

Abstract
Pemphigus vulgaris is an autoimmune disease caused by IgG antibodies against desmoglein 3 (Dsg3). Previously, we isolated a pathogenic mAb against Dsg3, AK23 IgG, which induces a pemphigus vulgaris-like phenotype characterized by blister formation. In the present study, we generated a transgenic mouse expressing AK23 IgM to examine B-cell tolerance and the pathogenic role of IgM. Autoreactive transgenic B cells were found in the spleen and lymph nodes, whereas anti-Dsg3 AK23 IgM was detected in the cardiovascular circulation. The transgenic mice did not develop an obvious pemphigus vulgaris phenotype, however, even though an excess of AK23 IgM was passively transferred to neonatal mice. Similarly, when hybridoma cells producing AK23 IgM were inoculated into adult mice, no blistering was observed. Immunoelectron microscopy revealed IgM binding at the edges of desmosomes or interdesmosomal cell membranes, but not in the desmosome core, where AK23 IgG binding has been frequently detected. Furthermore, in an in vitro dissociation assay using cultured keratinocytes, AK23 IgG and AK23 IgM F(ab′)2 fragments, but not AK23 IgM, induced fragmentation of epidermal sheets. Together, these observations indicate that antibodies must gain access to Dsg3 integrated within desmosomes to induce the loss of keratinocyte cell-cell adhesion. These findings provide an important framework for improved understanding of B-cell tolerance and the pathophysiology of blister formation in pemphigus.
Pemphigus vulgaris (PV) is a life-threatening, organ-specific autoimmune blistering disease of the skin and mucous membranes. It is characterized clinically by painful oral erosions and flaccid skin blisters, histologically by suprabasal acantholysis (ie, loss of cell-cell adhesion between suprabasal keratinocytes), and immunopathologically by IgG autoantibodies against desmoglein 3 (Dsg3), a cadherin-type cell-cell adhesion molecule found in desmosomes.1,2 Compelling evidence indicates that IgG autoantibodies against Dsg3 are pathogenic and play a primary role in inducing blister formation in pemphigus. IgGs affinity-purified from the sera of PV patients using the extracellular domain of Dsg3 cause suprabasal acantholysis when injected into neonatal mice.3 When anti-Dsg3 IgG is immunoadsorbed from the sera of PV patients using the same Dsg3 domain, those sera lose their ability to cause blister formation in neonatal mice.4 Furthermore, monoclonal antibodies (mAbs) against Dsg3 from a model mouse and from PV patients induce the formation in mice of blisters with typical PV histology.5,6 The pathogenic roles of autoantibodies against nondesmoglein molecules remain to be clarified.7,8
We previously developed a PV model mouse by the adoptive transfer of lymphocytes from Dsg3−/− mice immunized with rDsg3 to Rag2−/− mice that express Dsg3.9 Recipient mice showed stable anti-Dsg3 IgG production and developed a PV phenotype characterized by mucosal erosions and acantholytic blisters, similar to those seen in PV patients. We subsequently isolated AK series of anti-Dsg3 IgG monoclonal antibodies from the PV model mice and demonstrated their pathogenic heterogeneity.5 The pathogenic AK23 IgG mAb binds to the adhesive interface of Dsg3, the functionally important part of the molecule, whereas other nonpathogenic mAbs, such as AK7 IgG, react with the central or carboxyl-terminal extracellular regions of Dsg3, where no direct intermolecular interactions are predicted to occur.10
In humoral immune responses, IgM is the Ig isotype secreted during the primary immune response, and its production precedes that of IgG. IgM is a surface marker of immature and mature B cells. Nevertheless, approximately 20% of mature naïve B cells in the peripheral blood of healthy donors produce low-affinity self-reactive antibodies and approximately 5% antibodies with low levels of polyreactivity.11 Although IgM autoantibodies are not found in the sporadic form of pemphigus, high levels of IgM autoantibodies against desmoglein 1 (Dsg1) were recently detected in sera from patients with fogo selvagem, a form of pemphigus foliaceus endemic in certain areas of Brazil (notably in Limão Verde), as well as healthy individuals.12 Nonetheless, the pathogenic relevance of IgM autoantibodies in PV remains to be elucidated.
To explore mechanisms of B-cell tolerance to Dsg3, we first generated anti-Dsg3 IgM transgenic mice using cDNAs encoding the variable regions of the H and L chains of AK7 IgG mAb.13 In AK7-IgM transgenic mice, functionally competent Dsg3-reactive B cells were readily detected in peripheral lymphoid organs such as the spleen, as well as in lymph nodes, whereas anti-Dsg3 AK7 IgM was found in the cardiovascular circulation and on keratinocyte cell surfaces. These results indicate that autoreactive B cells against Dsg3 are able to develop in the presence of Dsg3 but are ignored by the immune system. We speculated that this was probably because the AK7 IgM mAb is nonpathogenic. However, when the pathogenic AK23 IgG mAb was injected into AK7-IgM transgenic mice and blisters were formed, AK7 B cells were eliminated from the bone marrow and spleen via a Fas-mediated process in a CD4+ T cell-dependent manner.14 These findings suggest that autoreactive B cells persist as long as they are not harmful, but that once damaging events such as tissue destruction are sensed, some danger signals, whose mechanisms were not fully understood, are induced and mature autoreactive B cells are eliminated in the periphery.
To further evaluate B-cell tolerance to B-cells produced pathogenic antibodies, we generated mice transgenic for the anti-Dsg3 AK23 IgM mAb. Unexpectedly, AK23-IgM B-cell transgenic mice did not develop a PV-like phenotype, although AK23-IgM-producing B cells were found in the periphery. To exclude the possibility that this finding was the result of insufficient AK23-IgM production, we established AK23-IgM hybridoma cells from the transgenic mice and administered an excess of AK23 IgM to neonatal and adult mice. Blister formation was not observed in any of the treated mice. We investigated why AK23 IgG, but not AK23 IgM, is able to induce blistering in vitro and in vivo and found that antibodies must be able to gain access to Dsg3 integrated within desmosomes to induce the blister formation that characterizes pemphigus.
Materials and Methods
Mice
Dsg3−/− mice were generated by mating female Dsg3+/− and male Dsg3−/− mice (Jackson Laboratory, Bar Harbor, ME). These mice have a mixed 129/Sv(H-2b)/C57BL/6J(H-2b) genetic background. C57BL/6J mice were purchased from Taconic Farms (Germantown, NY). To generate the PV model mouse, splenocytes from nonimmunized Dsg3−/− mice were adoptively transferred to C57BL/6 Rag2−/− mice via the tail vein. Anti-Dsg IgM and IgG titers were examined by enzyme-linked immunosorbent assay (ELISA) and live keratinocyte staining.5 All mice were maintained under specific pathogen-free conditions at our animal facility. All animal experiments were performed in accordance with our institutional guidelines.
B-Cell Transgenic Mice
cDNA fragments encoding the variable regions of the H and L chains (VH and VL) of AK23 IgG were cloned from an AK23 IgG hybridoma by RT-PCR. In brief, total RNA was prepared from AK23 hybridoma cells and cDNAs for the H and L chains amplified by one-step RT-PCR (Qiagen, Valencia, CA). Subsequently, the H and L chains were linked by nested PCR and subcloned into a modified pCANTAB5E vector (GE Healthcare, Piscataway, NJ). The resulting plasmids were transformed into XL1-Blue (Stratagene, La Jolla, CA), in which single-chain variable-region fragments were expressed as phage surface molecules. For the rDsg3 ELISA, positive clones were obtained and their sequences were determined. The AK23.4 clone was used in the present study. The H-chain signal, variable region, and 3′ sequences were assembled by PCR. The signal sequence and 3′ region were amplified from an anti-TNP H-chain EcoRI-EcoRI fragment (kindly provided by Hajime Karasuyama) using the DN956-DN959 primer pair. The variable region sequence was amplified from AK23.4 using the DN958-DN961 primer pair. The 3′ sequence was amplified from the anti-TNP H-chain fragment using the DN960-DN957 primer pair. All three resulting PCR fragments were then reamplified with the DN956-DN957 primer pair. AK23-H was generated by the replacement of the XbaI-XbaI fragment of AK7-H. The primers used had the following sequences: (DN956) 5′-CTAGTCTAGATGGACTAGGTTCTTATGGA-3′, (DN957) 5′-CTAGTCTAGACAGCAACTACCCTTTTGA-3′, (DN958) 5′-TCTCTTCACAGGTGTCCACTCTCAGGTCCAACTGCAGCAGTCT-3′, (DN959) 5′-AGACTGCTGCAGTTGGACCTGAGAGTGGACACCTGTGAAGAGA-3′, (DN960) 5′-ACCACGGTCACCGTTTCCTCAGGTAAGAATGGCTTCTCCA-3′, and (DN961) 5′-TGGAGAAGCCATTCTTACCTGAGGAAACGGTGACCGTGGT-3′.
The L-chain signal and variable region sequences were also assembled by PCR. The signal sequence and 3′ region were amplified from an anti-TNP L-chain HindIII-HindIII fragment using DN950-DN951 and DN954-DN955 primer pairs: (DN950) 5′-CACCAAGCTTCAGCTGTCTTGTTTCAGTGACTGAT-3′ and (DN951) 5′-GACCTGGGTCTGTGACTTCATTTTGAGGAGGCAAC-3′; (DN954) 5′-TTCGGTGGAGGCACCAAGCTGGAGCTGAAACGTAAGTACACTTTTCTCATC-3′ and (DN955) 5′-CACCAAGCTTCTGCAGTCAGACCCAGATCTCAATAAC-3′. The AK7 L-chain variable region was amplified from AK23-4 using a DN952-DN953 primer pair, as follows: (DN952) 5′-AGGTTGCCTCCTCAAAATGAAGTCACAGACCCAGGTC-3′ and (DN953) 5′-GATGAGAAAAGTGTACTTACGTTTCAGCTCCAGCTTGGTGCCTCCACCGAA-3′. These PCR fragments were then fused through amplification using the DN950-DN955 primer pair. AK23-L was generated by replacing the PvuII-PstI region of the AK7-L plasmid with the amplified PCR products. Transcription of the transgenes was under the control of native immunoglobulin gene promoters and enhancers. AK23-H and AK23-L plasmids were microinjected into fertilized eggs of C57BL/6 mice. Viable zygotes were then transferred into the oviducts of pseudopregnant C57BL/6 mice. Two H-chain transgenic mouse founder lines and three L-chain transgenic mouse founder lines were established. The lines H20 and L35, which stably express H-chain and L-chain transgenes, respectively, were used in the present study. AK23-IgM transgenic mice were obtained by mating H20 and L35 mice.
Generation and Isolation of Anti-Dsg3 IgM Hybridoma Cells
Splenocytes were isolated from AK23 IgM transgenic mice and were fused with P3 mouse myeloma cells at a ratio of 5:1 using polyethylene glycol (PEG 4000; Merck, Darmstadt, Germany). Selection was then performed using hypoxanthine-aminopterin-thymidine medium and 10% hybridoma cloning factor (IGEN, Gaithersburg, MD), as described previously.5 Initially, hybridoma cells were screened by mDsg3 ELISA. Live keratinocytes were also screened. Positive clones were subcloned three times using the limiting dilution method. The mAb isotype was determined using an isotyping kit (Roche Diagnostics, Mannheim, Germany). The mAb was then purified using a HiTrap IgM purification HP chromatography column (GE Healthcare) and dialyzed against PBS.
Characterization of Anti-Dsg3 Antibodies by ELISA and Live Keratinocyte Staining
Anti-Dsg3 IgM titer was measured by ELISA using rDsg3, as described previously.5 ELISA plates were coated with 5 μg/mL rDsg3 and blocked with 3% skim milk. To confirm the calcium dependency of AK23 IgM, the ELISA plates were next treated with 5 mmol/L EDTA for 30 minutes at room temperature, and then incubated with AK23 IgM. Live staining of the mouse PAM212 keratinocyte was performed as described previously.5 Alexa Fluor 488-conjugated goat anti-mouse IgM and Alexa Fluor 488-conjugated goat anti-mouse IgG (Invitrogen, Carlsbad, CA) were used to detect the binding of AK23 IgM mAb. Stained cells were visualized by fluorescence microscopy.
Immunofluorescence Study on Colocalization with Desmoplakin
Primary human epidermal keratinocytes were seeded onto glass coverslips and allowed to proliferate to the desired confluence. Cells were switched to medium containing 0.6 mmol/L calcium chloride for 18 hours before treatment with either AK23 IgG or AK23 IgM at 37°C for 1 hour. Antibodies were added to the cell culture medium at a final concentration of 20 to 30 μg/mL. Cells were then washed three times with PBS and fixed through incubation on ice with −20°C methanol for 2 minutes. Desmoplakin (DP) was detected using a rabbit anti-DP antibody, NW6 (provided by Dr. Kathleen Green, Northwestern University Feinberg School of Medicine, Evanston, IL). Primary antibody was detected using a goat anti-rabbit IgG antibody conjugated to Alexa Fluor 555, a goat anti-mouse IgG antibody conjugated to Alexa Fluor 488, and a goat anti-mouse IgM μ-chain antibody conjugated to fluorescein isothiocyanate (Invitrogen). Stained cells were mounted on coverslips using ProLong Gold antifade reagent (Invitrogen).
Confocal immunofluorescence microscopy was performed using a DMI-6000B microscope (Leica Microsystems, Wetzlar, Germany) equipped with two solid-state lasers (491 and 561 nm) and a VT Infinity two-dimensional-array scanner (VisiTech International, Sunderland, UK). Images were captured using an electron multiplier deep-cooled CCD camera (C9100-12; Hamamatsu, Sewickley, PA). Image acquisition was driven by Simple PCI software version 6.5 (Hamamatsu). Colocalization was quantified using MetaMorph software version 6.0r1 (Universal Imaging, West Chester, PA). For each sample, 20 single-plane images were analyzed for the colocalization of AK23 IgG and AK23 IgM with DP. Statistical comparisons were performed using Student's t-test.
Flow Cytometric Analysis
Two-color flow cytometry was performed using a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA), and the resulting data were analyzed using CellQuest Pro software. Directly conjugated APC-anti-IgM (R6-60.2; BD Biosciences), fluorescein isothiocyanate-conjugated anti-IgMa (DS-1; BD Biosciences), fluorescein isothiocyanate-conjugated anti-IgMb (AF6-78; BD Biosciences), and phycoerythrin-conjugated anti-mouse CD19 (1D3; BD Biosciences) antibodies were used. To detect the AK23 idiotype, cells were incubated with 2.5 μg/mL rDsg3, washed, and stained with Alexa Fluor 488-conjugated anti-E-Tag antibody (GE Healthcare) after washing.
Immunoelectron Microscopy
Samples were isolated from the hard palate of mice inoculated with AK23 IgM hybridoma cells for immuno-electron microscopy (immuno-EM) analysis. Postembedding immuno-EM was performed as described previously.15 In brief, hard palate tissue was cut into small pieces (<1 mm2), which were cryofixed through rapid immersion in liquid propane (−190°C). Cryofixed tissue was then cryosubstituted in pure methanol for 48 hours at −80°C and then embedded in Lowicryl K11M (Ladd Research Industries, Williston, VT) at −60°C. Specimens were polymerized through exposure to UV radiation at −60°C for 48 hours and at room temperature for a further 48 hours. Ultrathin sections were cut perpendicular to the skin surface, collected on nickel grids coated with a polyvinyl resin Formvar support film, and processed for immunogold labeling. The ultrathin sections were immunolabeled with an affinity-purified antibody specific for rabbit anti-mouse IgM (μ-chain, diluted 1:50; Invitrogen) and a 15-nm colloidal gold-conjugated goat anti-rabbit IgG (diluted 1:40) (GE Healthcare). As a control, oral mucous membrane from Rag2−/− mice was used as a substrate in place of equivalent samples from mAb hybridoma-inoculated mice. To assess the localization of gold particles, the number of cell membrane-associated particles was counted in each immuno-EM image. Overall, 25 immuno-EM images were analyzed from AK23 IgM hybridoma-inoculated mice and 29 images from AK23 IgG hybridoma-inoculated mice (n = 4). To quantify differences in particle deposition, labeling percentages were calculated as follows: Labeling Percentage = (No. of desmosome-associated particles/No. of cell membrane-associated particles) × 100. Statistical comparisons were performed using Student's t-test.
Passive Transfer Assay Using Neonatal Mice
To evaluate the pathogenic activities of the AK23 IgM mAb, a passive transfer study was performed using neonatal mouse. ICR (Institute of Cancer Research) mice were used, age 12 to 24 hours, body weight 1.5 to 2.0 g (Japan SLC, Shizuoka, Japan). Purified AK23 IgM (150 μg) was injected alone or together with a small amount of exfoliative toxin A (ETA), which specifically digests Dsg1. The dose of ETA used (1 μg/mouse) was 50% of the minimum dose required to induce gross blistering. The skin was evaluated macroscopically and microscopically at 18 to 24 hours after injection of ETA. Blistering was evaluated microscopically, using 3-mm sections obtained from the whole body.
Ascites Formation Assay Using Adult Mice
To evaluate the pathogenic activity of AK23 IgM mAb in adult mice, we performed an ascites formation assay using hybridoma cells, as described previously.5 Hybridoma cells producing AK23 IgM mAb were inoculated (5 × 106 to 1 × 107 cells per mouse, i.p.) into Rag2−/− immunodeficient mice that had been primed with 2,6,10,14-tetramethylpentadecane (Wako Pure Chemical Industries, Osaka, Japan). Ascites formation and the development of PV-like characteristics, such as weight loss and patchy hair loss, were monitored. Oral mucosa biopsies were obtained from inoculated mice when they developed a PV-like phenotype. Ascites formation was observed after day 14.
Preparation of AK23 IgM mAb F(ab′)2 Fragments
AK23 IgM mAb was digested using a Pierce ImmunoPure IgM fragmentation kit (Thermo Scientific, Rockford, IL) to generate F(ab′)2 fragments. Purified AK23 IgM mAb was transferred to digestion buffer (50 mmol/L Tris, 150 mmol/L NaCl, 10 mmol/L CaCl2, 0.05% NaN3) using a dextran desalting column. It was then applied to a pepsin column equilibrated with digestion buffer and incubated at 37°C for 90 minutes. Digested F(ab′)2 fragments were collected and transferred to PBS.
In Vitro Dissociation Assay Using Normal Human Epidermal Keratinocytes
Primary normal human epidermal keratinocytes were seeded into 12-well plates in calcium-free MCDB 153 medium as described previously.16 After reaching confluence, cells were incubated for 12 hours in keratinocyte growth medium supplemented with 0.8 mmol/L calcium chloride and containing various concentrations of AK23 IgM. To detect the binding of AK23 IgM to Dsg3 alone, 1 μg ETA was added to the culture medium for the final 2 hours to inactivate Dsg1. Monolayers were washed twice with PBS and then incubated with 1.2 units of dispase II (Roche Diagnostics) for 30 minutes at 37°C. Floating monolayers were washed twice with PBS and gently pipetted through a 1-mL pipette tip five times to mechanically dissociate nonadherent cells. Dissociation scores were calculated based on the number of fragmented cell sheets (N) using the following formula: dissociation score = [(Nwith mAb − Nwithout mAb)/(Nwith AK23 − Nwithout mAb)] × 100. Cell sheets treated with 1 mg/mL AK23 were included in each assay to adjust for interassay variability. Fragmented cell sheets were stained with crystal violet, and measurements were made using Image-Pro Plus software version 4.5.1 (Media Cybernetics, Bethesda, MD).
Results
Dsg3-Specific AK23-IgM-Producing B Cells in Peripheral Lymphoid Organs
Among the AK series mAbs isolated from PV model mice, AK23 IgG mAb is pathogenic and able to induce a PV-like phenotype characterized histologically by mucosal erosion and suprabasal acantholysis.5,16 To isolate cDNAs encoding the H and L chains of AK23 IgG mAb, which reacts with human and mouse Dsg3, total RNA from AK23-producing hybridoma cells was used as a template for PCR amplification applying mixed primers targeted to the H and L chains. The resulting PCR products were sequenced (Figure 1A). Our attempts to generate AK23 single-chain variable-region fragments were unsuccessful, because they were produced as an insoluble protein (T. Ota and M. Amagai, unpublished observation). Transgenic constructs encoding the H and L chains of the AK23 mAb were generated on an IgM backbone (Figure 1B). Transcription of these transgenes was under the control of native immunoglobulin gene promoters and enhancers. The H-chain transgene contains exons encoding both membrane-bound and secreted forms of the AK23 mAb derived from an IgMa allotype, such that both forms of IgM should be generated in mice carrying the transgene. Transgene-derived heavy chain can detect with IgMa antibody (C57BL/6 endogenous heavy chain is IgMb). Five transgenic mouse founder lines were established (two H chain and three L chain), and AK23-IgM transgenic mice were generated by mating the H20 and L35 lines for further characterization.
Figure 1.
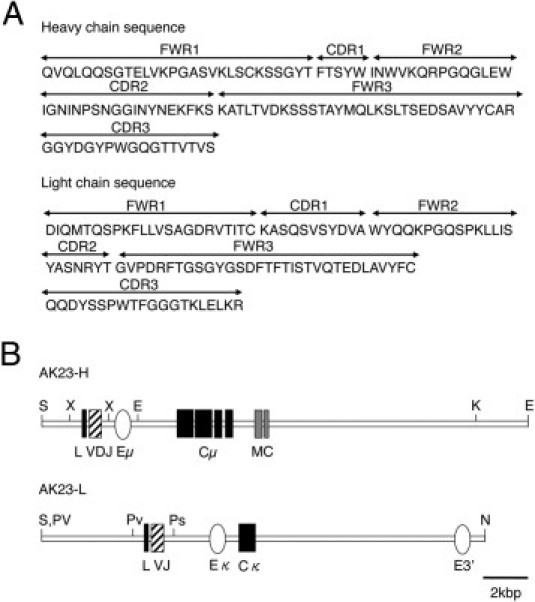
Structures of the AK23 H and L immunoglobulin transgene constructs. A: The amino acid sequences of the H and L chains of AK23 were determined from the DNA sequence of AK23 single-chain variable-region fragment. The FWR and CDR domains were annotated accordingly.17B: The AK23 heavy-chain variable region (contained in an XbaI-XbaI fragment) was linked with an H-chain promoter and a μ-isotype constant region (Cμ) that included a transmembrane domain (MC). The AK23 L-chain (κ isotype) variable region (contained in a PvuII-PstI fragment was linked to a κ-chain promoter and constant region. E, EcoRI; Eμ, μ-region enhancer; Eκ, κ-region enhancer; K, KpnI; N, NotI; Ps, PstI; Pv, PvuII; S, SalI; X, XbaI.
We expected to detect IgMa-positive B cells in bone marrow, where Dsg3 is not produced, but not in the periphery, where Dsg3 is expressed. However, significant numbers of IgMa B cells were detected in the spleen and lymph nodes of AK23-IgM mice (Figure 2A). To exclude the possibility that IgMa B cells lose their reactivity to Dsg3 because of incomplete allelic exclusion, we used rDsg3 to detect anti-Dsg3 IgM composed of transgenic H and L chains. Again, substantial numbers of CD19+ B cells expressed surface IgM, which reacted with rDsg3 (Figure 2A). In contrast, in wild-type mice, no such Dsg3-reactive B cells were detected. Furthermore, IgM reactive to Dsg3 was also observed in the blood of AK23-IgM mice. These findings collectively indicate that AK23-IgM B cells are not deleted or inactivated in the periphery.
Figure 2.
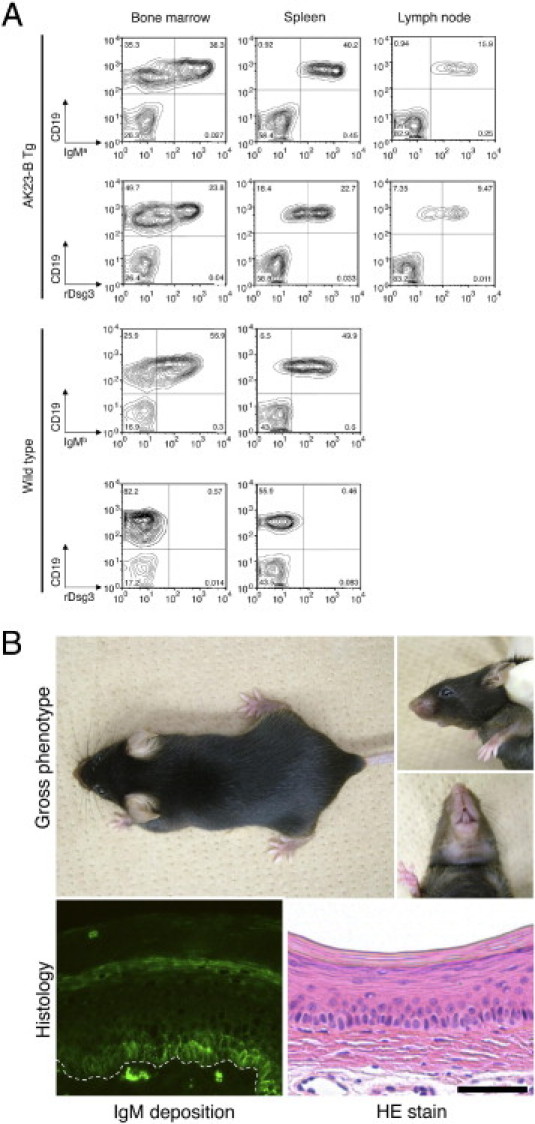
Dsg3-specific AK23 IgM transgenic B cells escape tolerance. A: Fluorescence-activated cell sorting analysis of AK23 IgM transgenic mice detected IgMa-positive B cells in lymphoid organs, including bone marrow (38.3%), spleen (40.2%), and lymph nodes (15.9%). Dsg3-reactive IgM-expressing B cells were also detected in bone marrow (23.8%), spleen (22.7%), and lymph nodes (9.47%). In wild-type mice, IgMb-positive B cells, but not Dsg3-reactive B cells, were detected in lymphoid organs. B: Macroscopic and microscopic phenotype of AK23 IgM transgenic mice. AK23 IgM transgenic mice exhibited no obvious PV-like characteristics, such as patchy hair loss or weight loss. Although IgM was found deposited on the cell surfaces of basal layer keratinocytes in vivo, no blisters were formed. The dotted line indicates the basement membrane. Scale bar = 50 μm.
Absence of PV-Like Phenotype in AK23-IgM Transgenic Mice
Because AK23 IgG mAb is pathogenic and AK23-IgM B cells were detected in the periphery and AK23 IgM in the circulation, we expected AK23-IgM transgenic mice to exhibit a PV-like phenotype. Contrary to our expectation, however, AK23-IgM mice did not display any obvious PV characteristics (Figure 2B). The mice did not undergo the weight loss or patchy hair loss that has been described in PV model mice or mice inoculated with AK23-IgG hybridoma cells.5,9 Histological analysis of the oral mucosa, esophagus, and skin revealed no evidence of suprabasal acantholysis (Figure 2B). Direct immunofluorescence showed the deposition of IgM on the surfaces of keratinocytes in the basal layers of the oral mucosa in vivo (Figure 2B). However, the IgM staining pattern was quite different from that of IgG.5 IgM staining was essentially restricted to the basal cell layer, appearing as coarse dots along the cell membranes (Figure 2B).
Isolation and Characterization of an Anti-Dsg3 IgM Monoclonal Antibody from AK23 IgM Transgenic Mice
To explain why AK23 IgM mice failed to display a PV-like phenotype, we considered the possibility that circulating AK23 IgM levels are insufficient to cause blisters to form in AK23 IgM mice. To explore this possibility, we isolated AK23 IgM hybridoma cells from AK23 IgM mice. The IgM mAb they produced comprised a μ heavy-chain isotype and a κ light-chain isotype. To confirm that AK23 IgM and AK23 IgG recognize the same epitope, we performed a blocking ELISA using AK9 and AK18 antibodies that recognize several different Dsg3 epitopes. The binding of AK23 IgM to mouse Dsg3 was significantly reduced by AK23 IgG, but not by AK9 or AK18 IgGs (Figure 3A). The calcium dependency of AK23 IgG binding was shared with AK23 IgM (Figure 3B).
Figure 3.
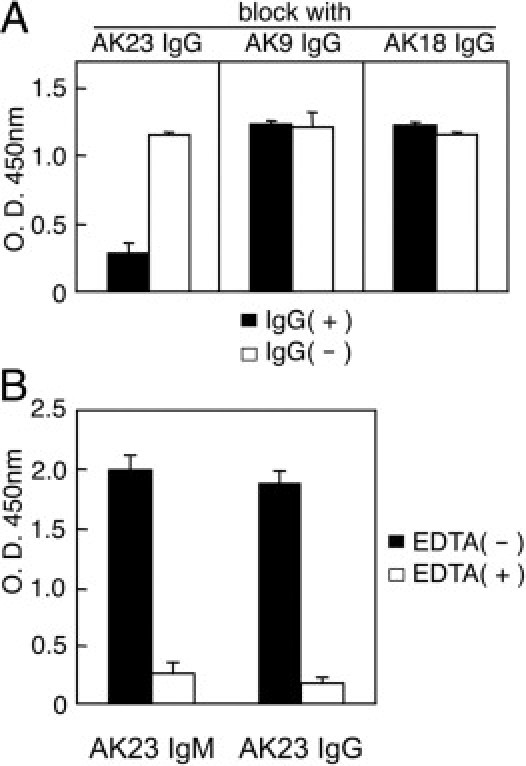
AK23 IgM recognizes the same Dsg3 epitope as AK23 IgG. A: The binding of AK23 IgM in a mouse Dsg3 ELISA was significantly blocked by AK23 IgG, but not by AK9 IgG or AK18 IgG. IgG(+), with AK IgG pretreatment; IgG(−), without AK IgG pretreatment. B: AK23 IgM reactivity was tested by mouse Dsg3 ELISA performed with and without EDTA (5 mmol/L) pretreatment.
Indirect immunofluorescence studies using mouse hard palate tissue revealed quite different staining patterns for AK23 IgM and AK23 IgG (Figure 4A). Sera from AK23-IgM mice and AK23 IgM mAb showed no apparent reactivity toward keratinocyte cell surfaces, whereas AK23 IgG mAb yielded clear staining. However, F(ab′)2 fragments from AK23 IgM mAb clearly bound to keratinocyte cell surfaces. Similarly, AK23 IgM failed to react with normal human skin, whereas AK23 IgG yielded a strong signal (data not shown). When these same antibodies were applied to mouse PAM212 keratinocytes, AK23 IgM did stain keratinocyte cell surfaces, but its staining pattern differed from that of AK23 IgG (Figure 4B). Like AK23 IgG, AK23 IgM bound to the cell-cell borders, but it also reacted with cell border-free areas, yielding a dotted pattern. To further characterize this dotted IgM staining, AK23 IgG-stained and AK23 IgM-stained primary human epidermal keratinocytes were costained for DP (Figure 4C). DP is a component of the cytoplasmic plaque of desmosomes. Thus, any Dsg3 that is not colocalized with DP on the plasma membrane should not be integrated within desmosomes. Quantification of the colocalization of AK23 and DP indicated that approximately 45% of total IgG colocalized with DP, compared with only 25% of IgM (P < 0.005; Figure 4D).
Figure 4.
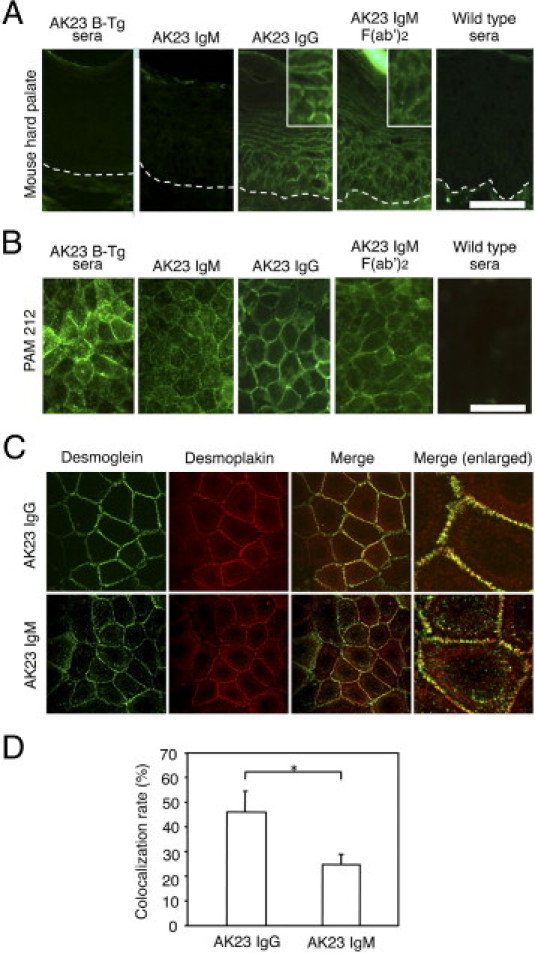
Comparisons of the immunoreactivities of AK23 IgM, AK23 IgG, and F(ab′)2 fragments of AK23 IgM. A: Indirect immunofluorescence (IIF) staining of hard palate tissue from wild-type mice. Sera from AK23 IgM transgenic mice and AK23 IgM mAb yielded no signal, whereas AK23 IgG and AK23 IgM F(ab′)2 yielded linear cell surface staining. Insets, digitally enlarged. Dotted lines indicate the basement membrane. B: Live staining of mouse PAM212 keratinocytes. Both AK23 IgM and AK23 IgG stained keratinocyte cell surfaces, but the staining patterns were quite different. AK23 IgM appeared as coarse dots on cell border-free areas. C: AK23 IgM and AK23 IgG (green) were costained with desmoplakin (red). Although AK23 IgG largely colocalized with desmoplakin, AK23 IgM did not. D: Quantification of colocalization indicated significantly lower colocalization of DP with AK23 IgM than with AK23 IgG. *P < 0.005. Scale bars = 50 μm.
These findings indicate that, although AK23 IgM and AK23 IgG recognize the same Dsg3 epitope, their ability to gain access to Dsg3 differs, and suggest that the dotted staining of cultured keratinocytes by AK23 IgM may have been caused by the binding of antibody to Dsg3 clustered on the cell membrane and not by integration within desmosomes.
AK23 IgM mAb Is Unable To Induce Blisters
To further evaluate the pathogenic activity of the AK23 IgM mAb in vivo, we performed a passive transfer assay using neonatal mice.5,18 Identical doses of AK23 IgM and AK23 IgG (150 μg/mouse) were injected subcutaneously into neonatal mice (n = 5). Macroscopic analysis failed to detect blisters in either treatment group at 18 to 24 hours after injection, whereas microscopic blisters were found in mice injected with AK23 IgG but not AK23 IgM (Figure 5). Although equivalent amounts of immunoglobulin were injected, greater amounts of bound AK23 IgG were detected than of bound AK23 IgM in vivo. Moreover, AK23 IgM staining exhibited a dotted pattern, whereas AK23 IgG staining was relatively even. To eliminate Dsg1, whose cell-adhesive function may compensate for impaired Dsg3 function, we used ETA, a Dsg1-specific serine protease produced by Staphylococcus aureus.19 Mice were coinjected with AK23 IgM or AK23 IgG and half the minimum effective dose of ETA. Mice receiving AK23 IgG and ETA showed extensive blistering, as well as suprabasal acantholysis in histological analyses, whereas animals receiving AK23 IgM and ETA exhibited no macroscopic or microscopic evidence of blistering (n = 5) (Figure 5).
Figure 5.
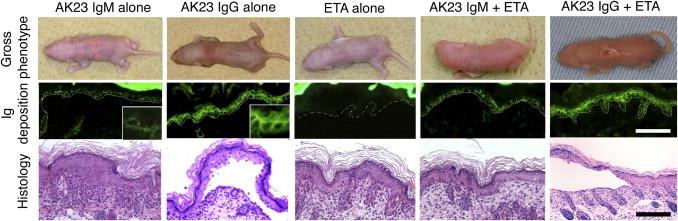
Evaluation of the pathogenic activity of AK23 IgM by passive transfer assay using neonatal mice. Neonatal mice injected with AK23 IgG (150 μg), but not AK23 IgM (150 μg), developed microscopic blisters. The staining pattern of in vivo bound AK23 IgM was characterized by low-intensity coarse dots, whereas that of AK23 IgG was linear. Mice in neither treatment group developed macroscopic blisters, because of functional compensation by Dsg1 in the skin. When coinjected with half of the minimum effective dose of exfoliative toxin A (ETA), a Dsg1-specific serine protease produced by Staphylococcus aureus, AK23 IgG induced the formation of blisters and yielded typical PV histological features, whereas AK23 IgM produced no obvious signs of blister formation. Dotted lines indicate basement membrane. Scale bar = 50 μm (insets, digitally enlarged).
We further tested the pathogenic activity of the AK23 IgM mAb by inoculating AK23 IgM-producing hybridoma cells into Rag2−/− immunodeficient mice.5 Circulating AK23 IgM titers in mice inoculated with hybridoma cells were approximately six times higher than those in AK23-IgM transgenic mice (Figure 6A). However, mice that received AK23 IgM hybridoma cells did not exhibit macroscopic or microscopic blistering, despite IgM being deposited on the cell surfaces of oral mucosal keratinocytes (n = 3) (Figure 6B). In contrast, mice receiving AK23 IgG hybridoma cells developed a PV-like phenotype characterized histologically by oral erosions and suprabasal acantholysis, patchy telogen hair loss, and weight loss due to reduced food intake (n = 3) (Figure 6, B and C). Although mice in both treatment groups displayed antibody deposition in vivo, IgM was essentially restricted to the basal layer of the oral mucosa, appearing as coarse dots, whereas IgG was detected in multiple mucosal cell layers, yielding strong and more uniform staining of keratinocyte cell surfaces.
Figure 6.
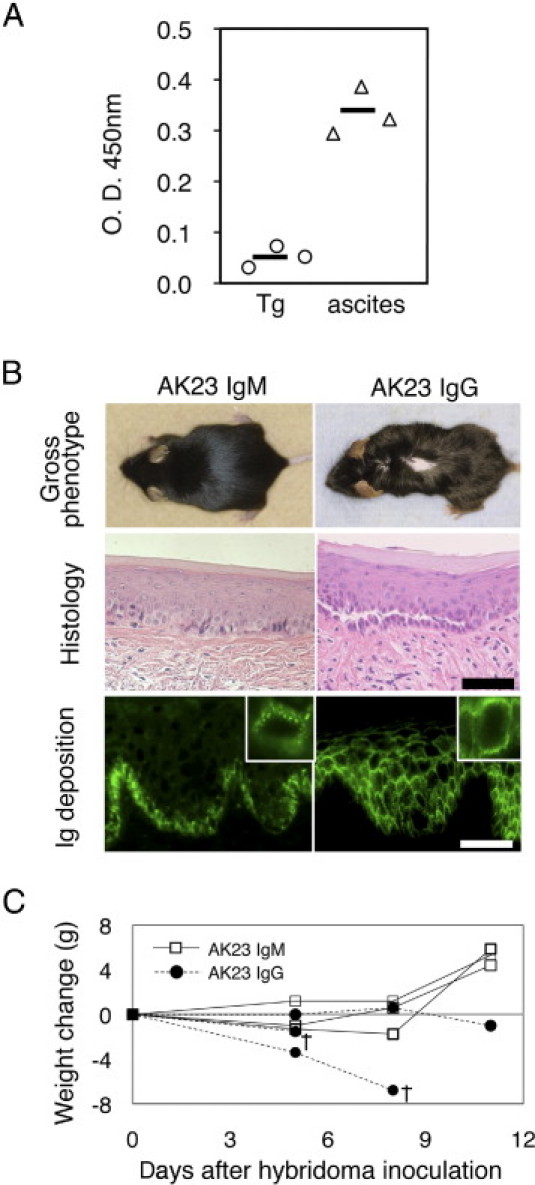
Evaluation of the pathogenic activity of AK23 IgM by ascites formation assay using adult mice. A: Circulating AK23 IgM titers were approximately six times higher in mice inoculated with AK23 IgM-producing hybridoma cells (triangles) than in AK23 IgM transgenic mice (circles). Sera were diluted 1:5 and analyzed by mouse Dsg3 ELISA. B: Phenotypes of mice injected with AK23 IgM- and AK23 IgG-producing hybridoma cells. Although mice injected with AK23 IgG hybridoma cells exhibited patchy hair loss and suprabasal acantholysis, those that received AK23 IgM hybridoma cells developed no apparent PV phenotype. Although both mice displayed in vivo deposition of AK23 antibodies, IgM was essentially restricted to the basal cell layer of the oral mucosa, appearing as coarse dots, whereas IgG strongly and evenly stained the cell surfaces of keratinocytes in multiple mucosal layers. Images are representative of 18 recipient mice that received AK23 IgM hybridoma cells. Insets, digitally enlarged. C: Changes in body weight were monitored after the injection of hybridoma cells, because the development of oral erosions has the potential to impair food intake. Whereas body weight increased in mice injected with AK23 IgM hybridoma cells, it declined in animals injected with AK23 IgG hybridoma cells, and some of the animals died (n = 3). Scale bars = 50 μm. O.D., optical density; Tg, AK23 IgM transgenic; ascites, mice with AK23 IgM hybridoma cells; †, day of death.
Together, these findings suggest that AK23 IgM is not pathogenic and is unable to induce blister formation.
F(ab′)2 Fragments of AK23 IgM, but Not Intact AK23 IgM, Induce Keratinocyte Dissociation in Vitro
To further measure the pathogenic activity of AK23 IgM mAb, we performed an in vitro dissociation assay16 (Figure 7). A minimum dose of ETA was applied to cells, to prevent compensation by Dsg1. We first compared pathogenic strength between AK23 IgM and AK23 IgG according to molecular stoichiometry (mol. wt. approximately 980 kDa for IgM and 150 kDa for IgG). We used 2 μg/mL of AK23 IgG as a positive control in this assay. We added 10, 35, 70, and 140 μg/mL of AK23 IgM, but no apparent dissociation activity was observed. In molar ratio, the 140 μg/mL AK23 IgM was equivalent to 10.8 times the 2 μg/mL AK23 IgG.
Figure 7.
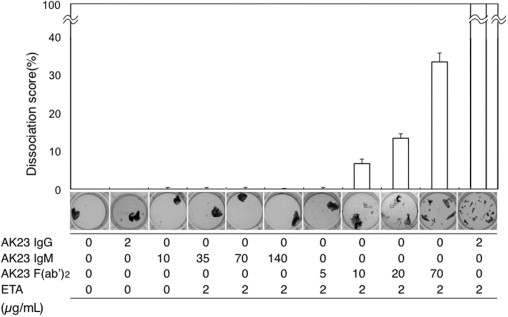
Quantitative evaluation of the pathogenic activity of AK23 IgM by in vitro dissociation assay using cultured human epidermal keratinocytes. To quantitatively evaluate the pathogenic activity of AK23 IgM, we performed an in vitro dissociation assay using NHEK cells. Relative dissociation scores were calculated relative to the dissociation score for AK23 IgG plus ETA. Whereas AK23 IgM exhibited no dissociation activity at concentrations of 35 or 140 μg/mL, AK23 IgM F(ab′)2 fragments yielded dissociation scores of >6.7% and >33.3% at concentrations of 10.0 and 70.0 μg/mL, respectively. Doses of monoclonal antibodies and of ETA are indicated along the x axis.
We then tested whether F(ab′)2 (mol. wt. approximately 100 kDa) derived from AK23 IgM shows dissociation activity. We tested 5, 10, 20, and 70 μg/mL of F(ab′)2 and found that F(ab′)2 indeed had pathogenic activity at 20 μg/mL or more. The dissociation activity was not as high as 2 μg/mL of AK23 IgG even with 70 μg/mL of F(ab′)2, probably because some of the binding activity of F(ab′)2 was lost by pepsin digestion.
Although AK23 IgM failed to bind the cell surfaces of keratinocytes in mouse hard palate, as assessed by indirect immunofluorescence, F(ab′)2 fragments yielded positive staining (Figure 4). When F(ab′)2 fragments were added to the culture medium, this did not produce a dotted staining pattern, but instead stained the cell-cell borders (Figure 4). Thus, although intact AK23 IgM did not show pathogenic activity and was unable to block Dsg3 function, F(ab′)2 fragments of AK23 did display such activity. Together, these finding suggest that the lack of pathogenic activity with AK23 IgM may be a consequence of its large molecular size (it occurs as a pentamer).
AK23 IgM Labels the Edges of Desmosomes or Interdesmosomal Cell Membranes, as Shown by Immuno-EM Microscopy
To determine the precise localization of AK23 IgM and AK23 IgG on keratinocyte cell surfaces in vivo, we performed immuno-EM studies using hard palate tissue from mice inoculated with AK23 IgM-producing or AK23 IgG-producing hybridoma cells (n = 4) (Figure 8A). Notably, AK23 IgM and AK23 IgG showed distinct patterns of binding. AK23 IgG labeling was predominantly detected on desmosomes.20 In marked contrast, AK23 IgM labeled primarily the cell membranes between desmosomes (Figure 8A). When AK23 IgM labeling was detected on desmosomes, it was restricted to the edges of the desmosomes, with no labeling observed in the desmosome core. When quantitative analysis was applied to localization of IgG and IgM labeling in relation to desmosomes, approximately 50% of IgG labeling on cell membranes was found on desmosomes, whereas only <5% of IgM labeling was detected on desmosomes (P < 0.001, n = 2) (Figure 8B).
Figure 8.
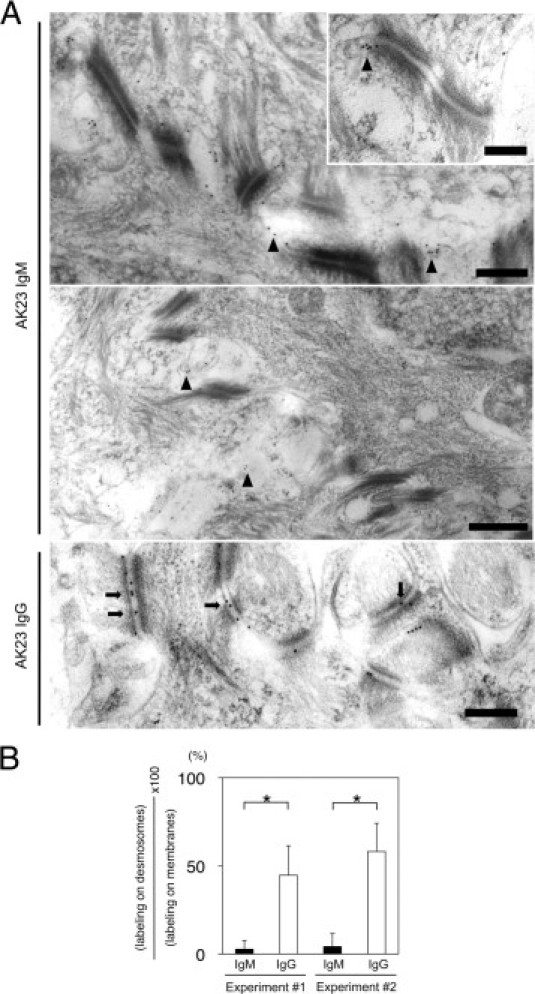
Ultrastructural localization of AK23 IgM and AK23 IgG in vivo in hard palate of adult mice receiving hybridoma cells. A: AK23 IgM was deposited largely to the cell membranes between desmosomes or the edges of desmosomes in vivo (arrowheads), but not in the desmosome core. Inset, representative image of AK23 IgM deposition. AK23 IgG was heavily deposited in the midlines of desmosomes (arrows). B: Quantitative analysis indicates significantly stronger labeling of desmosomes in mice injected with AK23 IgG-producing hybridoma cells than in those injected with AK23 IgM-producing cells. Scale bars: 500 nm (main images); 200 nm (inset). *P < 0.01, t-test.
The results of our immuno-EM study indicate that AK23 IgM binds primarily to Dsg3 that is not integrated to desmosomes, and is unable to gain access to Dsg3 integrated within the desmosome core.
Discussion
IgM antibodies are produced at an early stage in humoral immune responses (before B cells undergo somatic hypermutation and isotype switching) and play an important role in acquired immunity. In the tissue-specific autoimmune disease pemphigus vulgaris, the initial immune response to Dsg3 should occur as a normal immune response. In pemphigus model mice injected with naïve Dsg3−/− splenocytes, anti-Dsg3 IgM is produced at early stage in the immune response, before anti-Dsg3 IgG is subsequently produced (Tomoaki Yokoyama and M.A., unpublished observation). Recently, an anti-Dsg1 IgM autoantibody typically found in fogo selvagem patients was also detected in several healthy individuals.12 Nonetheless, the pathophysiological role of anti-Dsg3 IgM in PV remains unclear. In the present study, we used transgenic techniques to generate an IgM isotype of an IgG mAb that recognizes a critical Dsg3 epitope located on the functionally important amino-terminal adhesive interface and then examined its pathogenic role in PV.
We used cDNAs encoding the variable regions of the AK23 IgG mAb to generate AK23-IgM transgenic mice. Contrary to our expectations, Dsg3-reactive AK23 IgM-producing B cells were detected in the periphery and anti-Dsg3 IgM in the cardiovascular circulation (Figure 2A). B cells recognizing the pathogenic epitope of Dsg3 were not deleted or inactivated by tolerance mechanisms, as occurs in AK7-IgM transgenic mice.13 Furthermore, AK23-IgM transgenic mice displayed no apparent PV phenotype (Figure 2B). In our search for possible explanations, we initially considered the possibility that circulating AK23 IgM levels might be insufficient to cause blistering in AK23 IgM mice. To test this postulate, we isolated AK23 IgM mAb-producing hybridoma cells. Although an excess of AK23 IgM was passively transferred to neonatal mice, no blisters were formed (Figure 5). When AK23 IgM hybridoma cells were inoculated into the peritoneum, circulating AK23 IgM levels increased to a level approximately six times higher than those detected in AK23 IgM transgenic mice, but no blisters were formed (Figure 6). To evaluate the pathogenic activity of AK23 IgM in a more quantitative fashion, we performed an in vitro dissociation assay.16 Although AK23 IgG and F(ab′)2 fragments induced fragmentation of epidermal sheets, intact AK23 IgM did not (Figure 7). Collectively, these findings clearly indicate that, although it recognizes the same critical epitope of Dsg3 as the pathogenic AK23 IgG, AK23 IgM is itself not pathogenic.
Although the molecular weight of the IgM monomer is only 190 kDa, IgM normally forms a pentamer known as macroglobulin (hence the M in IgM), with a molecular weight of approximately 980 kDa, much higher than that of IgG (150 kDa). When IgM is digested by pepsin, the molecular weight of the resulting F(ab′)2 fragments is approximately 100 kDa, smaller than that of IgG.21 Because monovalent Fab′ fragments from the sera of PV patients are capable of inducing blister formation, one can conclude that the Fc portion of IgG does not directly contribute to the pathogenesis of blister formation in pemphigus.22 AK23 IgG and F(ab′)2 fragments of AK23 IgM produced essentially identical linear cell-cell border staining patterns in human skin sections and cultured keratinocytes, whereas AK23 IgM yielded no staining of human skin and a coarse dotted staining pattern in cultured keratinocytes, and the dots did not necessarily colocalize with desmoplakin (Figure 4). More convincingly, immuno-EM demonstrated that AK23 IgG predominantly labeled desmosomes in vivo, as previously found in recipient mice with AK23 IgG hybridoma cells,20 whereas AK23 IgM labeling was detected mostly in the cell membranes between desmosomes, with weaker staining occurring at the edges of desmosomes (Figure 8). These findings indicate differential accessibility of AK23 IgG and AK23 IgM to Dsg3 in vivo and suggest that the large size of AK23 IgM does not allow it to bind to Dsg3 integrated within the desmosome core, although sufficient amounts of IgM are within the intercellular space. We concluded, therefore, that AK23 IgM does not stimulate blister formation because it is unable to bind to Dsg3 integrated within the desmosome core.
The precise mechanism behind blister formation after the binding of pathogenic anti-Dsg3 IgG to Dsg3 in vivo remains controversial. The principal source of contention concerns whether direct functional inhibition of Dsg3 by IgG alone is sufficient to induce blistering, or whether subsequent cytoplasmic signaling is also necessary.23,24 Polyclonal anti-Dsg3 IgG antibodies in sera from pemphigus patients and pemphigus model mice are now widely accepted to comprise mixtures of both pathogenic and nonpathogenic IgGs, based on the results of studies using monoclonal antibodies from patients and mice.5,6 A growing number of studies suggest that inhibition of signaling pathways such as MAPK and c-Myc,25–27 as well as endocytic processing of Dsg3,28,29 can prevent loss of keratinocyte adhesion in PV IgG-treated cells. Overall, these studies suggest that PV IgG may modulate the assembly and disassembly kinetics of desmosomes, and that modifying keratinocyte signaling pathways can blunt keratinocyte sensitivity to PV IgG.
Based on the present results, however, we conclude that the direct inhibition of Dsg3 adhesive function by AK23 IgG may be required to initiate the loss of keratinocyte cell-cell adhesion. For example, the binding of AK23 IgM to free nondesmosomal Dsg3 on cell membranes does not cause blisters or loss of adhesion strength. Therefore, binding to nondesmosomal Dsg3 is apparently insufficient to trigger blister formation. Dsg3-depleted desmosomes have been shown to be formed in cultured normal human keratinocytes and DJM1 cells through the aggregation and internalization of free-floating nondesmosomal Dsg3.30,31 However, the findings of our in vivo studies instead indicate that the binding of AK23 to Dsg3 integrated within desmosomes is essential to disrupt adhesion. Alternatively, it is possible that different isotypes of immunoglobulin differentially affect Dsg3 endocytosis or cellular signaling pathways implicated in acantholysis. Our research group has studies underway to address these additional possibilities.
Although IgMs against Dsg are not found in the sporadic form of pemphigus, anti-Dsg1 IgM was recently detected in as many as 58% of fogo selvagem patients tested, as well in 42% of healthy donors from a rural region of Brazil.12 This anti-Dsg1 IgM was shown, by ELISA and immunoprecipitation, to bind to recombinant Dsg1, but it did not bind to human skin sections in indirect immunofluorescence studies, in which AK23 IgM also failed to yield a positive signal. Our findings suggest that the anti-Dsg1 IgM is most likely nonpathogenic and unable to cause blisters, because of the inaccessibility of Dsg1 integrated within desmosomes. Why then are IgM autoantibodies produced in these patients? The role of IgM in autoimmunity is a hot topic. Healthy individuals are known to produce low-affinity, natural IgM antibodies that are polyreactive to toxins and bacteria to which they have not apparently been exposed and that presumably act as the first line of defense against infection.32,33 IgM antibodies compensate for their low antigen affinity by forming pentameric structures with relatively high avidities. Serum IgMs provide an initial response to foreign antigen and play a regulatory role in the subsequent production of high-affinity IgG antibodies. In the context of autoimmunity, IgM autoantibodies have often been considered to be simply nonpathogenic, but they appear to be actually protective.34,35 Mice deficient in serum IgM exhibit spontaneous autoimmunity (eg, the production of anti-DNA IgG), indicating the role of IgM in protecting against the development of autoimmunity.36 Antigen microarray informatics study, however, has demonstrated that the IgM antibodies produced by neonatal humans recognize mostly self molecules that are associated with major autoimmune diseases later in life.37 Dissecting the role of autoreactive IgM antibodies is an important step in furthering our understanding of the pathophysiology of autoimmune diseases.
In the present study, we generated Dsg3-reactive B-cell transgenic mice expressing IgM derived from both nonpathogenic AK7 IgG, as described previously,13 and pathogenic AK23 IgG. In both types of mouse, functionally competent Dsg3-reactive B cells escaped deletion or inactivation and persisted in the periphery. Moreover, anti-Dsg3 IgM was detected in the circulation. However, in both types of animal, the IgM produced was nonpathogenic and did not induce blistering of the skin or mucous membranes, nor were B cells producing these IgMs harmful to the mice. In contrast, AK7 IgM transgenic mice injected with pathogenic AK23 IgG developed blisters, and the AK7 B cells in their spleens were eliminated.14 This elimination of autoimmune B cells was CD4+ T cell-dependent and involved apoptosis.14 Thus, autoreactive B cells persist as long as they cause no harm, but when damaging or dangerous events such as tissue destruction are sensed, mature autoreactive B cells in the periphery are eliminated. The IgM B-cell transgenic mice used in the present study do not undergo class-switching from IgM to IgG due to the transgenic construct used. B-cell knock-in mice expressing AK23 variable regions should help to prove this concept and may represent an important tool in the further dissection of tolerance and autoimmune mechanisms involving B cells.
Acknowledgments
We thank Dr. Shigeo Koyasu for helpful discussion, Dr. Shoichiro Tsukita for the mouse myeloma P3 cells, Dr. Koji Hashimoto and Dr. Yuji Shirakata (Ehime University) for providing the NHEK cells, Ms. Minae Suzuki for performing the immunofluorescence staining, and Ms. Hiromi Itoh for her assistance with the care of our study animals.
Footnotes
Supported by grants-in-aid for scientific research from the Ministry of Education, Culture, Sports, Science and Technology of Japan; by Health and Labor Sciences research grants for research on measures for intractable diseases from the Ministry of Health, Labor and Welfare of Japan; and by a research subsidy from the Uehara Memorial Foundation.
References
- 1.Amagai M., Klaus-Kovtun V., Stanley J.R. Autoantibodies against a novel epithelial cadherin in pemphigus vulgaris, a disease of cell adhesion. Cell. 1991;67:869–877. doi: 10.1016/0092-8674(91)90360-b. [DOI] [PubMed] [Google Scholar]
- 2.Amagai M. Pemphigus. In: Bolognia J.L., Jorizzo J.L., Rapini R.P., editors. ed 2. vol 1. Mosby Elsevier; London: 2008. pp. 417–429. (Dermatology). [Google Scholar]
- 3.Amagai M., Karpati S., Prussick R., Klaus-Kovtun V., Stanley J.R. Autoantibodies against the amino-terminal cadherin-like binding domain of pemphigus vulgaris antigen are pathogenic. J Clin Invest. 1992;90:919–926. doi: 10.1172/JCI115968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Amagai M., Hashimoto T., Shimizu N., Nishikawa T. Absorption of pathogenic autoantibodies by the extracellular domain of pemphigus vulgaris antigen (Dsg3) produced by baculovirus. J Clin Invest. 1994;94:59–67. doi: 10.1172/JCI117349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tsunoda K., Ota T., Aoki M., Yamada T., Nagai T., Nakagawa T., Koyasu S., Nishikawa T., Amagai M. Induction of pemphigus phenotype by a mouse monoclonal antibody against the amino-terminal adhesive interface of desmoglein 3. J Immunol. 2003;170:2170–2178. doi: 10.4049/jimmunol.170.4.2170. [DOI] [PubMed] [Google Scholar]
- 6.Payne A.S., Ishii K., Kacir S., Lin C., Li H., Hanakawa Y., Tsunoda K., Amagai M., Stanley J.R., Siegel D.L. Genetic and functional characterization of human pemphigus vulgaris monoclonal autoantibodies isolated by phage display. J Clin Invest. 2005;115:888–899. doi: 10.1172/JCI24185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nguyen V.T., Ndoye A., Shultz L.D., Pittelkow M.R., Grando S.A. Antibodies against keratinocyte antigens other than desmogleins 1 and 3 can induce pemphigus vulgaris-like lesions. J Clin Invest. 2000;106:1467–1479. doi: 10.1172/JCI10305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grando S.A. Autoimmunity to keratinocyte acetylcholine receptors in pemphigus. Dermatology. 2000;201:290–295. doi: 10.1159/000051540. [DOI] [PubMed] [Google Scholar]
- 9.Amagai M., Tsunoda K., Suzuki H., Nishifuji K., Koyasu S., Nishikawa T. Use of autoantigen-knockout mice in developing an active autoimmune disease model for pemphigus. J Clin Invest. 2000;105:625–631. doi: 10.1172/JCI8748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boggon T.J., Murray J., Chappuis-Flament S., Wong E., Gumbiner B.M., Shapiro L. C-cadherin ectodomain structure and implications for cell adhesion mechanisms. Science. 2002;296:1308–1313. doi: 10.1126/science.1071559. [DOI] [PubMed] [Google Scholar]
- 11.Wardemann H., Yurasov S., Schaefer A., Young J.W., Meffre E., Nussenzweig M.C. Predominant autoantibody production by early human B cell precursors. Science. 2003;301:1374–1377. doi: 10.1126/science.1086907. [DOI] [PubMed] [Google Scholar]
- 12.Diaz L.A., Prisayanh P.S., Dasher D.A., Li N., Evangelista F., Aoki V., Hans-Filho G., dos Santos V., Qaqish B.F., Rivitti E.A., Cooperative Group on Fogo Selvagem Research The IgM anti-desmoglein 1 response distinguishes Brazilian pemphigus foliaceus (fogo selvagem) from other forms of pemphigus. J Invest Dermatol. 2007;128:667–675. doi: 10.1038/sj.jid.5701121. [DOI] [PubMed] [Google Scholar]
- 13.Ota T., Aoki-Ota M., Tsunoda K., Simoda K., Nishikawa T., Amagai M., Koyasu S. Auto-reactive B cells against peripheral antigen, desmoglein 3, escape from tolerance mechanism. Int Immunol. 2004;16:1487–1495. doi: 10.1093/intimm/dxh150. [DOI] [PubMed] [Google Scholar]
- 14.Ota T., Aoki-Ota M., Tsunoda K., Nishikawa T., Koyasu S., Amagai M. Autoreactive B-cell elimination by pathogenic IgG specific for the same antigen: implications for peripheral tolerance. Int Immunol. 2008;20:1351–1360. doi: 10.1093/intimm/dxn095. [DOI] [PubMed] [Google Scholar]
- 15.Shimizu H., McDonald J.N., Kennedy A.R., Eady R.A. Demonstration of intra- and extracellular localization of bullous pemphigoid antigen using cryofixation and freeze substitution for postembedding immunoelectron microscopy. Arch Dermatol Res. 1989;281:443–448. doi: 10.1007/BF00510078. [DOI] [PubMed] [Google Scholar]
- 16.Ishii K., Harada R., Matsuo I., Shirakata Y., Hashimoto K., Amagai M. In vitro keratinocyte dissociation assay for evaluation of the pathogenicity of anti-desmoglein 3 IgG autoantibodies in pemphigus vulgaris. J Invest Dermatol. 2005;124:939–946. doi: 10.1111/j.0022-202X.2005.23714.x. [DOI] [PubMed] [Google Scholar]
- 17.Kabat E.A., Wu T.T., Bilofsky H. Evidence indicating independent assortment of framework and complementarity-determining segments of the variable regions of rabbit light chains: Delineation of a possible J minigene. J Exp Med. 1980;152:72–84. doi: 10.1084/jem.152.1.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anhalt G.J., Labib R.S., Voorhees J.J., Beals T.F., Diaz L.A. Induction of pemphigus in neonatal mice by passive transfer of IgG from patients with the disease. N Engl J Med. 1982;306:1189–1196. doi: 10.1056/NEJM198205203062001. [DOI] [PubMed] [Google Scholar]
- 19.Amagai M., Matsuyoshi N., Wang Z.H., Andl C., Stanley J.R. Toxin in bullous impetigo and staphylococcal scalded-skin syndrome targets desmoglein 1. Nat Med. 2000;6:1275–1277. doi: 10.1038/81385. [DOI] [PubMed] [Google Scholar]
- 20.Shimizu A., Ishiko A., Ota T., Saito H., Oka H., Tsunoda K., Amagai M., Nishikawa T. In vivo ultrastructural localization of the desmoglein 3 adhesive interface to the desmosome mid-line. J Invest Dermatol. 2005;124:984–989. doi: 10.1111/j.0022-202X.2005.23706.x. [DOI] [PubMed] [Google Scholar]
- 21.Jones R.G., Landon J. Enhanced pepsin digestion: a novel process for purifying antibody F(ab′)(2) fragments in high yield from serum. J Immunol Methods. 2002;263:57–74. doi: 10.1016/s0022-1759(02)00031-5. [DOI] [PubMed] [Google Scholar]
- 22.Rock B., Labib R.S., Diaz L.A. Monovalent Fab′ immunoglobulin fragments from endemic pemphigus foliaceus autoantibodies reproduce the human disease in neonatal Balb/c mice. J Clin Invest. 1990;85:296–299. doi: 10.1172/JCI114426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Amagai M., Ahmed A.R., Kitajima Y., Bystryn J.C., Milner Y., Gniadecki R., Hertl M., Pincelli C., Kurzen H., Fridkis-Hareli M., Aoyama Y., Frusić-Zlotkin M., Müller E., David M., Mimouni D., Vind-Kezunovic D., Michel B., Mahoney M., Grando S. Are desmoglein autoantibodies essential for the immunopathogenesis of pemphigus vulgaris, or just “witnesses of disease”? Exp Dermatol. 2006;15:815–831. doi: 10.1111/j.1600-0625.2006.00499_1.x. [DOI] [PubMed] [Google Scholar]
- 24.Kitajima Y., Aoyama Y. A perspective of pemphigus from bedside and laboratory-bench. Clin Rev Allergy Immunol. 2007;33:57–66. doi: 10.1007/s12016-007-0036-5. [DOI] [PubMed] [Google Scholar]
- 25.Berkowitz P., Chua M., Liu Z., Diaz L.A., Rubenstein D.S. Autoantibodies in the autoimmune disease pemphigus foliaceus induce blistering via p38 mitogen-activated protein kinase-dependent signaling in the skin. Am J Pathol. 2008;173:1628–1636. doi: 10.2353/ajpath.2008.080391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mao X, Sano Y, Park JM, Payne AS: p38 MAPK activation is downstream of the loss of intercellular adhesion in pemphigus vulgaris. J Biol Chem 286:1283–1291 [DOI] [PMC free article] [PubMed]
- 27.Caldelari R., de Bruin A., Baumann D., Suter M.M., Bierkamp C., Balmer V., Müller E. A central role for the armadillo protein plakoglobin in the autoimmune disease pemphigus vulgaris. J Cell Biol. 2001;153:823–834. doi: 10.1083/jcb.153.4.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Calkins C.C., Setzer S.V., Jennings J.M., Summers S., Tsunoda K., Amagai M., Kowalczyk A.P. Desmoglein endocytosis and desmosome disassembly are coordinated responses to pemphigus autoantibodies. J Biol Chem. 2006;281:7623–7634. doi: 10.1074/jbc.M512447200. [DOI] [PubMed] [Google Scholar]
- 29.Aoyama Y., Nagai M., Kitajima Y. Binding of pemphigus vulgaris IgG to antigens in desmosome core domains excludes immune complexes rather than directly splitting desmosomes. Br J Dermatol. 2010;162:1049–1055. doi: 10.1111/j.1365-2133.2010.09672.x. [DOI] [PubMed] [Google Scholar]
- 30.Aoyama Y., Kitajima Y. Pemphigus vulgaris-IgG causes a rapid depletion of desmoglein 3 (Dsg3) from the Triton X-100 soluble pools, leading to the formation of Dsg3-depleted desmosomes in a human squamous carcinoma cell line, DJM-1 cells. J Invest Dermatol. 1999;112:67–71. doi: 10.1046/j.1523-1747.1999.00463.x. [DOI] [PubMed] [Google Scholar]
- 31.Shu E., Yamamoto Y., Aoyama Y., Kitajima Y. Intraperitoneal injection of pemphigus vulgaris-IgG into mouse depletes epidermal keratinocytes of desmoglein 3 associated with generation of acantholysis. Arch Dermatol Res. 2007;299:165–167. doi: 10.1007/s00403-007-0754-9. [DOI] [PubMed] [Google Scholar]
- 32.Hurez V., Dietrich G., Kaveri S.V., Kazatchkine M.D. Polyreactivity is a property of natural and disease-associated human autoantibodies. Scand J Immunol. 1993;38:190–196. doi: 10.1111/j.1365-3083.1993.tb01712.x. [DOI] [PubMed] [Google Scholar]
- 33.Manson J.J., Mauri C., Ehrenstein M.R. Natural serum IgM maintains immunological homeostasis and prevents autoimmunity. Springer Semin Immunopathol. 2005;26:425–432. doi: 10.1007/s00281-004-0187-x. [DOI] [PubMed] [Google Scholar]
- 34.Melero J., Tarragó D., Núñez-Roldán A., Sánchez B. Human polyreactive IgM monoclonal antibodies with blocking activity against self-reactive IgG. Scand J Immunol. 1997;45:393–400. doi: 10.1046/j.1365-3083.1997.d01-418.x. [DOI] [PubMed] [Google Scholar]
- 35.Li Q.Z., Xie C., Wu T., Mackay M., Aranow C., Putterman C., Mohan C. Identification of autoantibody clusters that best predict lupus disease activity using glomerular proteome arrays. J Clin Invest. 2005;115:3428–3439. doi: 10.1172/JCI23587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ehrenstein M.R., Cook H.T., Neuberger M.S. Deficiency in serum immunoglobulin (Ig)M predisposes to development of IgG autoantibodies. J Exp Med. 2000;191:1253–1258. doi: 10.1084/jem.191.7.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Merbl Y., Zucker-Toledano M., Quintana F.J., Cohen I.R. Newborn humans manifest autoantibodies to defined self molecules detected by antigen microarray informatics. J Clin Invest. 2007;117:712–718. doi: 10.1172/JCI29943. [DOI] [PMC free article] [PubMed] [Google Scholar]