion product was electrophoresed on 10% SDS-PAGE gels and stained with InstantBlue (Expedeon, San Diego, CA). The gels were scanned using a Bio-Rad GS-800 densitometer, and the band density was quantified using ImageJ software.
Quantitative RT-PCR Analysis
To study gene expression in fibrous tissue in the allograft lumen, a microdissection technique was used to extract mRNA from the tissue interior to the tracheal cartilage, and then gene expression was analyzed using real-time RT-PCR. Frozen pieces of tracheal sections (20 μm thick) were cut onto glass slides and counterstained lightly with hematoxylin. The tissue interior to the tracheal cartilage was microdissected using an 18-gauge needle under stereomicroscopic visualization and was immediately put into the appropriate buffer from the RNeasy fibrous tissue mini kit (Qiagen, Mississauga, ON, Canada; Valencia, CA). RNA was extracted from 10 to 20 sections for each sample, according to manufacturer's instructions. An ABI reverse transcriptase reaction kit (Applied Biosystems, Foster City, CA) was used for the reverse transcription reaction, and real-time PCR was performed on an ABI-7900HT system (Applied Biosystems). The geometric mean of the gene expression of actin-β (ACTB) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH), which were selected as the most stable set of house keeping genes among eight tested candidate genes using GeNorm software (http://medgen.ugent.be/∼jvdesomp/genorm") and using the method of Vandesompele24 for data normalization.
Statistical Analysis
Data are reported as means ± SEM. A one-way analysis of variance test and a post hoc Tukey's test were performed to compare multiple groups. In the analysis of procollagen gene expression, regression analysis was used to determine the contribution of doses and administration timing to the response. All statistical analyses were performed using JMP version 5.0 software (SAS Institute, Cary, NC). P < 0.05 is reported as significant.
Results
MMP-2 and MMP-14 Are Localized to Myofibroblasts in Fibrotic Active OB Lesions after Lung Transplantation in Patients
We first examined lung tissue specimens obtained from lung transplant recipients affected with BOS. All examined lungs contained completely obliterated active fibrotic OB lesions, occupied by dense collagen deposition in trichrome staining (Figure 1A), a number of α-SMA+ myofibroblasts, and a relatively small number of CD3+ T cells in immunofluorescence (Figure 1B). Double immunofluorescence labeling localized both MMP-2 and MMP-14 to myofibroblasts in OB lesions, whereas MMP-9 was not localized to myofibroblasts (Figure 1C), but instead to a relatively small number of infiltrating cells among myofibroblasts (Figure 1B, inset).
Figure 1.
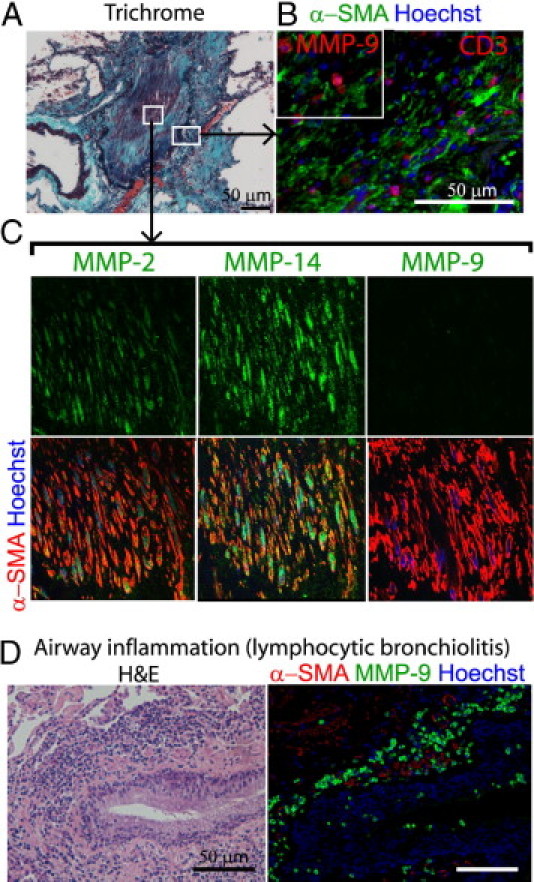
Localization of MMP-2 and MMP-14 in active fibrotic human OB lesions after lung transplantation. A: Masson trichrome staining demonstrates collagen deposition in the lumen of a small airway affected by active OB. Boxed areas correspond to images at higher magnification in B and C. B: Immunofluorescence labeling for CD3 (T cells) and α-SMA (myofibroblasts) demonstrates a number of myofibroblasts and a relatively small number of infiltrating T cells. Inset: Immunofluorescence labeling for MMP-9 and α-SMA, demonstrating expression of MMP-9 by infiltrating cells among myofibroblasts. Same original magnification as the main image. C: Immunofluorescence labeling for MMP-2, −14, or −9 and α-SMA for myofibroblasts demonstrates a number of myofibroblasts in the lumen positive for MMP-2 and MMP-14 but not MMP-9. The orange color of myofibroblasts indicates colocalization of α-SMA with MMP-2 or MMP-14. Original magnification, 600×. D: A lesion of lymphocytic bronchiolitis, showing inflammatory infiltration around a bronchiole under H&E staining (left) and inflammatory cells positive for MMP-9 around the airway (right). Note that smooth muscle layers positive for α-SMA (red) around the airway are negative for MMP-9 (green). Scale bars = 50 μm.
In addition, four of the seven specimens contained lesions of airway inflammation or lymphocytic bronchiolitis (Figure 1D), which is considered to be a precursor lesion of OB3 and is often found in association with airway neutrophilia.1 We observed numerous cells positive for MMP-9 in such inflammatory airways (Figure 1D).
MMP-2, MMP-14, and Gelatinolytic Activity in Allograft Airway Fibrosis in a Rat Intrapulmonary Tracheal Transplant Model
We then examined allograft airway fibrosis of an intrapulmonary tracheal transplant model. Consistent with our previous observation,20 allograft airways developed almost complete luminal fibrous obliteration by post-transplant day 21, whereas isografts recovered airway epithelium with luminal patency by day 21 (Figure 2A). The fibrous tissue in the allograft lumen at day 21 was occupied by a number of α-SMA positive myofibroblasts and a relatively small number of CD3+ T cells (Figure 2A). MMP-2 and MMP-14 were localized mainly to myofibroblasts (Figure 2B). To examine the ECM-degrading capacity of these MMPs, we performed in situ fluorescence gelatin zymography, in which fluorescein isothiocyanate-conjugated gelatin emits fluorescence on degradation. Indeed, green fluorescence was observed particularly in the pericellular area of myofibroblasts (Figure 2C). Additional MMP inhibitors such as EDTA and SC080 eradicated the gelatin degradation capacity (Figure 2C), demonstrating MMP-dependent gelatin degradation.
Figure 2.
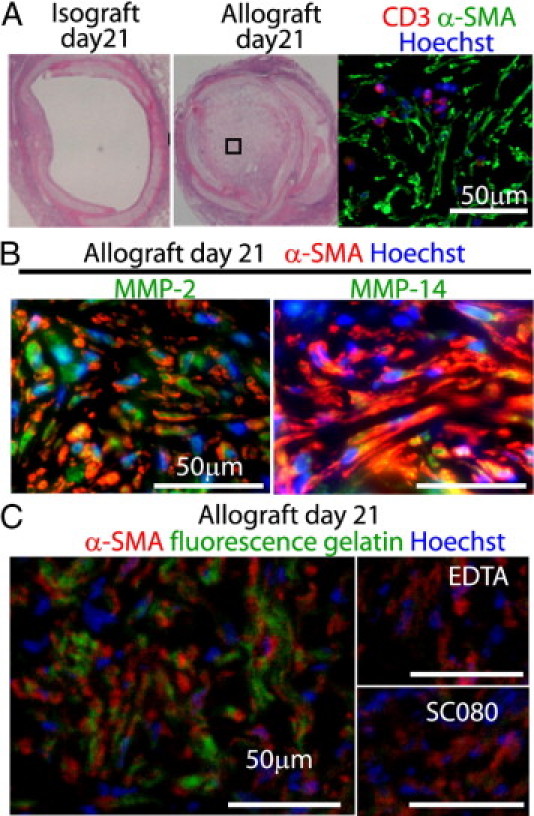
MMP-2, MMP-14, and gelatinolytic activity in allograft airway fibrosis in an intrapulmonary tracheal transplant model. A: An isograft and an allograft at day 21 after intrapulmonary tracheal transplantation. Under H&E staining, isografts showed complete recovery of the epithelium with an open lumen (left), but the allografts showed complete luminal obliteration with fibrous tissue (middle). Immunofluorescence labeling for CD3 and α-SMA demonstrates a number of myofibroblasts and a relatively small number of infiltrating T cells (right). B: Localization of MMP-2 and MMP-14 to α-SMA+ myofibroblasts in allograft airway fibrosis at day 21. Graft-infiltrating immune cells are also positively stained for these MMPs. C: Fluorescence in situ zymography for an established allograft airway fibrosis at day 21. In situ gelatin zymography using fluorescence-quenched DQ-gelatin in combination with immunofluorescence labeling for α-SMA in a frozen tissue section demonstrates gelatinolytic activity in allograft airway fibrosis. Note the pericellular green fluorescence indicating active matrix degradation at the periphery of the cells. Negative controls including EDTA, which chelates Zn2+ necessary for MMP activity, or direct inactivation of MMPs with SC080 in vitro demonstrate that the gelatinolytic activity is mediated by MMPs. Scale bars = 50 μm.
SC080 Treatment for Established Fibrosis
To examine the role of MMPs in inflammation and tissue remodeling of allograft airway fibrosis, we next treated allograft recipient animals with a broad-spectrum MMP inhibitor, SC080, from post-transplant day 21, when allograft obliteration was completed (Figure 3A). Five different regimens (doses and timings) were used to achieve different in vivo concentrations and kinetics. In general, plasma concentrations of SC080 peaked 1 hour after administration, followed by a rapid decline by 50% to 75% over the next 2 hours. Among the five regimens, 5 mg/kg b.i.d. (hereafter referred to as the intensive SC080 treatment) showed the most stable and highest plasma concentration: Cmax = 5.3 ± 0.69 μmol/L (mean ± SEM), Cmin = 1.1 ± 0.09 μmol/L, area under the curve AUC = 70.0, and calculated Cmean = 2.9 μmol/L. The lowest dose of 2.5 mg/kg q.d. (hereafter referred to as the low-dose SC080 treatment) showed the lowest plasma drug levels: Cmax = 2.0 ± 0.25 μmol/L, Cmin = 0.05 ± 0.00 μmol/L, AUC = 16.9, and calculated Cmean = 0.7 μmol/L. The highest dose was selected based on reported maximum tolerable dose in rats (T. Sunyer, personal communication) with a musculoskeletal-syndrome-like symptom.25 The lowest dose was selected based on our previous study.20
Figure 3.
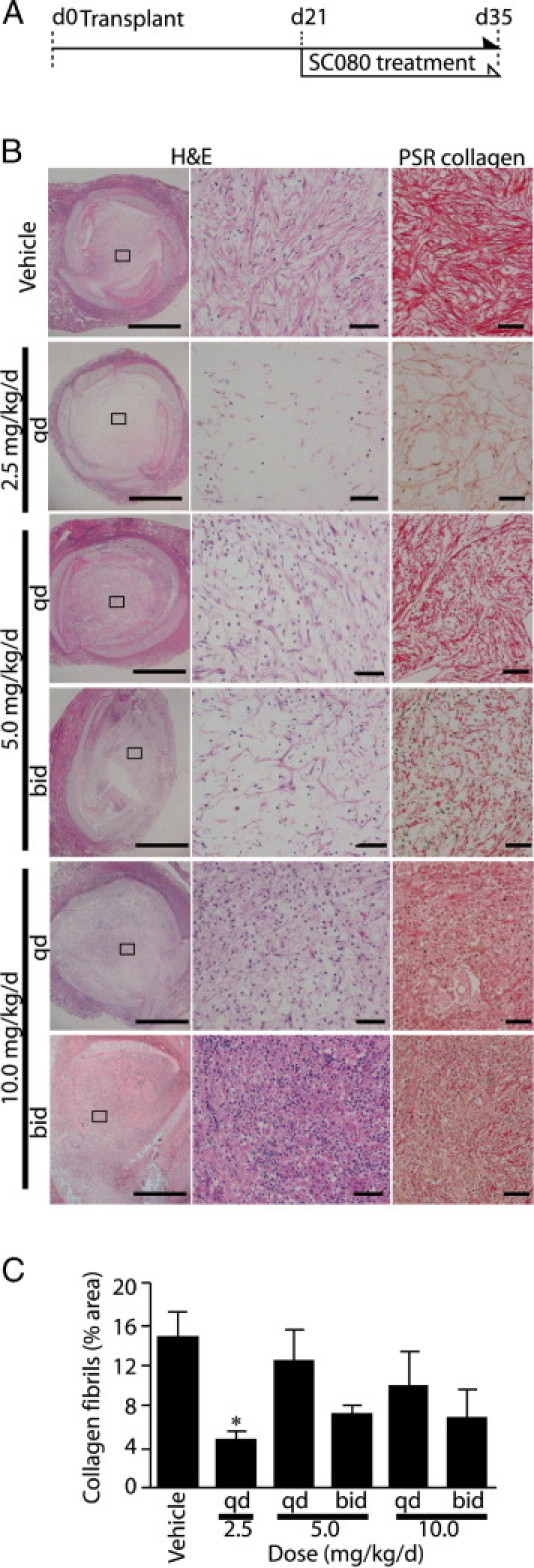
The lowest dose of SC080 most effectively reduces extracellular matrix and myofibroblasts in the lumen. A: Five different regimens of SC080 (2.5 to 10 mg/kg day, administered q.d. or b.i.d.) were tested on allograft recipient animals from day 21 to day 35 after transplantation. B: Allograft tracheae were histologically examined at day 35 using H&E and PSR collagen staining. Boxed areas in the left column correspond to images at higher magnification in the middle and right columns. Both treated and control allografts showed luminal obliteration. Animals treated with a low dose of SC080 (2.5 mg/kg per day, q.d.) showed the lowest amount of ECM and the least cellularity. Animals treated with intensive SC080 (10 mg/kg per day, b.i.d.) showed increased mononuclear cell infiltration. Treatment with intermediate doses resulted in moderate changes in cellularity and ECM deposition. Scale bars: 500 μm (left column); 50 μm (middle and right columns). C: Morphometric quantification of PSR staining under polarized light demonstrated significantly reduced total collagen in low-dose SC080 treatment. *P < 0.05 versus vehicle control, Tukey's test, n = 5 for each group.
Low-Dose SC080 Reduces Interstitial Collagen in the Lumen of Established Fibrosis
After 2 weeks of SC080 treatment of allograft recipients, the different regimens of SC080 treatment changed airway fibrosis in different ways (Figure 3B). The lumens of control allografts were occupied by a dense deposition of ECM with a number of spindle-shaped myofibroblast-like cells. PSR collagen staining showed dense depositions and organized alignment of collagen fibers (Figure 3B). Surprisingly, allografts after low-dose SC080 treatment showed the lowest cellularity and decreased density in ECM deposition in the lumen (Figure 3B). Conversely, allografts of intensive SC080 treatment showed massive infiltration of mononuclear cells under H&E staining, greater even than for control allografts (Figure 3B). Intermediate doses showed moderated dose-dependent changes in cellularity and collagen deposition (Figure 3B). Observation of PSR collagen staining under polarized light is a special technique to visualize intact interstitial collagen fibers by taking advantage of their birefringence characteristics to exclude denatured collagen.22 Morphometric quantification of PSR staining under polarized light demonstrated significantly smaller amounts of collagen with the low-dose SC080 treatment (Figure 3C).
Low-Dose SC080 Treatment Increases MMP-Dependent Collagenolytic Activity
Given that the balance between the production and degradation of collagen determines total collagen, we further examined procollagen gene expression and collagen degradation. Gene expression of procollagen α1(I) and α1(III), precursors of collagen type I and type III respectively, did not differ between the low-dose treatment and control; expression levels of these genes then decreased in a dose-dependent manner (P < 0.01; Figure 4A).
Figure 4.
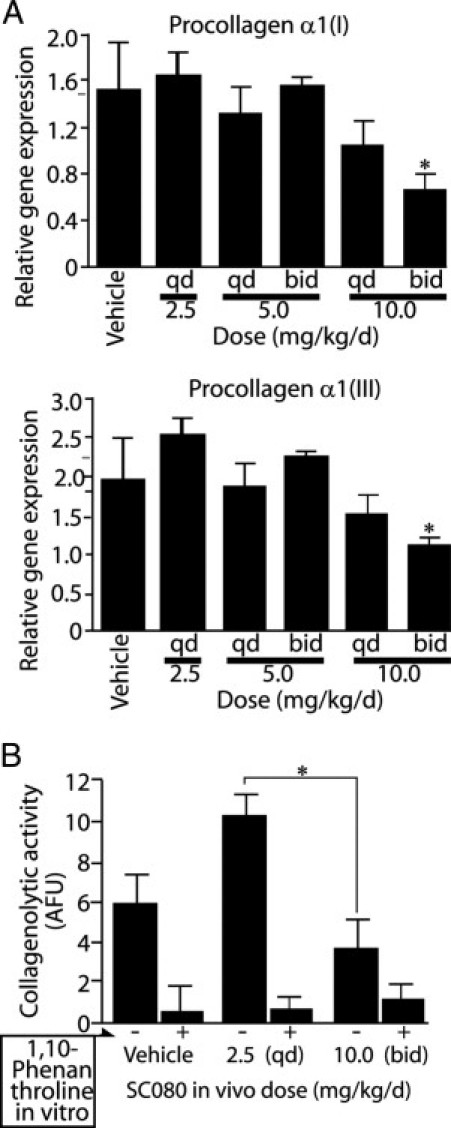
Low-dose SC080 treatment increases MMP-dependent collagenolytic activity without changing procollagen gene expression. A: Quantitative real-time RT-PCR analysis for gene expression of procollagen α1(I) and α1(III), the precursors for type I and type III collagen, respectively. Both genes showed dose-dependent decreases in expression. P < 0.01, regression analysis. The high-dose treatment showed a significant decrease in procollagen α1(I) and α1(III) gene expression. *P < 0.05, analysis of variance followed by a Tukey post hoc analysis. B: Collagenolytic activity in tissue homogenates of allograft tracheae for control, low-dose SC080 treatment (2.5 mg/kg per day, q.d.) and intensive SC080 treatment (10 mg/kg per day, b.i.d.). Collagenolytic activity was significantly higher in the low-dose SC080 group than in intensive SC080 group. *P < 0.05, n = 5 for each group.
Conversely, in collagenolytic activity analysis using fluorescein isothiocyanate-conjugated collagen type I that emits fluorescence on degradation, the low-dose SC080 treatment group showed significantly higher collagenolytic activity than the intensive treatment group (P < 0.05; Figure 4B). The decreased fluorescence observed with the in vitro addition of a pan-MMP inhibitor, 1,10-phenanthroline, demonstrated that most of the collagen degradation is dependent on MMP activity. Transgenic deletion and chemical inhibition of MMPs are known to induce feedback up-regulation of collagenolytic MMPs26–28; thus, it is possible that low-dose SC080 was insufficient to eradicate all of the MMP activity, or it may be that low-dose SC080 preferentially inhibited some MMPs more than others and paradoxically increased MMP-dependent collagenolytic activity.
SC080 Treatment Modifies Cellular Components of Allograft Airway Fibrosis
We also examined the effects of SC080 treatment on the cellular components of allograft airway fibrosis. We compared the numbers of myofibroblasts, macrophages, T cells, and B cells among the low-dose and intensive SC080 treatment groups and the control group. Notably, both low-dose and intensive treatments of SC080 showed significant effects in reducing the number of myofibroblasts (P < 0.01; Figure 5, A and D), but with different effects on their morphology. In the low-dose SC080 treatment, small irregular-shaped myofibroblasts showed a scattered distribution; in the intensive SC080 treatment, large, well-stretched myofibroblasts were aligned in an organized manner (Figure 5B). Apoptosis was a speculated mechanism leading to the decreased number of myofibroblasts. Although a small number of TUNEL-positive myofibroblasts were observed in intensive SC080 treatment (2 to 5 cells per graft section on average; Figure 5B, inset), we observed almost no TUNEL-positive myofibroblasts in the low-dose SC080 group.
Figure 5.
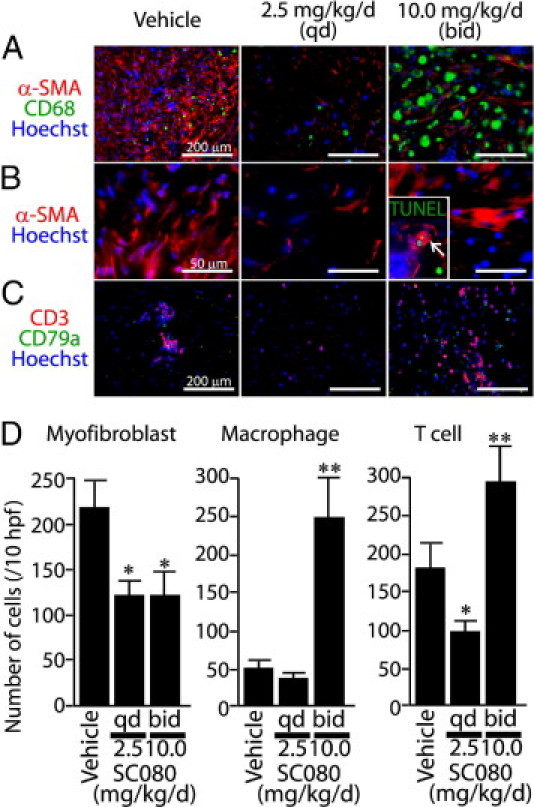
SC080 treatment modifies cellular components of allograft airway fibrosis. A: Immunofluorescence labeling for myofibroblasts (α-SMA) and macrophages (CD68). B: Immunofluorescence labeling for myofibroblasts (α-SMA). Note the irregular-shaped myofibroblasts with scattered distribution in the low-dose group and the large, well-stretched myofibroblasts in the intensive treatment group compared with those in controls. Inset: Immunofluorescence with TUNEL; the arrow indicates a TUNEL-positive myofibroblast. Same original magnification as the main image. C: Immunofluorescence labeling for T cells (CD3) and B cells (CD79a). The groups of low-dose (2.5 mg/kg per day, q.d.) and intensive (10 mg/kg per day, b.i.d.) SC080 treatment and vehicle control were examined. D: Semiquantitative analysis of the numbers of myofibroblasts, macrophages, and T cells in the fibrotic area of the graft lumen. The number of myofibroblasts was smaller in the low-dose and intensive SC080 treatments (*P < 0.05). T-cell numbers in the low-dose group were significantly smaller than in the control group (*P < 0.05), whereas the numbers of macrophages and T cells were significantly larger in the intensive SC080 treatment group than in the control group (**P < 0.05). n = 5 for each group. Scale bars: 200 μm (A and C); 50 μm (B).
The numbers of CD68+ macrophages (Figure 5, A and D) and CD3+ T cells (Figure 5, C and D) were significantly increased in the intensive SC080 treatment group. T-cell infiltration was decreased in the low-dose SC080 treatment group, compared with control (Figure 5D). CD79a+ B-cell infiltration was minimal in all groups (Figure 5C; quantitative data not shown).
Increased MMP-2 Gene Expression and Reduced Levels of Inflammation-Related MMPs in Low-Dose SC080 Treatment
In general, collagen degradation depends mostly on MMPs,25 and only a limited number of MMP family members (including MMP-1, although not in rodents, and MMP-2, -8, -13, and -14) can degrade interstitial collagen.29,30 MMPs can also be expressed by inflammatory cells in this animal model20 and under many other conditions.31 Using real-time RT-PCR, we examined gene expression of MMPs that might be involved in collagen degradation and/or inflammation. Among these, MMP-2 was the only one whose expression in the low-dose SC080 treatment was significantly higher than that of control (Figure 6A). In immunofluorescence staining, MMP-2 was localized mainly to myofibroblasts in the low-dose SC080 treatment (Figure 6B). Conversely, gene expression of MMP-8, -9, and -13 in this group was significantly decreased, compared with the vehicle control (Figure 6A). We speculated that the change in the MMP profile was associated with increased collagen degradation (Figure 4B) and decreased T-cell infiltration (Figure 5) by the low-dose SC080 treatment.
Figure 6.
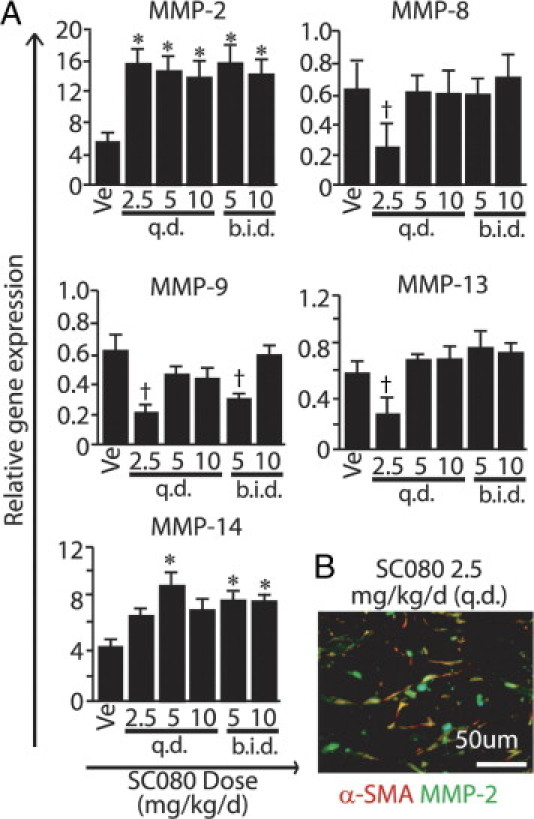
Increased MMP-2 gene expression and reduced inflammation-related MMPs in low-dose SC080 treatment. A: Gene expression analysis of MMPs by real-time RT-PCR. Gene expression of MMP-2 in the low-dose SC080 group was significantly higher than that in controls (*P < 0.05) and gene expression of MMP-14 was not significantly different, whereas gene expression of MMP-8, −9, and −13 in the low-dose SC080 group was significantly lower than in controls (†P < 0.05). B: Double immunofluorescence labeling for α-SMA and MMP-2. MMP-2 was localized mainly to myofibroblasts positive for α-SMA in the fibrous tissue of allograft airway lumen in the low-dose SC080 group and in vehicle controls. Scale bar = 50 μm.
Effect of SC080 Treatment for Cultured Myofibroblasts
To explore the mechanism whereby low-dose SC080 treatment affected established fibrosis, we conducted a series of experiments in vitro. Rat pulmonary fibroblasts were primarily cultured and incubated with recombinant TGF-β1 to induce myofibroblasts. Incubation with TGF-β1 for 48 hours was sufficient to induce the α-SMA+ myofibroblast phenotype in >80% of the cells (Figure 7, A and B). The following in vitro experiments were conducted using these myofibroblasts induced by TGF-β1 pretreatment.
Figure 7.
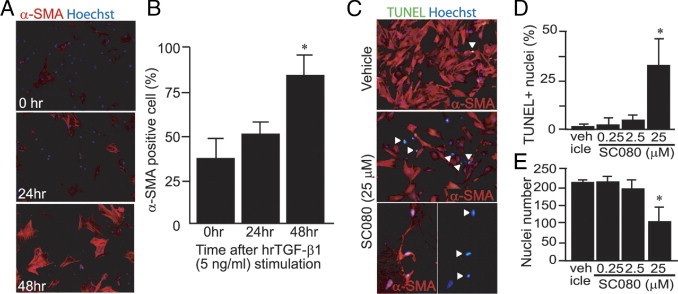
Apoptosis was induced in myofibroblasts by a high concentration of SC080. A: Immunofluorescence labeling of primary cultured rat pulmonary fibroblasts for α-SMA with stimulation with human recombinant TGF-β1 (5 ng/mL) for 0, 24, and 48 hours. B: Semiquantification of the percentage of α-SMA+ myofibroblasts among all fibroblasts. After 48 hours of stimulation, >80% of the cells showed the phenotype of myofibroblasts (*P < 0.05, n = 4). C: Double immunofluorescence labeling for α-SMA and TUNEL after incubation of myofibroblasts with a high concentration of SC080 (25 μmol/L) or vehicle for 48 hours. Arrowheads indicate positive TUNEL staining. The two images at the bottom of C show, at higher magnification, a shrunken morphology of myofibroblasts (left) and the corresponding TUNEL-positive nuclei (right). D: Semiquantitative analysis of the percentage of apoptotic cells. TUNEL-positive cells were significantly increased only in the group treated with the highest concentration of SC080 (*P < 0.05, n = 5). E: Semiquantitative analysis of the number of nuclei. After treatment with the highest concentration of SC080, the number of cells was significantly reduced (*P < 0.05, n = 5). Original magnification, 200×, except for the bottom pictures of C (400×).
Myofibroblasts were treated in vitro with different concentrations of SC080 (0.25 to 250 μmol/L) for 72 hours. Treatment of myofibroblasts with the highest concentration of SC080 (250 μmol/L) resulted in detachment of almost all of the cells from the plate, and staining was not performed. The second highest concentration of SC080 (25 μmol/L) showed an increase in TUNEL-positive apoptotic cells (Figure 7, C and D) and a significant decrease in the cell number (Figure 7E). This effect was not obvious at lower concentrations of SC080. The result demonstrated that SC080 induces apoptosis in myofibroblasts in a concentration-dependent manner in vitro. This finding may be relevant to TUNEL-positive myofibroblasts in the intensive SC080 treatment group in vivo (Figure 5A); however, considering the low concentration of SC080 in vivo in the low-dose SC080 treatment, this is unlikely to explain the effect of low-dose SC080 on the numbers of myofibroblasts.
Effect of SC080 on MMP-2-Mediated Collagen Degradation by Myofibroblasts
Treatment of the same cultured myofibroblasts with a relatively low concentration of SC080 (250 nmol/L) resulted in an increase in the levels of both pro- and active forms of MMP-2 (Figure 8, A and B), but had no effect on MMP-9 levels (Figure 8, A and C). Collagenolytic activity of the cell culture supernatant from cultured myofibroblasts was examined. The culture supernatant alone did not contain a significant amount of collagen type I (Figure 8D). SC080-treated myofibroblasts degraded exogenous collagen type I more efficiently than vehicle-treated myofibroblasts (Figure 8, D and E). Notably, additional treatment of the culture supernatant with a high concentration of SC080 (250 μmol/L) inhibited collagen degradation almost completely (Figure 8D), demonstrating that SC080 works as a general MMP inhibitor if the concentration is sufficiently high. Importantly, coincubation of the cell culture supernatant with an MMP-2 specific inhibitor prevented degradation of type-I collagen to the same level as did retreatment with a high concentration of SC080 (Figure 8, D and E). These results demonstrate that a low concentration of SC080 increases MMP-2 expression and activity in myofibroblasts and contributes to degradation of type-I collagen. Notably, this concentration of SC080 was similar to the average plasma concentration of the low-dose SC080 treatment in vivo.
Figure 8.
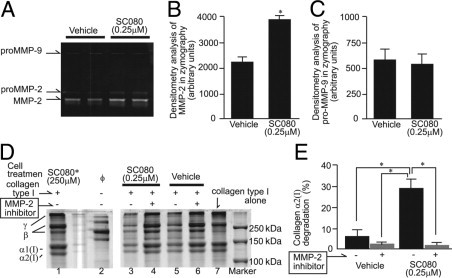
The effect of SC080 on MMP-2-mediated collagen degradation by myofibroblasts. A: A representative SDS-PAGE gelatin zymograph of cell culture supernatant. Treatment of myofibroblasts with a relatively low concentration of SC080 (250 nmol/L) increased pro- and active forms of MMP-2 but did not affect pro-MMP-9. B: Quantification of band densities of active MMP-2 in SDS-PAGE gelatin zymography (*P < 0.01, n = 8). C: Quantification of band densities of pro-MMP-9 in SDS-PAGE gelatin zymography (n = 8). D: A representative result from a collagenolytic activity assay. Concentrated culture supernatant of myofibroblasts pretreated with SC080 (0.25 μmol/L) or vehicle was incubated with collagen type I. Supernatant from SC080-treated cells show higher collagenolytic activity (lane 3) than that from vehicle-treated cells (lane 5), whereas collagenolytic activity was abolished by additional MMP-2-specific inhibitor (lanes 4 and 6) to the same level as an additional high concentration of SC080 (250 μmol/L, lane 1). Note that culture supernatant from myofibroblasts without any additional treatment (lane 2) contains no detectable collagen type I (lane 7). E: Quantified band densities of collagenolytic activity assay (*P < 0.01, n = 6).
Coculture with T Cells Supports Persistence of Myofibroblasts
Considering the reduced T-cell infiltration after low-dose SC080 treatment in vivo, we speculated that the infiltration of T cells plays an important role in the persistence of myofibroblasts. To examine this possibility, we cocultured T cells with myofibroblasts at a lower serum concentration. We isolated T cells from the peripheral blood of healthy Lewis rats and then cocultured them with myofibroblasts. T cells were mostly CD45RC− effector T cells (> 90%) with an almost equal CD4/CD8 ratio. To examine the effect of direct and indirect contact between myofibroblasts and T cells, a Transwell support was used. The number of α-SMA positive myofibroblasts was largest in the directly cocultured group, whereas the coculture group across a Transwell support showed a moderate effect, compared with the myofibroblast monoculture (Figure 9, A and B). Notably, in the myofibroblast monoculture, the number of cells was significantly lower, compared with the number of cells at the beginning of the coculture experiment (Figure 9B). The result suggests that coexisting T cells play a role in the persistence of myofibroblasts and that the effect is in part mediated in a contact-dependent manner. Thus, the reduced numbers of infiltrating T-cells after the low-dose SC080 treatment in vivo may explain the reduced numbers of myofibroblasts.
Figure 9.
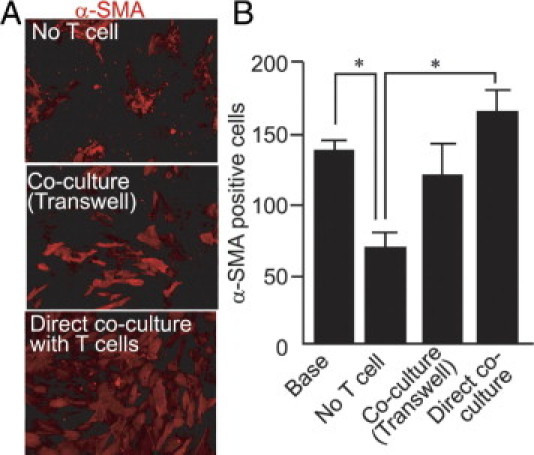
Coculture with T cells supports persistence of myofibroblasts. A: Myofibroblasts cultured in 1% fetal bovine serum for 96 hours alone or with T cells across a 0.4-μm-pore Transwell support show clumping and loss of α-SMA expression, whereas myofibroblasts cultured with T cells show preserved phenotype and numbers. Direct coculture of myofibroblasts with T cells facilitates the persistence of the myofibroblast phenotype. B: Cells with α-SMA expression without clumping were morphometrically quantified. Myofibroblasts directly cocultured with T cells show significantly larger numbers than those cultured without T cells (*P < 0.01). Original magnification, 200×.
Low-Dose SC080 Combined with Immunosuppression Partially Reverses Established Allograft Airway Obliteration in Vivo
To further reinforce the hypothesis that T-cell infiltration supports myofibroblast persistence, we further tested the effect of cyclosporine, a conventional calcineurin-inhibiting immunosuppressive drug, with or without low-dose SC080 treatment in our in vivo model. Using the same animal model, we previously demonstrated that cyclosporine treatment after the initiation of fibrotic process (post-transplant day 14) does not reduce fibrosis, although it can effectively reduce T-cell infiltration in allograft airways.23 All treatment was initiated at post-transplant day 28, when allograft lumens were almost completely obliterated (Figure 10A). Indeed, cyclosporine treatment alone reduced both T-cell infiltration and numbers of myofibroblasts (Figure 10) at day 42. As we expected, cyclosporine treatment alone was not effective enough to reverse fibrosis. SC080 treatment alone also showed significant effects on the numbers of T cells and myofibroblasts and a trend toward decreased collagen content (Figure 10), similar to those of low-dose SC080 treatment from day 21 to day 35.
Figure 10.
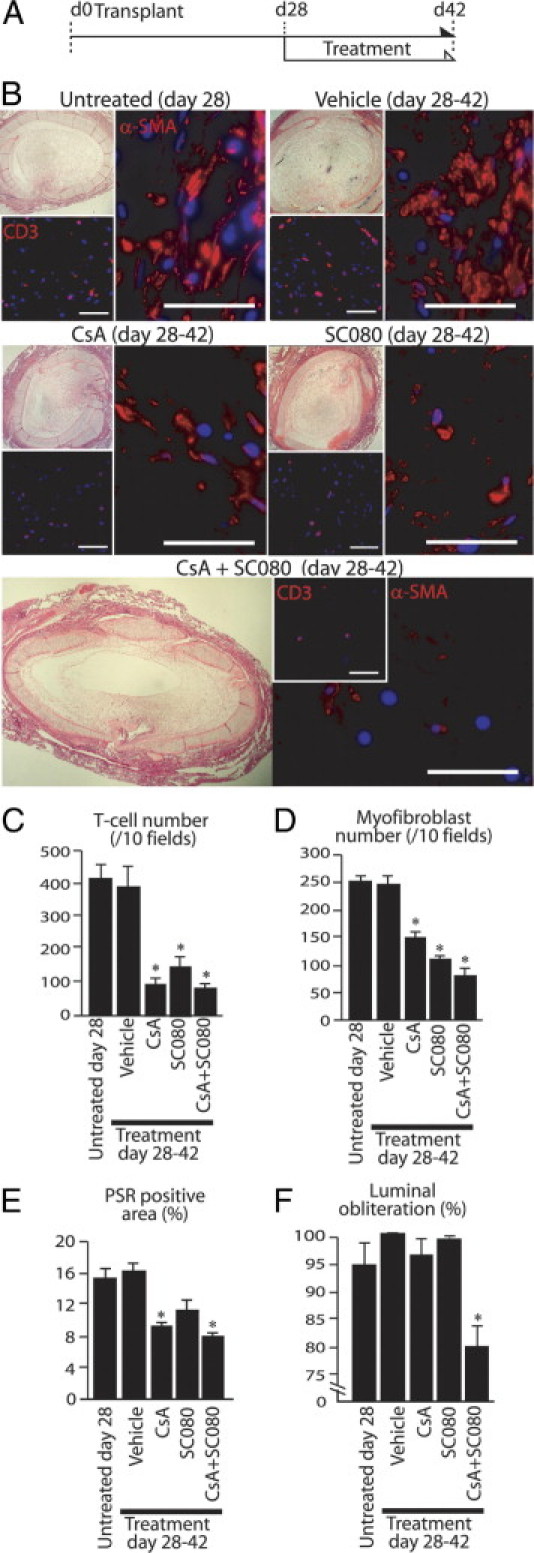
Low-dose SC080 combined with immunosuppression partially reverses obliterative allograft airway fibrosis. A: Low-dose SC080 (2.5 mg/kg per day, q.d.), cyclosporine (10 mg/kg per day, q.d.), or their combination was administered to allograft recipient animals from day 28 to day 42. B: Allograft airways under H&E staining and immunofluorescence labeling for CD3 (T cell) and α-SMA (myofibroblasts). Control groups at days 28 and 42, cyclosporine alone, and low-dose SC080 alone show complete obliteration of allograft airways (top left of each group), whereas the numbers of T cells (bottom left) and myofibroblasts (right) was smaller in both the cyclosporine and the SC080 treatment groups. Partial opening of the allograft airway lumen was observed in the combined treatment group (bottom, left), with a small number of myofibroblasts and T cells (bottom, right). C: Semiquantitative analysis of T cells demonstrated significantly reduced numbers of T cells in obliterative fibrosis of allograft airways in the cyclosporine, low-dose SC080, and combined cyclosporine and low-dose SC080 treatment groups, compared with control groups, at days 28 and 42 (*P < 0.01). D: Semiquantitative analysis of myofibroblasts demonstrated significantly reduced numbers of myofibroblasts in obliterative fibrosis of allograft airways in the cyclosporine, low-dose SC080, and combined cyclosporine and low-dose SC080 treatment groups, compared with control groups, at days 28 and 42 (*P < 0.01). E: Morphometric quantification of PSR staining under polarized light demonstrated significantly reduced total collagen among groups (*P < 0.05). F: Morphometric quantification demonstrated significantly reduced luminal obliteration in the combined cyclosporine and low-dose SC080 treatment group, compared with other treatment groups (including untreated controls) at day 28 (*P < 0.01, n = 6 for each group). Scale bars = 50 μm. CsA, cyclosporine.
Strikingly, the allografts of the combined treatment group showed a partial opening in the lumen (Figure 10B). Epithelial regeneration was not observed in any of the groups. Morphometric quantification demonstrated a significantly reduced obliteration in the combined treatment group, even in comparison with day 28 controls (P < 0.01; Figure 10F).
Finally, to rule out the possibility that cyclosporine has a direct toxic effect on myofibroblasts, cultured myofibroblasts were treated with various concentrations of cyclosporine (from 50 ng/mL to 5 μg/mL) for 96 hours. Although we previously demonstrated that 50 ng/mL of cyclosporine is sufficient to inhibit T-cell proliferation almost completely,20 none of the treatments affected the numbers of myofibroblasts (data not shown). These results suggest that T-cell infiltration plays a role in the maintenance of established fibrosis, although immunosuppression alone is not effective enough to reverse fibrosis; additional SC080 treatment that further modifies tissue remodeling and inflammation synergistically induced partial regression of established fibrosis.
Discussion
In human lung transplantation, up-regulation of MMP-8 and MMP-9 has been documented to be associated with neutrophilic airway inflammation and the later development of BOS.32–34 We also found a high expression of MMP-9 in inflammatory cells in lymphocytic bronchiolitis (Figure 1). In contrast, MMP expression profiles in the fibrous stages of OB/BOS have been relatively unknown. In the present study, we demonstrated localization of MMP-2 and MMP-14 to myofibroblasts and a relatively small number of MMP-9+ infiltrating cells in active fibrous human lung OB lesions (Figure 1). Because different stages of OB/BOS may respond to different treatment strategies, this novel finding might lead to the development of a biomarker for the fibrous stage of OB. Further investigation (using, for example, bronchoalveolar lavage to measure the levels of MMP-2 and MMP-14) would be important in translating the present findings into clinical practice.
The spatial and temporal expression pattern of MMPs in human OB lesions was similar to our previous findings in the rat intrapulmonary tracheal transplant model of OB.20 Allografts in this model first develop lymphocyte aggregates combined with infiltration of neutrophils by approximately post-transplant day 7, the process of which is analogous to lymphocytic bronchiolitis. The inflammatory cells express high levels of MMP-9 at day 7 after allograft (but not isograft) transplantation, and then MMP-9 is down-regulated after day 14. MMP-2 and MMP-14 are up-regulated from day 21, when myofibroblasts infiltrate and the subepithelial fibrosis extends to almost completely obliterate the lumen. Notably, we consistently found low-grade T-cell infiltration after day 21 in the present study, suggesting a persistent immune response to allografts even after the establishment of fibrosis. These analogies encouraged us to further investigate MMP-mediated inflammation and tissue remodeling in allograft airway fibrosis using this animal model.
We hypothesized that allograft airway fibrosis is a dynamic and potentially reversible process involving MMPs. To test this hypothesis, we used various regimens of a broad-spectrum MMP inhibitor, SC080. Notably, the low-dose SC080 treatment showed a significant beneficial effect on fibrosis, but the medium-dose and high-dose treatments did not (Figure 3).
The roles of MMPs in inflammation and fibrosis are generally complex. Although MMPs play important roles in the migration of immune responsive cells,35 recent studies have revealed complex immunomodulatory effects of MMPs.36 For example, MMP-2 plays an important role in terminating airway inflammation by degrading chemokines, and lack of this effect results in persistent inflammation.37 In addition, physiological levels of MMP-8 exhibit anti-inflammatory properties.38 MMPs can also be both profibrotic and antifibrotic.39 MMPs are profibrotic by releasing and activating profibrotic growth factors (eg, TGF-β1), which in turn activate migration, proliferation, and survival of fibroblasts and myofibroblasts.11,40,41 Conversely, MMPs can also be anti-fibrotic by degrading extracellular matrix to prevent its excessive accumulation in the tissue.29 Thus, we carefully analyzed the mechanism of the effect of low-dose SC080 treatment on established fibrosis in vivo and in vitro.
Given that myofibroblasts are the major source of interstitial collagen in fibrosis,20 the reduced numbers of myofibroblasts after low-dose SC080 treatment seemed to explain the decrease in collagen content in fibrosis. Of note, however, we found no difference in procollagen gene expression. Rather, tissue collagenolytic activity in allografts treated with low-dose SC080 was increased (Figure 4). This suggests that the reduced numbers of myofibroblasts does not by itself well explain the reduced collagen matrix. Among collagenolytic MMPs, only MMP-2 showed increased gene expression in vivo (Figure 6). Indeed, in the following in vitro experiments, a low concentration of SC080 increased MMP-2 in myofibroblasts and significantly contributed to the degradation of collagen type I (Figure 8). Supporting our results, Mizuno et al42 reported a beneficial effect of MMP-2 in an animal model of pulmonary fibrosis, in which hepatocyte growth factor up-regulated MMP-2 and reduced the numbers of myofibroblasts and the amount of collagen in the lung after bleomycin treatment.
We also attempted to explain the mechanism whereby the numbers of myofibroblasts were decreased by SC080 treatment. We first speculated that apoptosis of myofibroblasts might be involved, because of a previously reported apoptosis-inducing effect of SC080 on smooth muscle cells.43 However, we detected TUNEL-positive myofibroblasts only when a high concentration of SC080 was administered in vitro (Figure 7). Thus, apoptosis directly induced by SC080 was unlikely to be the major mechanism involved in the reduced numbers of myofibroblasts in low-dose SC080 treatment.
We previously demonstrated that SC080 treatment does not directly affect activation of T cells; rather, it significantly reduces trafficking of T cells into the allograft lumen.20 Because unresolved inflammation can continuously stimulate resident stromal cells12,13 to provide surviving signals to myofibroblasts,14 we next examined the possibility that reduced T-cell infiltration (Figure 5C) could have an effect on myofibroblasts. Indeed, direct contact between T cells and myofibroblasts contributed to the persistence of myofibroblasts in vitro (Figure 9). This possibility was further supported by an in vivo experiment, in which cyclosporine treatment for established fibrosis reduced the numbers of both myofibroblasts and T cells (Figure 10). These results demonstrated that persistent T-cell infiltration is a contributing factor to persistence of myofibroblasts in allograft airway fibrosis.
Importantly, it has been suggested that low-dose or incomplete MMP inhibition is more beneficial in reducing inflammation than complete blocking of MMPs, because MMPs can play both pro- and anti-inflammatory roles.44 Complex roles of MMPs in the alloimmune response have also been suggested in an animal model of OB.45 The benefit of incomplete MMP inhibition is represented by the fact that a subantimicrobial dose of doxycycline is the only MMP inhibitor approved by the U.S. Food and Drug Administration.44 Moreover, the harm of excessive MMP inhibition is also supported by the massive infiltration of T cells and macrophages in the intensive SC080 treatment in the present study (Figure 5) and by similar exaggerated inflammatory responses reported in other transplant models using potent MMP inhibitors.46,47 Alternatively, the intensive SC080 treatment may have elicited an inflammatory response secondary to significant pharmacologically induced cell injury or necrosis due to a direct toxic effect of SC080.
Taken together, the beneficial effect of low-dose SC080 treatment was explained by both an increase in the degradation of collagen matrix mediated by MMP-2 and a decrease in T-cell infiltration with an associated reduction in the numbers of myofibroblasts. The low-dose SC080 treatment alone, however, was not effective enough to reverse fibrosis, and neither was cyclosporine alone. Nevertheless, when these two treatments were combined, we found a reduced obliteration of allograft airway lumen in established allograft airway fibrosis (Figure 10).
These observations suggest the following principles to note when intervening in persistent fibrosis. First, an underlying unresolved inflammation is important, even though treatment for inflammation alone is unlikely to reverse fibrosis. Second, modulation of ECM may be an important part of the treatment. For example, if myofibroblasts were simply eradicated by some means, this would only leave an acellular scar tissue that is unlikely to resolve.10 Third, regeneration of normal tissue components, such as the epithelium, is ultimately necessary to replace fibrosis. Preservation of the original scaffold, such as the basement membrane, is also an important component for the complete regeneration of tissue.10 These principles are likely to apply for many chronic inflammatory and fibrotic disorders. Further investigation is needed, particularly in the areas of interaction between continuous inflammation and persistent myofibroblasts, as well as the role of myofibroblasts in resolution of fibrous ECM.
The role of tissue inhibitors of metalloproteinases (TIMPs) was beyond the scope of the present study. Nonetheless, because the MMP system is closely associated with TIMPs, we are aware that these molecules may also play roles in regression of fibrosis. For example, TIMP-2 is known to form a complex with MMP-2 and MMP-14 on the cell membrane and plays an important role in activation of MMP-2.48,49 Thus, MMP-14 and TIMP-2 may also contribute to collagen degradation. Moreover, evidence suggests that TIMPs are not simply endogenous inhibitors of MMPs, but also play roles as growth factors for some cell types.50 TIMP-1 has been demonstrated to support survival of myofibroblasts.51 Transgenic deletion of TIMP-1 showed a protective effect on allograft airway fibrosis.52 The complex effect of the MMP-TIMP system in fibrosis calls for further investigation in this field.
In the present study, intrapulmonary tracheal transplantation served as an ideal model for investigating the mechanism of fibrosis involving persistent alloimmune responses. This model has multiple advantages over orthotopic lung transplant models in investigating the basic mechanisms of allograft fibrosis, because of the simplicity and reproducibility of obliterative airway fibrosis.53 We do acknowledge the limitations of this animal model of lung transplantation, including the use of a large airway, the lack of an interface with an ambient air, and lack of a vascular anastomosis.53 Blood circulation is critical for normal and aberrant wound repair. We previously reported that blood circulation to the graft recovers within 7 days after transplantation.21 The initial ischemia and subsequent angiogenesis may be relevant to the pathogenesis of OB after lung transplantation.18,54 Conversely, the role of ambient air in this animal model remains unclear. Although the transplanted airway is not directly connected to the recipient airway, the graft is placed in lung parenchyma that is filled with air. We previously tested the role of air in the lung by closing both ends of a tracheal graft; this did not change the result in either allografts or isografts (unpublished data). At the time of fibrosis regression, however, it is still possible that the air around the graft could easily come into the lumen and play a role in the regression of fibrosis. Ambient air may also be important in the pathogenesis of OB, in that small airways receive air without intervening filtration in association with aspiration or infectious microorganisms. To address these questions, other animal models such as orthotopic tracheal transplantation and orthotopic lung transplantation may have useful complementary roles in the future.
In conclusion, we demonstrated multiple roles of MMPs in tissue remodeling and inflammation, and modulation of the MMP system led to regression of established fibrosis in combination with immunosuppression. Because lung transplant recipients receive immunosuppression, addition of the novel low-dose SC080 treatment or other treatment that modifies the MMP system would be clinically relevant. More importantly, this finding encourages further investigation into the fibrotic stage of OB, which hitherto has been assumed to be a static and irreversible condition.
Footnotes
Supported by the Canadian Cystic Fibrosis Foundation. M.S. was a recipient of the Canadian Cystic Fibrosis Foundation postdoctoral fellowship (2007–2009).
References
- 1.Stewart S., Fishbein M.C., Snell G.I., Berry G.J., Boehler A., Burke M.M., Glanville A., Gould F.K., Magro C., Marboe C.C., McNeil K.D., Reed E.F., Reinsmoen N.L., Scott J.P., Studer S.M., Tazelaar H.D., Wallwork J.L., Westall G., Zamora M.R., Zeevi A., Yousem S.A. Revision of the 1996 working formulation for the standardization of nomenclature in the diagnosis of lung rejection. J Heart Lung Transplant. 2007;26:1229–1242. doi: 10.1016/j.healun.2007.10.017. [DOI] [PubMed] [Google Scholar]
- 2.Nicod L.P. Mechanisms of airway obliteration after lung transplantation. Proc Am Thorac Soc. 2006;3:444–449. doi: 10.1513/pats.200601-007AW. [DOI] [PubMed] [Google Scholar]
- 3.Boehler A., Estenne M. Post-transplant bronchiolitis obliterans. Eur Respir J. 2003;22:1007–1018. doi: 10.1183/09031936.03.00039103. [DOI] [PubMed] [Google Scholar]
- 4.Kumar V., Abbas A.K., Fausto N. Robbins & Cotran Pathologic Basis of Disease. ed 7. Elsevier Saunders; Philadelphia: 2005. Tissue renewal and repair: regeneration, healing and fibrosis; pp. 87–118. [Google Scholar]
- 5.Iimuro Y., Nishio T., Morimoto T., Nitta T., Stefanovic B., Choi S.K., Brenner D.A., Yamaoka Y. Delivery of matrix metalloproteinase-1 attenuates established liver fibrosis in the rat. Gastroenterology. 2003;124:445–458. doi: 10.1053/gast.2003.50063. [DOI] [PubMed] [Google Scholar]
- 6.Siller-López F., Sandoval A., Salgado S., Salazar A., Bueno M., Garcia J., Vera J., Gálvez J., Hernández I., Ramos M., Aguilar-Cordova E., Armendariz-Borunda J. Treatment with human metalloproteinase-8 gene delivery ameliorates experimental rat liver cirrhosis. Gastroenterology. 2004;126:1122–1133. doi: 10.1053/j.gastro.2003.12.045. discussion 949. [DOI] [PubMed] [Google Scholar]
- 7.Watanabe T., Niioka M., Hozawa S., Kameyama K., Hayashi T., Arai M., Ishikawa A., Maruyama K., Okazaki I. Gene expression of interstitial collagenase in both progressive and recovery phase of rat liver fibrosis induced by carbon tetrachloride. J Hepatol. 2000;33:224–235. doi: 10.1016/s0168-8278(00)80363-3. [DOI] [PubMed] [Google Scholar]
- 8.Watanabe T., Niioka M., Ishikawa A., Hozawa S., Arai M., Maruyama K., Okada A., Okazaki I. Dynamic change of cells expressing MMP-2 mRNA and MT1-MMP mRNA in the recovery from liver fibrosis in the rat. J Hepatol. 2001;35:465–473. doi: 10.1016/s0168-8278(01)00177-5. [DOI] [PubMed] [Google Scholar]
- 9.Zhou X., Hovell C.J., Pawley S., Hutchings M.I., Arthur M.J., Iredale J.P., Benyon R.C. Expression of matrix metalloproteinase-2 and −14 persists during early resolution of experimental liver fibrosis and might contribute to fibrolysis. Liver Int. 2004;24:492–501. doi: 10.1111/j.1478-3231.2004.0946.x. [DOI] [PubMed] [Google Scholar]
- 10.Wallace W.A., Fitch P.M., Simpson A.J., Howie S.E. Inflammation-associated remodelling and fibrosis in the lung - a process and an end point. Int J Exp Pathol. 2007;88:103–110. doi: 10.1111/j.1365-2613.2006.00515.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thannickal V.J., Horowitz J.C. Evolving concepts of apoptosis in idiopathic pulmonary fibrosis. Proc Am Thorac Soc. 2006;3:350–356. doi: 10.1513/pats.200601-001TK. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sivakumar P., Das A.M. Fibrosis, chronic inflammation and new pathways for drug discovery. Inflamm Res. 2008;57:410–418. doi: 10.1007/s00011-008-7166-y. [DOI] [PubMed] [Google Scholar]
- 13.Filer A., Pitzalis C., Buckley C.D. Targeting the stromal microenvironment in chronic inflammation. Curr Opin Pharmacol. 2006;6:393–400. doi: 10.1016/j.coph.2006.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chung M.J., Liu T., Ullenbruch M., Phan S.H. Antiapoptotic effect of found in inflammatory zone (FIZZ)1 on mouse lung fibroblasts. J Pathol. 2007;212:180–187. doi: 10.1002/path.2161. [DOI] [PubMed] [Google Scholar]
- 15.Fallowfield J.A., Kendall T.J., Iredale J.P. Reversal of fibrosis: no longer a pipe dream? Clin Liver Dis. 2006;10:481–497. doi: 10.1016/j.cld.2006.08.022. viii. [DOI] [PubMed] [Google Scholar]
- 16.Benyon R.C., Iredale J.P. Is liver fibrosis reversible? Gut. 2000;46:443–446. doi: 10.1136/gut.46.4.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Serrano-Mollar A., Nacher M., Gay-Jordi G., Closa D., Xaubet A., Bulbena O. Intratracheal transplantation of alveolar type II cells reverses bleomycin-induced lung fibrosis. Am J Respir Crit Care Med. 2007;176:1261–1268. doi: 10.1164/rccm.200610-1491OC. [DOI] [PubMed] [Google Scholar]
- 18.Sato M., Keshavjee S. Bronchiolitis obliterans syndrome: alloimmune-dependent and -independent injury with aberrant tissue remodeling. Semin Thorac Cardiovasc Surg. 2008;20:173–182. doi: 10.1053/j.semtcvs.2008.05.002. [DOI] [PubMed] [Google Scholar]
- 19.Bröcker V., Länger F., Fellous T.G., Mengel M., Brittan M., Bredt M., Milde S., Welte T., Eder M., Haverich A., Alison M.R., Kreipe H., Lehmann U. Fibroblasts of recipient origin contribute to bronchiolitis obliterans in human lung transplants. Am J Respir Crit Care Med. 2006;173:1276–1282. doi: 10.1164/rccm.200509-1381OC. [DOI] [PubMed] [Google Scholar]
- 20.Sato M., Liu M., Anraku M., Ogura T., D'Cruz G., Alman B.A., Waddell T.K., Kim E., Zhang L., Keshavjee S. Allograft airway fibrosis in the pulmonary milieu: a disorder of tissue remodeling. Am J Transplant. 2008;8:517–528. doi: 10.1111/j.1600-6143.2007.02106.x. [DOI] [PubMed] [Google Scholar]
- 21.Dutly A.E., Andrade C.F., Verkaik R., Kugathasan L., Trogadis J., Liu M., Waddell T.K., Stewart D.J., Keshavjee S. A novel model for post-transplant obliterative airway disease reveals angiogenesis from the pulmonary circulation. Am J Transplant. 2005;5:248–254. doi: 10.1111/j.1600-6143.2004.00680.x. [DOI] [PubMed] [Google Scholar]
- 22.Whittaker P., Kloner R.A., Boughner D.R., Pickering J.G. Quantitative assessment of myocardial collagen with picrosirius red staining and circularly polarized light. Basic Res Cardiol. 1994;89:397–410. doi: 10.1007/BF00788278. [DOI] [PubMed] [Google Scholar]
- 23.Sato M., Hirayama S., Lara-Guerra H., Anraku M., Waddell T.K., Liu M., Keshavjee S. MMP-dependent migration of extra-pulmonary myofibroblast progenitors contributing to post-transplant airway fibrosis in the lung. Am J Transplant. 2009;9:1027–1036. doi: 10.1111/j.1600-6143.2009.02605.x. [DOI] [PubMed] [Google Scholar]
- 24.Vandesompele J., De Preter K., Pattyn F., Poppe B., Van Roy N., De Paepe A., Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-7-research0034. research0034.1–research0034.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peterson J.T. Matrix metalloproteinase inhibitor development and the remodeling of drug discovery. Heart Fail Rev. 2004;9:63–79. doi: 10.1023/B:HREV.0000011395.11179.af. [DOI] [PubMed] [Google Scholar]
- 26.Ducharme A., Frantz S., Aikawa M., Rabkin E., Lindsey M., Rohde L.E., Schoen F.J., Kelly R.A., Werb Z., Libby P., Lee R.T. Targeted deletion of matrix metalloproteinase-9 attenuates left ventricular enlargement and collagen accumulation after experimental myocardial infarction. J Clin Invest. 2000;106:55–62. doi: 10.1172/JCI8768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peterson J.T., Hallak H., Johnson L., Li H., O'Brien P.M., Sliskovic D.R., Bocan T.M., Coker M.L., Etoh T., Spinale F.G. Matrix metalloproteinase inhibition attenuates left ventricular remodeling and dysfunction in a rat model of progressive heart failure. Circulation. 2001;103:2303–2309. doi: 10.1161/01.cir.103.18.2303. [DOI] [PubMed] [Google Scholar]
- 28.Levitt N.C., Eskens F.A., O'Byrne K.J., Propper D.J., Denis L.J., Owen S.J., Choi L., Foekens J.A., Wilner S., Wood J.M., Nakajima M., Talbot D.C., Steward W.P., Harris A.L., Verweij J. Phase I and pharmacological study of the oral matrix metalloproteinase inhibitor, MMI270 (CGS27023A), in patients with advanced solid cancer. Clin Cancer Res. 2001;7:1912–1922. [PubMed] [Google Scholar]
- 29.Lauer-Fields J.L., Juska D., Fields G.B. Matrix metalloproteinases and collagen catabolism. Biopolymers. 2002;66:19–32. doi: 10.1002/bip.10201. [DOI] [PubMed] [Google Scholar]
- 30.Holmbeck K., Bianco P., Yamada S., Birkedal-Hansen H. MT1-MMP: a tethered collagenase. J Cell Physiol. 2004;200:11–19. doi: 10.1002/jcp.20065. [DOI] [PubMed] [Google Scholar]
- 31.Mott J.D., Werb Z. Regulation of matrix biology by matrix metalloproteinases. Curr Opin Cell Biol. 2004;16:558–564. doi: 10.1016/j.ceb.2004.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hübner R.H., Meffert S., Mundt U., Böttcher H., Freitag S., El Mokhtari N.E., Pufe T., Hirt S., Fölsch U.R., Bewig B. Matrix metalloproteinase-9 in bronchiolitis obliterans syndrome after lung transplantation. Eur Respir J. 2005;25:494–501. doi: 10.1183/09031936.05.00091804. [DOI] [PubMed] [Google Scholar]
- 33.Taghavi S., Krenn K., Jaksch P., Klepetko W., Aharinejad S. Broncho-alveolar lavage matrix metalloproteases as a sensitive measure of bronchiolitis obliterans. Am J Transplant. 2005;5:1548–1552. doi: 10.1111/j.1600-6143.2005.00865.x. [DOI] [PubMed] [Google Scholar]
- 34.Smith G.N., Jr, Mickler E.A., Payne K.K., Lee J., Duncan M., Reynolds J., Foresman B., Wilkes D.S. Lung transplant metalloproteinase levels are elevated prior to bronchiolitis obliterans syndrome. Am J Transplant. 2007;7:1856–1861. doi: 10.1111/j.1600-6143.2007.01850.x. [DOI] [PubMed] [Google Scholar]
- 35.Cataldo D.D., Tournoy K.G., Vermaelen K., Munaut C., Foidart J.M., Louis R., Noël A., Pauwels R.A. Matrix metalloproteinase-9 deficiency impairs cellular infiltration and bronchial hyperresponsiveness during allergen-induced airway inflammation. Am J Pathol. 2002;161:491–498. doi: 10.1016/S0002-9440(10)64205-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sternlicht M.D., Werb Z. How matrix metalloproteinases regulate cell behavior. Annu Rev Cell Dev Biol. 2001;17:463–516. doi: 10.1146/annurev.cellbio.17.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Corry D.B., Rishi K., Kanellis J., Kiss A., Song Lz L.Z., Xu J., Feng L., Werb Z., Kheradmand F. Decreased allergic lung inflammatory cell egression and increased susceptibility to asphyxiation in MMP2–deficiency. Nat Immunol. 2002;3:347–353. doi: 10.1038/ni773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Owen C.A., Hu Z., Lopez-Otin C., Shapiro S.D. Membrane-bound matrix metalloproteinase-8 on activated polymorphonuclear cells is a potent, tissue inhibitor of metalloproteinase-resistant collagenase and serpinase. J Immunol. 2004;172:7791–7803. doi: 10.4049/jimmunol.172.12.7791. [DOI] [PubMed] [Google Scholar]
- 39.Han Y.P. Matrix metalloproteinases, the pros and cons, in liver fibrosis. J Gastroenterol Hepatol. 2006;21(Suppl 3):S88–S91. doi: 10.1111/j.1440-1746.2006.04586.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Horowitz J.C., Rogers D.S., Sharma V., Vittal R., White E.S., Cui Z., Thannickal V.J. Combinatorial activation of FAK and AKT by transforming growth factor-beta1 confers an anoikis-resistant phenotype to myofibroblasts. Cell Signal. 2007;19:761–771. doi: 10.1016/j.cellsig.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Greenberg R.S., Bernstein A.M., Benezra M., Gelman I.H., Taliana L., Masur S.K. FAK-dependent regulation of myofibroblast differentiation [Erratum appeared in FASEB J 2006, 20:1573] FASEB J. 2006;20:1006–1008. doi: 10.1096/fj.05-4838fje. [DOI] [PubMed] [Google Scholar]
- 42.Mizuno S., Matsumoto K., Li M.Y., Nakamura T. HGF reduces advancing lung fibrosis in mice: a potential role for MMP-dependent myofibroblast apoptosis. FASEB J. 2005;19:580–582. doi: 10.1096/fj.04-1535fje. [DOI] [PubMed] [Google Scholar]
- 43.Merklinger S.L., Jones P.L., Martinez E.C., Rabinovitch M. Epidermal growth factor receptor blockade mediates smooth muscle cell apoptosis and improves survival in rats with pulmonary hypertension. Circulation. 2005;112:423–431. doi: 10.1161/CIRCULATIONAHA.105.540542. [DOI] [PubMed] [Google Scholar]
- 44.Sorsa T., Golub L.M. Is the excessive inhibition of matrix metalloproteinases (MMPs) by potent synthetic MMP inhibitors (MMPIs) desirable in periodontitis and other inflammatory diseases?: That is: ‘Leaky' MMPIs vs excessively efficient drugs. Oral Dis. 2005;11:408–409. doi: 10.1111/j.1601-0825.2005.01160.x. [DOI] [PubMed] [Google Scholar]
- 45.Fernández F.G., Campbell L.G., Liu W., Shipley J.M., Itohara S., Patterson G.A., Senior R.M., Mohanakumar T., Jaramillo A.P. Inhibition of obliterative airway disease development in murine tracheal allografts by matrix metalloproteinase-9 deficiency. Am J Transplant. 2005;5:671–683. doi: 10.1111/j.1600-6143.2005.00751.x. [DOI] [PubMed] [Google Scholar]
- 46.Yoshida S., Iwata T., Chiyo M., Smith G.N., Foresman B.H., Mickler E.A., Heidler K.M., Cummings O.W., Fujisawa T., Brand D.D., Baker A., Wilkes D.S. Metalloproteinase inhibition has differential effects on alloimmunity, autoimmunity, and histopathology in the transplanted lung. Transplantation. 2007;83:799–808. doi: 10.1097/01.tp.0000258600.05531.5d. [DOI] [PubMed] [Google Scholar]
- 47.Lutz J., Yao Y., Song E., Antus B., Hamar P., Liu S., Heemann U. Inhibition of matrix metalloproteinases during chronic allograft nephropathy in rats. Transplantation. 2005;79:655–661. doi: 10.1097/01.tp.0000151644.85832.b5. [DOI] [PubMed] [Google Scholar]
- 48.Sato H., Takino T., Okada Y., Cao J., Shinagawa A., Yamamoto E., Seiki M. A matrix metalloproteinase expressed on the surface of invasive tumour cells. Nature. 1994;370:61–65. doi: 10.1038/370061a0. [DOI] [PubMed] [Google Scholar]
- 49.Zigrino P., Drescher C., Mauch C. Collagen-induced proMMP-2 activation by MT1-MMP in human dermal fibroblasts and the possible role of alpha2beta1 integrins. Eur J Cell Biol. 2001;80:68–77. doi: 10.1078/0171-9335-00134. [DOI] [PubMed] [Google Scholar]
- 50.Arthur M.J. Fibrogenesis II: Metalloproteinases and their inhibitors in liver fibrosis. Am J Physiol Gastrointest Liver Physiol. 2000;279:G245–G249. doi: 10.1152/ajpgi.2000.279.2.G245. [DOI] [PubMed] [Google Scholar]
- 51.Murphy F.R., Issa R., Zhou X., Ratnarajah S., Nagase H., Arthur M.J., Benyon C., Iredale J.P. Inhibition of apoptosis of activated hepatic stellate cells by tissue inhibitor of metalloproteinase-1 is mediated via effects on matrix metalloproteinase inhibition: implications for reversibility of liver fibrosis. J Biol Chem. 2002;277:11069–11076. doi: 10.1074/jbc.M111490200. [DOI] [PubMed] [Google Scholar]
- 52.Chen P., Farivar A.S., Mulligan M.S., Madtes D.K. Tissue inhibitor of metalloproteinase-1 deficiency abrogates obliterative airway disease after heterotopic tracheal transplantation. Am J Respir Cell Mol Biol. 2006;34:464–472. doi: 10.1165/rcmb.2005-0344OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sato M., Keshavjee S., Liu M. Translational research: animal models of obliterative bronchiolitis after lung transplantation. Am J Transplant. 2009;9:1981–1987. doi: 10.1111/j.1600-6143.2009.02770.x. [DOI] [PubMed] [Google Scholar]
- 54.Luckraz H., Goddard M., McNeil K., Atkinson C., Sharples L.D., Wallwork J. Is obliterative bronchiolitis in lung transplantation associated with microvascular damage to small airways? Ann Thorac Surg. 2006;82:1212–1218. doi: 10.1016/j.athoracsur.2006.03.070. [DOI] [PubMed] [Google Scholar]