

Abstract
Deep infiltrating endometriosis (DIE) is a particular clinical and histological entity of endometriosis responsible for chronic pelvic pain and infertility. Here we characterize the proliferative phenotype of DIE cells, to explore the cellular and molecular mechanisms that could explain their aggressive potential. In addition, the inhibition of mTOR/AKT pathway was tested, as a potential treatment of DIE. Included were 22 patients with DIE and 12 control patients without endometriosis. Epithelial and stromal cells were extracted from biopsies of eutopic endometrium and deep infiltrating endometriotic nodules from patients with DIE. Cell proliferation was determined by thymidine incorporation. Oxidative stress was assayed by spectrofluorometry. The ERK and mTOR/AKT pathways were analyzed in vitro by Western blot and for AKT in vivo in a mouse model of DIE. The proliferation rate of eutopic endometrial cells and of deep infiltrating endometriotic cells from DIE patients was higher than that of endometrial cells from controls. The hyperproliferative phenotype of endometriotic cells was associated with an increase in endogenous oxidative stress, and with activation of the ERK and mTOR/AKT pathways. mTOR/AKT inhibition by temsirolimus decreased endometriotic cell proliferation both in vitro and in vivo in a mouse model of DIE. Blocking the mTOR/AKT pathway offers new prospects for the treatment of DIE.
Endometriosis, a common disease that affects approximately 6% to 10% of women of childbearing age,1 is characterized by the presence of endometrial tissue outside the uterine cavity. Little is known about the factors that could explain the clinical and histological heterogeneity of endometriosis and, particularly, the development of deep infiltrating endometriosis (DIE). DIE is an aggressive disease responsible for chronic pelvic pain,2 in its intensity inducing severe disability3 and infertility.4 Surgical resection of DIE lesions is the main curative treatment available,5 but surgery often needs to be extensive6 and is associated with significant morbidity.7 Effectiveness of medical treatments, currently based on hormonal therapy that blocks ovarian function, is only transient. New drugs have recently been considered for the treatment of endometriosis; the use of antiaromatases,8,9 antioxidant molecules,10 or more recently anti-metabolites such as 5-FU11 has been suggested. The effectiveness of those treatments in vitro and in animal models relies on the modulation of cellular mechanisms responsible for invasion, unrestrained growth, neoangiogenesis, and distant spreading of endometriotic cells.12 In line with those observations, it has recently been shown that ovarian endometriotic cells display a high endogenous oxidative stress, with profound alteration of the reactive oxygen species (ROS) detoxification pathways associated with increased cellular proliferation and activation of the MAP kinase ERK 1/2 pathway.10 Activation of the ERK pathway has been found in eutopic endometrium and in ovarian endometrioma of a large number of women with ovarian endometriosis.13,14
Little is known about the role of other potentially proproliferative pathways in endometriosis. The PI3K/mTOR/AKT pathway has been found to be activated in ovarian endometriosis15–17 and has been implicated in the pathogenesis of ovarian cancer.17 To date, however, no study has explored this pathway in DIE, and the involvement of this pathway has never been explored in vivo.
In the present study, we explored the proliferative phenotype of deep infiltrating endometriotic cells and highlighted the cellular and molecular mechanisms that could explain their aggressive potential. Based on our in vitro results showing a constitutive activation of AKT/mTOR pathway in deep infiltrating endometriotic cells, we evaluated the therapeutic potential of an mTOR inhibitor in vitro and in vivo in DIE.
Materials and Methods
Sample Collection
Biopsies of eutopic endometrium and deep infiltrating endometriotic nodules were obtained from 22 patients undergoing surgical treatment for DIE with rectal involvement defined by muscularis involvement.18 Low rectal endometriosis was defined preoperatively, based on the following clinical and endoscopic ultrasonographic criteria: rectal invasion of the infraperitoneal rectum (located within 8 cm of the dentate line, reachable on rectal examination) and full-thickness invasion of the muscular layer (>15 mm on rectal endoscopic ultrasonography).7 DIE was confirmed in all cases by a pathologist experienced in endometriosis (S.A.). All of the patients had been treated by luteinizing-hormone releasing hormone (LHRH) agonists for a minimum of 1 month before surgery. Control endometrial specimens were obtained from 12 patients without macroscopic endometriosis undergoing laparoscopy for other reasons (tubal obstruction infertility, nonendometriotic ovarian cyst, myoma). Ethics approval for the present study was obtained from the ethics committee at Cochin Hospital (no. 05-2006 “génomique et protéomique de l'endométriose”). Written informed consent was obtained from each patient and control subject.
Specimens were collected under sterile conditions and immediately transported to the laboratory in Dulbecco's modified Eagle's medium (Gibco Invitrogen, Cergy Pontoise, France) with 10% fetal calf serum. The addition of 10% fetal calf serum affected neither the ERK nor the ARK pathways (Velarde et al19 and unpublished data). No steroid hormones were added in cell culture. Estrogen (e2) and progesterone (p4) were undetectable in cell culture supernatants as determined by an immunodiagnostic system (Advia Centaur XP immunoassay system; Siemens Healthcare Diagnostics, Saint-Denis, France). The absence of steroid hormones in culture supernatants reproduces clinical conditions in patients who receive treatment with LHRH agonists. The time that elapsed between the biopsy and the procedure of cell isolation never exceeded 1 hour. The same culture medium was used throughout the study.
Cell Isolation and Culture
Primary endometrial and deep endometriotic cell cultures were prepared from biopsies using an adaptation of the method described by Velarde et al.19 Biopsy specimens were rinsed and minced into small pieces, then digested with dispase 5‰ and collagenase (2 mg/mL, Gibco Invitrogen, Cergy Pontoise, France) for 1 hour at 37°C and separated using serial filtration. Red blood cells were removed by hypotonic lysis (0.15 mol/L NH4Cl, 1 mmol/L KHCO3, 0.1 mmol/L Na2EDTA). Debris was removed by filtration through 100-μm aperture sieves. Epithelial cells were retained on 40-μm aperture sieves, with stromal cells remaining in the filtrate. Both types of cell were plated onto Primaria flasks (Becton Dickinson Labware, Le Pont de Claix, France) and were cultured in Dulbecco's modified Eagle's medium (Gibco Invitrogen) with 10% fetal calf serum.14 For each patient with DIE, four cell populations were obtained: eutopic endometrial stromal cells (Es), eutopic endometrial epithelial cells (Ee), deep infiltrating endometriotic stromal cells (Ds), and deep infiltrating endometriotic epithelial cells (De). For each control, we used two cell populations: control endometrial stromal cells (Cs) and control endometrial epithelial cells (Ce).
The purification of stromal and epithelial cells was assessed by staining with 1:100 fluorescein isothiocyanate-labeled anti-cytokeratin and Cy3-labeled anti-vimentin antibodies (Sigma-Aldrich, St. Louis, MO). Fluorescence was analyzed using an Olympus fluorescent microscope (Hamburg, Germany) and images were captured using an Olympus cell imaging station. Both populations were negative for CD3 (T cell), CD45 (leukocytes), and CD11b (monocytes and granulocytes) staining. All experiments were performed on primary cultures of each cell population, and the various tests were performed in triplicate.
Cell Proliferation
The cells that reached confluence were harvested after trypsin treatment. Then cells were counted with a Trypan Blue method and 104 cells well were seeded onto 96-well plates (Nunc, Roskilde, Denmark). Cellular viability was approximately 95% to 100%, regardless of cell type. Cells were incubated for 48 hours in Dulbecco's modified Eagle's medium with 10% fetal calf serum alone or with the addition of 6.4 mmol/L N-acetyl-L-cysteine (NAC), or 200 μmol/L ERK inhibitor (A77-1726), or various concentrations of the mTOR/AKT inhibitor temsirolimus (0.3 to 24 μmol/L) at 37°C under 5% CO2. Cell proliferation was determined by pulsing the cells with [3H]thymidine (Amersham, 1 μCi per well; GE Healthcare) during the last 18 hours of culture20 and measuring the radioactivity incorporated by liquid scintillation counting. Results are expressed as counts per minute (cpm).
Cellular Production of ROS
Cells (104 per well) were seeded into 96-well plates (Nunc, Roskilde, Denmark) and were incubated for 18 hours alone or with 6.4 mmol/L NAC. Cellular levels of O2•−, H2O2, and NO were assessed by spectrofluorometry using 125 μmol/L dihydroethidium (DHE; Interchim, Montluçon, France), 100 μmol/L 2′,7′-dichlorohydrofluorescein diacetate (H2DCFDA; Molecular Probes, Eugene, OR), and 4′, 5′-diaminofluorescein diacetate (DAF2DA; Sigma-Aldrich), respectively, during 5 hours. The levels of O2•−, H2O2, and NO were calculated in each sample as follows. The ROS rate in arbitrary units per minute was calculated by subtracting initial fluorescence intensity from final fluorescence intensity and dividing the difference by the final number of minutes. Conditions in the present study were 300 minutes and 104 cells per well.
Immunoblotting of Cell Lysates
Cells were lysed in ice-cold radioimmunoprecipitation assay buffer (10 mmol/L TrisHCl, pH 7.5, 5 mol/L NaCl, 1% Triton X-100, 0.1% SDS) supplemented with 25 mmol/L sodium fluoride, 0.5 mmol/L sodium orthovanadate, and anti-protease 1%. Equal amounts of protein (30 μg) were loaded and separated by 10% SDS-PAGE. Transfer and blocking were performed. For ERK/pERK staining, polyvinylidene difluoride membrane was saturated with 5% skim milk for 1 hour at room temperature, then incubated with rabbit anti-human ERK IgG antibodies or with rabbit anti-human pERK IgG antibodies (Santa Cruz Biotechnology, Santa Cruz, CA) overnight at 4°C. For AKT/pAKT staining, polyvinylidene difluoride membrane was saturated with TBSA (BSA-TBST: 1× Tris-buffered saline, 0.1% Tween-20 with 5% bovine serum albumin) overnight at 4°C with rabbit anti-human AKT IgG antibodies and rabbit anti-human pAKT IgG antibodies (Cell Signaling Technology, Danvers, MA) for 1 hour at room temperature. For phospho-p70S6K (serine 371/threonine 389) staining, polyvinylidene difluoride membrane was saturated with TBSA (BSA-TBST: 1× Tris-buffered saline, 0.1% Tween-20 with 5% bovine serum albumin) for 1 hour at room temperature, then incubated with rabbit anti-human phospho-p70S6K IgG antibodies (Ozyme, Saint-Quentin-en-Yvelines, France) overnight at 4°C. β-Actin was used as housekeeping protein. Specific antibodies were detected using horseradish peroxidase-conjugated goat anti-rabbit IgG Ab and visualized by an enhanced chemoluminescence system (Pierce ECL; Perbio Sciences, Berbières, France; Thermo Fisher Scientific, Rockford, IL).21
IHC Analysis of pERK and pAKT
Paraffin-embedded tissue sections (4-μm thick) were deparaffinized and antigen retrieval was performed by microwave boiling of the sections in 10 mmol/L citrate buffer (pH 6.0) twice for 10 minutes each. Sections were incubated in 20% (v/v) normal goat serum in PBS with 4% (w/v) bovine serum albumin for 30 minutes at room temperature, and then the tissue sections were stained overnight at 4°C with antibodies against phospho-p44/42 MAP kinase (clone 20G11, diluted 1:100; Cell Signaling Technology) or with antibodies against phospho-AKT (clone 736E11, diluted 1:25; Cell Signaling Technology). Horseradish peroxidase-conjugated secondary antibodies were detected using the 3,3′-diaminobenzidine peroxidase substrate kit (Vector Laboratories, Burlingame, CA). Cells were considered positive only when a clear staining of the cell nucleus and/or the cytoplasm was observed.
Mouse Model
Nude mice were grafted with two fragments of a nodule from one patient with DIE. Female nude mice between 6 and 8 weeks of age weighing 20 to 22 g were used. Animals received humane care in compliance with institutional guidelines. Mice were anesthetized with intraperitoneal injections of tribromoethanol. An incision was made on the ventral midline and two fragments of human endometriotic nodules were sutured to the parietal peritoneum with 7/0 prolene. Each fragment was measured using a rule caliper. An additional fragment was fixed with 10% formaldehyde and set in paraffin for initial histological analysis. The abdominal wall was sutured with a 6/0 nylon thread.22 A subcutaneous injection of 0.5 μg β-estradiol was performed on day 1 and day 2 to facilitate the implantation of endometriotic nodules.23
On day 7 after implantation, surgery was performed on the mice to confirm viability of the implant. Treatment started on day 7: the treated group received intraperitoneal injections of temsirolimus (3 mg/kg in 100 μL PBS, 5 days per week for 2 weeks) and the control group received intraperitoneal injections of 100 μL PBS under the same conditions. After 2 weeks of treatment, animals were sacrificed by cervical dislocation. Implants were extracted and fixed with 10% formaldehyde and set in paraffin. Serial 4-μm sections were prepared and stained with H&E and were examined histologically by pathologists experienced in endometriosis (S.A., F.B., and M.L.). Pathology scores were on a scale of 0 (receded lesion with stromal fibrosis, hemosiderin and absence of glandular structure) to 3 (active lesion with fresh blood, profuse stromal cellular infiltration and developed glandular organization).10 Scoring was performed by three different scientists unaware of the treatments received in each group (S.A., F.B., and M.L.).
Statistical Analysis
Data are reported as means of independent triplicate experiments for each cell population. Means were compared by Student's t-test. A level of P < 0.05 was accepted as significant.
Results
Biopsies and Characteristics of Population
The study population comprised 22 patients. Twenty biopsies of eutopic endometrium and 21 biopsies of deep infiltrating endometriotic nodule were obtained from those patients. Initially, 20 Ee and 20 Es cell lines were extracted from 20 samples of eutopic endometrium, and 20 De and 21 Ds cell lines were extracted from 21 samples from deep infiltrating endometriotic nodules. Nine cell lines were eliminated for failure of culture (n = 6) or bacterial contamination (n = 3). A final total of 18 Ee, 18 Es, 17 De, and 18 Ds cell lines were included for study. As control subjects, 12 women were included, and 12 biopsies of healthy endometrium were collected. Two of the control subjects had a preoperative hormonal treatment (estroprogestatives in one case for contraception and LHRH agonists to reduce operative bleeding during uterine myoma resection in the other case). The 12 samples of endometrium provided 12 epithelial and 12 stromal control endometrial cell lines. Of these, 3 cell lines were eliminated for failure of culture, yielding a final total of 11 Ce and 10 Cs cell lines. The clinical characteristics of the population are shown in Table 1.
Table 1.
Clinical Characteristics of DIE and Control Patients
| DIE | Control | |
|---|---|---|
| Sample size | n = 22 | n = 12 |
| Age (years)⁎ | 32 ± 1.5 (21–47) | 31.9 ± 1.09 (19–36) |
| Treatments before surgery (no.) | 22 | 2 |
| None | 0 | 9 |
| Estroprogestative agents | – | 1 |
| LHRH agonists | 22 | 1 |
| Duration of treatment (months)† | 11 (4–60) | 6 (2–32) |
| Infertility | 8 | 3 |
| Duration of symptoms (months)† | 48 (12–168) | – |
| Gynecologic symptoms | 22 | – |
| Bowel symptoms | 22 | – |
| Cyclic exacerbation of symptoms | 20 | – |
| C-reactive protein (mg/L)⁎ | 7.55 ± 2.78 (0.09–36.7) | – |
| Biopsy | ||
| Endometrium | 20 | 12 |
| Deep lesion | 21 | – |
Data are presented as mean ± SD (range).
Data are presented as median (range).
Purification of Endometrial and Endometriotic Cells
The cells were checked by optical microscopy and the patterns of expression of cytokeratin and vimentin were characterized by direct immunofluorescence using specific antibodies (Figure 1A). The epithelial cells showed positive staining for cytokeratin and negative staining for vimentin. The stromal cells showed positive staining for vimentin and negative staining for cytokeratin.
Figure 1.
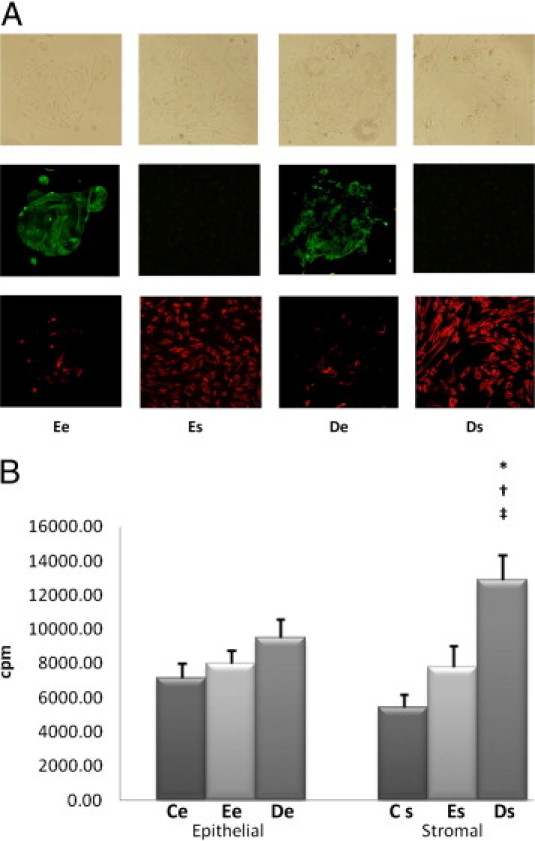
Purification and proliferative rates of endometriotic cells. A: Eutopic endometrial epithelial cells (Ee), eutopic endometrial stromal cells (Es), deep infiltrating endometriotic epithelial cells (De), and deep infiltrating endometriotic stromal cells (Ds) examined by contrast phase microscopy (top row), immunofluorescence with fluorescein isothiocyanate anti-cytokeratin antibodies (middle row), or Cy3 anti-vimentin antibodies (bottom row). Original magnification, ×100. B: Basal proliferative rate was assessed in the various cell lines: control endometrial epithelial cell lines (Ce), n = 11; Ee cell lines, n = 18; De cell lines, n = 17; control endometrial stromal cell lines (Cs), n = 10; Es cell lines, n = 18; and Ds cell lines, n = 18. Cell proliferation was determined by thymidine incorporation. Results are expressed as counts per minute (cpm). *P < 0.001, Es versus Ce cells; †P < 0.01, De versus Ee cells; ‡P < 0.05, stromal versus epithelial cells within the same endometrial eutopic or deep infiltrating lesion.
Proliferation of Deep Infiltrating Endometriotic Cells
There were no significant differences among De, Ee, and Ce cells in terms of proliferative rates. The proliferative rate of Es cells was not higher than that of Cs cells; however, the proliferative rate of Ds cells was greater by 39% than that of Es cells (P < 0.01) and by 68% than that of Cs cells (P < 0.001) (Figure 1B).
Within deep nodules, the proliferative rate of Ds cells was increased by 26% compared with De cells (P < 0.05) (Figure 1B).
There were no differences in terms of cellular proliferation between the group of fertile patients (n = 14) and the group of infertile patients (n = 8) (data not shown), nor between the group with C-reactive protein of <5 mg/L (n = 13) and the group with C-reactive protein of >5 mg/L (n = 9) (data not shown).
Cellular Production of ROS
Production of Superoxide Anions
The spontaneous production of superoxide anions (O2•−) in Ee cells was increased by 28% compared with Ce cells (P < 0.05). The production of O2•− in De cells was not increased significantly compared with Ee cells, but was increased by 36% compared with Ce cells (P < 0.05). The production of O2•− in Es cells was not increased significantly compared with Cs cells. In contrast, the production of O2•− in Ds cells was increased by 35% compared with Es cells (P < 0.001) and by 43% compared with Cs cells (P < 0.001) (Figure 2A).
Figure 2.
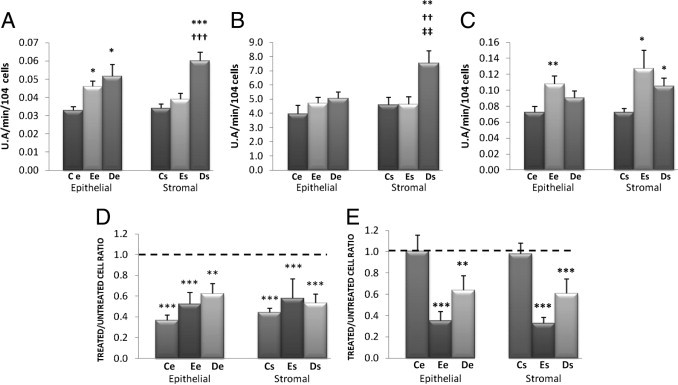
A–C: Cellular production of ROS and detoxification of H2O2 by N-acetyl-L-cysteine (NAC). Basal intracellular levels of O2•− (A), H2O2 (B), and NO (C) were assessed by spectrofluorometry in the various cell lines (11 Ce, 18 Ee, 17 De, 10 Cs, 18 Es, and 18 Ds). U.A., arbitrary fluorescence intensity units. *P < 0.05, **P < 0.01, and ***P < 0.001, Es versus Ce cells. ††P < 0.01, †††P < 0.001, De versus Ee cells. ‡‡P < 0.01, stromal versus epithelial cells within the same endometrial eutopic or deep infiltrating lesion. D and E: Intracellular levels of H2O2 (D) and proliferative rate (E) were assessed in cell lines treated with 6.4 mmol/L NAC versus untreated cells. The level in untreated cells is indicated by a horizontal dashed line. **P < 0.01, ***P < 0.001.
Within deep nodules, the production of O2•− did not differ significantly between Ds and De cells (Figure 2A).
Production of Hydrogen Peroxide
There were no significant differences among De, Ee, and Ce cells in terms of H2O2 production. The production of H2O2 in Es cells was not increased significantly compared with Cs cells. In contrast, the production of H2O2 in Ds cells was increased by 39% compared with Es cells (P < 0.01) and by 39% compared with Cs cells (P < 0.01) (Figure 2B).
Within deep nodules, the production of H2O2 in Ds cells was increased by 23% compared with De cells (P < 0.01) (Figure 2B).
Production of Nitric Oxide
The production of NO in Ee cells was increased by 36% compared with Ce cells (P < 0.01). In contrast, the production of NO in De cells did not differ significantly from that in Ee and Ce cells. The production of NO in Es cells was increased by 46% compared with Cs cells (P < 0.05). The production of NO in Ds cells was not increased significantly compared with Es cells, but was increased by 30% compared with Cs cells (P < 0.05) (Figure 2C).
Within deep nodules, production of NO did not differ significantly between Ds and De cells (Figure 2C).
Cellular Detoxification of H2O2 by N-Acetyl-L-Cysteine
Treatment of Ce, Ee, and De cells with 6.4 mmol/L NAC decreased their production of H2O2 by 64%, 48%, and 38%, respectively, compared with untreated cells (P < 0.001 for all comparisons). Treatment of Cs, Es, and Ds cells with 6.4 mmol/L NAC decreased the production of H2O2 by 56%, 42%, and 47% compared with untreated cells (P < 0.001 for all comparisons) (Figure 2D).
Treatment of Ce and Cs cells with 6.4 mmol/L NAC did not decrease their proliferative rates, but for Ee and De cells the same treatment decreased their proliferative rates by 65% and by 36% compared with untreated cells (P < 0.001 for both comparisons). Treatment of Es and Ds cells with 6.4 mmol/L NAC decreased their proliferative rates by 77% and by 39% compared with untreated cells (P < 0.001 for both comparisons) (Figure 2E).
Exploration of the ERK Pathway
The ERK protein was equally present in the epithelial and stromal cell lines derived from control endometrium (Ce, Cs), eutopic endometrium (Ee, Es), and deep infiltrating endometriotic nodules (De, Ds). Levels of pERK, the phosphorylated activated form of ERK, were low in the Ce and Cs cell lines; however, pERK was expressed in the epithelial and stromal cell lines in both eutopic endometrium (Ee, Es) and deep infiltrating endometriotic nodules (De, Ds) (Figure 3A).
Figure 3.
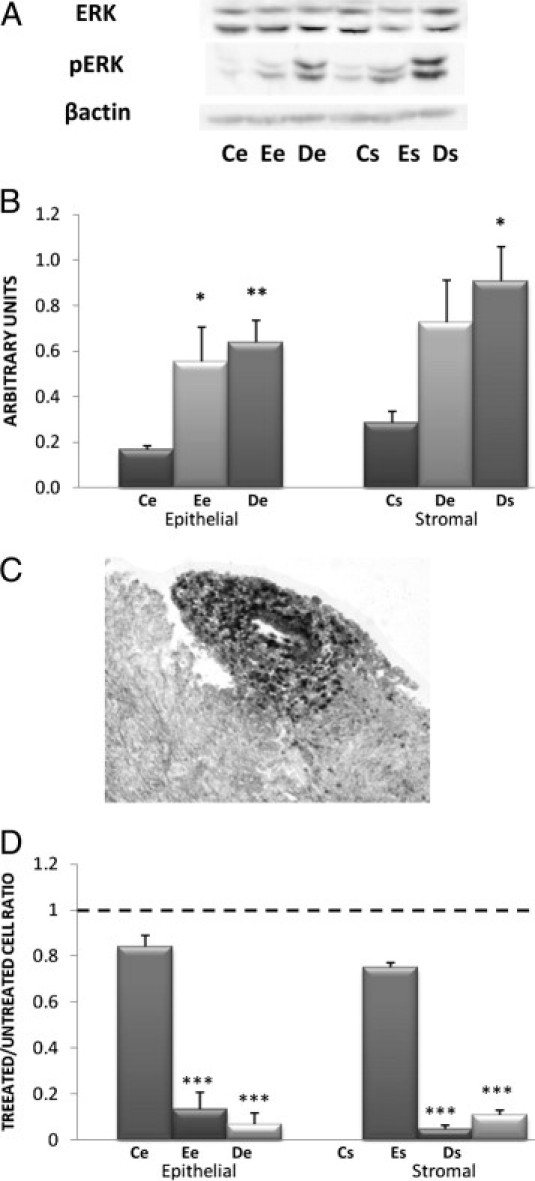
Exploration of the ERK pathway. A: Determination of ERK and pERK by Western blot in cell lysates. Western blotting of ERK and pERK were performed on lysates of the various cell lines (11 Ce, 18 Ee, 17 De, 10 Cs, 18 Es, and 18 Ds) using specific anti-ERK and anti-pERK antibodies. A representative Western blot is shown, obtained with all cell lines extracted from one patient and from one control. B: Quantitative analysis of ERK and pERK. Quantitative analysis of ERK and pERK was performed in the various cell lines (11 Ce, 18 Ee, 17 De, 10 Cs, 18 Es, and 18 Ds) by Western blot analysis. The mean optical density ratio for pERK/ERK was calculated in each endometriotic cell type and compared with control endometrial cells: *P < 0.05, **P < 0.01. C: IHC expression of pERK on deep infiltrating endometriotic tissue. A representative specimen is shown, from one of three patients. pERK-positive cells appear black. Original magnification, ×400. D: Effect of A77-1726 on endometriotic cell proliferation. Proliferative rates were determined by thymidine incorporation in the various cell lines (11 Ce, 18 Ee, 17 De, 10 Cs, 18 Es, and 18 Ds) treated with 200 μmol/L A77-1726 and were compared with untreated cells. The level in untreated cells is indicated by a horizontal dashed line. ***P < 0.001.
ERK activation, reflected by an elevated pERK/ERK ratio, was increased by 69% in Ee cells compared with Ce cells (P < 0.05). ERK activation in De cells was not increased compared with Ee cells, but was increased by 73% compared with Ce cells (P < 0.01) (Figure 3B).
ERK activation in Es cells was not increased significantly compared with Cs cells, nor in Ds cells compared with Es cells, but was increased by 68% compared with Cs cells (P < 0.05) (Figure 3B).
Within deep nodules, ERK activation did not differ significantly between Ds and De cells (Figure 3B).
Activation of the ERK pathway was also demonstrated by immunohistochemistry (IHC) on deep infiltrating tissue (Figure 3C).
Inhibition of the ERK Pathway by the Selective Protein Tyrosine Kinase Inhibitor A77-1726
Treatment of Ce cells with 200 μmol/L of the selective ERK inhibitor A77-1726 did not significantly decrease their proliferative rate. Treatment of Ee and De cells with 200 μmol/L of the selective ERK inhibitor A77-1726 decreased their proliferative rates by 87% and by 93%, respectively, compared with untreated cells (P < 0.001 for both comparisons) (Figure 3D).
Treatment of Cs cells with 200 μmol/L of the selective ERK inhibitor A77-1726 did not significantly decrease their proliferative rate. Treatment of Es and Ds cells with 200 μmol/L A77-1726 decreased their proliferative rate by 95% and 89%, respectively, compared with untreated cells (P < 0.001 for both comparisons) (Figure 3D).
Exploration of the mTOR/AKT Pathway
The AKT protein and its phosphorylated activated form, pAKT, were present in all cell lines, but were present in higher amounts in the epithelial and stromal cell lines derived from eutopic endometrium (Ee, Es) and deep infiltrating endometriotic nodules (De, Ds) than in the epithelial and stromal cell lines derived from control endometrium (Ce, Cs). The phosphorylated form of p70S6K (serine 371/threonine 389) was present mostly in the epithelial and stromal cell lines derived from deep infiltrating endometriotic nodules (De, Ds), as opposed to the epithelial and stromal cell lines derived from eutopic (Ee, Es) and control endometrium (Ce, Cs) (Figure 4A).
Figure 4.
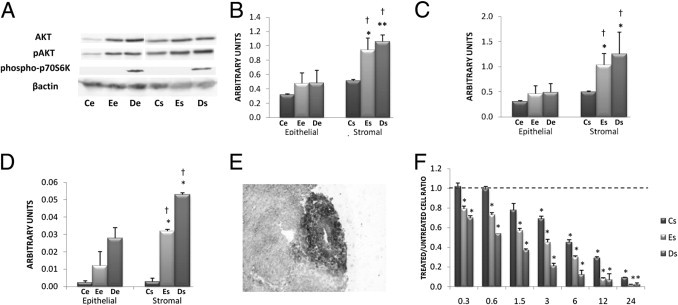
Exploration of the mTOR/AKT pathway. A: Determination of AKT, pAKT, and phospo-p70S6K (serine 371/threonine 389) by Western blot in cell lysates. Western blotting of AKT, pAKT, phospo-p70S6K, and β-actin was performed on lysates of the various cell lines (11 Ce, 18 Ee, 17 De, 10 Cs, 18 Es, and 18 Ds) using specific anti-AKT, anti-pAKT, anti-phospo-p70S6K, and anti-β-actin antibodies. The figure represents one representative Western blot obtained with all cell lines extracted from one patient and from one control. B and C: Quantitative analysis of AKT (B) and pAKT (C) was performed in the various cell lines (11 Ce, 18 Ee, 17 De, 10 Cs, 18 Es, and 18 Ds) by Western blot analysis. The mean optical density ratios AKT/β-actin and pAKT/β-actin were calculated in the various endometriotic cell lines. *P < 0.05 and **P < 0.001, endometriotic versus control cells; †P < 0.05, stromal versus epithelial cells within the same endometrial eutopic or deep infiltrating lesion. D: Quantitative analysis of phospo-p70S6K was performed in the various cell lines (11 Ce, 18 Ee, 17 De, 10 Cs, 18 Es, and 18 Ds) by Western blot analysis. The mean optical density ratio phospo-p70S6K/β-actin was calculated in the various endometriotic cell lines. *P < 0.05, endometriotic versus control cells. Stromal versus epithelial cells within the same endometrial eutopic or deep infiltrating lesion: †P < 0.05. E: IHC expression of pAKT on deep infiltrating endometriotic tissue. A representative specimen is shown, from one of three patients. pAKT-positive cells appear black. Original magnification, ×400. F: Effect of temsirolimus on proliferation. Proliferative rate was determined by thymidine incorporation for the various stromal cell lines (10 Cs, 18 Es, and 18 Ds) treated in vitro with increasing concentrations (0.3 to 24 μmol/L) of temsirolimus. The basal level of proliferation of untreated endometriotic cells is indicated by a horizontal dashed line. *P < 0.05, treated versus untreated cells.
AKT expression in Ee cells was increased by 33% compared with Ce cells. AKT expression in De cells was not increased compared with Ee cells, but was increased by 33% compared with Ce cells (Figure 4B). AKT expression in Es cells was increased by 46% compared with Cs cells (P < 0.05), and AKT expression in Ds cells was increased by 52% compared with Cs cells (P < 0.001) (Figure 4B). Within deep nodules, AKT expression in Ds cells was increased by 55% compared with De cells (P < 0.05) (Figure 4B).
The expression of pAKT in Ee cells was increased by 33% compared with Ce cells and pAKT expression in De cells was increased by 37% compared with Ce cells (Figure 4C). The expression of pAKT in Es cells was increased by 52% compared with Cs cells (P < 0.05) and pAKT expression in Ds cells was increased by 60% compared with Cs cells (P < 0.05) (Figure 4C). Within deep nodules, the expression of pAKT in Ds cells was increased by 61% compared with De cells (P < 0.05) (Figure 4C).
The expression of phospho-p70S6K in Ee cells was increased by 81% compared with Ce cells and phospho-p70S6K expression in De cells was increased by 92% compared with Ce cells (P < 0.05) (Figure 4D). The expression of phospho-p70S6K in Es cells was increased by 92% compared with Cs cells (P < 0.05), and phospho-p70S6K expression in Ds cells was increased by 95% compared with Cs cells (P < 0.05) (Figure 4D). Within deep nodules, the expression of phospho-p70S6K in Ds cells was increased by 48% compared with De cells (P < 0.05) (Figure 4D).
Activation of the AKT pathway was also demonstrated by IHC on deep infiltrating tissue (Figure 4E).
Inhibition of the AKT Pathway by the mTOR Inhibitor Temsirolimus
With 24 μmol/L temsirolimus, the proliferative rate of Es and Ds cells was decreased by 94% and by 98% respectively (P < 0.001 for both comparisons). The proliferative rate was dose-dependently decreased and significant for all concentrations. The proliferative rate of Cs cells was dose-dependently decreased and significant from 3 μmol/L (Figure 4F).
Mouse Model
Twelve nude mice were grafted with two fragments of nodules from two patients with DIE. Two mice died on day 8, which was 1 day after laparotomy to confirm the viability of the implant. The control group comprised 4 mice with 8 implants and the treated group comprised 6 mice with 12 implants. On day 0, the appearance of the implanted deep infiltrating endometriotic tissue was typical, with endometriotic glands, simple endometrial epithelium with stroma surrounded by fibrosis, and hyperplastic smooth muscle fibers. At 3 weeks after the implantation of deep endometriotic infiltrating endometriotic tissue and only 2 weeks after the beginning of the treatment, the 8 control implants showed persistence of active lesions with glandular organization (score 2.44 ± 0.18), whereas the 12 treated implants displayed fibrotic and avascular lesions with hemosiderin (score 1.19 ± 0.25, P < 0.01) (Figure 5, A and B).
Figure 5.
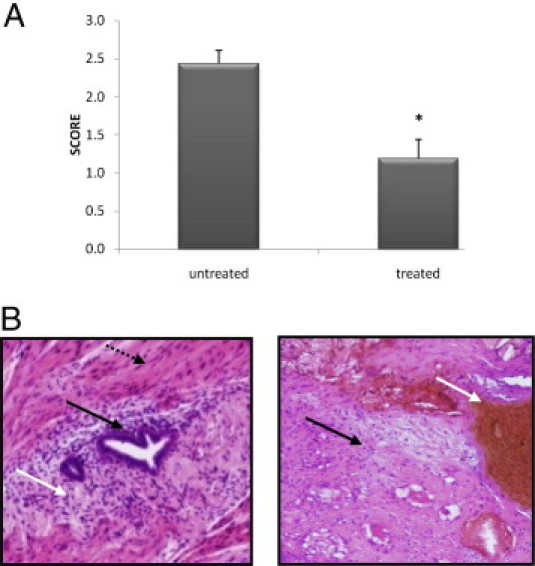
Effect of temsirolimus on deep infiltrating endometriotic tissue in a mouse model. A: Pathology scores were calculated on day 21 on untreated and treated mice: *P < 0.01. B: Histological views of deep infiltrating implant on day 21 in untreated mice (left) and in treated mice (right). In untreated mice, the appearance of the implanted deep infiltrating endometriotic tissue was typical, with endometriotic glands (black arrow) and with stroma (white arrow) surrounded by fibrosis and hyperplastic smooth muscle fibers (dotted black arrow). By contrast, treated mice (right) displayed fibrotic and avascular lesions (black arrow) with hemosiderin (white arrow).
Discussion
Here, we show for the first time the hyperproliferative phenotype of deep infiltrating endometriotic cells and the direct relationship between an increased endogenous oxidative stress and the activation of the ERK and the mTOR/AKT pathway. Moreover, we demonstrate that the pharmacological inhibition of the mTOR/AKT pathway abrogates the proliferation of endometriotic cells both in vitro and in vivo and is therefore a potential new treatment for endometriosis in humans.
The population of patients studied was homogeneous for symptoms. In our institution, all DIE patients are preoperatively treated with LHRH agonists. Indeed, this treatment is not only effective on the rAFS score,24,25 but also blocks the menstrual cycle, so that all patients can be compared in terms of hormonal menstrual variation. Even if LHRH agonists modulate endogenous oxidative stress or the ERK and AKT pathways as they do for IL-8,26 eutopic endometrium clearly differs from ectopic endometrium in each patient in terms of activation of these processes. This observation suggests that our data can be interpreted independently of LHRH treatment. Even though two control subjects had hormonal treatment, the control group was homogeneous for all of the experiments.
The yield of the isolation procedure was satisfactory, in that more than 17 lines of each cell type were derived from endometrial and deep infiltrating endometriotic biopsies (>75% success). Although cell-cell interactions between epithelial and stromal cells may play a role in the initiation and development of endometriosis,27 we chose to culture and test the two cell types separately, to determine the specific molecular abnormalities in each of them.
We first investigated the relationship between the cell proliferative rate and alterations in intracellular oxidative stress. In contrast to De cells, Ds cells displayed an increased proliferative rate and oxidative stress. Ovarian endometriotic cells are also characterized by a hyperproliferative rate in association with an up-regulated oxidative stress.10 However, the disorders predominantly affect epithelial cells in ovarian endometriosis and predominantly affect stromal cells in DIE, suggesting that ovarian endometriosis and deep infiltrating endometriosis result from the dysregulation of two different cell types.
The increase in oxidative stress results from the imbalance between production of ROS and their detoxification by antioxidant systems.28 Reinforcing catalase and glutathione reductase activities using the antioxidant molecule NAC significantly decreased the intracellular concentration of H2O2. The subsequent decrease in the cellular proliferative rate is reminiscent of our previous observations in cancer cells.20 Although the effects of NAC on the intracellular concentration of H2O2 were similar in both eutopic and deep infiltrating epithelial and stromal cells, the inhibition of proliferation predominated in eutopic epithelial and stromal cells. These results suggest that other mechanisms or amplified reactions act within deep infiltrating lesions to maintain the cells in a hyperproliferative state. Moreover, decreasing the amounts of H2O2 generated in control endometrial epithelial and stromal cells NAC does not decrease their proliferation rates. Thus, in normal cells, the levels of such pro-oxidative molecules as O2•− and H2O2 are not high enough to down-regulate cell proliferation as the ERK pathway does. In those cells, therefore, the inhibition of basal level of H2O2 has no influence on the proliferation rate. Taken together, those results suggest that antioxidant molecules could be used as a new treatment for DIE.
Oxidative stress, particularly the overproduction of O2•− and of H2O2, is known to activate the MAP kinase ERK 1/2 pathway in several cell types.29 Indeed, we found the ERK pathway activated through the phosphorylation of ERK, especially in deep infiltrating endometriotic stromal cells. This is in agreement with the known role of the ERK pathway in survival and proliferation of endometriotic cells,30 namely through expression of c-Fos and c-Jun.31 Those data have been confirmed by observations that the ERK pathway was hyperactivated10,13,19 and the related genes were overexpressed32–34 in patients with endometriosis. Furthermore, a relationship between activation of the ERK pathway and proliferation of deep infiltrating endometriotic cells could be established using a specific inhibitor of phosphorylation of the protein tyrosine kinase ERK (A77-1726).
This is the first time that the activation of the ERK pathway has been shown in DIE and that a relationship has been established between up-regulation of oxidative stress and increased cell proliferation. Although the same observations have been made in ovarian endometriosis, the distribution of the molecular abnormalities differs between ovarian endometriosis and DIE, in that they predominate in epithelial cells in the ovarian endometriosis and in stromal cells in DIE. This suggests that ovarian endometriosis is an epithelial disease and DIE, a stromal disease. On the other hand, oxidative stress is probably not the only factor responsible for activation of the ERK pathway. Because proinflammatory cytokines such as IL-1β and tumor necrosis factor-α are also increased in the endometrium of endometriosis patients.35–37 Thus, abrogating the activation of the ERK pathway in deep endometriotic cells using inhibitors of protein tyrosine kinase can be considered for a new therapeutic approach.
On the other hand, the phosphatidylinositol-3 kinases (PI3Ks) as well as the mammalian target of rapamycin (mTOR) pathways are two key cellular signaling pathways that affect broad aspects of cellular functions, including metabolism, growth, and survival.38,39 Although initially viewed as two separate pathways, the PI3K and mTOR pathways are connected via the serine/threonine kinase AKT.40 AKT, also termed PKB (protein kinase B), is involved in metabolism regulation, growth, proliferation, and apoptosis through PI3K and mTOR.41 The PI3K/mTOR pathway is activated by a broad array of different stimuli via specific receptors, including antigen receptors, cytokine receptors, insulin receptor, or the insulin-like growth factor I receptor. After the engagement of the receptors, PI3K phosphorylates phosphatidylinositol 4,5-bisphosphate (PIP2) to generate phosphatidylinositol-3,4,5-trisphosphate (PIP3) as a second messenger to recruit and activate downstream targets, including AKT. One main effector of PI3K and AKT is the high molecular weight kinase mTOR.42 mTOR controls protein synthesis through phosphorylation and inactivation of the repressor of mRNA translation, eukaryotic initiation factor 4E-binding protein 1, and through phosphorylation and activation of S6 kinase.43 The loss of mTOR function leads to an arrest in the G1 phase of the cell cycle, along with a severe reduction in protein synthesis.
Several observations have pointed to the potential role of the mTOR/AKT pathway in endometriosis. First, the P3KCA mutation responsible for the constitutive activation of AKT is the most frequently encountered mutation in ovarian clear cell carcinoma,44 a tumor that is associated with endometriosis.12,45 Moreover, AKT activity is higher in ovarian endometriosis than in normal endometrium,46 and it has been postulated that estrogens might be one of the factors responsible for the high AKT activation in endometriotic cells.17 However, if estrogens activate AKT in epithelial endometriotic cells,17 this phenomenon is less clear in stromal endometriotic cells.15 Indeed, in the stromal cells, a specific estrogen receptor antagonist fails to antagonize the effects of estradiol and exerts a stimulatory effect on AKT phosphorylation.15 We found that AKT is hyperactivated in endometriotic lesions from patients with DIE, as it is in ovarian endometriosis; however, ERK and AKT activation predominates in endometriotic stromal cells in DIE, but in endometriotic epithelial cells in ovarian endometriosis.
Because patients with DIE had been treated with an LHRH agonist, and because no estrogen could be detected in culture supernatants of endometriotic cells, our results suggest a constitutive estrogen-independent mechanism of AKT activation in DIE. This hypothesis is in line with the predominant involvement of endometriotic stromal cells in DIE. Moreover, the constitutive activation of the AKT pathway in endometriotic cells could be explained by the overproduction of endogenous ROS, as already observed in cancer cell lines for AKT activation and in endometriotic cells for ERK activation.47,48 Because AKT affects cell proliferation and survival, the phenotype of stromal cells in DIE may be linked to increased AKT activation. The decrease in the proliferative rate of eutopic endometrial epithelial cells and deep infiltrating endometriotic stromal cells after treatment with temsirolimus, a selective inhibitor of mTOR, establishes for the first time a link between activation of the AKT pathway and the hyperproliferative phenotype of those cells. Moreover, the inhibitory effect of temsirolimus on the survival and development of endometriotic implants in a mouse model of DIE suggests this molecule for treatment of patients with DIE, as in patients with kidney cancer.49 This molecule could be rapidly tested in women with DIE.
Acknowledgment
We thank Ms. Agnes Colle for editing the manuscript.
Footnotes
Supported by grants from Paris Descartes University.
B.D. and F.B. contributed equally to this work.
References
- 1.Giudice L.C., Kao L.C. Endometriosis. Lancet. 2004;364:1789–1799. doi: 10.1016/S0140-6736(04)17403-5. [DOI] [PubMed] [Google Scholar]
- 2.Chapron C., Fauconnier A., Dubuisson J.B., Barakat H., Vieira M., Bréart G. Deep infiltrating endometriosis: relation between severity of dysmenorrhoea and extent of disease. Hum Reprod. 2003;18:760–766. doi: 10.1093/humrep/deg152. [DOI] [PubMed] [Google Scholar]
- 3.Laursen B.S., Bajaj P., Olesen A.S., Delmar C., Arendt-Nielsen L. Health related quality of life and quantitative pain measurement in females with chronic non-malignant pain. Eur J Pain. 2005;9:267–275. doi: 10.1016/j.ejpain.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 4.de Ziegler D., Borghese B., Chapron C. Endometriosis and infertility: pathophysiology and management. Lancet. 2010;376:730–738. doi: 10.1016/S0140-6736(10)60490-4. [DOI] [PubMed] [Google Scholar]
- 5.Chopin N., Vieira M., Borghese B., Foulot H., Dousset B., Coste J., Mignon A., Fauconnier A., Chapron C. Operative management of deeply infiltrating endometriosis: results on pelvic pain symptoms according to a surgical classification. J Minim Invasive Gynecol. 2005;12:106–112. doi: 10.1016/j.jmig.2005.01.015. [DOI] [PubMed] [Google Scholar]
- 6.Chapron C., Chiodo I., Leconte M., Amsellem-Ouazana D., Chopin N., Borghese B., Dousset B. Severe ureteral endometriosis: the intrinsic type is not so rare after complete surgical exeresis of deep endometriotic lesions. Fertil Steril. 2010;93:2115–2120. doi: 10.1016/j.fertnstert.2009.01.102. [DOI] [PubMed] [Google Scholar]
- 7.Dousset B., Leconte M., Borghese B., Millischer A.E., Roseau G., Arkwright S., Chapron C. Complete surgery for low rectal endometriosis: long-term results of a 100-case prospective study. Ann Surg. 2010;251:887–895. doi: 10.1097/SLA.0b013e3181d9722d. [DOI] [PubMed] [Google Scholar]
- 8.Bulun S.E. Endometriosis. N Engl J Med. 2009;360:268–279. doi: 10.1056/NEJMra0804690. [DOI] [PubMed] [Google Scholar]
- 9.Fedele L., Somigliana E., Frontino G., Benaglia L., Vigano P. New drugs in development for the treatment of endometriosis. Expert Opin Investig Drugs. 2008;17:1187–1202. doi: 10.1517/13543784.17.8.1187. [DOI] [PubMed] [Google Scholar]
- 10.Ngô C., Chéreau C., Nicco C., Weill B., Chapron C., Batteux F. Reactive oxygen species controls endometriosis progression. Am J Pathol. 2009;175:225–234. doi: 10.2353/ajpath.2009.080804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ngô C., Nicco C., Leconte M., Chéreau C., Weill B., Batteux F., Chapron C. Antiproliferative effects of anastrozole, methotrexate, and 5-fluorouracil on endometriosis in vitro and in vivo. Fertil Steril. 2009;94 doi: 10.1016/j.fertnstert.2009.09.031. 1632–1638.e1. [DOI] [PubMed] [Google Scholar]
- 12.Varma R., Rollason T., Gupta J.K., Maher E.R. Endometriosis and the neoplastic process. Reproduction. 2004;127:293–304. doi: 10.1530/rep.1.00020. [DOI] [PubMed] [Google Scholar]
- 13.Murk W., Atabekoglu C.S., Cakmak H., Heper A., Ensari A., Kayisli U.A., Arici A. Extracellularly signal-regulated kinase activity in the human endometrium: possible roles in the pathogenesis of endometriosis. J Clin Endocrinol Metab. 2008;93:3532–3540. doi: 10.1210/jc.2007-2051. [DOI] [PubMed] [Google Scholar]
- 14.Ngô C., Nicco C., Leconte M., Chéreau C., Arkwright S., Vacher-Lavenu M.C., Weill B., Chapron C., Batteux F. Protein kinase inhibitors can control the progression of endometriosis in vitro and in vivo. J Pathol. 2010;222:148–157. doi: 10.1002/path.2756. [DOI] [PubMed] [Google Scholar]
- 15.Cinar O., Seval Y., Uz Y.H., Cakmak H., Ulukus M., Kayisli U.A., Arici A. Differential regulation of Akt phosphorylation in endometriosis. Reprod Biomed Online. 2009;19:864–871. doi: 10.1016/j.rbmo.2009.10.001. [DOI] [PubMed] [Google Scholar]
- 16.Laudanski P., Szamatowicz J., Kowalczuk O., Kuźmicki M., Grabowicz M., Chyczewski L. Expression of selected tumor suppressor and oncogenes in endometrium of women with endometriosis. Hum Reprod. 2009;24:1880–1890. doi: 10.1093/humrep/dep175. [DOI] [PubMed] [Google Scholar]
- 17.Zhang H., Zhao X., Liu S., Li J., Wen Z., Li M. 17betaE2 promotes cell proliferation in endometriosis by decreasing PTEN via NFkappaB-dependent pathway. Mol Cell Endocrinol. 2010;317:31–43. doi: 10.1016/j.mce.2009.11.009. [DOI] [PubMed] [Google Scholar]
- 18.Chapron C., Bourret A., Chopin N., Dousset B., Leconte M., Amsellem-Ouazana D., de Ziegler D., Borghese B. Surgery for bladder endometriosis: long-term results and concomitant management of associated posterior deep lesions. Hum Reprod. 2010;25:884–889. doi: 10.1093/humrep/deq017. [DOI] [PubMed] [Google Scholar]
- 19.Hornung D., Ryan I.P., Chao V.A., Vigne J.L., Schriock E.D., Taylor R.N. Immunolocalization and regulation of the chemokine RANTES in human endometrial and endometriosis tissues and cells. J Clin Endocrinol Metab. 1997;82:1621–1628. doi: 10.1210/jcem.82.5.3919. [DOI] [PubMed] [Google Scholar]
- 20.Laurent A., Nicco C., Chéreau C., Goulvestre C., Alexandre J., Alves A., Lévy E., Goldwasser F., Panis Y., Soubrane O., Weill B., Batteux F. Controlling tumor growth by modulating endogenous production of reactive oxygen species. Cancer Res. 2005;65:948–956. [PubMed] [Google Scholar]
- 21.Song J.S., Kang C.M., Yoo M.B., Kim S.J., Yoon H.K., Kim Y.K., Kim K.H., Moon H.S., Park S.H. Nitric oxide induces MUC5AC mucin in respiratory epithelial cells through PKC and ERK dependent pathways. Respir Res. 2007;8:28. doi: 10.1186/1465-9921-8-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bruner-Tran K.L., Webster-Clair D., Osteen K.G. Experimental endometriosis: the nude mouse as a xenographic host. Ann N Y Acad Sci. 2002;955:328–339. doi: 10.1111/j.1749-6632.2002.tb02793.x. discussion 340–342, 396–406. [DOI] [PubMed] [Google Scholar]
- 23.Grümmer R., Schwarzer F., Bainczyk K., Hess-Stumpp H., Regidor P.A., Schindler A.E., Winterhager E. Peritoneal endometriosis: validation of an in-vivo model. Hum Reprod. 2001;16:1736–1743. doi: 10.1093/humrep/16.8.1736. [DOI] [PubMed] [Google Scholar]
- 24.Donnez J., Nisolle M., Gillerot S., Anaf V., Clerckx-Braun F., Casanas-Roux F. Ovarian endometrial cysts: the role of gonadotropin-releasing hormone agonist and/or drainage. Fertil Steril. 1994;62:63–66. doi: 10.1016/s0015-0282(16)56816-2. [DOI] [PubMed] [Google Scholar]
- 25.Shaw R.W. An open randomized comparative study of the effect of goserelin depot and danazol in the treatment of endometriosis: Zoladex Endometriosis Study Team. Fertil Steril. 1992;58:265–272. doi: 10.1016/s0015-0282(16)55205-4. [DOI] [PubMed] [Google Scholar]
- 26.Sakamoto Y., Harada T., Horie S., Iba Y., Taniguchi F., Yoshida S., Iwabe T., Terakawa N. Tumor necrosis factor-alpha-induced interleukin-8 (IL-8) expression in endometriotic stromal cells, probably through nuclear factor-kappa B activation: gonadotropin-releasing hormone agonist treatment reduced IL-8 expression. J Clin Endocrinol Metab. 2003;88:730–735. doi: 10.1210/jc.2002-020666. [DOI] [PubMed] [Google Scholar]
- 27.Cooke P.S., Buchanan D.L., Young P., Setiawan T., Brody J., Korach K.S., Taylor J., Lubahn D.B., Cunha G.R. Stromal estrogen receptors mediate mitogenic effects of estradiol on uterine epithelium. Proc Natl Acad Sci USA. 1997;94:6535–6540. doi: 10.1073/pnas.94.12.6535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sies H. Oxidative Stress: Oxidants and Antioxidants. Academic Press; New York: 1991. Oxidative Stress: Introduction; pp. pv–pxxii. [Google Scholar]
- 29.McCubrey J.A., Lahair M.M., Franklin R.A. Reactive oxygen species-induced activation of the MAP kinase signaling pathways. Antioxid Redox Signal. 2006;8:1775–1789. doi: 10.1089/ars.2006.8.1775. [DOI] [PubMed] [Google Scholar]
- 30.Raman M., Chen W., Cobb M.H. Differential regulation and properties of MAPKs. Oncogene. 2007;26:3100–3112. doi: 10.1038/sj.onc.1210392. [DOI] [PubMed] [Google Scholar]
- 31.Luo X., Ding L., Chegini N. Gonadotropin-releasing hormone and TGF-beta activate MAP kinase and differentially regulate fibronectin expression in endometrial epithelial and stromal cells. Am J Physiol Endocrinol Metab. 2004;287:E991–E1001. doi: 10.1152/ajpendo.00200.2004. [DOI] [PubMed] [Google Scholar]
- 32.Borghese B., Mondon F., Noël J.C., Fayt I., Mignot T.M., Vaiman D., Chapron C. Gene expression profile for ectopic versus eutopic endometrium provides new insights into endometriosis oncogenic potential. Mol Endocrinol. 2008;22:2557–2562. doi: 10.1210/me.2008-0322. [DOI] [PubMed] [Google Scholar]
- 33.Matsuzaki S., Canis M., Vaurs-Barrière C., Boespflug-Tanguy O., Dastugue B., Mage G. DNA microarray analysis of gene expression in eutopic endometrium from patients with deep endometriosis using laser capture microdissection. Fertil Steril. 2005;84(Suppl 2):1180–1190. doi: 10.1016/j.fertnstert.2005.04.041. [DOI] [PubMed] [Google Scholar]
- 34.Wu Y., Kajdacsy-Balla A., Strawn E., Basir Z., Halverson G., Jailwala P., Wang Y., Wang X., Ghosh S., Guo S.W. Transcriptional characterizations of differences between eutopic and ectopic endometrium. Endocrinology. 2006;147:232–246. doi: 10.1210/en.2005-0426. [DOI] [PubMed] [Google Scholar]
- 35.Donnez J., Smoes P., Gillerot S., Casanas-Roux F., Nisolle M. Vascular endothelial growth factor (VEGF) in endometriosis. Hum Reprod. 1998;13:1686–1690. doi: 10.1093/humrep/13.6.1686. [DOI] [PubMed] [Google Scholar]
- 36.Sugimura K., Okizuka H., Imaoka I., Kaji Y., Takahashi K., Kitao M., Ishida T. Pelvic endometriosis: detection and diagnosis with chemical shift MR imaging. Radiology. 1993;188:435–438. doi: 10.1148/radiology.188.2.8327693. [DOI] [PubMed] [Google Scholar]
- 37.Yoshino O., Osuga Y., Hirota Y., Koga K., Hirata T., Harada M., Morimoto C., Yano T., Nishii O., Tsutsumi O., Taketani Y. Possible pathophysiological roles of mitogen-activated protein kinases (MAPKs) in endometriosis. Am J Reprod Immunol. 2004;52:306–311. doi: 10.1111/j.1600-0897.2004.00231.x. [DOI] [PubMed] [Google Scholar]
- 38.Deane J.A., Fruman D.A. Phosphoinositide 3-kinase: diverse roles in immune cell activation. Annu Rev Immunol. 2004;22:563–598. doi: 10.1146/annurev.immunol.22.012703.104721. [DOI] [PubMed] [Google Scholar]
- 39.Wullschleger S., Loewith R., Hall M.N. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 40.Sekulić A., Hudson C.C., Homme J.L., Yin P., Otterness D.M., Karnitz L.M., Abraham R.T. A direct linkage between the phosphoinositide 3-kinase-AKT signaling pathway and the mammalian target of rapamycin in mitogen-stimulated and transformed cells. Cancer Res. 2000;60:3504–3513. [PubMed] [Google Scholar]
- 41.Brazil D.P., Yang Z.Z., Hemmings B.A. Advances in protein kinase B signalling: AKTion on multiple fronts. Trends Biochem Sci. 2004;29:233–242. doi: 10.1016/j.tibs.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 42.Brown E.J., Beal P.A., Keith C.T., Chen J., Shin T.B., Schreiber S.L. Control of p70 s6 kinase by kinase activity of FRAP in vivo. Nature. 1995;377:441–446. doi: 10.1038/377441a0. [Erratum appeared in Nature 1995;378:644] [DOI] [PubMed] [Google Scholar]
- 43.Hay N., Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 44.Kuo K.T., Mao T.L., Jones S., Veras E., Ayhan A., Wang T.L., Glas R., Slamon D., Velculescu V.E., Kuman R.J., Shih IeM. Frequent activating mutations of PIK3CA in ovarian clear cell carcinoma. Am J Pathol. 2009;174:1597–1601. doi: 10.2353/ajpath.2009.081000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Somigliana E., Vigano' P., Parazzini F., Stoppelli S., Giambattista E., Vercellini P. Association between endometriosis and cancer: a comprehensive review and a critical analysis of clinical and epidemiological evidence. Gynecol Oncol. 2006;101:331–341. doi: 10.1016/j.ygyno.2005.11.033. [DOI] [PubMed] [Google Scholar]
- 46.Yagyu T., Tsuji Y., Haruta S., Kitanaka T., Yamada Y., Kawaguchi R., Kanayama S., Tanase Y., Kurita N., Kobayashi H. Activation of mammalian target of rapamycin in postmenopausal ovarian endometriosis. Int J Gynecol Cancer. 2006;16:1545–1551. doi: 10.1111/j.1525-1438.2006.00625.x. [DOI] [PubMed] [Google Scholar]
- 47.Clerkin J.S., Naughton R., Quiney C., Cotter T.G. Mechanisms of ROS modulated cell survival during carcinogenesis. Cancer Lett. 2008;266:30–36. doi: 10.1016/j.canlet.2008.02.029. [DOI] [PubMed] [Google Scholar]
- 48.Dong-Yun S., Yu-Ru D., Shan-Lin L., Ya-Dong Z., Lian W. Redox stress regulates cell proliferation and apoptosis of human hepatoma through Akt protein phosphorylation. FEBS Lett. 2003;542:60–64. doi: 10.1016/s0014-5793(03)00338-7. [DOI] [PubMed] [Google Scholar]
- 49.Hudes G., Carducci M., Tomczak P., Dutcher J., Figlin R., Kapoor A., Staroslawska E., Sosman J., McDermott D., Bodrogi I., Kovacevic Z., Lesovoy V., Schmidt-Wolf I.G., Barbarash O., Gokmen E., O'Toole T., Lustgarten S., Moore L., Motzer R.J., Global ARCC Trial Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med. 2007;356:2271–2281. doi: 10.1056/NEJMoa066838. [DOI] [PubMed] [Google Scholar]