

Abstract
Toll-like receptors (TLRs) 7 and 9 recognize nucleic acid determinants from viruses and bacteria and elicit the production of type I interferons and proinflammatory cytokines. TLR7 and TLR9 are similar regarding localization and signal transduction mechanisms. However, stimulation of these receptors has differing effects in modulating viral pathogenesis and in direct toxicity in the central nervous system (CNS). In the present study, we examined the potential of the TLR7 agonist imiquimod and the TLR9 agonist cytosine-phosphate-guanosine oligodeoxynucleotide (CpG-ODN) to induce neuroinflammation after intracerebroventricular inoculation. CpG-ODN induced a more robust inflammatory response than did imiquimod after inoculation into the CNS, with higher levels of several proinflammatory cytokines and chemokines. The increase in cytokines and chemokines correlated with breakdown of the blood–cerebrospinal fluid barrier and recruitment of peripheral cells to the CNS in CpG-ODN–inoculated mice. In contrast, TLR7 agonists induced a strong interferon β response in the CNS but only low levels of other cytokines. The difference in response to these agonists was not due to differences in distribution or longevity of the agonists but rather was correlated with cytokine production by choroid plexus cells. These results indicate that despite the high similarity of TLR7 and TLR9 in binding nucleic acids and inducing similar downstream signaling, the neuroinflammation response induced by these receptors differs dramatically due, at least in part, to activation of cells in the choroid plexus.
Neuroinflammation is a common response to infection or injury of the central nervous system (CNS) and includes the induction of proinflammatory cytokines, the activation of glial cells, and the recruitment of inflammatory cells to the CNS.1–5 Neuroinflammatory responses may depend on the initiation of innate immune responses triggered by the stimulation of intrinsic brain cells with pathogen-associated molecular patterns, the repeated structural motifs generated by microbes that are not normally found in the host, or by debris from apoptotic or necrotic cells after injury. The recognition of pathogen-associated molecular patterns is mediated by several families of germline-encoded receptors, called pattern recognition receptors.
Toll-like receptors (TLRs) 7 and 9 are two important pattern recognition receptors that can initiate innate immune responses after viral or bacterial infection. TLR7 recognizes single-stranded RNA from multiple viruses and bacteria,6–8 whereas TLR9 recognizes bacterial and viral cytosine-phosphate-guanosine (CpG) DNA motifs (unmethylated DNA motifs containing CpG nucleotides flanked by 5′ purines and two 3′ pyrimidines).9–11 Both receptors share many common characteristics. They are transported to the endolysosome from the endoplasmic reticulum by the protein Unc93b1, require endosomal acidification for activation, and signal through the same adaptor molecule, myeloid differentiation factor. TLR7 and TLR9 can influence the outcome of viral infection in the CNS. TLR7 regulates the innate immune response to retrovirus and West Nile virus infections of the CNS, and TLR9 has been implicated in mediating the innate immune response to herpes simplex virus infection in the brain.12–16
TLR7 and TLR9 agonists have been analyzed for their potential use in the therapeutic treatment of CNS maladies from glioblastomas to viral infection. However, responses to stimulation of these receptors in the CNS often differ, particularly in neonates. For example, peripheral administration of TLR9 agonists protected neonatal mice from a lethal challenge with neurotropic Tacaribe arenavirus, whereas TLR7 agonists did not.17,18 Intracerebroventricular (ICV) inoculation of TLR9 agonists was lethal in newborn mice, but ICV inoculation of TLR7 agonists was not.17–19 Understanding differences in the neuroinflammatory capabilities of TLR7 and TLR9 agonists will be important for using the agonists in the treatment of neurologic diseases, including neurovirulent viral infections. This is particularly true in the developing brain, where viral infection can lead to severe neurologic damage or death.
In the present study, we directly compared innate immune responses to TLR7 and TLR9 agonist stimulation in the CNS using ICV inoculation of the TLR7 agonist imiquimod and the TLR9 agonist type B CpG oligodeoxynucleotide (ODN) 1826 in neonatal mice. We examined the proinflammatory cytokine response and the recruitment of peripheral immune cells after TLR7 and TLR9 agonist stimulation. We also examined which cells in the CNS responded to TLR7 and TLR9 agonist stimulation and how activation of these cells affected the blood–cerebrospinal fluid (CSF) barrier.
Materials and Methods
TLR Agonists
The TLR7 agonists imiquimod (R837) and fluorescein isothiocyanate (FITC)–labeled CL264 and the TLR9 agonists type A CpG-ODN 1585 (5′-GGGGTCAACGTTGAGGGGGG-3′), type B CpG-ODN 1826 (5′-TCCATGACGTTCCTGACGTT-3′), FITC-labeled ODN 1826, and type C CpG-ODN 2395 (5′-TCGTCGTTTTCGGCGC:GCGCCG-3′) were purchased from InvivoGen (San Diego, CA). All the agonists were suspended in endotoxin-free water, aliquoted, and stored at −20°C until use.
Mice
Inbred Rocky Mountain White (IRW) mice, TLR7-deficient IRW mice, C57BL/6 mice, and TLR9-deficient C57BL/6 mice were used 48 hours after birth for the present study. TLR7-deficient C57BL/6 mice20 and TLR9-deficient C57BL/6 mice11 were provided by Shizuo Akira (Osaka University). TLR7-deficient C57BL/6 mice were backcrossed with IRW mice for at least 10 generations.12 All the animal procedures were conducted in accordance with Louisiana State University Institutional Animal Care and Use Committee or Rocky Mountain Laboratories Animal Care and Use Committee guidelines.
ICV Inoculations of Newborn Mice
Agonists were inoculated ICV as described previously.19 In brief, 2-day-old mice were anesthetized by hypothermia, and 10 μL of the appropriate TLR agonist in PBS/0.2% trypan blue was inoculated into the lateral ventricles (5 μL per ventricle) using a 33-gauge needle and a Hamilton syringe. PBS with 0.2% trypan blue was used for mock ICV inoculations. Imiquimod was tested at a range of 20 nmol [∼2.5 μg/g of body weight (bw)] to 500 nmol (∼62.5 μg/g bw) per mouse. CpG-ODN 1826 was tested at a range from 77.5 pmol (∼0.25 μg/g bw) to 3100 pmol (∼10 μg/g bw) per mouse.
Preparation of Brain Tissue for Histologic and Molecular Analysis
At the end of the experimental protocol, mice were anesthetized by deep inhalation anesthesia followed by axillary incision and cervical dislocation. Brains were removed and immediately cut into two halves by midsagittal dissection, snap frozen in liquid nitrogen, and stored at −80°C for molecular analysis. One half was used for RNA extraction and the other half was used for multiplex analysis of cytokine and chemokine production. For in situ hybridization and/or immunohistochemical (IHC) analysis, brains were fixed in 10% neutral buffered formalin for 48 hours and were divided into three coronal sections before processing for histologic analysis. Slides were read in a blinded study and were scored for severity of infiltrate in the meninges or ventricles or for perivascular cuffing on a scale of none, mild, moderate, or severe.
RNA Extraction, Reverse Transcription, and Real-Time PCR
Total RNA was isolated from brain tissue using TRIzol reagent (Invitrogen, Carlsbad, CA). Total RNA was treated with DNase I (Ambion, Austin, TX) for 30 minutes at 37°C and was purified over RNA cleanup columns (Zymo Research, Irvine, CA) following the manufacturer's instructions. RNA samples were converted to cDNA using the iScript reverse transcription kit (Bio-Rad Laboratories, Hercules, CA) following the manufacturer's instructions and included DNA contamination controls that did not undergo reverse transcription. cDNA samples were diluted fivefold in RNase-free water before analysis by quantitative real-time PCR using SYBR Green SuperMix with ROX (Bio-Rad Laboratories). The primers to detect Ccl2, Cd3e, Cxcl10, F4/80, Gapdh, Gfap, Ifnb1, and Tnf cDNA were described previously.19 Other primers were as follows: Il6 (5′-CCGGAGAGGAGACTTCACAG-3′ forward and 5′-TCCACGATTTCCCAGAGAAC-3′ reverse), Il12 (5′-CCTGAAGTGTGAAGCACCAA-3′ forward and 5′-TCAGGGGAACTGCTACTGCT-3′ reverse), Irf7 (5′-CCAGTTGATCCGCATAAGGT-3′ forward and 5′-AGCATTGCTGAGGCTCACTT-3′ reverse), MyD88 (5′-CATGGTGGTGGTTGTTTCTG-3′ forward and 5′-CTGTTGGACACCTGGAGACA-3′ reverse), Tlr7 (5′-GGCATTCCCACTAACACCAC-3′ forward and 5′-TTGGACCCCAGTAGAACAGG-3′ reverse), Tlr9 (5′-ACTTCGTCCACCTGTCCAAC-3′ and 5′-TCATGTGGCAAGAGAAGTGC-3′ reverse), and Ttr (5′-GCTTCCCTTCGACTCTTCCT-3′ forward and 5′-GCATCCAGGACTTTGACCAT-3′ reverse). All the primers were used at a concentration of 0.5 μmol/L. Untranscribed controls and water were used as negative controls. All the samples were run in triplicate on a 384-well plate using an ABI PRISM 7900 machine (Applied Biosystems, Foster City, CA) with an automatically set baseline and a manually set CT of 0.19, which intersects the mid-log phase of curves for all the PCR pairs. Dissociation curves were used to confirm amplification of a single product for each primer pair per sample. Data for each sample were initially calculated as the percentage difference in CT value with the housekeeping gene Gapdh (ΔCT = CT Gapdh − CT gene of interest).
For analysis of interferon (IFN)-related genes, a PCR gene array was used. RNA was purified from brain tissue of mice at 2 hours postinoculation (hpi) as described previously herein. First-strand cDNA was synthesized using 100 ng of RNA. The PCR array was completed according to the manufacturer's instructions (SABiosciences, Frederick, MD) using an ABI PRISM 7900 sequence detection system (Applied Biosystems). A total of 84 genes related to the PCR array were analyzed in a 384-well format. The CT values were analyzed using an RT2 Profiler PCR array data analysis template (SABiosciences). The samples were analyzed only if the test passed all the quality controls, including real-time efficiency and lack of DNA contamination.
Multiplex Analysis of Cytokine and Chemokine Proteins
Cytokine and chemokine production in brain tissue after TLR7 and TLR9 stimulation was analyzed by multiplex bead assay as described previously.19 In brief, brain tissues were homogenized using Kontes disposable pellet pestles (Fisher Scientific, Hampton, NH) in 200 μL of Bio-Plex cell lysis solution (Bio-Rad Laboratories) containing complete Mini protease inhibitors (Roche Applied Science, Indianapolis, IN) and 2 mmol/L phenylmethylsulfonyl fluoride. Final volumes were adjusted to 300 mg/mL of tissue with lysis buffer, and cellular debris was removed by centrifugation at 4500 × g for 15 minutes at 4°C. Samples were analyzed for cytokine and chemokine protein expression using a BioSource 20-plex assay (Invitrogen) on a Luminex 100 instrument (Bio-Rad Laboratories) following the manufacturer's instructions. Samples were calculated as picograms per milliliter using a standard curve from in-plate standards and were subsequently converted to femtograms per milligram of brain tissue.
Isolation of Choroid Plexus From Neonatal Brain
The brain was removed from neonatal mice within 72 hours of birth with the base of the spinal cord attached. It was then placed in Dulbecco's modified Eagle's medium/Ham's F12 containing 10% fetal bovine serum and 1% penicillin/streptomycin (Invitrogen). The cerebellum was peeled from the base of the spinal cord, exposing the fourth ventricle. The choroid plexus was detached from the ventricle and placed into HBSS. The remaining brain tissue was divided through the midline, the choroid plexus was detached from the lateral ventricles and placed in HBSS. For RNA analysis, choroid plexus cells were placed in zRNA buffer (Zymo Research) and were lysed by trituration using a 27-gauge needle. RNA from choroid plexus cells was compared with RNA from neonatal astrocytes and microglia, which were isolated as previously described.21
Antigen Retrieval and IHC Analysis
IHC analysis was completed using an automated Discovery XT staining module and reagents (Ventana Medical Systems Inc., Tucson, AZ). For detection of microglia/macrophages, a 1:2000 dilution of polyclonal rabbit anti-Iba1 (Wako Chemicals USA Inc., Richmond, VA) was used. For detection of CD3, a 1:100 dilution of polyclonal rabbit anti-human CD3 (Dako, Carpinteria, CA) was used. Both antibodies were detected using a goat biotinylated anti-rabbit IgG (BioGenex Laboratories Inc., Fremont, CA) and appropriate substrate.
Detection of FITC-Labeled Agonists
Mice were inoculated ICV with 80 pmol of FITC-labeled CpG-ODN 1826 (InvivoGen) as the TLR9 agonist or with 5 nmol of FITC-labeled CL264 (InvivoGen), a TLR7 agonist that is similar in size and structure to imiquimod, as described previously herein. Brain tissues were removed at 1, 4, or 12 hpi; were cut into three coronal sections; and were processed for histologic analysis. Tissue sections on slides were deparaffinized in xylene; rehydrated in 100%, 95%, and 70% ethanol; and washed in PBS. Antigen retrieval was performed with citrate target retrieval solution (0.018 mol/L citric acid, 8.2 μmol/L sodium citrate dihydrate, pH 6.0) in a decloaking chamber (Biocare Medical, Concord, CA) at 120°C for 20 minutes following the manufacturer's instructions. Tissue sections were brought to room temperature and washed in 0.5% fish skin gelatin in PBS twice for 10 minutes, and then slides were incubated with donkey serum blocking solution [2% donkey serum, 1% bovine serum albumin, 0.05% fish skin gelatin, 0.1% Triton X-100 (Roche Diagnostics GmbH, Mannheim, Germany), and 0.05% Tween 20 (Promega Corp., Madison, WI) in PBS] for 30 minutes at room temperature. FITC-labeled agonists were detected using a 1:100 dilution of rabbit anti-fluorescein primary antibody (Invitrogen) and a 1:200 dilution of goat anti-rabbit Alexa Fluor 488 secondary antibody (Invitrogen). A nonlabeled CpG-ODN 1826 (InvivoGen)–inoculated mouse was used as a negative control for nonspecific staining.
Blood-Brain Barrier Permeability
Blood-brain barrier (BBB) permeability changes after TLR7 or TLR9 stimulation in the brain were determined by measuring Evans blue diffusion into the brain. Mice were inoculated ICV with imiquimod, CpG-ODN, or vehicle control but without trypan blue. One hundred microliters of 1% (w/v) Evans blue solution was injected i.p. 4 hours before testing BBB permeability (ie, at 8 or 20 hpi for testing BBB permeability at 12 and 24 hpi, respectively). Mice were anesthetized by deep inhalation anesthesia and were perfused via intracardiac puncture with 1 mL of PBS. Brains were subsequently removed and analyzed for the blue color using a stereomicroscope.
In Situ Hybridization and IHC Analysis
Expression of cytokine mRNA inside individual cells was determined by in situ hybridization followed by IHC analysis as previously described.19 In brief, the tissue sections were fixed in 4% paraformaldehyde, and proteins were denatured in 200 mmol/L HCl followed by treatment with 5 µg/mL of proteinase K. The sections were incubated overnight with digoxigenin-labeled RNA antisense or sense probes. Tissue sections were treated with RNase A and then were incubated with alkaline phosphatase antidigoxigenin antibody (Roche Diagnostics) for 60 minutes. Sections were washed and incubated with Fast Red solution (Dako) until sufficient color had developed. Slides were washed in blocking buffer (10X blocking buffer diluted in 1X maleic acid solution; Roche Diagnostics) and then were incubated overnight at 4°C with polyclonal rabbit anti–glial fibrillary acidic protein antibodies (Dako) or rabbit anti-Iba1 antibodies (Wako Chemicals USA Inc.). Slides were then incubated with goat anti-rabbit IgG secondary antibodies labeled with horseradish peroxidase conjugate (Zymed Laboratories, San Francisco, CA) and were developed with diaminobenzidine substrate (Pierce Biotechnology, Rockford, IL). Slides were then counterstained with hematoxylin. No staining was observed in mock controls or with nonspecific probes.
Results
Distinct Responses to TLR7 and TLR9 Stimulation in the Neonatal Brain
Studies with TLR9 agonists demonstrated that TLR9 agonists administered ICV were fatal in neonatal BALB/c mice.17 This response was not observed in neonatal IRW mice inoculated with TLR7 agonists,19 despite the fact that both receptors mediate similar signaling pathways. To directly compare stimulation of these two receptors in the CNS, we inoculated neonatal IRW mice with varying concentrations of both TLR7 and TLR9 agonists. For TLR9 stimulation, CpG-ODN 1826, a type B ODN, which differs from type A and type C ODNs based on primary sequence motif and secondary and tertiary structures,22,23 was chosen because it induced a stronger cytokine response in the CNS than either type A or type C agonists after either ICV inoculation in vivo (Figure 1A) or stimulation of astrocytes and microglia in vitro (data not shown). Specificity of ODN 1826 for TLR9 in the CNS was demonstrated using TLR9-deficient mice (Figure 1B). For TLR7 stimulation, imiquimod was chosen because it induces a slightly stronger inflammatory response in the CNS compared with other TLR7 agonists.19 Furthermore, studies with TLR7-deficient mice demonstrated that imiquimod-induced neuroinflammatory responses required TLR7.19 Similar to previous reports, ICV inoculation of TLR9 agonists in neonates induced unresponsiveness or death within 4 hours in all inoculated mice at 3100 pmol (10 μg/g bw) (data not shown). Dose analysis indicated that 155 to 2325 pmol (0.5 to 7.5 μg/g bw) of CpG-ODN–inoculated ICV induced clinical signs of unresponsiveness within 24 hours, whereas 80 pmol (0.25 μg/g bw) of CpG-ODN did not induce any clinical signs (Figure 2). In contrast to CpG-ODN stimulation, various amounts of the TLR7 agonist imiquimod ranging from 20 nmol (∼2.5 μg/g bw) to 500 nmol (∼62.5 μg/g bw) injected ICV did not induce any detectable signs of discomfort in neonatal mice.21 Thus, stimulation of TLR7 and TLR9 in the neonatal brain induced different clinical responses after direct inoculation in the CNS.
Figure 1.
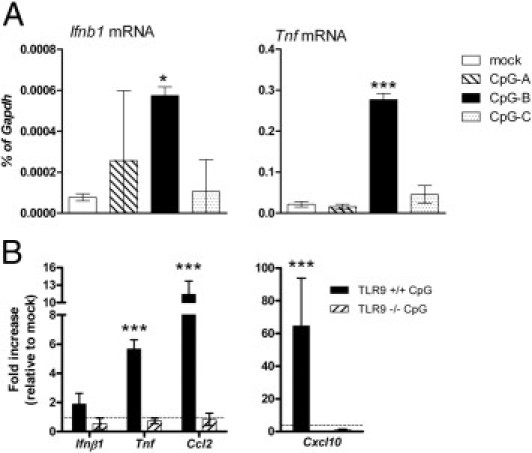
Comparison of cytokine responses induced by different types of CpG-ODN. A: Wild-type IRW mice were inoculated ICV with vehicle control or 80 pmol of CpG-ODN type A, B, or C, as described in Materials and Methods. Brain tissues were removed at 12 hpi, processed, and analyzed for mRNA expression. B: Wild-type and TLR9-deficient C57BL/6 mice were inoculated with vehicle control or 80 pmol of CpG-ODN type B. At 12 hpi, tissues were removed and processed as described previously herein. Data are the mean ± SD for four to five mice per group and are representative of two replicate experiments. Statistical analysis was completed by one-way analysis of variance with Newman-Keuls posttest. *P < 0.05, ***P < 0.001.
Figure 2.
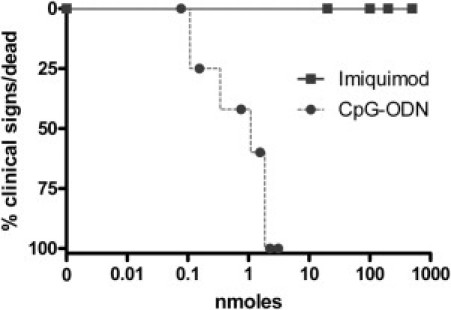
Survival curve analysis after imiquimod or CpG-ODN inoculation in the neonatal brain. Mice were inoculated with the indicated concentration of the TLR7 agonist imiquimod or the TLR9 agonist CpG-ODN 1826 and were observed for clinical signs of substantial distress or death within the first 12 hpi. Each symbol represents the percentage of mice with clinical signs or death for each dose with five to seven mice per dose.
Comparison of Cytokine Responses to TLR7 and TLR9 Agonist Administration in the CNS
To examine the difference in responses to TLR7 and TLR9 agonist stimulation in the CNS, we compared the proinflammatory cytokine response after TLR7 and TLR9 stimulation. For this comparison, we used the concentration of imiquimod that induced the strongest cytokine response after ICV inoculation (100 nmol, 12.5 μg/g bw)19 and the highest concentration of CpG-ODN 1826 (80 pmol, 0.25 μg/g bw) that did not induce distress or death in neonatal pups but still induced high levels of cytokines (data not shown). Inoculation of 100 nmol of the TLR7 agonist imiquimod or 80 pmol of the TLR9 agonist CpG-ODN induced similar expression levels of several cytokines, including IL-5, CXCL9, and CXCL10, in the CNS at 12 hpi (Figure 3). However, the cytokines IL-1α, IL-1β, IL-6, IL-12p40, and TNF and the chemokines CCL2, CCL3, and CXCL1 were induced substantially higher in the TLR9 agonist–inoculated mice (Figure 3). Low to undetectable levels of cytokine protein expression were observed at 24, 48, and 72 hpi for both agonists, indicating that the inflammatory response subsides within 24 hpi (data not shown). Analysis of different concentrations of type B CpG-ODN showed no substantial difference in cytokine levels in response to higher concentrations (data not shown). Thus, ICV inoculation of TLR9 agonists induced a substantially higher proinflammatory cytokine response than did TLR7 agonists. This in vivo response differs from in vitro comparisons in glial cells and B cells, where TLR7 and TLR9 agonists induced similar levels of cytokine production.21,24
Figure 3.
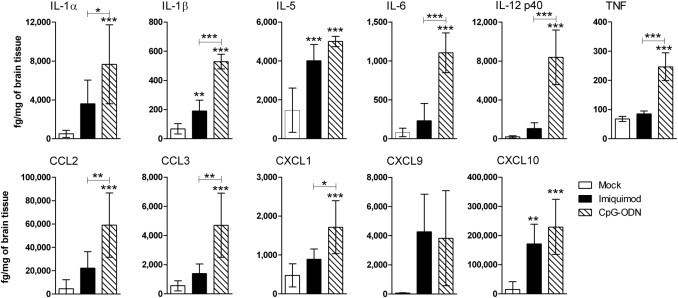
Comparison of proinflammatory cytokine responses after imiquimod or CpG-ODN inoculation in the brain. Mice were inoculated ICV with 100 nmol imiquimod or 80 pmol of CpG-ODN as described in Materials and Methods, and brain tissues were removed at 12 hpi. One half of the sagittally divided brain was homogenized in lysis buffer containing protease inhibitors and was analyzed for protein expression using a multiplex bead assay. Samples were calculated as picograms per milliliter using a standard curve from in-plate standards and were subsequently converted to femtograms per milligram of brain tissue. Data are the mean ± SD for four to eight mice per group. Statistical analysis was completed by one-way analysis of variance with Newman-Keuls posttest. *P < 0.05, **P < 0.01, and ***P < 0.001. Asterisks above bars indicate a significant up-regulation compared with mock-treated controls. Horizontal lines beneath asterisks indicate the difference between the indicated groups.
Kinetic analysis of mRNA expression of the proinflammatory cytokines responding to TLR7 or TLR9 stimulation in the CNS indicated peak up-regulation of cytokine mRNA between 2 and 12 hpi, with down-regulation by 24 to 48 hpi (Figure 4). Similar to protein levels, mRNA expression of proinflammatory cytokines Il6, Il12, Ccl2, and Tnf was significantly higher in CpG-ODN–inoculated mice compared with imiquimod-inoculated mice (Figure 4; data not shown). Some cytokine mRNA up-regulation was observed in imiquimod-inoculated mice, but these levels were not significant relative to the levels induced by CpG-ODN inoculation (Figure 4). The exception to this was the expression of Ifnb1, which was strongly induced at 2 hpi after imiquimod inoculation but was induced only at low levels in CpG-ODN–inoculated mice at 6 hpi (Figure 4). We further studied the extent of the type I IFN response after TLR7 or TLR9 stimulation in the CNS. Mice inoculated with TLR7 agonists had heighted mRNA expression of Ifnb1 but not Ifna2 or Ifna4 mRNA (Figure 5). The expression of Ifnb1 mRNA correlated with an increase in IFN-induced gene expression, including the up-regulation of IFN-induced protein with tetratricopeptide repeats 1 (Ifit1), Ifit3, myxovirus resistance 2 (Mx2), IFN regulatory factor 1 (Irf1), and IFN-induced 15-kDa protein (Isg15) mRNA (Figure 5). This was not observed in mice inoculated with TLR9 agonists. Thus, TLR7 and TLR9 stimulation results in different proinflammatory cytokine responses in the CNS. TLR7 stimulation leads to a pronounced Ifnb1 response and low-level production of other cytokines, whereas TLR9 stimulation leads to a pronounced proinflammatory cytokine response with limited type I IFN expression.
Figure 4.
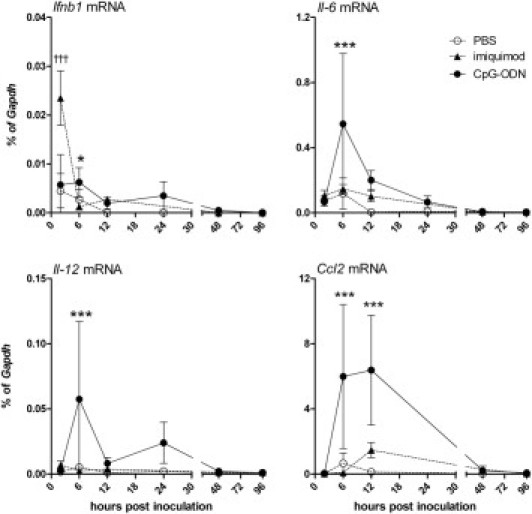
CpG-ODN and imiquimod inoculation induce different cytokine responses. Mice at 48 hours of age were ICV inoculated with 100 nmol of imiquimod or 80 pmol of CpG-ODN as described in Materials and Methods; brain tissues were removed at 2, 6, 12, 24, 48, or 96 hpi and were snap frozen, processed, and analyzed for mRNA expression. Some of the TLR7 agonist–stimulated samples were used in a previous study.19 Data are the mean ± SD for four to five mice per group. Statistical analysis was completed by one-way analysis of variance with Newman-Keuls posttest. *P < 0.05, ***P < 0.001 for CpG-ODN–inoculated mice compared with PBS controls; †††P < 0.001 for imiquimod-inoculated mice compared with PBS controls.
Figure 5.
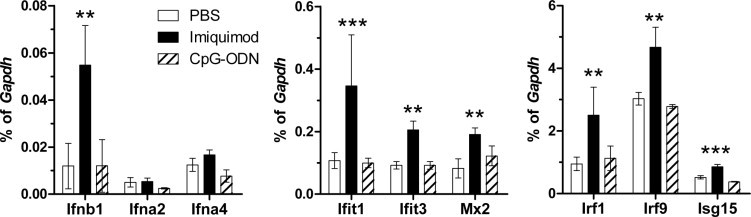
Imiquimod, but not CpG-ODN, inoculation induces up-regulation of interferon stimulated genes mRNA. mRNA from the 2 hpi time point described in Figure 4 were analyzed by real-time PCR analysis of 84 different IFN-related genes using a super array as described in Materials and Methods. Data of the array genes that were induced by either imiquimod or CpG-ODN stimulation are shown. Data are the mean ± SD for four mice per group. Statistical analysis was completed by one-way analysis of variance with Dunnett's posttest to compare the imiquimod- and CpG-ODN–treated groups with PBS controls. **P < 0.01, ***P < 0.001.
To examine whether the differences in the innate immune response induced by TLR7 versus TLR9 agonists were related to their location or longevity in the CNS after inoculation, FITC-labeled agonists were used to track the spread of agonists in the CNS and the length of time the agonists were detectable in the CNS. For this analysis, we used CL264 as the TLR7 agonist instead of imiquimod because a labeled form of this agonist was readily available. CL264 induces similar responses to imiquimod stimulation (data not shown), and both molecules are purine derivatives. Analysis of FITC-labeled TLR7 and TLR9 agonists in the CNS demonstrated that these agonists were widespread throughout the brain, including the brain parenchyma, and throughout the meninges by 1 hpi, with a similar pattern of spread (Figure 6). Both agonists were also readily detectable at 4 hpi, although not as strong as at 1 hpi (data not shown). By 12 hpi, both agonists were almost completely removed from the brain, with rare detection of agonist near blood vessels or in ventricles or meninges (data not shown). This indicates that both agonists were either degraded or removed from the CNS by 12 hpi. Thus, TLR7 and TLR9 agonists were widespread throughout the brain early after inoculation but did not remain in the CNS for longer than 12 hours, indicating similar longevity and spread throughout the CNS.
Figure 6.
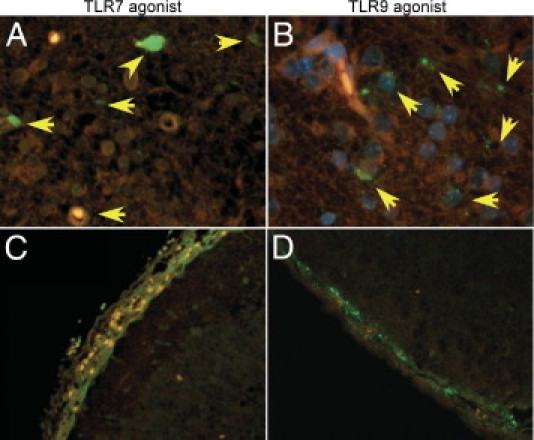
Detection of agonist in the CNS after inoculation. Mice were inoculated with 10 nmol of FITC-labeled TLR7 agonist CL264 (A and C), 80 pmol of FITC-labeled CpG-ODN (B and D), or unlabeled CpG-ODN (data not shown). Brain tissue was removed at 1 hpi, cut into three coronal sections, and processed for histologic analysis. Tissue sections were stained for FITC using rabbit antifluorescein primary antibody and goat anti-rabbit Alexa Fluor 488 secondary antibody. Original magnification: ×1000 (A and B); ×400 (C and D). A and B: Yellow arrows indicate FITC-labeled agonists taken up by cells in the brain parenchyma. C and D: FITC-labeled agonists in the meninges of inoculated mice at 1 hpi.
Source of Cytokine Expression After in Vivo Inoculation of TLR Agonists
One explanation for the differences in the cytokine responses to TLR7 and TLR9 stimulation could be the cell types activated by each receptor. To examine which cells were responsible for cytokine production, we used in situ hybridization analysis for detection of Tnf and Il6 mRNA, two cytokines that were differentially induced by TLR9 and TLR7 agonist stimulation. The primary cell types expressing Tnf and Il6 mRNA after TLR7 or TLR9 agonist stimulation were cells of the ventricles and choroid plexus (Figure 7). Cells in the lateral ventricles were positive for Tnf mRNA in both TLR7 agonist– and TLR9 agonist–inoculated mice (Figure 7, A and B). However, staining was more widespread in TLR9 agonist–inoculated mice with cells in the third ventricle positive for Tnf mRNA (Figure 7D), which was not observed in imiquimod-inoculated mice (Figure 7C). Il6 mRNA expression was also more readily detected in CpG-ODN–inoculated mice compared with imiquimod-inoculated mice (Figure 7, E and F). Il6 mRNA-positive cells were also occasionally observed in the brain parenchyma in tissues from both TLR7 agonist– and TLR9 agonist–inoculated mice (Figure 7F; data not shown). Neither Tnf nor Il6 mRNA-expressing cells were detected in mock-inoculated mice (data not shown). Thus, most cells expressing Tnf and Il6 mRNA were in the ventricles and choroid plexus of the brain, with higher numbers of cells expressing Tnf and Il6 mRNA in TLR9-agonist–inoculated mice compared with TLR7 agonist–inoculated mice.
Figure 7.
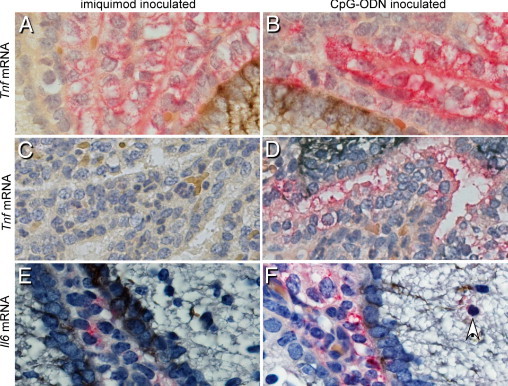
In situ hybridization and IHC analysis of coronal sections of the brain tissues from imiquimod- and CpG-ODN–inoculated mice at 12 hpi. Tissue sections from imiquimod-inoculated (A, C, and E) or CpG-ODN–inoculated (B, D, and F) mice were analyzed for Tnf (A–D) or Il6 (E and F) mRNA expression using a digoxigenin-labeled RNA probe and detected with Fast Red substrate (red color). Sections were also analyzed for the astrocyte marker glial fibrillary acidic protein by IHC analysis using a peroxidase antibody and diaminobenzidine (brown black) to discern between ependymal cells and astrocytes. Tnf mRNA was strongly detected in the lateral ventricle (B) and third ventricle (D) of CpG-ODN–inoculated mice, whereas only some of the cells were positive for Tnf mRNA in imiquimod-inoculated mice (A and C). Il6 mRNA was also detected more strongly in the lateral ventricles of CpG-ODN–inoculated mice (F) compared with imiquimod-inoculated mice (E). The arrow points to Il6 mRNA-positive infiltrating cell outside the ventricle. Original magnification: ×1000 (A and B); ×400 (C–F). All the images were taken using a digital camera attached to a Nikon scope. Images are representative of the tissue. Nonspecific RNA probes and no–primary antibody controls were used as negative controls for all the experiments.
The response of choroid plexus cells in TLR7 agonist– and TLR9 agonist–inoculated mice could be a direct response by choroid plexus cells to TLR stimulation or could be a secondary response to microglia or astrocytes activated by TLR7 or TLR9. Expression of TLR9 on choroid epithelia or on ependymal cells has not been reported, and only low-level expression of TLR7 on ependymal cells has been observed.25 We, therefore, examined choroid plexus from neonatal mice for their expression of Tlr7 and Tlr9 mRNA and compared this expression with that of purified astrocytes, which readily respond to TLR7 or TLR9 stimulation despite low levels of TLR7 or TLR9 expression, as well as microglia, which express higher levels of both receptors. Analysis of cell-specific mRNA expression of Ttr, Gfap, and F4/80 was used to verify the population of choroid plexus cells, astrocytes, and microglia (Figure 8, A–C). Tlr9 mRNA expression was comparable between astrocytes and choroid plexus cells, whereas Tlr7 mRNA expression was lower in choroid plexus cells (Figure 8, D and E). Analysis of the mRNA expression of downstream signaling molecules indicated that choroid plexus cells expressed Myd88 and Irf7 mRNA at similar levels to those found in astrocytes (Figure 8, F and G). As expected, mRNA expression of all four genes was lower in astrocytes and choroid plexus cells compared with microglia, although mRNA expression of Tlr9, Myd88,and Irf7 was only twofold to threefold lower in choroid plexus cells compared with microglia. The expression of Tlr9, Myd88, and Irf7 mRNA by choroid plexus cells suggests that the cytokine production by these cells could be a primary response to TLR9 stimulation.
Figure 8.
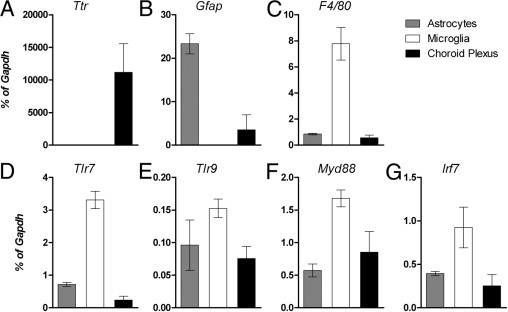
Choroid plexus cells express Tlr9, Myd88, and Irf7 mRNA at comparable levels to astrocytes. Choroid plexus from the ventricles was removed as described in Materials and Methods. mRNA from purified astrocytes, microglia, and choroid plexus cells were analyzed for expression of Ttr (A), Gfap (B), and F4/80 (C) mRNA by real-time PCR to confirm the purity of each population. mRNA from each cell population was then examined for the expression of Tlr7 (D), Tlr9 (E), Myd88 (F), and Irf7 (G) mRNA. Data are the mean ± SD for five to six samples per group.
TLR9 Stimulation Compromises the Blood-CSF Barrier and Results in Significant Cellular Infiltrate in the CNS
The cytokine response by cells of the choroid plexus to CpG-ODN stimulation suggested that activation of these cells through TLR9 may affect the integrity of the blood-CSF barrier. To examine whether CpG-ODN inoculation compromised the blood-CSF barrier or the BBB, we inoculated mice i.p. with Evans blue dye after agonist inoculation. No Evans blue staining was detectable by gross examination of brain tissue at 12 hpi. However, localization of Evans blue was observed in brain tissue from four of five CpG-ODN–inoculated mice at 24 hpi (Figure 9C). Evans blue staining was principally detected in the areas of the ventricles (Figure 9C) and was most readily observed in the tissue rather than on the surface of the brain tissue. This suggests that the blood-CSF barrier was compromised rather than widespread breakdown of the BBB. Faint to undetectable levels of Evans blue dye was observed in brain tissue from imiquimod-inoculated mice (Figure 9B), whereas no Evans blue was observed in mock-inoculated mice (Figure 9A). Thus, the blood-CSF barrier seems to be compromised by TLR9 agonists by 24 hpi, although there does not seem to be a complete breakdown of the BBB at this time point.
Figure 9.
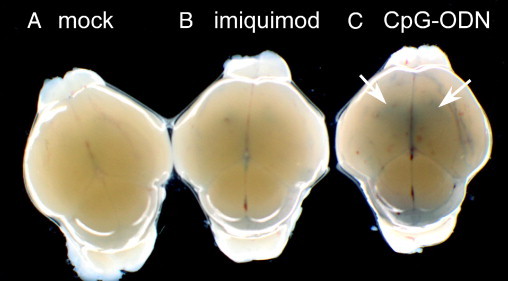
CpG-ODN induces breakdown of the blood-CSF barrier. Mice were inoculated with mock control (A), 100 nmol of imiquimod (B), or 80 pmol of CpG-ODN (C). A–C: At 20 hpi, mice were inoculated with Evans blue dye i.p. Four hours later, mice were perfused with PBS and brain tissue was removed and analyzed using a stereomicroscope. Blue dye was observed in ventricle areas (arrows) from CpG-ODN–inoculated mice compared with either mock- or imiquimod-inoculated mice. Data are representative of four to five mice per group from two replicate experiments. All the mock-inoculated brains were negative for Evans blue. Weak staining was observed in the imiquimod-inoculated brains. Of the CpG-ODN–inoculated mouse brains, one showed weak to no staining, whereas four of five were clearly positive for Evans blue. The dark line in C on the right side of the CpG-ODN–inoculated brain is a scissor cut.
Increased mRNA levels of Cd3ε and F4/80 were also observed in brain tissue from CpG-ODN–inoculated mice, suggesting recruitment of T cells and recruitment/activation of macrophages/microglia to the CNS, respectively. Increased mRNA expression for both molecules peaked at 48 hpi but remained elevated for 4 days after stimulation (Figure 10). In contrast, limited to no increase in Cd3ε or F4/80 mRNA expression was observed in TLR7 agonist–inoculated mice, as previously reported.19 Histologic analysis of brain tissue sections indicated that the cellular infiltrates were primarily in the meninges and ventricles of inoculated mice (Figure 11). All five of the CpG-ODN–inoculated mice had severe meningitis, whereas only one of four imiquimod-inoculated mice had severe meningitis. In addition, imiquimod- and CpG-ODN–inoculated mice had moderate to severe suppurative ventriculitis, which was characterized by mild hemorrhage and cellular infiltration of the lateral ventricles. This was not observed in control-treated mice. Only minimal perivascular cuffing was observed in CpG-ODN– or imiquimod-inoculated mice at 48 hpi. IHC analysis indicated increased staining for the microglia/macrophage marker Iba1 and CD3, primarily around the ventricles and in the meninges in TLR9 agonist–inoculated mice compared with TLR7 agonist–inoculated mice (Figure 12; data not shown). Analysis of Iba1 demonstrated positive cells with elongated processes, suggesting an increase in reactivity of microglia cells and cells without elongated process, which may be either activated microglia or activated macrophages (Figure 12E). These results suggest that activation of cells in the choroid plexus through TLR9, but not TLR7, results in damage to the blood-CSF barrier and in the recruitment of peripheral cells in the brain. Thus, a single inoculum of CpG-ODN in the neonatal brain is sufficient to induce a pronounced inflammatory response in the CNS, including the production of high levels of proinflammatory cytokines, damage to the blood-CSF barrier, and recruitment of inflammatory cells into the CNS. This strong neuroinflammatory response may explain the clinical unresponsiveness of neonatal mice after inoculation with CpG-ODN.
Figure 10.
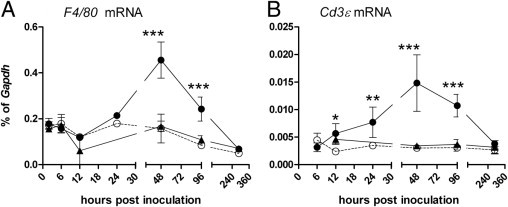
Kinetics of cellular responses in the CNS after imiquimod or CpG-ODN inoculation in the neonatal brain. The RNA samples described in Figure 4 were processed for real-time quantitative RT-PCR analysis using primers specific for F4/80 (A) and Cd3e (B) mRNA. Values were calculated relative to the expression of Gapdh controls. Data are the mean ± SD for four to seven mice per group per time point and represent the combined data from two independent experiments. Statistical analysis was completed by two-way analysis of variance with Bonferroni posttest. *P < 0.05, **P < 0.01, and ***P < 0.001.
Figure 11.
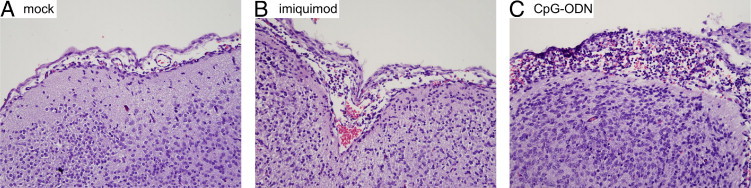
Meningitis associated with TLR9 agonist inoculation. H&E-stained tissue sections from mock (A), imiquimod (B), or CpG-ODN (C) inoculated mice were analyzed in a blinded study for histologic changes. Substantial inflammation was observed in the meninges of CpG-ODN–inoculated mice (C) compared with mock-treated (A) or imiquimod-treated (B) mice. Original magnification, ×100.
Figure 12.
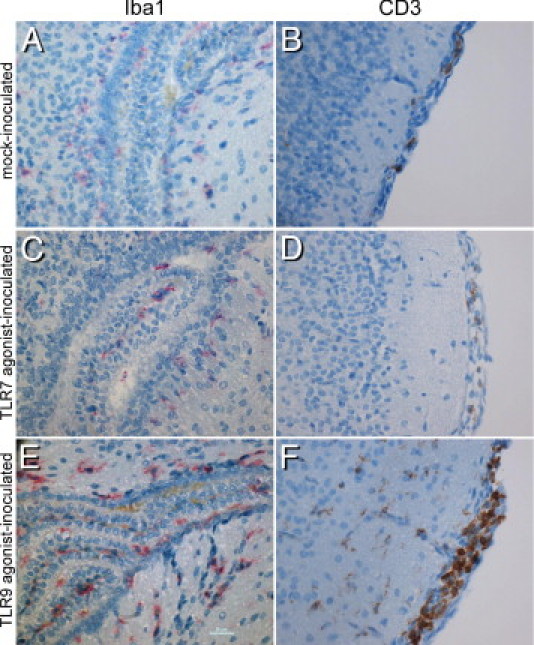
Increased presence of Iba1+ cells and CD3+ cells in CpG-ODN–inoculated mice. Brain tissue from mock- (A and B), imiquimod- (C and D), and CpG-ODN– (E and F) inoculated mice were stained for the microglia/macrophage marker Iba1 (red) (A, C, and E) or CD3 (brown) (B, D, and F). Increased staining for Iba1 and CD3 was detected in brain tissue from CpG-ODN–inoculated mice compared with either mock- or imiquimod-inoculated mice. A, C, and E: Iba1+ cells in and near the ventricles. B, D, and F: CD3+ cells in the meninges of inoculated mice, noting the higher number of CD3 cells in CpG-ODN–inoculated mice.
Influence of TLR7 on TLR9-Mediated Neuroinflammatory Responses
Multiple studies have suggested interplay between TLR7 and TLR9 in regulating innate immune responses.21,26,27 To examine whether TLR7 influenced TLR9-mediated unresponsiveness in mice, we inoculated wild-type and TLR7-deficient mice with a range of CpG-ODN concentrations. TLR7-deficient mice were more sensitive to CpG-ODN–induced unresponsiveness, with low amounts of CpG-ODN inducing unresponsiveness or death in TLR7-deficient mice but not in wild-type mice (Figure 13A). This correlated with increased production of several cytokines in TLR7-deficient mice compared with wild-type controls (Figure 13, C–E), although not all cytokines were up-regulated in TLR7-deficient animals (Figure 13B). Thus, TLR7 deficiency affects CpG-ODN–induced neuroinflammation and unresponsiveness in neonatal mice, demonstrating TLR7 regulation of TLR9-mediated responses in the CNS.
Figure 13.
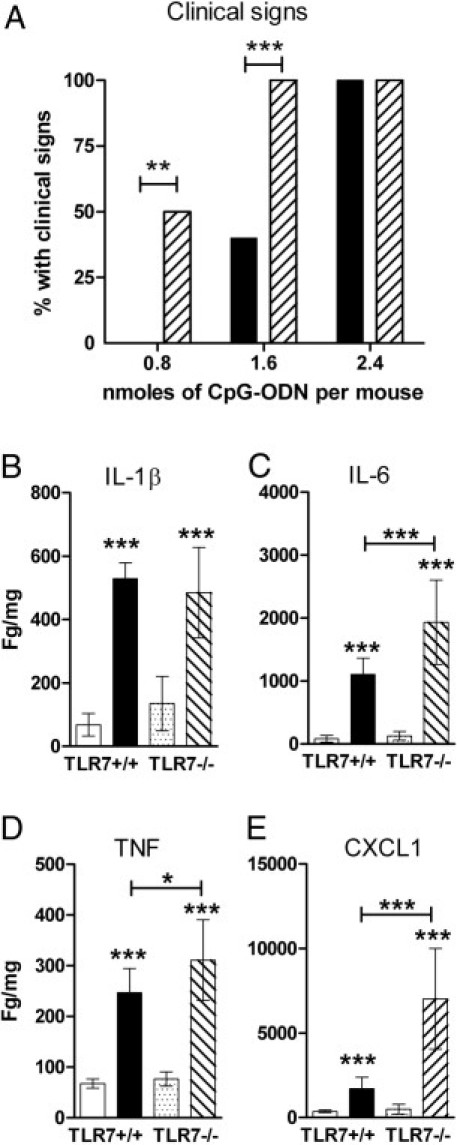
TLR7-deficient mice are more susceptible to CpG-ODN–induced clinical signs and induced heightened cytokine responses compared with wild-type mice. A: Wild-type or TLR7-deficient mice were inoculated with the indicated concentration of CpG-ODN 1826 and were observed for clinical signs of substantial distress or death within the first 12 hpi. Data are presented as the percentage of mice that developed clinical signs for each amount of CpG-ODN with 5 to 12 mice per group per strain. For statistical analysis, mice were scored as 0 for nonclinical and 1 for clinical and were analyzed by two-way analysis of variance with a Bonferroni posttest to determine significance. Black bars indicate TLR7+/+; hashed bars, TLR7−/− B–E: Brain tissue from wild-type or TLR7-deficient mice were removed at 12 hpi with PBS or CpG-ODN and were analyzed for cytokine levels as described in Figure 3. Statistical analysis was completed by one-way analysis of variance with Newman-Keuls posttest. *P < 0.05, **P < 0.01, and ***P < 0.001. Asterisks above bars indicate a significant up-regulation compared with mock-treated controls. Horizontal lines beneath asterisks indicate the difference between the indicated groups. White bars indicate PBS, TLR7+/+; black bars, CpG-ODN, TLR7+/+; dotted bars, PBS, TLR7−/−; hashed bars, CpG-ODN, TLR7−/−.
Discussion
In the present study, we found that stimulation of TLR7 and TLR9 in the CNS lead to distinct neuroinflammatory responses. Stimulation of TLR9 resulted in a strong proinflammatory cytokine response with recruitment of inflammatory cells to the CNS and, at higher concentrations, induced unresponsiveness or death in mice. In contrast, stimulation through TLR7 leads to a strong IFNβ response but minimal inflammation and no observable clinical signs. The dissimilarity in the neuroinflammatory response elicited by TLR7 and TLR9 signaling may explain some of the differences observed in the ability of TLR7 and TLR9 agonists to provide protection in the CNS during viral infection and in the toxic nature of CpG-ODNs in the CNS.17,18 This in vivo result differs substantially from in vitro stimulation of these receptors, which elicited similar cytokine responses in astrocytes, microglia, and B cells.21,24
The difference in the proinflammatory responses between TLR7 and TLR9 agonist stimulation were due, at least in part, to their ability to activate cells of the choroid plexus to produce proinflammatory cytokines, such as TNF and IL-6. The difference in this response was surprising because TLR9 is not readily detectable on choroid plexus cells by IHC analysis of mouse brain tissue.25 However, analysis of gene expression of isolated choroid plexus demonstrated expression of Tlr9, Myd88, and Irf7 mRNA at comparable levels as those observed with astrocytes (Figure 8), indicating that these cells may be directly responding to CpG-ODN stimulation. A similar phenomenon is observed with lipopolysaccharide induction of TNF expression by ependymal cells, despite the lack of detectable TLR4 on these cells.25,28,29 The strong response of choroid plexus cells to CpG-ODN in this study may be due, in part, to the route of CpG-ODN. Analysis of agonists after ICV inoculation showed widespread distribution at 1 and 4 hpi, but staining was also intense in the ventricles themselves. Thus, cells in the ventricles would be in contact with a higher concentration of CpG-ODNs than other cells in the CNS and, thereby, have a greater response to CpG-ODNs. The ability of choroid plexus epithelial cells to respond to pathogen-associated molecular patterns would allow these cells to play an important role in allowing recruitment of inflammatory cells during the early stages of an infection in the CNS. However, prolonged administration of CpG-ODNs in the adult mouse brain resulted in loss of cilia and swelling of ependymal cells and in damage to the ventricle lining,30 suggesting that continued activation via TLR9 may lead to ependymal death, either through direct stimulation or by activation of surrounding cell types.
Other cell types may also play a contributing role in the proinflammatory cytokine response. Il6-expressing cells were observed close to blood vessels in TLR7- and TLR9-stimulated brain tissues, indicating that infiltrating cells might be the source of the cytokines induced by TLR7 or TLR9 agonist stimulation (Figure 7F). In addition, high numbers of microglia/macrophages were detected in the ventricles of CpG-ODN–inoculated mice and may contribute to activating choroid plexus cells (Figure 12E). In vitro stimulation of microglia or astrocytes with imiquimod or CpG-ODN induces a robust proinflammatory cytokine profile.21 However, no substantial differences in proinflammatory cytokine responses were observed between CpG-ODN and imiquimod stimulation of either cell type,21 suggesting that astrocytes and microglia may not be the primary mediators of the heightened proinflammatory cytokine response to CpG-ODN inoculation. TLR7 agonists did induce twofold to threefold higher levels of Ifnb1 mRNA than did TLR9 agonist stimulation in primary astrocytes and microglia in vitro.21 Therefore, it is possible that glial cells are responsible for the heightened levels of IFNβ to TLR7 stimulation in vivo. However, repeated attempts to detect Ifnb1 mRNA in tissue sections by in situ hybridization analysis were not successful, so we were unable to confirm whether these cell types were the primary source of Ifnb1.
The high amount of imiquimod needed to elicit neuroinflammatory responses relative to CpG-ODN may simply be due to a lower affinity of imiquimod for TLR7 than of CpG-ODN for TLR9. In addition, the molar amounts calculated assume imiquimod to be an individual molecule rather than a complex of stacked molecules, which imiquimod is known to form. Thus, the true molar amounts of imiquimod may be substantially lower. However, the low response to imiquimod stimulation is not due to a concentration effect because the amount of imiquimod in this study has been previously shown to be optimal for proinflammatory cytokine induction in the CNS.19 Other TLR7 agonists, including single-stranded RNA, did not induce as strong of proinflammatory cytokine responses in the CNS as imiquimod (data not shown).19 Thus, the neuroinflammatory response to TLR7 agonist in this study was the optimal response to TLR7 observed in the CNS, despite the low level compared with CpG-ODN-induced neuroinflammation.
The enhanced response to TLR9 agonist stimulation in TLR7-deficient mice, both in clinical response and heighted cytokine response, indicates that TLR7 has a modulatory role in regulating TLR9-mediated responses in the CNS (Figure 13). We previously showed that TLR7 deficiency in microglia, but not in astrocytes, also affects the cytokine response to TLR9 agonist stimulation.21 Cross-regulation between TLR7 and TLR9 has also been observed in systemic lupus erythematosus.26,27 Furthermore, deficiency in murine TLR8, a molecule that is considered not to be fully functional in mice, enhances TLR7-mediated responses.31 It is possible that the regulation is due to competition for shared proteins among TLR7, TLR8, and TLR9. The lack of one receptor enhances the availability of accessory proteins for the other receptors. UNC93b1, an accessory protein that binds to TLR7 and TLR9, preferentially binds TLR9, which limits TLR7 signaling. Because TLR7 and TLR9 share many adaptor proteins, TLR7 may preferentially bind certain adaptor proteins and, thus, limit the potential for TLR9-mediated signaling.
TLR9 agonists are currently being studied as immunomodulatory compounds for the treatment of a variety of illnesses, including cancer, viral infections, and autoimmune diseases, and as immune response modifiers to enhance vaccines.32–36 For example, treatment with TLR9 agonists has been recently shown to ablate glioblastomas in the CNS.37 In the present study, TLR9 agonists induced substantially higher proinflammatory responses in the CNS compared with TLR7 agonists. A single low-dose inoculum of TLR9 agonist induced pronounced inflammation in the brain, including severe meningitis, that remained up to 4 days after inoculation. Although this strong response may be responsible for the ability of TLR9 agonists to affect virus-induced neurologic disease, it also suggests that TLR9 activation, either by insult or by immunomodulatory molecules, may have damaging effects on the CNS as well.
Acknowledgments
We thank Sue Priola, Sonja Best, Byron Caughey, and Deborah Tribouillard-Tanvier for critical reading of the manuscript and Dan Long and Rebecca Rosenke for technical assistance with IHC analysis.
Footnotes
Supported in part by the Intramural Research Program of the NIH and in part by National Center for Research Resources grant IP20RR020159.
References
- 1.Asensio V.C., Campbell I.L. Chemokine gene expression in the brains of mice with lymphocytic choriomeningitis. J Virol. 1997;71:7832–7840. doi: 10.1128/jvi.71.10.7832-7840.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dickson D.W., Lee S.C., Mattiace L.A., Yen S.H., Brosnan C. Microglia and cytokines in neurological disease, with special reference to AIDS and Alzheimer's disease. Glia. 1993;7:75–83. doi: 10.1002/glia.440070113. [DOI] [PubMed] [Google Scholar]
- 3.Griffin D.E. Immune responses to RNA-virus infections of the CNS. Nat Rev Immunol. 2003;3:493–502. doi: 10.1038/nri1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kelder W., McArthur J.C., Nance-Sproson T., McClernon D., Griffin D.E. Beta-chemokines MCP-1 and RANTES are selectively increased in cerebrospinal fluid of patients with human immunodeficiency virus-associated dementia. Ann Neurol. 1998;44:831–835. doi: 10.1002/ana.410440521. [DOI] [PubMed] [Google Scholar]
- 5.McCoig C., Castrejon M.M., Saavedra-Lozano J., Castano E., Baez C., Lanier E.R., Saez-Llorens X., Ramilo O. Cerebrospinal fluid and plasma concentrations of proinflammatory mediators in human immunodeficiency virus-infected children. Pediatr Infect Dis J. 2004;23:114–118. doi: 10.1097/01.inf.0000109247.67480.7a. [DOI] [PubMed] [Google Scholar]
- 6.Diebold S.S., Kaisho T., Hemmi H., Akira S., Reis e Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 2004;303:1529–1531. doi: 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- 7.Heil F., Hemmi H., Hochrein H., Ampenberger F., Kirschning C., Akira S., Lipford G., Wagner H., Bauer S. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science. 2004;303:1526–1529. doi: 10.1126/science.1093620. [DOI] [PubMed] [Google Scholar]
- 8.Lund J.M., Alexopoulou L., Sato A., Karow M., Adams N.C., Gale N.W., Iwasaki A., Flavell R.A. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc Natl Acad Sci U S A. 2004;101:5598–5603. doi: 10.1073/pnas.0400937101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barton G.M. Viral recognition by Toll-like receptors. Semin Immunol. 2007;19:33–40. doi: 10.1016/j.smim.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 10.Bauer S., Kirschning C.J., Hacker H., Redecke V., Hausmann S., Akira S., Wagner H., Lipford G.B. Human TLR9 confers responsiveness to bacterial DNA via species-specific CpG motif recognition. Proc Natl Acad Sci U S A. 2001;98:9237–9242. doi: 10.1073/pnas.161293498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hemmi H., Takeuchi O., Kawai T., Kaisho T., Sato S., Sanjo H., Matsumoto M., Hoshino K., Wagner H., Takeda K., Akira S. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–745. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 12.Lewis S.D., Butchi N.B., Khaleduzzaman M., Morgan T.W., Du M., Pourciau S., Baker D.G., Akira S., Peterson K.E. Toll-like receptor 7 is not necessary for retroviral neuropathogenesis but does contribute to virus-induced neuroinflammation. J Neurovirol. 2008;1 doi: 10.1080/13550280802345723. [DOI] [PubMed] [Google Scholar]; –11
- 13.Lima G.K., Zolini G.P., Mansur D.S., Freire Lima B.H., Wischhoff U., Astigarraga R.G., Dias M.F., Silva M.G., Bela S.R., do Valle Antonelli L.R., Arantes R.M., Gazzinelli R.T., Bafica A., Kroon E.G., Campos M.A. Toll-like receptor (TLR) 2 and TLR9 expressed in trigeminal ganglia are critical to viral control during herpes simplex virus 1 infection. Am J Pathol. 2010;177:2433–2445. doi: 10.2353/ajpath.2010.100121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sorensen L.N., Reinert L.S., Malmgaard L., Bartholdy C., Thomsen A.R., Paludan S.R. TLR2 and TLR9 synergistically control herpes simplex virus infection in the brain. J Immunol. 2008;181:8604–8612. doi: 10.4049/jimmunol.181.12.8604. [DOI] [PubMed] [Google Scholar]
- 15.Town T., Bai F., Wang T., Kaplan A.T., Qian F., Montgomery R.R., Anderson J.F., Flavell R.A., Fikrig E. Toll-like receptor 7 mitigates lethal West Nile encephalitis via interleukin 23-dependent immune cell infiltration and homing. Immunity. 2009;30:242–253. doi: 10.1016/j.immuni.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Welte T., Reagan K., Fang H., Machain-Williams C., Zheng X., Mendell N., Chang G.J., Wu P., Blair C.D., Wang T. Toll-like receptor 7-induced immune response to cutaneous West Nile virus infection. J Gen Virol. 2009;90:2660–2668. doi: 10.1099/vir.0.011783-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pedras-Vasconcelos J.A., Goucher D., Puig M., Tonelli L.H., Wang V., Ito S., Verthelyi D. CpG oligodeoxynucleotides protect newborn mice from a lethal challenge with the neurotropic Tacaribe arenavirus. J Immunol. 2006;176:4940–4949. doi: 10.4049/jimmunol.176.8.4940. [DOI] [PubMed] [Google Scholar]
- 18.Pedras-Vasconcelos J.A., Puig M., Sauder C., Wolbert C., Ovanesov M., Goucher D., Verthelyi D. Immunotherapy with CpG oligonucleotides and antibodies to TNF-α rescues neonatal mice from lethal arenavirus-induced meningoencephalitis. J Immunol. 2008;180:8231–8240. doi: 10.4049/jimmunol.180.12.8231. [DOI] [PubMed] [Google Scholar]
- 19.Butchi N.B., Pourciau S., Du M., Morgan T.W., Peterson K.E. Analysis of the neuroinflammatory response to TLR7 stimulation in the brain: comparison of multiple TLR7 and/or TLR8 agonists. J Immunol. 2008;180:7604–7612. doi: 10.4049/jimmunol.180.11.7604. [DOI] [PubMed] [Google Scholar]
- 20.Hemmi H., Kaisho T., Takeuchi O., Sato S., Sanjo H., Hoshino K., Horiuchi T., Tomizawa H., Takeda K., Akira S. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat Immunol. 2002;3:196–200. doi: 10.1038/ni758. [DOI] [PubMed] [Google Scholar]
- 21.Butchi N.B., Du M., Peterson K.E. Interactions between TLR7 and TLR9 agonists and receptors regulate innate immune responses by astrocytes and microglia. Glia. 2010;58:650–664. doi: 10.1002/glia.20952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krug A., Rothenfusser S., Hornung V., Jahrsdorfer B., Blackwell S., Ballas Z.K., Endres S., Krieg A.M., Hartmann G. Identification of CpG oligonucleotide sequences with high induction of IFN-α/β in plasmacytoid dendritic cells. Eur J Immunol. 2001;31:2154–2163. doi: 10.1002/1521-4141(200107)31:7<2154::aid-immu2154>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 23.Marshall J.D., Fearon K.L., Higgins D., Hessel E.M., Kanzler H., Abbate C., Yee P., Gregorio J., Cruz T.D., Lizcano J.O., Zolotorev A., McClure H.M., Brasky K.M., Murthy K.K., Coffman R.L., Nest G.V. Superior activity of the type C class of ISS in vitro and in vivo across multiple species. DNA Cell Biol. 2005;24:63–72. doi: 10.1089/dna.2005.24.63. [DOI] [PubMed] [Google Scholar]
- 24.Hanten J.A., Vasilakos J.P., Riter C.L., Neys L., Lipson K.E., Alkan S.S., Birmachu W. Comparison of human B cell activation by TLR7 and TLR9 agonists. BMC Immunol. 2008;9:39. doi: 10.1186/1471-2172-9-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mishra B.B., Mishra P.K., Teale J.M. Expression and distribution of Toll-like receptors in the brain during murine neurocysticercosis. J Neuroimmunol. 2006;181:46–56. doi: 10.1016/j.jneuroim.2006.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nickerson K.M., Christensen S.R., Shupe J., Kashgarian M., Kim D., Elkon K., Shlomchik M.J. TLR9 regulates TLR7- and MyD88-dependent autoantibody production and disease in a murine model of lupus. J Immunol. 2010;184:1840–1848. doi: 10.4049/jimmunol.0902592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Santiago-Raber M.L., Dunand-Sauthier I., Wu T., Li Q.Z., Uematsu S., Akira S., Reith W., Mohan C., Kotzin B.L., Izui S. Critical role of TLR7 in the acceleration of systemic lupus erythematosus in TLR9-deficient mice. J Autoimmun. 2010;34:339–348. doi: 10.1016/j.jaut.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 28.Kim Y.S., Honkaniemi J., Sharp F.R., Tauber M.G. Expression of proinflammatory cytokines tumor necrosis factor-α and interleukin-1β in the brain during experimental group B streptococcal meningitis. Brain Res Mol Brain Res. 2004;128:95–102. doi: 10.1016/j.molbrainres.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 29.Tarlow M.J., Jenkins R., Comis S.D., Osborne M.P., Stephens S., Stanley P., Crocker J. Ependymal cells of the choroid plexus express tumour necrosis factor-α. Neuropathol Appl Neurobiol. 1993;19:324–328. doi: 10.1111/j.1365-2990.1993.tb00447.x. [DOI] [PubMed] [Google Scholar]
- 30.Tauber S.C., Ebert S., Weishaupt J.H., Reich A., Nau R., Gerber J. Stimulation of Toll-like receptor 9 by chronic intraventricular unmethylated cytosine-guanine DNA infusion causes neuroinflammation and impaired spatial memory. J Neuropathol Exp Neurol. 2009;68:1116–1124. doi: 10.1097/NEN.0b013e3181b7fde5. [DOI] [PubMed] [Google Scholar]
- 31.Demaria O., Pagni P.P., Traub S., de G.A., Branzk N., Murphy A.J., Valenzuela D.M., Yancopoulos G.D., Flavell R.A., Alexopoulou L. TLR8 deficiency leads to autoimmunity in mice. J Clin Invest. 2010;120:3651–3662. doi: 10.1172/JCI42081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Broomfield S.A., van der Most R.G., Prosser A.C., Mahendran S., Tovey M.G., Smyth M.J., Robinson B.W., Currie A.J. Locally administered TLR7 agonists drive systemic antitumor immune responses that are enhanced by anti-CD40 immunotherapy. J Immunol. 2009;182:5217–5224. doi: 10.4049/jimmunol.0803826. [DOI] [PubMed] [Google Scholar]
- 33.Krieg A.M. Therapeutic potential of Toll-like receptor 9 activation. Nat Rev Drug Discov. 2006;5:471–484. doi: 10.1038/nrd2059. [DOI] [PubMed] [Google Scholar]
- 34.Murad Y.M., Clay T.M. CpG oligodeoxynucleotides as TLR9 agonists: therapeutic applications in cancer. BioDrugs. 2009;23:361–375. doi: 10.2165/11316930-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 35.Panter G., Kuznik A., Jerala R. Therapeutic applications of nucleic acids as ligands for Toll-like receptors. Curr Opin Mol Ther. 2009;11:133–145. [PubMed] [Google Scholar]
- 36.Zaks K., Jordan M., Guth A., Sellins K., Kedl R., Izzo A., Bosio C., Dow S. Efficient immunization and cross-priming by vaccine adjuvants containing TLR3 or TLR9 agonists complexed to cationic liposomes. J Immunol. 2006;176:7335–7345. doi: 10.4049/jimmunol.176.12.7335. [DOI] [PubMed] [Google Scholar]
- 37.Alizadeh D., Zhang L., Brown C.E., Farrukh O., Jensen M.C., Badie B. Induction of anti-glioma natural killer cell response following multiple low-dose intracerebral CpG therapy. Clin Cancer Res. 2010;16:3399–3408. doi: 10.1158/1078-0432.CCR-09-3087. [DOI] [PMC free article] [PubMed] [Google Scholar]