

Abstract
Opiates have long been used as analgesics to relieve pain associated with various medical conditions. Here, we evaluated the effect and mechanism of mu opioid signaling on the intestinal wound healing response and assessed downstream pathways known to be protective against intestinal injury. Mice (C57BL/6) were exposed to 3% dextran sodium sulfate (DSS) for 7 days or 4% DSS for 5 days followed by 7 days of water. The mu opioid receptor (MOR)-specific agonist [D-Arg2,Lys4]dermorphin-(1,4)-amide (DALDA) and the antagonist cyprodime were injected s.c. daily for in vivo studies or used for in vitro analysis. We found that MOR activation attenuated DSS-induced histologic and gross intestinal injury and weight loss; diminished Ifng, Tnf, and Il6 mRNA expression; and promoted intestinal healing during recovery. DALDA also enhanced colonocyte proliferation (Ki-67 staining) by 350%. MOR activation increased Stat3 phosphorylation in both DALDA-treated mice and the CMT-93 cell line. Importantly, DALDA-induced colonocyte migration was completely ablated by shStat3 knockdown. Together, this work shows that MOR activation protects against and enhances recovery from DSS-induced intestinal injury. This is associated with an increase in Stat3 activation. Furthermore, Stat3 is required for DALDA-induced colonocyte migration. Consequently, manipulation of MOR signaling may represent a novel means to promote mucosal healing and to maintain intestinal homeostasis after intestinal injury.
A critical component of intestinal homeostasis is the presence of an intestinal mucosal barrier, comprised of a single monolayer of intestinal epithelial cells (IECs) that isolates highly antigenic luminal contents from an immunologically rich and underlying stromal compartment.1 A wide body of research has shown that loss of intestinal barrier function leads to the development of various gastrointestinal inflammatory disorders, including inflammatory bowel diseases (IBD).1,2 Conditions leading to an impaired mucosal barrier function are diverse and include genetic predisposition, medications (nonsteroidal anti-inflammatory drugs,3 antibiotics), radiation exposure,4 and ischemic episodes.5 Overt damage to the epithelial barrier caused by these injuries triggers a host response, termed restitution/wound healing.6 In this response, cells at the edge of the wound undergo a de-differentiation process and migrate into the wounded area. They then undergo cytoskeletal rearrangement,7 re-differentiate, and finally re-establish tight junction barriers with their neighboring cells.6 This process does not require epithelial proliferation, but renewal of cells are needed to replenish the decreased enterocyte pool after injury.7 Numerous proliferative signals such as epidermal growth factor,7 transforming growth factor β,8,9 and cytokines such as IL-2210 are implicated in the maintenance of the mucosal barrier. At the molecular level, these proliferative signals use various pathways, including NF-κB, mitogen-activated protein kinase, Stat3, and PI3K/Akt6 pathways. Identifying factors that engage these signaling pathways and promote wound healing is of great therapeutic interest for diseases such as IBD.
Factors that promote wound healing include the various neuropeptides produced by the enteric nervous system. Of strong relevance to IBD, several neuropeptides modulate immune cell function and could consequently affect the development of intestinal inflammation.11 Opioids are a class of neuropeptides that have traditionally been used as analgesics; however, emerging literature associates this class of neuropeptide with intestinal proliferation and inflammation.12 Vertebrates traditionally express three types of opioid receptors: mu, delta, and kappa, all of which are G-protein–coupled receptors that preferentially bind to Gi proteins. Binding of ligand to these receptors leads to the prototypical inactivation of neural pain fibers.13–15 Interestingly, recent studies have shown opioids to protect against ischemia/reperfusion-induced cardiac injury.16,17 Furthermore, the peripheral mu-specific agonist [D-Arg2,Lys4]dermorphin-(1,4)-amide (DALDA)18 reduces inflammation in two experimental models of murine colitis: 2,4,6-trinitrobenzene sulfonic acid (TNBS) and adoptive transfer of CD45RBhiCD4+ T cells.19 Conversely, experimental colitis is exacerbated in mu opioid receptor (MOR) knockout mice,19 and MOR expression is up-regulated in mucosal samples from human patients with IBD compared with controls.20 Together, these data implicate an important role for MOR signaling in regulating gut homeostasis. Although these studies have not fully addressed the mechanism by which MOR signaling ameliorates intestinal inflammation, it appears that down-regulation of cytokine production and modulation of T-cell function contributes to MOR's beneficial effect.
On the basis of its beneficial effect in other models of colitis,19 as well as in ischemia-induced cardiac injury,16,17 we hypothesized that MOR signaling is cytoprotective in the context of intestinal barrier damage. With the use of a chemical model of acute injury and wound healing that is independent of T and B cells,21 we observed that the mu opioid agonist DALDA protects against dextran sodium sulfate (DSS)-induced intestinal injury and promotes healing through activation of Stat3 and induction of cytoprotective factors regulated in part by Stat3. Our findings suggest that MOR agonists could represent a novel means to modulate tissue injury and enhance intestinal restitution and proliferation.
Materials and Methods
DSS-Induced Acute Injury
Wild-type and transgenic NF-κBEGFP reporter mice (C57BL/6 background) were maintained in standard housing cages in specific pathogen-free conditions. Mice (N = 7), were given 3% DSS (MP Biomedicals, Aurora, OH) in their drinking water for 7 days, whereas control mice received water alone. Water consumption was comparable between the different experimental groups. Throughout DSS administration, vehicle (10% dimethyl sulfoxide/90% saline), the peripheral (does not cross the blood-brain barrier) MOR agonist DALDA18 (100% saline, 50 μg/kg; US Biological, Swampscott, MA), or the MOR antagonist cyprodime22 (10% dimethyl sulfoxide /90% saline, 10 mg/kg; Tocris Bioscience, Ellisville, MI) was injected s.c. daily on the mid-dorsal surface of the mice. Mice were monitored daily for weight loss and visible signs of rectal bleeding. Occult bleeding was evaluated at day 4 of the experiment (Hemoccult; Beckmann Coulter Inc., Fullerton, CA), in accordance with internal pilot studies that showed occult bleeding to be reliably observable in this model from day 4 onward. Clinical score, assessing weight loss, occult blood, and stool consistency were calculated as previously described.23 All animal experiments were approved by the Institutional Animal Care and Use Committee of the University of North Carolina at Chapel Hill.
DSS Recovery Studies
C57BL/6 mice (N = 6) were administered 4% DSS in their drinking water for 5 days, followed by water for a recovery period of 7 days. DALDA or vehicle was injected s.c. daily during the water recovery phase, as described earlier. Occult bleeding was evaluated at day 12. Mice were euthanized at days 8 and 12. Colonoscopies were performed as described previously,24 on days 5, 8, and 12.
Sample Collection and Histologic Evaluation
Mice were sacrificed at the indicated time points by CO2 asphyxiation followed by cervical separation. The colon was dissected and flushed with ice-cold PBS, longitudinally splayed, Swiss rolled, fixed in 10% formalin for 24 hours, and then embedded in paraffin. Colitis severity was evaluated with H&E-stained sections by a blinded investigator on a scale from 0 to 40, as described previously.2,25,26
Immunohistochemistry (IHC) staining for pStat3(Y705) (Cell Signaling Technology Inc., Beverly, MA) was performed according to the manufacturer's specifications at a 1:50 dilution as previously described.24 Ki-67 staining was performed as previously described.24 All sections were counterstained with hematoxylin. Two different sections (per animal), approximately 200 μm apart, were assessed for staining, and representative images are shown.
For protein analysis, distal colons were dissected, flushed with ice-cold PBS, and homogenized for 15 seconds with a polytron (IKA Works Inc., Wilmington, NC) in cold RIPA buffer containing proteinase inhibitors (Complete Mini; Roche Diagnostics GmbH, Penzberg, Germany) and phosphatase inhibitor cocktail no. 2 at a 1:100 dilution (Sigma-Aldrich, St. Louis, MO). After incubation at 4°C for 30 minutes, samples were sonicated and cleared. Lysates were then diluted 1:1 with 2× Laemmli buffer.
Confocal Analysis of EGFP Expression
Colons were fixed in 4% paraformaldehyde for 40 minutes at room temperature and permeabilized in 2% Triton X-100 for 18 to 22 hours at 4°C. Tissues were rinsed in 1× PBS, counterstained with propidium iodide (Molecular Probes, Eugene, OR) for 30 minutes, and then placed in custom imaging chambers and immersed in Focus Clear (Cedar Lane Labs, Burlington, NC). Tissues were imaged with an Olympus Fluoview FV1000MPE microscope, with a 25× long-working distance water immersion objective. Enhanced green fluorescent protein (EGFP) was excited with a 495-nm laser, and propidium iodide was excited with a 559-nm laser. Tissue was imaged from the surface to a depth of 120 μm with a standard scale for laser power, offset, and gain on the basis of depth that was maintained between samples. Images were analyzed with BioImage XD and Imaris (Bitplane AG, Zurich, Switzerland).
CMT-93 Cell Culture and Stimulation
Murine rectal carcinoma cells (passage 33 to 59; American Type Culture Collection, Manassas, VA) were cultured in Dulbecco's modified Eagle's medium-high glucose as described previously.27 Cells were starved for 18 to 22 hours in Opti-MEM and then treated with DALDA (10 μmol/L) or naloxone (10 mmol/L; Sigma-Aldrich) for the times indicated. Cells were directly lysed in 1× Laemmli buffer, and protein concentration was measured with Bio-Rad quantification assay (Bio-Rad Laboratories, Hercules, CA).
Luciferase Assays
CMT-93 cells were infected with Ad5κB-Luc vector28 for 12 hours in Opti-MEM and then were stimulated with lipopolysaccharide (LPS; 10 μg/mL) in the presence or absence of 10 μmol/L DALDA. Luciferase assay was performed on a Monolight 2010 luminometer for 20 seconds (Analytical Luminescence, San Diego, CA).
Western Blot Analysis
Proteins (30 μg) were separated with the use of 10% SDS-PAGE and transferred to nitrocellulose membranes and then probed with antibodies to pStat3(Tyr705) (1:2000), extracellular signal-related kinase 1 and 2 (ERK1/2; 1:1000), pERK1/2 (1:2000; Cell Signaling Technology Inc.), Stat3 (1:1000; Santa Cruz Biotechnologies, Santa Cruz, CA), and β-actin (1:1000; MP Biomedical, Solon, OH) and were used with 5% milk in Tris-buffered saline (TBS)-Tween (0.1%) or 5% bovine serum albumin in TBS-Tween (0.1%; for Cell Signaling Technology Inc. antibodies), followed by the appropriate horseradish peroxidase–conjugated secondary antibody (GE Healthcare, Piscataway, NJ) and detected by chemiluminescence. Densitometry analysis for pStat3 was performed with NIH ImageJ software version 1.43 (http://rsbweb.nih.gov/ij), to determine the ratio of pStat3 to Stat3, when indicated.
Immunofluorescence
CMT-93 cells were grown to 50% confluence in Opti-MEM media on glass cover slides. Cells were fixed in ice-cold methanol, blocked with normal goat serum, and incubated with 1:100 anti-pStat3(Y705) (Cell Signaling Technology Inc.) followed by anti-rabbit tetramethylrhodamine isothiocyanate (1:100; Jackson Immunoresearch Laboratories, West Grove, PA) for 30 minutes. Cells were then counterstained with Hoechst dye and mounted with Fluoromount-G (Southern Biotech, Birmingham, AL). Images were captured with a Zeiss LSM 710 confocal microscope (Zeiss, Jena, Germany).
RNA Isolation and Real-Time PCR
RNA isolation from colonic tissues and CMT-93 cells and subsequent mRNA analysis were performed as previously described,24 using an ABI Prism HT7700. Specificity and linearity of amplification for each primer set were determined by melting curve analysis and calculation of the slope from serial diluted samples. Relative fold changes were determined with the ΔΔCT calculation method. Values were normalized to the internal control glyceraldehyde-3-phosphate dehydrogenase. Primers were Tnf (5′-ATGAGCACAGAAAGCATGATC-3′ and 5′-TACAGGCTTGTCACTCGAATT-3′),24 Il6 (5′-CGGAGGCTTAATTACACATGTT-3′ and 5′-CTGGCTTTGTCTTTCTTGTTATC-3′),29 Il1b (5′-GCCCATCCTCTGTGACTCAT-3′ and 5′-AGGCCACAGGTATTTTGTCG-3′), GAPDH (5′-GGTGAAGGTCGGAGTCAACGGA-3′ and 5′-GAGGGATCTCGCTCCTGGAAGA-3′),29 and Cox2 (Ptgs2) (5′-TGAGTACCGCAAACGCTTCTC-3′ and 5′-TGCAGCCATTTCCTTCTCTCCT-3′).30 Cyclin D (Ccnd1), Ifng, Myc, and Reg3b are QuantiTect primers (Qiagen, Valencia, CA).
Migration and Proliferation Assays
Migration assays were performed as described previously.31 Briefly, CMT-93 cells were plated to confluence in Opti-MEM and then were scratched with a P1000 pipette tip. DALDA (10 μmol/L) in PBS or PBS alone was then immediately added. Four different regions per plate were demarked, and images were taken at the specified time points with the use of an Olympus IX7 microscope (Center Valley, PA). Images were analyzed with NIH ImageJ software. Each experiment was measured at four individual points and was repeated independently at least two times.
Reproductive proliferation assays32 were performed by plating CMT-93 cells at a known cellular density, and, 2 hours after plating, cells were switched to 1% serum media and stimulated for 8 hours with 10 μmol/L DALDA in PBS or PBS alone. Cells were then trypsinized and counted with the use of trypan blue. Each plate was counted four different times, and the experiment was repeated independently at least two times.
shRNA Knockdown
A stable shStat3 knockdown CMT-93 cell line was created with a lentivirus provided by the University of North Carolina at Chapel Hill Lentiviral shRNA (short hairpin RNA) Core with a mixture of four different shStat3 constructs. Open Biosystems TC1 library numbers TCRN0000071453 to TCRN0000071457 were used (pLKO.1 vector).33 Control shRNA virus SHC-002 (Sigma-Aldrich) was used to create a stable control knockdown line. Cells were maintained in selection media containing 2 μg/mL puromycin, to which the lentiviral vectors conferred resistance but completely killed uninfected cells.
Statistical Analyses
Statistical analyses were performed with GraphPad Prism version 5.0a (GraphPad, La Jolla, CA). Comparisons of weight between mouse groups and over time were done with a two-way analysis of variance. Weight posttests and further comparisons made between mice were analyzed with a Mann-Whitney U-test at a 95% confidence interval. In vitro data and Ki-67 and pStat3 cell counting were compared with Student's t-tests, at a 95% confidence interval. All graphs depict mean ± SEM. Experiments were considered statistically significant if P < 0.05.
Results
DALDA Ameliorates DSS-Induced Intestinal Injury
To investigate the effect of MOR signaling in intestinal injury response, we used the mu-specific agonist DALDA in a DSS model of acute intestinal injury. Mice were administered 3% DSS for 7 days, along with sterile s.c. injections of saline (vehicle), mu-specific agonist DALDA (50 μg/kg),18 or the mu-specific antagonist cyprodime (10 mg/kg).22 We found that DALDA significantly attenuated DSS-induced weight loss (Figure 1A). Clinical scores, which factors in weight loss, diarrhea, and bloody stool, showed that DALDA-treated mice displayed a significant reduction in disease compared with untreated mice (mean, 5.92 versus 3.93; P < 0.01), whereas MOR antagonism exacerbated disease (mean, 5.92 versus 7.92; P < 0.01; Figure 1B). Histologic scores (Figure 1C) showed that DALDA significantly reduced intestinal inflammation compared with vehicle-treated mice (mean, 24.2 to 10.8; P < 0.01), whereas cyprodime worsened inflammation (mean, 24.2 to 33.5; P < 0.05). In addition, DSS-induced histologic crypt damage was attenuated by DALDA treatment, whereas cyprodime exacerbated damage (Figure 1D). The extent of the pathologic changes of DALDA and cyprodime on DSS-induced intestinal injury is shown in Figure 1E.
Figure 1.
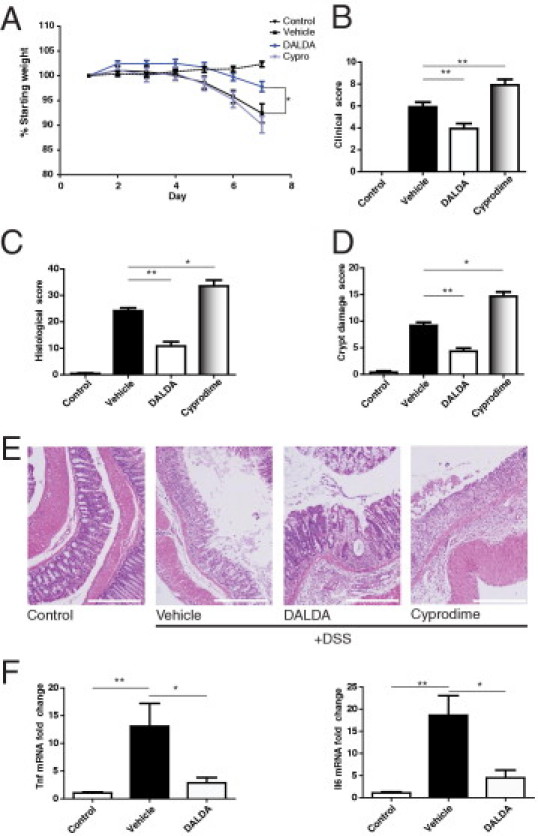
MOR activation protects against DSS-induced intestinal injury. Mice were given 3% DSS for 7 days concurrent with opioid treatment. DALDA (50 μg/kg), cyprodime (10 mg/kg), or vehicle was injected s.c. daily. All graphs depict mean ± SEM. A: Percentage of weight loss from starting weight. N = 7 for all groups. P < 0.001 × 2-way analysis of variance between treatment groups and over time; P < 0.05 for DALDA-treated versus vehicle-treated at day 7. B: Clinical score. N = 7 for all groups. P < 0.01 for DALDA-treated versus vehicle-treated. C: Histologic score. N ≥ 5 for all groups. D: Crypt damage score. N > 4 for all groups. E: Representative H&E photomicrographs. Scale bar = 100 μm for all sections. F: Fold change Tnf and Il6 distal colonic mRNA expression. N ≥ 4 for all groups. *P < 0.05, **P < 0.01.
After establishing that MOR activation protected against DSS-induced injury, we assessed the effects of DALDA on inflammatory cytokines. We found that DALDA significantly reduced colonic Tnf and Il6 mRNA expression (Figure 1F). Because these cytokines are induced in part through immune-derived NF-κB signaling during experimental colitis,34 we next explored the effect of daily s.c. injections of DALDA on NF-κB activity with the use of fresh-fixed tissue sections obtained from NF-κBEGFP reporter mice and confocal microscopy imaging. As previously reported, EGFP expression in the lamina propria increased in DSS-exposed NF-κBEGFP reporter mice.29,35 Interestingly, lamina propria-derived EGFP expression (NF-κB activity) was strongly reduced in DALDA-treated mice (see Supplemental Figure S1 at http://ajp.amjpathol.org). Moreover, DALDA failed to block LPS-induced NF-κB transcriptional activity in CMT-93 cells (see Supplemental Figure S1 at http://ajp.amjpathol.org). These findings indicate that DALDA protects against DSS-induced acute intestinal injury in mice and is associated with reduced immune cell-derived NF-κB activation and pro-inflammatory gene expression without directly inhibiting NF-κB signaling.
MOR Activation Enhances Recovery from DSS-Induced Intestinal Injury
Because wound healing is a critical process during tissue repair, we evaluated the effect of DALDA on recovery from DSS-induced intestinal injury. Mice were administered 4% DSS for 5 days, followed by 7 days of s.c. injections of saline or 50 μg/kg DALDA. Gross mucosal damage visualized by colonoscopy showed attenuation of macroscopic lesions by day 7 in DALDA-treated mice (Figure 2B). Furthermore, DALDA enhanced weight loss recovery, and DALDA-treated mice returned to near-normal weight by day 7 of the recovery phase (Figure 2A). DALDA similarly reduced clinical scores after 7 days of recovery (mean, 5.3 versus 2.3; P < 0.05; Figure 2C), whereas histologic scores were noticeably lower after only 3 days of therapy (mean, 29.2 versus 15.6, P < 0.01 at day 3; 23.2 versus 11.3, P < 0.01 at day 7; Figure 2D), with initial improvements associated with decreased crypt damage (mean, 9.7 versus 3.7; P < 0.05 at day 3, Figure 2E). In accordance with histologic and clinical decreases in disease, Tnf, Il6, Il1b, and Ifng mRNA levels were significantly reduced in DALDA-treated than in vehicle-treated mice (Figure 2F). These findings indicate that DALDA protects against DSS-induced intestinal injury and promotes wound healing.
Figure 2.
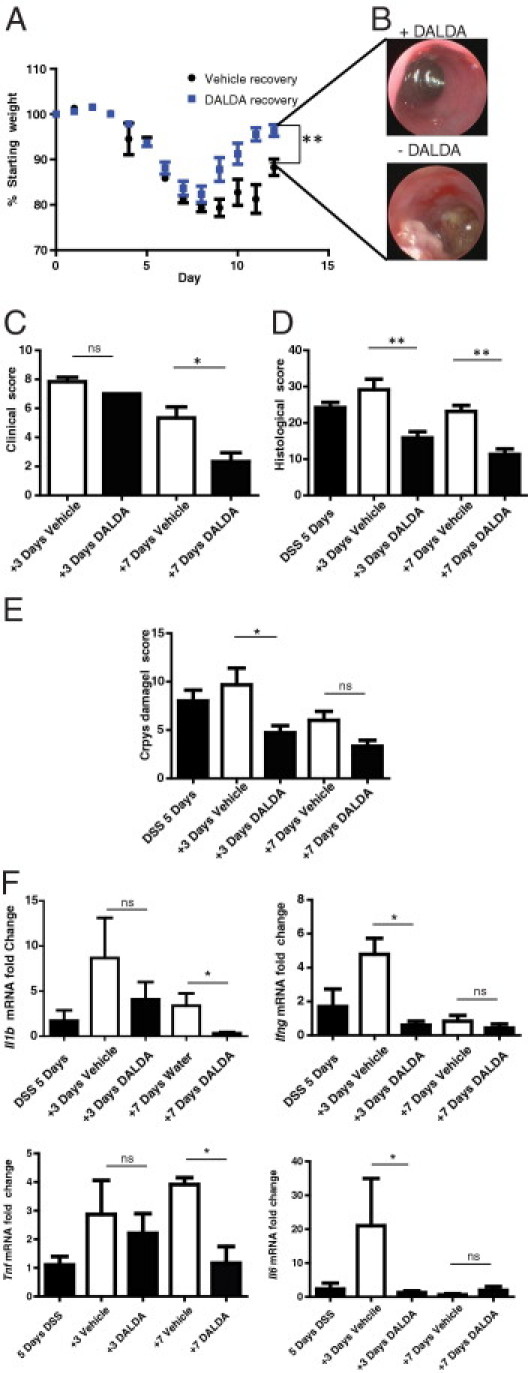
MOR activation enhances recovery from DSS-induced intestinal injury. Mice were administered 4% DSS for 5 days, followed by 7 days of water recovery concurrent with treatment: 50 μg/kg DALDA or vehicle was injected s.c. daily. All graphs depict mean ± SEM. A: Percentage of weight loss from starting weight. N > 6 for all groups. B: Representative colonoscopies for each group on the final day of the experiment. C: Clinical score. N = 6 for all groups. D: Histologic score. N > 4 for all groups. E: Crypt damage score. N > 4 for all groups. F: Fold change Tnf, Il6, Il1b, and Ifng mRNA accumulation was determined by RT-PCR. N ≥ 4 for all groups. ns, not significant. *P < 0.05, **P < 0.01.
DALDA Enhances Colonocyte Proliferation and Stat3 Activation in Vivo
Because proliferation is an important feature of the healing response that occurs after migration/restitution, we measured proliferation in DALDA-treated mice by Ki-67 staining. We observed a modest 38% increase in proliferative activity (P < 0.01) in DALDA-treated mice compared with control (Figure 3). Importantly, proliferation index increased by almost twofold (P < 0.05) in DSS-exposed, DALDA-treated mice compared with DSS alone (Figure 3).
Figure 3.
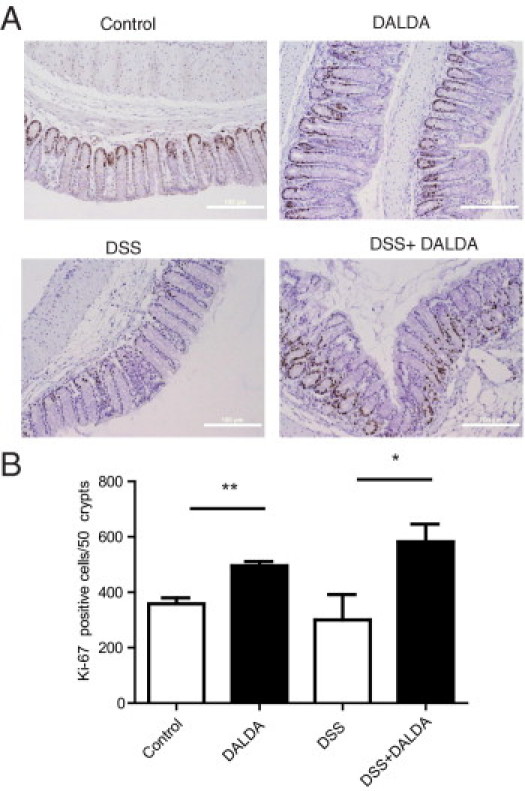
Enhanced colonocyte proliferation in DALDA-treated mice. All graphs depict mean ± SEM. A: Representative photomicrographs. Scale bar = 100 μm. B: Ki-67 positive cells were counted in 50 crypts across multiple fields of view. N ≥ 4 for all groups except DALDA alone, for which N = 3. *P < 0.05, **P < 0.01.
Because Stat3 signaling is also a critical proliferative response in the DSS acute model of intestinal injury,10 we next determined the effect of MOR agonists on Stat3 activation. Compared with untreated controls, DSS-exposed mice showed a fivefold increase in colonic pStat3. DALDA therapy alongside DSS injury increased pStat3 10-fold (P < 0.001) over that of untreated controls (Figure 4A); surprisingly, mice given DALDA alone without DSS injury did not have increased levels of pStat3 over controls (N = 4; data not shown), suggesting that Stat3 activation by MOR is tightly regulated and that MOR-mediated activation occurs only in the presence of injury. IHC indicated that DALDA-induced Stat3 phosphorylation appears predominately in colonocytes (Figure 4, B and C), resulting in a 2.7-fold increase in pStat3-positive colonocytes per 20 crypts (P < 0.05) in DALDA-treated compared with vehicle-treated mice exposed to DSS. We next assessed the expression of cytoprotective genes regulated in part by Stat3, using real-time PCR. As anticipated, DALDA significantly enhanced expression of cyclooxygenase 2 (Ptgs2), Myc, Reg3b, and cyclin D (Ccnd1) mRNA levels in DSS-exposed mice compared with vehicle-treated mice (Figure 4D). These specific cytoprotective genes were selected because their expression decreased in DSS-exposed Stat3ΔIEC mice compared with wild-type mice,10 showing that they are regulated by Stat3 in colonocytes.
Figure 4.
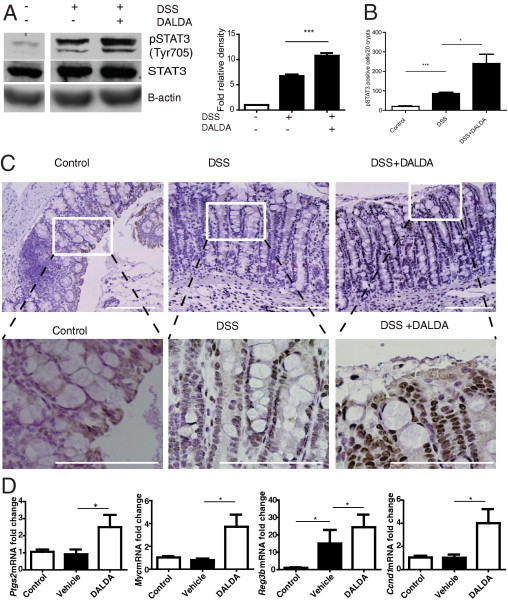
DALDA enhances Stat3 activation in DSS-induced intestinal injury. Mice were administered 3% DSS + vehicle or 3% DSS + DALDA (50 μg/kg, injected s.c. daily) for 7 days. All graphs depict mean ± SEM. A: Levels of Stat3 phosphorylation (Y705) in distal colonic tissue measured by Western blot analysis. Relative level of pStat3 over total Stat3 was determined by densitometry. Representative of four individual mice. B and C: pStat3 IHC analysis. Representative images of four mice per group. Scale bars = 100 μm in all images. White boxes outline higher magnification images of each section. Bar graph shows pStat3-positive colonocytes per 20 crypts. D: Expression of Stat3-dependent genes (Ptgs2, Myc, Reg3b, Ccnd1) in distal colonic tissue. N ≥ 3 per group. *P < 0.05, **P < 0.01.
DALDA Enhances Stat3 Phosphorylation in Vitro
To gain more insight into the role of Stat3 in MOR-mediated protective effect, we used an in vitro cell system. CMT-93 cells were exposed to DALDA (10 μmol/L) for 1 hour after which time Stat3 phosphorylation was measured by Western blot analysis. Densitometric analysis showed that pStat3 levels increased by approximately threefold in DALDA-treated cells compared with unstimulated cells (P < 0.05 compared with an assumption of a zerofold change; Figure 5A). In addition, immunofluorescence assays showed an increased level of pStat3 in DALDA-treated cells compared with unstimulated cells (Figure 5B). We determined whether the general opioid antagonist naloxone could inhibit DALDA-induced pStat3 activation, and indeed we see that 30 minutes of pretreatment with naloxone (Figure 6C) blocks Stat3 phosphorylation. We then investigated the effect of DALDA on cytoprotective genes regulated in part by Stat3 with the use of real-time PCR. Expression of Myc, cyclooxygenase 2 (Ptgs2), cyclin D (Ccnd1), and Reg3b mRNA were significantly induced in DALDA-stimulated CMT-93 cells (Figure 5C). These studies indicate that colonocytes respond to a MOR agonist by activating Stat3 signaling and downstream cytoprotective mediators.
Figure 5.
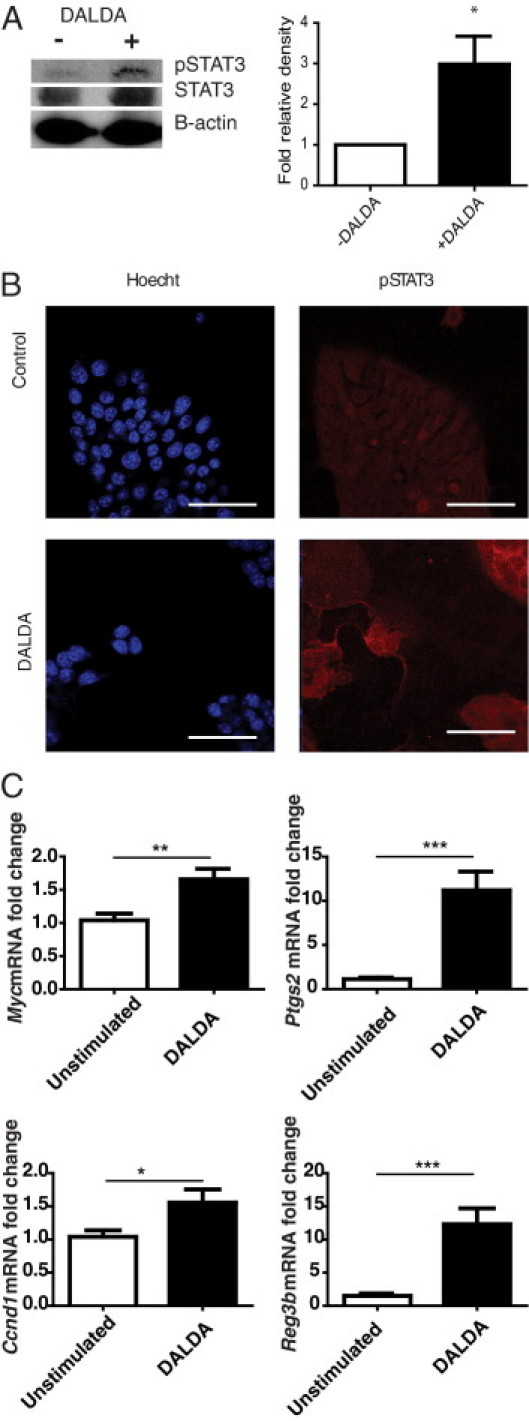
DALDA induces Stat3 phosphorylation in CMT-93 cells. A–C: CMT-93 cells were treated for 1 hour with 10 μmol/L DALDA or vehicle. A: Western blot analysis and densitometry compare pStat3 with Stat3 within the same sample. Representative of eight independent experiments. B: pStat3 immunofluorescence staining, representative of triplicate samples, from two independent experiments. C: Activation of Stat3-dependent genes (Ptgs2, Myc, Ccnd1, Reg3b). N ≥ 4 for both groups, in two independent experiments. *P < 0.05, **P < 0.01, and ***P < 0.001.
Figure 6.
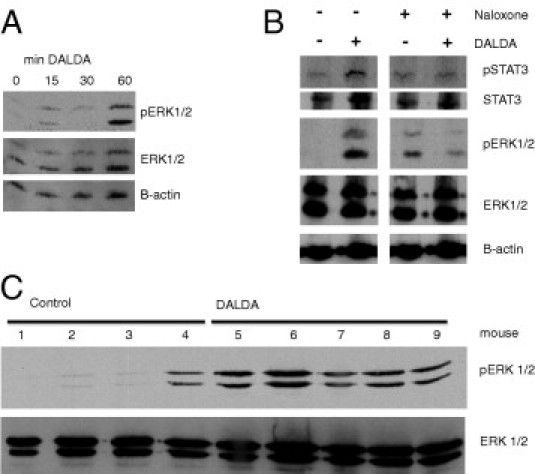
DALDA induces ERK1/2 phosphorylation by an opioid-dependent pathway. A: Western blot analysis for pERK1/2 in CMT-93 cells stimulated with 10 μmol/L DALDA for 0 to 60 minutes. Representative of three individual experiments. B: Western blot analysis for pStat3 and pERK1/2 in CMT-93 cells stimulated with DALDA for 1 hour with or without 30-minute pretreatment with 1 mmol/L of the opioid antagonist naloxone. Representative of three individual experiments. C: DALDA induces colonic ERK1/2 phosphorylation in vivo. Western blot analysis for pERK1/2 from distal colonic protein extracts from individual control mice or mice injected with 50 μg/kg DALDA daily s.c. for 7 days.
ERK phosphorylation is a well-established downstream consequence of Gi-coupled G protein-coupled receptor activation and a classic effector of MOR activation.36 We detected ERK1/2 phosphorylation in the distal colonic tissue of DALDA-treated mice and in DALDA-stimulated CMT-93 cells (Figure 6, A and C). To confirm the specificity of DALDA-induced ERK1/2 phosphorylation, CMT-93 cells were pretreated (30 minutes) with the MOR competitive antagonist naloxone (10 mmol/L) and then stimulated with DALDA. Interestingly, levels of pERK1/2 are slightly augmented in naloxone-treated cells, a phenomenon previously reported.37,38 Importantly, naloxone attenuated DALDA-induced pERK1/2 in CMT-93 cells (Figure 6B).
DALDA Enhances Colonocyte Migration in a Stat3-Dependent Manner
Because DALDA promoted proliferation in vivo and activated protective/proliferative signaling in vitro, we next explored the effect of MOR activation on cellular restitution/migration with the use of an in vitro wound healing/scratch-assay model. Immediately after wounding, cells were incubated with DALDA (10 μmol/L), and 8 hours later the distance migrated into the scratch was measured with ImageJ software. Migration increased by 67% (P < 0.001) in DALDA-treated compared with untreated cells (Figure 7, A and B). Furthermore, reproductive cellular proliferation assays showed that DALDA enhanced CMT-93 proliferation by 45% during the same time period (Figure 7C; P < 0.05). Because DALDA enhanced Stat3 phosphorylation in vivo and in vitro, we determined the functional effect of this signaling event on CMT-93 migration with the use of a shRNA knockdown approach. CMT-93 cells were infected with a lentiviral vector containing shStat3 and a puromycin-resistance gene, allowing for the selection and maintenance of a stable knockdown line. Stat3 expression was efficiently inhibited in shStat3-infected cells compared with scramble controls (Figure 7D). Moreover, DALDA-induced pStat3 was strongly blocked in shStat3-infected cells compared with the short hairpin control (Figure 7E). Most importantly, DALDA-induced cellular migration was completely abrogated in shStat3-infected cells compared with control-infected cells (Figure 7F). These findings show that DALDA drives colonocyte migration in a Stat3-dependent manner and, combined with our in vivo studies, show that MOR activation enhances intestinal wound healing by both enhancing restitution/migration and cellular proliferation, in a manner involving Stat3 signaling.
Figure 7.
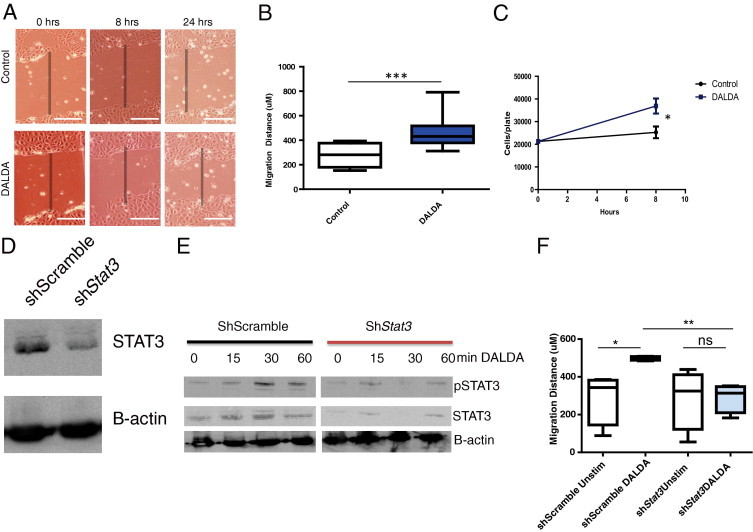
DALDA enhances colonocyte migration in a Stat3-dependent manner. A: CMT-93 monolayers were scratched and treated with 10 μmol/L DALDA or vehicle. Migration was observed over a 24-hour period. A: Representative photomicrographs of cell migration. Shaded bar shows initial wound area. Scale bar = 100 μm. B: Quantification of migration distance after 8 hours. Mean results of three3 independent experiments; N = 4 for each experiment. C: Reproductive cellular proliferation assays. CMT-93 cells were treated with 10 μmol/L DALDA or vehicle for 8 hours, and direct cell quantification was performed. Mean results of two independent experiments; N = 4 for each experiment. D–F: Cells were infected with a shStat3 lentivirus to reduce Stat3 expression or control scrambled shRNA (shScramble). D: Western blot analysis representative of three independent experiments show Stat3 knockdown in shStat3 cells. E: Western blot analyses show reduced DALDA-induced Stat3 phosphorylation in shStat3-infected CMT-93 cells. Representative of two individual experiments. F: Quantification of migration over an 8-hour period. Representative of two separate experiments; N = 4 per experiment. ns, not significant. *P < 0.05, **P < 0.01.
Discussion
As a result of their ability to promote intestinal repair, increasing clinical interest has focused on the use of trophic factors to treat diseases associated with chronic epithelial injury such as IBD.2,39,40 Here, we have demonstrated that peripheral activation of the mu opioid pathway protects against DSS-induced intestinal injury and enhances wound healing. We have shown that the MOR-specific agonist DALDA activates proliferative signaling in the intestine, a response associated with enhanced colonocyte-derived Stat3 activation and induction of cytoprotective genes. In contrast, the MOR-specific antagonist cyprodime worsens DSS-induced intestinal injury, suggesting that this opioid pathway plays an essential role in the intestinal wound-healing response. Interestingly, previous reports have shown that low-dose therapy of the nonspecific opioid antagonist naltrexone protects against both DSS-induced injury in mice and ameliorates Crohn's disease in a small, open-label patient study.41,42 In these studies, naltrexone was deliberately administered at levels below the dose needed to pharmacologically block receptor function. Because the MOR-specific antagonist cyprodime exacerbates DSS-induced colitis, it is probable that low-dose naltrexone therapy acts to potentiate endogenous opiate signaling.41–43 However, such a mechanism remains controversial and has never been truly established.
Our work provides significant new insights into the mechanisms behind DALDA-mediated amelioration of intestinal injury and inflammation. Chronic opiate administration is known to be immunosuppressive clinically and specifically inhibits T-cell, B-cell, and natural killer-cell function.11,44,45 Moreover, DALDA has been shown to be protective in two T-cell driven models of murine colitis: the TNBS and adoptive transfer of CD45RBhiCD4+ T-cell models.19 This implies that MOR-mediated immunosuppression may provide a potential mechanism for MOR-mediated protection from intestinal injury. However, DSS-induced acute colitis has been shown to be independent of T cells, B cells, and natural killer cells21; hence, DALDA cannot be solely operating through inhibition of adaptive immune responses. Although we observed decreased activation of NF-κB (EGFP expression) and inflammatory gene expression in DALDA-treated mice, these observations are probably a secondary effect of the drug because genetic ablation of IEC-derived NF-κB signaling46–50 and blockade of the pathway29,35,51 are both known to exacerbate injury response in the intestine. In addition, in vitro NF-κB transcriptional activity in IECs was not inhibited by DALDA treatment (data not shown).
We show that DALDA operates in part through the enhancement of wound healing and cellular proliferation in response to DSS-induced epithelial damage. Indeed, an early effect of DALDA therapy is improvement of the crypt damage score and enhanced epithelial proliferative responses (Ki-67 staining). These observations suggest that DALDA acts by enhancing both epithelial proliferation and damage repair responses. This concurs with our in vitro data showing that DALDA enhanced IEC proliferation/migration after wounding. We postulate that the enhanced wound healing afforded by DALDA limits luminal antigenic activation of lamina propria immune cells (NF-κB activation, cytokine expression) and inflammation through epithelial restitution/migration and enhanced colonocyte proliferation.
The novel finding that DALDA induced IEC Stat3 phosphorylation both in vivo and in vitro is particularly relevant to the wound-healing response. IEC-derived Stat3 signaling has been shown to confer protective responses against DSS-induced intestinal injury through the promotion of wound-healing responses10,52; furthermore, Stat3 signaling is a known driver of enterocyte proliferation.52,53 IHC staining showed that colonocytes are the predominant cells exhibiting pStat3 in DALDA-treated, DSS-exposed mice. DALDA enhanced expression of cytoprotective genes (Reg3b, Ccnd1, Cox2, myc) that are lost in Stat3ΔIEC mice known to be sensitive to DSS injury.10 The up-regulation of these genes only in the presence of DALDA suggests that, although DSS alone can up-regulate colonocyte Stat3 activation to an extent, only the presence of DALDA and subsequent exogenous MOR signaling is to generate protective responses, through higher elevated pStat3 levels and increased cytoprotective gene expression and proliferation (Ki-67 staining).
Our gene expression observations could also be recapitulated in vitro with CMT-93 colonocytes, whereby we also saw that DALDA increases pStat3 levels in CMT-93 cells and enhances cellular migration in response to a wound, as well as enhances cellular proliferation. Knockdown of Stat3 showed the importance of Stat3 signaling in DALDA-mediated enhanced migration, because this response was abrogated in shStat3-infected CMT-93 cells compared with control.
These observations strongly suggest that DALDA promotes protective function in the intestine through a mechanism involving Stat3 activation. Although IECs are the predominant cells exhibiting pStat3 in response to DALDA treatment, it is unclear whether these cells are the main target of the opioid. Tissue-specific MOR gene deletion will be necessary to evaluate the cellular compartment implicated in DALDA-induced protective response. In addition, although Stat3ΔIEC mice are available, previous work has shown that these mice are extremely sensitive to DSS colitis and have severe defects in epithelial restitution,10 making exploring the role of DALDA on such a sensitive background untenable.
ERK1/2 is a canonical downstream effector of Gi-coupled signaling and represents a classic read-out of opioid signaling. Interestingly, although we observed persistent in vivo activation of ERK1/2 in DALDA-injected mice, Stat3 phosphorylation was observed only in the context of DALDA administration during an injury response. This suggests that MOR-mediated Stat3-driven proliferation in vivo is tightly regulated, and activation depends on the injury status of the epithelium. A previous study has identified IL-22 as a key cytokine involved in DSS-induced pStat3 and injury response.10 Interestingly, we observed enhanced colonic IL-22 mRNA expression in DALDA-treated, DSS-exposed mice (J.R.G. and C.J., unpublished observation). Further studies will be required before establishing the functional role of IL-22 in DALDA-mediated cytoprotective effect in the intestine.
Our work highlights a potential new therapeutic class of compounds for the treatment of diseases involving intestinal barrier damage, including ischemic injury, radiation-induced damage, and IBD. One could envision the use of opioids as an adjunct therapy in IBD by helping to induce or maintain remission through promoting re-epithelialization of the intestine. In particular, opioids could be used as an adjunct in helping to induce remission in IBD by lowering the amount of prednisone needed or as a mainline therapy in steroid-refractory patients. Use of opioids for IBD therapy needs to be pursued cautiously, however, because our work shows that opioids probably act through increased colonocyte proliferation, and patients with IBD already have an increased risk of malignant transformation. Consequently, MOR-targeted therapy would probably be best suited toward short-term use for remission induction, similar to prednisone. Other, non-IBD applications could include enhancing recovery from radiotherapy-induced injury and use as a gut-saving agent in cases of ischemic injury.
In summary, this report establishes mu-specific opioids as promoters of colonocyte migration/restitution and proliferation in the intestine involving the activation of Stat3-dependent pathways. Because opioids are already available on the market and have well-known safety profiles, this class of compounds could represent a novel therapeutic approach to gastrointestinal therapy, once clinical trials validate their beneficial effect in patients.
Acknowledgments
We thank our colleagues from University of North Carolina at Chapel Hill: Joseph Galenko for his assistance with statistical analysis, Taku Kobayashi for his help generating the knockdown lines, Greg Gipson for technical assistance with mouse experiments, Janelle C. Arthur and Javier Rivera-Guzman for critical reading of the manuscript, Robert Bagnell and Robert Currin for their assistance with the fluorescence microscopy experiments, and Arlin Rogers for his blinded confirmation of our histologic assessment of intestinal injury.
Footnotes
Supported by National Institutes of Health grants DK047700 and DK073338, T32 GM008719, and 5T32-GM007040-34. The lentiviral constructs generated by the UNC Lenti-shRNA core were donated by Dr. Scott Plevy of UNC-CH.
Supplemental material for this article can be found at http://ajp.amjapthol.org or at doi:10.1016/j.path.2011.04.032.
Supplementary data
DALDA prevents DSS-induced EGFP expression in DSS-exposed NF-κBEGFP mice. NF-κBEGFP mice were administered 3% DSS for 7 days, concurrent with DALDA (50 μg/kg), cyprodime (10 mg/kg), or vehicle treatment. Colonic EGFP expression was assessed in whole-tissue mounts with the use of confocal microscopy. Propidium iodide (red) was used to visualize nuclei. Four mice per condition were visualized macroscopically (data not shown), and two per condition were visualized further by confocal microscopy. Original magnification: ×25 (A); ×60 (B) of the same tissues as shown in A. C: LPS-induced NF-κB transcriptional activity is not blocked by DALDA. CMT-93 cells were infected with Ad5κB-Luc for 12 hours and then stimulated with 10 μg/mL LPS in the presence or absence of 10 μM DALDA for 12 additional hours.
References
- 1.Sartor R.B. Microbial influences in inflammatory bowel diseases. Gastroenterology. 2008;134:577–594. doi: 10.1053/j.gastro.2007.11.059. [DOI] [PubMed] [Google Scholar]
- 2.Williams K.L., Fuller C.R., Dieleman L.A., DaCosta C.M., Haldeman K.M., Sartor R.B., Lund P.K. Enhanced survival and mucosal repair after dextran sodium sulfate-induced colitis in transgenic mice that overexpress growth hormone. Gastroenterology. 2001;120:925–937. doi: 10.1053/gast.2001.22470. [DOI] [PubMed] [Google Scholar]
- 3.Morteau O., Morham S.G., Sellon R., Dieleman L.A., Langenbach R., Smithies O., Sartor R.B. Impaired mucosal defense to acute colonic injury in mice lacking cyclooxygenase-1 or cyclooxygenase-2. J Clin Invest. 2000;105:469–478. doi: 10.1172/JCI6899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Packey C.D., Ciorba M.A. Microbial influences on the small intestinal response to radiation injury. Curr Opin Gastroenterol. 2009;26:88–94. doi: 10.1097/MOG.0b013e3283361927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kinross J., Warren O., Basson S., Holmes E., Silk D., Darzi A., Nicholson J.K. Intestinal ischemia/reperfusion injury: defining the role of the gut microbiome. Biomark Med. 2009;3:175–192. doi: 10.2217/bmm.09.11. [DOI] [PubMed] [Google Scholar]
- 6.Karrasch T., Jobin C. Wound healing responses at the gastrointestinal epithelium: a close look at novel regulatory factors and investigative approaches. Z Gastroenterol. 2009;47:1221–1229. doi: 10.1055/s-0028-1109766. [DOI] [PubMed] [Google Scholar]
- 7.Sturm A., Dignass A.U. Epithelial restitution and wound healing in inflammatory bowel disease. World J Gastroenterol. 2008;14:348–353. doi: 10.3748/wjg.14.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ciacci C., Lind S.E., Podolsky D.K. Transforming growth factor beta regulation of migration in wounded rat intestinal epithelial monolayers. Gastroenterology. 1993;105:93–101. doi: 10.1016/0016-5085(93)90014-4. [DOI] [PubMed] [Google Scholar]
- 9.Dignass A.U., Podolsky D.K. Cytokine modulation of intestinal epithelial cell restitution: central role of transforming growth factor beta. Gastroenterology. 1993;105:1323–1332. doi: 10.1016/0016-5085(93)90136-z. [DOI] [PubMed] [Google Scholar]
- 10.Pickert G., Neufert C., Leppkes M., Zheng Y., Wittkopf N., Warntjen M., Lehr H.-A., Hirth S., Weigmann B., Wirtz S., Ouyang W., Neurath M.F., Becker C. Stat3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J Exp Med. 2009;206:1465–1472. doi: 10.1084/jem.20082683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Borner C., Warnick B., Smida M., Hartig R., Lindquist J.A., Schraven B., Hollt V., Kraus J. Mechanisms of opioid-mediated inhibition of human T cell receptor signaling. J Immunol. 2009;183:882–889. doi: 10.4049/jimmunol.0802763. [DOI] [PubMed] [Google Scholar]
- 12.Mani A.R., Moore K.P. New insights into the role of endogenous opioids in the pathogenesis of gastrointestinal and liver disease. Gut. 2009;58:893–895. doi: 10.1136/gut.2007.141648. [DOI] [PubMed] [Google Scholar]
- 13.Gainetdinov R.R., Premont R.T., Bohn L.M., Lefkowitz R.J., Caron M.G. Desensitization of G protein-coupled receptors and neuronal functions. Annu Rev Neurosci. 2004;27:107–144. doi: 10.1146/annurev.neuro.27.070203.144206. [DOI] [PubMed] [Google Scholar]
- 14.Kieffer B.L., Gavériaux-Ruff C. Exploring the opioid system by gene knockout. Prog Neurobiol. 2002;66:285–306. doi: 10.1016/s0301-0082(02)00008-4. [DOI] [PubMed] [Google Scholar]
- 15.Law P.-Y., Wong Y.H., Loh H.H. Molecular mechanisms and regulation of opioid receptor signaling. Annu Rev Pharmacol Toxicol. 2000;40:389–430. doi: 10.1146/annurev.pharmtox.40.1.389. [DOI] [PubMed] [Google Scholar]
- 16.Gross E.R., Hsu A.K., Gross G.J. The JAK/STAT pathway is essential for opioid-induced cardioprotection: JAK2 as a mediator of Stat3, Akt, and GSK-3beta. Am J Physiol Heart Circ Physiol. 2006;291:H827–H834. doi: 10.1152/ajpheart.00003.2006. [DOI] [PubMed] [Google Scholar]
- 17.Li R., Wong G.T.C., Wong T.M., Zhang Y., Xia Z., Irwin M.G. Intrathecal morphine preconditioning induces cardioprotection via activation of delta, kappa, and mu opioid receptors in rats. Anesth Analg. 2009;108:23–29. doi: 10.1213/ane.0b013e3181884ba6. [DOI] [PubMed] [Google Scholar]
- 18.Schiller P.W., Dung N.T.M., Chung N.N., Lemieux C. Dermorphin analogs carrying an increased positive net charge in their “message” domain display extremely high mu-opioid receptor selectivity. J Med Chem. 1989;32:698–703. doi: 10.1021/jm00123a035. [DOI] [PubMed] [Google Scholar]
- 19.Philippe D., Dubuquoy L., Groux H., Brun V., Chuoi-Mariot M.T., Gaveriaux-Ruff C., Colombel J.F., Kieffer B.L., Desreumaux P. Anti-inflammatory properties of the mu opioid receptor support its use in the treatment of colon inflammation. J Clin Invest. 2003;111:1329–1338. doi: 10.1172/JCI16750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Philippe D., Chakass D., Thuru X., Zerbib P., Tsicopoulos A., Geboes K., Bulois P., Breisse M., Vorng H., Gay J., Colombel J.F., Desreumaux P., Chamaillard M. Mu opioid receptor expression is increased in inflammatory bowel diseases: implications for homeostatic intestinal inflammation. Gut. 2006;55:815–823. doi: 10.1136/gut.2005.080887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wirtz S., Neufert C., Weigmann B., Neurath M.F. Chemically induced mouse models of intestinal inflammation. Nat Protoc. 2007;2:541–546. doi: 10.1038/nprot.2007.41. [DOI] [PubMed] [Google Scholar]
- 22.Menéndez L., Lastra A., Meana Á A., Hidalgo A., Baamonde A. Analgesic effects of loperamide in bone cancer pain in mice. Pharmacol Biochem Behav. 2005;81:114–121. doi: 10.1016/j.pbb.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 23.Murthy S.N., Cooper H.S., Shim H., Shah R.S., Ibrahim S.A., Sedergran D.J. Treatment of dextran sulfate sodium-induced murine colitis by intracolonic cyclosporin. Dig Dis Sci. 1993;38:1722–1734. doi: 10.1007/BF01303184. [DOI] [PubMed] [Google Scholar]
- 24.Uronis J.M., Muhlbauer M., Herfarth H.H., Rubinas T.C., Jones G.S., Jobin C. Modulation of the intestinal microbiota alters colitis-associated colorectal cancer susceptibility. PLoS ONE. 2009;4:e6026. doi: 10.1371/journal.pone.0006026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cooper H.S., Murthy S.N., Shah R.S., Sedergran D.J. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab Invest. 1993;69:238–249. [PubMed] [Google Scholar]
- 26.Dieleman L.A., Palmen M.J., Akol H., Bloemena E., Pena A.S., Meuwissen S.G., Van Rees E.P. Chronic experimental colitis induced by dextran sulphate sodium (DSS) is characterized by Th1 and Th2 cytokines. Clin Exp Immunol. 1998;114:385–391. doi: 10.1046/j.1365-2249.1998.00728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haller D., Russo M.P., Sartor R.B., Jobin C. IKK-beta and phosphatidylinositol 3-kinase/Akt participate in non-pathogenic Gram-negative enteric bacteria-induced RelA phosphorylation and NF-kappa B activation in both primary and intestinal epithelial cell lines. J Biol Chem. 2002;277:38168–38178. doi: 10.1074/jbc.M205737200. [DOI] [PubMed] [Google Scholar]
- 28.Haller D., Holt L., Kim S.C., Schwabe R.F., Sartor R.B., Jobin C. Transforming growth factor-{beta}1 inhibits non-pathogenic Gram negative bacteria-induced NF-{kappa}B recruitment to the interleukin-6 gene promoter in intestinal epithelial cells through modulation of histone acetylation. J Biol Chem. 2003;278:23851–23860. doi: 10.1074/jbc.M300075200. [DOI] [PubMed] [Google Scholar]
- 29.Joo Y., Karrasch T., Muhlbauer M., Allard B., Narula A., Herfarth H., Jobin C. Tomato lycopene extract prevents lipopolysaccharide-induced NF-kB signaling but worsens dextran sulfate sodium-induced colitis in NF-kBEGFP mice. PLoS ONE. 2009;4:e4562. doi: 10.1371/journal.pone.0004562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Snider A.J., Kawamori T., Bradshaw S.G., Orr K.A., Gilkeson G.S., Hannun Y.A., Obeid L.M. A role for sphingosine kinase 1 in dextran sulfate sodium-induced colitis. FASEB J. 2009;23:143–152. doi: 10.1096/fj.08-118109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Karrasch T., Steinbrecher K.A., Allard B., Baldwin A.S., Jobin C. Wound-induced p38MAPK-dependent histone H3 phosphorylation correlates with increased COX-2 expression in enterocytes. J Cell Physiol. 2006;207:809–815. doi: 10.1002/jcp.20626. [DOI] [PubMed] [Google Scholar]
- 32.Cook J.A., Mitchell J.B. Viability measurements in mammalian cell systems. Anal Biochem. 1989;179:1–7. doi: 10.1016/0003-2697(89)90191-7. [DOI] [PubMed] [Google Scholar]
- 33.Kovacic J.C., Gupta R., Lee A.C., Ma M., Fang F., Tolbert C.N., Walts A.D., Beltran L.E., San H., Chen G., St. Hilaire C., Boehm M. Stat3-dependent acute Rantes production in vascular smooth muscle cells modulates inflammation following arterial injury in mice. J Clin Invest. 2010;120:303–314. doi: 10.1172/JCI40364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Karrasch T., Kim J.S., Muhlbauer M., Magness S.T., Jobin C. Gnotobiotic IL-10-/-;NF-kappa B(EGFP) mice reveal the critical role of TLR/NF-kappa B signaling in commensal bacteria-induced colitis. J Immunol. 2007;178:6522–6532. doi: 10.4049/jimmunol.178.10.6522. [DOI] [PubMed] [Google Scholar]
- 35.Karrasch T., Kim J.-S., Jang B.I., Jobin C. The flavonoid luteolin worsens chemical-induced colitis in NF-{kappa}B transgenic mice through blockade of NF-{kappa}B-dependent protective molecules. PLoS ONE. 2007;2:e596. doi: 10.1371/journal.pone.0000596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zheng H., Loh H.H., Law P.-Y. Beta-arrestin-dependent mu-opioid receptor-activated extracellular signal-regulated kinases (ERKs) translocate to nucleus in contrast to G protein-dependent ERK activation. Mol Pharmacol. 2008;73:178–190. doi: 10.1124/mol.107.039842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Almela P., Milanés M.V., Laorden M.L. Activation of the ERK signalling pathway contributes to the adaptive changes in rat hearts during naloxone-induced morphine withdrawal. Br J Pharmacol. 2007;151:787–797. doi: 10.1038/sj.bjp.0707301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jun-Li C., Jian-Hua H., Hai-Lei D., Yin-Ming Z. Activation of the spinal ERK signaling pathway contributes naloxone-precipitated withdrawal in morphine-dependent rats. Pain. 2005;118:336–349. doi: 10.1016/j.pain.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 39.Granata R., Trovato L., Gallo M.P., Destefanis S., Settanni F., Scarlatti F., Brero A., Ramella R., Volante M., Isgaard J., Levi R., Papotti M., Alloatti G., Ghigo E. Growth hormone-releasing hormone promotes survival of cardiac myocytes in vitro and protects against ischaemia-reperfusion injury in rat heart. Cardiovasc Res. 2009;83:303–312. doi: 10.1093/cvr/cvp090. [DOI] [PubMed] [Google Scholar]
- 40.Slonim A.E., Bulone L., Damore M.B., Goldberg T., Wingertzahn M.A., McKinley M.J. A preliminary study of growth hormone therapy for Crohn's disease. N Engl J Med. 2000;342:1633–1637. doi: 10.1056/NEJM200006013422203. [DOI] [PubMed] [Google Scholar]
- 41.Matters G.L., Harms J.F., McGovern C., Fitzpatrick L., Parikh A., Nilo N., Smith J.P. The opioid antagonist naltrexone improves murine inflammatory bowel disease. J Immunotoxicol. 2008;5:179–187. doi: 10.1080/15476910802131469. [DOI] [PubMed] [Google Scholar]
- 42.Smith J.P., Stock H., Bingaman S., Mauger D., Rogosnitzky M., Zagon I.S. Low-dose naltrexone therapy improves active Crohn's disease. Am J Gastroenterol. 2007;102:820–828. doi: 10.1111/j.1572-0241.2007.01045.x. [DOI] [PubMed] [Google Scholar]
- 43.Wang H.Y., Friedman E., Olmstead M.C., Burns L.H. Ultra-low-dose naloxone suppresses opioid tolerance, dependence and associated changes in mu opioid receptor-G protein coupling and Gβγ signaling. Neuroscience. 2005;135:247–261. doi: 10.1016/j.neuroscience.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 44.Ma J., Wang J., Wan J., Charboneau R., Chang Y., Barke R.A., Roy S. Morphine disrupts interleukin-23 (IL-23)/IL-17-mediated pulmonary mucosal host defense against Streptococcus pneumoniae infection. Infect Immun. 2010;78:830–837. doi: 10.1128/IAI.00914-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wei G., Moss J., Yuan C.-S. Opioid-induced immunosuppression: is it centrally mediated or peripherally mediated? Biochem Pharmacol. 2003;65:1761–1766. doi: 10.1016/s0006-2952(03)00085-6. [DOI] [PubMed] [Google Scholar]
- 46.Chen L.W., Egan L., Li Z.W., Greten F.R., Kagnoff M.F., Karin M. The two faces of IKK and NF-kappaB inhibition: prevention of systemic inflammation but increased local injury following intestinal ischemia-reperfusion. Nat Med. 2003;9:575–581. doi: 10.1038/nm849. [DOI] [PubMed] [Google Scholar]
- 47.Egan L.J., Eckmann L., Greten F.R., Chae S., Li Z.W., Myhre G.M., Robine S., Karin M., Kagnoff M.F. IkappaB-kinasebeta-dependent NF-kappaB activation provides radioprotection to the intestinal epithelium. Proc Natl Acad Sci U S A. 2004;101:2452–2457. doi: 10.1073/pnas.0306734101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Greten F.R., Eckmann L., Greten T.F., Park J.M., Li Z.W., Egan L.J., Kagnoff M.F., Karin M. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118:285–296. doi: 10.1016/j.cell.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 49.Nenci A., Becker C., Wullaert A., Gareus R., van Loo G., Danese S., Huth M., Nikolaev A., Neufert C., Madison B., Gumucio D., Neurath M.F., Pasparakis M. Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature. 2007;446:557–561. doi: 10.1038/nature05698. [DOI] [PubMed] [Google Scholar]
- 50.Steinbrecher K.A., Harmel-Laws E., Sitcheran R., Baldwin A.S. Loss of epithelial RelA results in deregulated intestinal proliferative/apoptotic homeostasis and susceptibility to inflammation. J Immunol. 2008;180:2588–2599. doi: 10.4049/jimmunol.180.4.2588. [DOI] [PubMed] [Google Scholar]
- 51.Joo Sung Kim C.J. The flavonoid luteolin prevents lipopolysaccharide-induced NF-{kappa};B signalling and gene expression by blocking I{kappa}B kinase activity in intestinal epithelial cells and bone-marrow derived dendritic cells. Immunology. 2005;115:375–387. doi: 10.1111/j.1365-2567.2005.02156.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Grivennikov S., Karin E., Terzic J., Mucida D., Yu G.Y., Vallabhapurapu S., Scheller J., Rose-John S., Cheroutre H., Eckmann L., Karin M. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009;15:103–113. doi: 10.1016/j.ccr.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yu H., Pardoll D., Jove R. STATs in cancer inflammation and immunity: a leading role for Stat3. Nat Rev Cancer. 2009;9:798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
DALDA prevents DSS-induced EGFP expression in DSS-exposed NF-κBEGFP mice. NF-κBEGFP mice were administered 3% DSS for 7 days, concurrent with DALDA (50 μg/kg), cyprodime (10 mg/kg), or vehicle treatment. Colonic EGFP expression was assessed in whole-tissue mounts with the use of confocal microscopy. Propidium iodide (red) was used to visualize nuclei. Four mice per condition were visualized macroscopically (data not shown), and two per condition were visualized further by confocal microscopy. Original magnification: ×25 (A); ×60 (B) of the same tissues as shown in A. C: LPS-induced NF-κB transcriptional activity is not blocked by DALDA. CMT-93 cells were infected with Ad5κB-Luc for 12 hours and then stimulated with 10 μg/mL LPS in the presence or absence of 10 μM DALDA for 12 additional hours.