

Abstract
Highly malignant human gliomas overexpress the G-protein–coupled chemoattractant receptor formyl peptide receptor (FPR1), which promotes tumor progression when activated. Our previous studies demonstrated that necrotic glioblastoma cells release chemotactic agonist(s) that activate FPR1 on viable tumor cells. In the present study, we identified an FPR1 agonist released by necrotic human glioblastoma cells. Necrotic tumor cell supernatant (NecSup) contained Annexin 1 (Anx A1), a chemotatic polypeptide agonist for FPR1. Immunoabsorption of Anx A1 with a specific antibody markedly reduced the chemotactic activity of NecSup for tumor cells and diminished its capacity to promote tumor cell growth, invasion, and colony formation on soft agar. In addition, Anx A1 was present in tumor xenografts formed by human glioblastoma cells in nude mice. Anx A1 knockdown significantly reduced the tumorigenicity of glioblastoma cells in nude mice, but FPR1/Anx A1 double knockdown diminished tumor growth even further. The clinical relevance of Anx A1 in gliomas was supported by the observation that Anx A1 was more highly expressed in poorly differentiated human primary gliomas compared with lower grade tumors. Our study implicates Anx A1 as a major component in necrotic tumor cell-derived stimulants of the growth of glioblastoma via the activation of FPR1.
Malignant glioma cells overexpress several cell surface receptors that, by sensing agonists present in the tumor microenvironment, promote tumor growth, invasion, and production of angiogenic factors. Previous studies have shown that tumor cells from highly malignant human glioma specimens express formyl peptide receptor 1 (FPR1),1 a seven transmembrane G-protein–coupled receptor (GPCR) that mediates leukocyte chemotaxis on activation by bacterial and host-derived agonists. FPR1 was originally identified in phagocytic leukocytes and plays an important role in host defense. Recently, FPR1 was reported to be also present in non-myeloid cells,2–4 suggesting additional pathophysiological functions of this GPCR. In glioblastoma (GBM) cell lines, FPR1 activation enhances the malignant phenotype of tumor cells, i.e., promoting tumor cell migration, survival, and production of angiogenic factors EGF and CXCL8.1,5 Although the first agonist identified for FPR1 was bacterial formylated peptide, there are also a number of host-derived agonists including mitochondrial formyl peptides,6 Annexin 1 (Anx A1)7 and a neutrophil granule protein cathepsin G.8 Therefore, FPR1 may be involved in pathophysiological processes in which its pathogen-derived or endogenous agonists are elevated. Studies further revealed that GBM cells undergoing necrosis release chemotactic agonist(s) that activate FPR1 in viable tumor cells.1 These observations suggest that GBM cells may use FPR1 to recognize agonists produced in the tumor microenvironment in a paracrine loop.9 However, the identity of FPR1 agonist(s) released by necrotic human GBM cells is unknown.
In this study, we determined the identity of a FPR1 agonist released by necrotic GBM cells. Because FPR1 is selectively expressed by more highly malignant gliomas, we chose to use a U87 cell line, which was derived from human GBM and expressed functional FPR1.1,2,5 Our results revealed that Anx A1 accounted for the majority of FPR1 agonist activity released by necrotic tumor cells.
Materials and Methods
Cells and Reagents
Human GBM cell lines U87 and SNB75 were obtained from ATCC (Manassas, VA). SHG-44 cells were established from a surgically removed Grade III human anaplastic astrocytoma (Suzhou University, Suzhou, China). FPR1-knocking down (FPR1 KD) U87 cells were established previously.1 Formyl-methionyl-leucyl-phenylalanine (fMLF) was from Sigma-Aldrich (St Louis, MO). The sources of antibodies were as follows: anti–Anx A1 from BD (Franklin Lakes, NJ); HRP-conjugated anti-mouse IgG from Cell Signaling Technology (Beverly, MA); FITC-labeled goat anti-mouse secondary antibody, anti-CD11b, anti-F4/80, anti-Ly6G and streptavidin-FITC antibodies from eBioscience (San Diego, CA). Protein G Dynabeads, Lipofectamine2000, Opti-MEMI media and DAPI were from Invitrogen (Carlsbad, CA). Stripping buffer and BCA assay kit were from Thermo Scientific (Rockford, IL). The ABC kit was from Maixin (Fuzhou, China).
NecSup and Removal of Anx A1
Necrotic tumor cell supernatant (NecSup) was generated as described.1 NecSup was mixed with the anti–Anx A1 antibody (1:50) and rotated at 4°C overnight. Protein G Dynabeads were added to the mixture followed by 2 hours rotation at 4°C. The supernatant was collected after centrifugation at 2000 rpm and stored at −70°C.
Western Blotting
NecSup with or without immunoabsorption was electrophoresed on 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) gel, transferred to blots, and reacted with the anti–Anx A1 antibody (1:5000) followed by an HRP-conjugated secondary antibody (1:1000). For detection of cellular Anx A1, tumor cells incubated in the presence or absence of stimulants for 20 hours were washed and lysed. The protein concentration was measured using BCA kit and 10 μg total proteins were electrophoresed.
RT-PCR
Total RNA was extracted from U87 GBM cells with an RNeasy mini kit (Qiagen, Valencia, CA). Human Anx A1 primers were 5′-GCAGGCCTGGTTTATTGAAA-3′ and 5′-GCTGTGCATTGTTTCGCTTA-3′. RT-PCR was performed with 0.3 μg total RNA for each sample consisting of a 15-minute reverse transcription at 42°C, 10-minute inactivation at 95°C, 40 cycles of denaturation at 95°C (15 seconds), annealing at 60°C (20 seconds), and extension at 72°C (30 seconds). PCR products were resolved by 1% agarose gel electrophoresis.
Ca2+ Mobilization
Intracellular Ca2+ mobilization was measured as previously described.10
Tumor Cell Proliferation
U87 GBM cells were seeded onto 96-well plates and allowed to adhere. The cells were incubated in different dilutions of NecSup in Dulbecco's modified Eagle's medium (DMEM; 1% fetal calf serum [FCS]) for 72 hours at 37°C. Cell proliferation was measured by CellTiter 96 Aqueous One Solution Assay kit (Promega, WI). Relative absorbance at 490 nm was obtained with a plate reader (FLUOstar Omega BMG Labtech).
Chemotaxis
Chemotaxis was measured with 48-well chemotaxis chambers.10 NecSup diluted with RPMI 1640 (1% bovine serum albumin [BSA]) was added to the lower compartment of the chamber (Neuro Probe Inc., Cabin John, MD). U87 GBM cells (1 × 106 cells/mL) were added to the upper compartment. Two compartments were separated by a 10-μm polycarbonate membrane (GE Osmonica Labstore, Minnetonka, MN) precoated with collagen I. After incubation for 4 hours at 37°C, cells that migrated across the membrane were stained and counted under light microscopy. Results were expressed as the chemotaxis index scored as the ratio of cell number responding to a chemoattractant gradient over the response to medium control in three high-power fields (HPF) in triplicate (×400).
Invasion
A 500-μl aliquot of diluted NecSup was added to each well of 24-well plates. Inserts (8-μm, Becton Dickinson Labware, Franklin Lakes, NJ) precoated with collagen I were lowered into the well, and 100 μl U87 GBM cells (1 × 106 cells) was added to the inserts for 4 hours' incubation at 37°C. The cells that migrated across the collagen I–coated membrane were stained and counted under light microscopy. Data are expressed by the invasion index scored as the ratio of cell number responding to stimulants over the response to medium control in three high-power fields (HPF) in triplicates (400 ×).
Colony Formation
Tumor colony formation was examined as described.10 A 600-μl aliquot of 0.6% agar was poured into each well of 24-well plates, followed by 200 μl of 0.3% agar containing U87 GBM cells and different dilutions of NecSup. Culture medium (500 μl) was added to wells for 2 week' incubation. The colonies were photographed, and the total numbers were determined under microscopy.
Knockdown of Anx A1 by siRNA
FPR1 knockdown (FPR1 KD) U87 GBM cells were generated as described previously.1,11 SiRNA pools targeting Anx A1 mRNA were from Dhamacon (Thermo Science). The target sequences are 5′-CAAGGUGGUCCCGGAUCA-3′, 5′-GAAGUGCGCCACAAGCAAA-3′, 5′-UGACCGAUCUGAGGACUUU-3′, and 5′-UAACUAAGCGAAACAAUGC-3′. Transfection was performed with Lipofectamine2000 in Opti–MEMI media with 40 nmol/L Anx A1 siRNA. The cells were harvested after 48 hours. U87 cells, Anx KD, FPR1 KD, or double KD cells (3 × 106) were s.c. injected into nude mice. Animal studies were approved by the NCI–Frederick Animal Care and Use Committee. Animal care was provided in accordance with the procedures outlined in the “Guide for Care and Use of Laboratory Animals.” Tumor volumes were calculated according to the formula V = 0.5 × length × width.2 Cryosections of tumors were stained with the anti–Anx A1 antibody followed by a FITC-labeled secondary antibody with DAPI counterstaining. Cryosections were also stained with antibodies to CD11b, F4/80, and Ly6G followed by a biotinylated secondary antibody and streptavidin–FITC to detect myeloid cells. The fluorescence images were taken by Olympus IX71 microscope with Image-Pro software.
Detection of Anx A1 in Human Primary Tumor Tissues
Human primary glioma specimens, including 33 grade II, 11 grade III, and 58 grade IV tumors, were examined for Anx A1 expression. Peritumor normal brain tissues were also retrieved as controls. Paraffin-embedded tissues were sliced into 6-μm sections, which were reacted with the anti–Anx A1 antibody overnight at 4°C. The positive staining was revealed by ABC kit reagents. The Anx A1 expression level was expressed by scores defining staining areas and intensity. The staining area was scored as follows: 0 = no Anx A1–positive cells; 1 = < 25% positive cells; 2 = 25% to 50% positive cells; and 3 = more than 50% positive cells. The staining intensity was score (1-3) according to the intensity of positive Anx A1 reaction. The total score was calculated by the formula: Score = area score × intensity score. The scoring of the specimens was performed by pictures of tumor sections with two independent pathologists. Representative Anx A1 expression scores are shown in Supplemental Figure S1 (available at http://ajp.amjpathol.org).
Statistical Analysis
The statistical significance was analyzed with t-test or two-way analysis of variance (Graphpad Prism Software) for chemotaxis, invasion, and cell growth experiments. Kendall's τ-b and Spearman in SPSS 10.0 software were used to analyze the correlation between Anx A1 expression and the grade of tumors. The correlation is considered to be significant at the value of 0.01.
Results
Anx A1 in Necrotic Tumor Cell Supernatant
We first determined the presence of FPR1 agonist activity in NecSup. The capacity of NecSup to cross-desensitize the response to a known FPR1 agonist in viable tumor cells was investigated. As shown in Figure 1A, stimulation of live GBM cell line U87 with fMLF, a well-established FPR1 ligand, elicited an increase of intracellular Ca2+ mobilization. Different concentrations of NecSup similarly induced Ca2+ flux in live tumor cells (Figure 1B). In cross-desensitization experiments, prior stimulation of GBM cells with fMLF diminished cell responses to subsequent stimulation with NecSup (Figure 1A). Conversely, stimulation with NecSup also reduced U87 GBM cell responses to subsequent stimulation with fMLF (Figure 1B). Thus, NecSup appeared to contain agonist(s) that share FPR1 with fMLF as demonstrated by the classical desensitization of a GPCR by its shared agonists.
Figure 1.
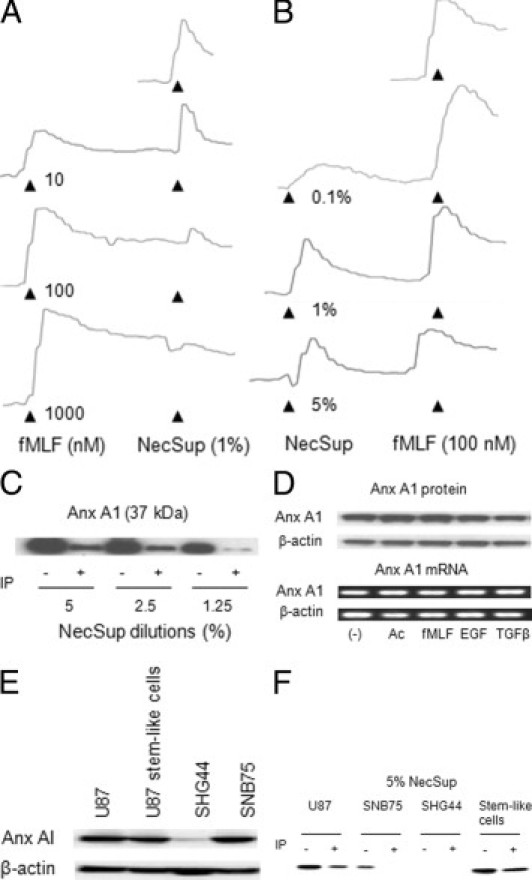
Detection of Anx A1 in NecSup. A: Desensitization of NecSup-induced calcium flux in U87 GBM cells by fMLF. Ca2+ mobilization in U87 GBM cells induced by fMLF and the effect on subsequent cell responses to NecSup were measured. B: Desensitization of FPR1 in U87 GBM cells by NecSup. The intensity of the fluorescence was expressed as the ratio of absorbance at 340 nm to 380 nm. C: Detection of Anx A1 in NecSup. NecSup was diluted with PBS. The anti–Anx A1 antibody and protein-G Dynabeads were used to immunoabsorb the Anx A1/antibody complex. The NecSup at different dilutions was then electrophoresed on a 10% SDS-PAGE and immunoblotted with an anti–Anx A1 antibody. D: Detection of Anx A1 expression in U87 GBM cells. U87 GBM cells were stimulated with Ac2-26 (Ac: 1 mmol/L), fMLF (100 nmol/L), EGF (10 ng/mL), or TGFβ (10 ng/mL) in DMEM without FCS. Upper panel: Equal amounts of total cellular proteins were subjected to SDS-PAGE and immunoblotting using an anti–Anx A1 antibody. β-Actin was used as a loading control. Lower panel: Stimulated U87 GBM cells were also examined for Anx A1 expression by RT-PCR. β-actin was used as a control. Experiments were performed two or three times. E: Detection of Anx A1 expression in U87 GBM cells, neurosphere cells, SHG44 anaplastic astrocytoma cells, and SNB75 GBM cells. F: Detection of Anx A1 expression in NecSup (at 5% concentration diluted in PBS) of SNB75, SHG44, and neurosphere cells by immunoblotting.
Because a reported FPR1 agonist protein Anx A1 has been shown to be produced by malignant tumor cells and may increase the invasiveness of tumor cells,7 we examined the presence of Anx A1 in NecSup by immunoblotting. Figure 1C shows that after U87 GBM cells were subjected to repeated freezing and thawing to cause necrosis, Anx A1 was released into the supernatant. An anti–Anx A1 antibody was able to bind Anx A1 in NecSup and the immune complex was largely removed by protein-G–coupled magnetic beads (Figure 1C). We further investigated whether the expression of Anx A1 in U87 GBM cells could also be induced with Ac2-26, which is another FPR1 agonist peptide3, and by fMLF, epidermal growth factor (EGF), or transforming growth factor–β (TGF-β). None of these stimulants increased Anx A1 protein (Figure 1D, upper panel) or mRNA content (Figure 1D, lower panel) in U87 GBM cells after 20-hour treatment. Thus, Anx A1 is constitutively expressed by U87 GBM cells. We also examined the Anx A1 expression in another two glioma cell lines, SHG44 and SNB75 (Figure 1E).1 Anx A1 was expressed in SNB75 cells, which were also derived from a human GBM. However, SHG44 cells, which were derived from a grade III human anaplastic astrocytoma, did not express Anx A1. Furthermore, we examined the expression of Anx A1 in CD133+ neurosphere forming stem-like cells isolated from U87 cells in serum-free stem cell culture. These cells have been found to possess multipotent differentiation capacity and markedly increased tumorigenicity in immunocompromised mice.9 The sphere cells also expressed Anx A1 (Figure 1E). Similar to the pattern of Anx A1 expression, Anx A1 protein was detected in the NecSup from SNB75 and stem-like cells, but not SHG44 cells (Figure 1F).
Anx A1 in Necrotic Tumor Cell Supernatant Induces U87 GBM Cell Chemotaxis and Invasion
We then examined whether Anx A1 might be responsible for the tumor cell chemotactic activity released by necrotic GBM cells. U87 GBM cells migrated in response to Ac2-26 and fMLF (Figure 2A), confirming their expression of functional FPR1. The NecSup also potently induced U87 GBM cell migration, and supernatant diluted with assay medium to as low as 1.25% was still capable of inducing significant tumor cell chemotaxis (Figure 2B). However, after the supernatant was absorbed with anti–Anx A1 antibody and protein-G–coupled microbeads, it lost most of the chemotactic activity for tumor cells (Figure 2B), indicating that Anx A1 was responsible for a considerable proportion of the FPR1 agonist activity contained in the NecSup. Immunoabsorption with anti–Anx A1 antibody did not affect the chemotactic activity of fMLF, confirming the specificity of the Anx A1 in the NecSup (data not shown).
Figure 2.
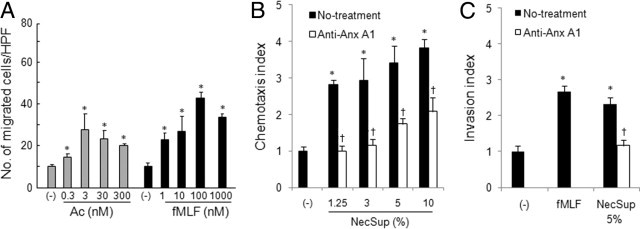
Induction of U87 GBM cell chemotaxis and invasion by NecSup. A: U87 GBM cell chemotaxis in response to Ac2-26 and fMLF. Cell chemotaxis was measured in response to different concentrations of Ac2-26 and fMLF. Data are mean ± SD. B: U87 GBM cell chemotaxis in response to NecSup. U87 cell chemotaxis was measured in response to different dilutions (% in assay medium) of NecSup. Data are mean ± SD.C: U87 GBM cell invasion. Different dilutions of NecSup were added to each well of 24-well cell culture plates. U87 GBM cells were added into the Transwell inserts (8 μm) pre-coated by collagen I, and the plates were incubated at 37°C for 4 hours. Data are mean ± SD *P < 0.05, significantly increased cell migration (A and B) or invasion (C) in response to stimulants or NecSup as compared with spontaneous migration in the presence of control medium (RPMI 1640, 1% BSA, [−]). †P < 0.05, compared with untreated NecSup. Data in this figure are representative of results from three independent experiments.
We further tested the effect of NecSup on U87 GBM cell invasion measured by their infiltration into the matrix proteins. In an in vitro invasion assay, necrotic tumor cell supernatant stimulated U87 GBM cell invasion into the matrix (Figure 2C). However, on removal of Anx A1 by immunoabsorption, the stimulating effects of the supernatant on tumor cell invasion were considerably reduced.
Stimulation of Tumor Cell Proliferation by Necrotic Tumor Cell Supernatant
The FPR1 agonist peptide fMLF has been shown to stimulate GBM cell proliferation.1,12 We therefore examined whether NecSup also possessed tumor cell growth stimulating activity. Figure 3A shows that NecSup enhanced U87 GBM cell proliferation. Immunoabsorption of NecSup with anti–Anx A1 antibody markedly reduced the proliferation-enhancing effects of NecSup in a 72-hour incubation period, indicating that Anx A1 in NecSup plays a significant role in increasing cell proliferation.
Figure 3.
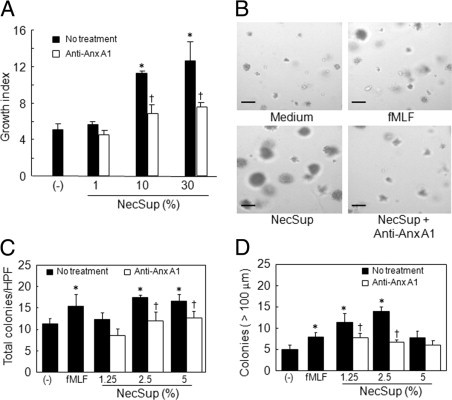
U87 GBM cell proliferation and tumor cell colony formation induced by NecSup. A: U87 GBM cell proliferation. U87 GBM cells were cultured with different dilutions of NecSup with or without anti–Anx A1 antibody immunoabsorption in DMEM (1% FCS). Cells were incubated for 72 hours. Relative absorbance at 490 nm was measured and cell proliferation was expressed as fold increase over the control in DMEM alone. *P < 0.05, significantly increased cell proliferation compared with DMEM (−). Data are mean ± SD. †P < 0.05, significantly reduced cell proliferation induced by anti–Anx A1 antibody immunoabsorbed NecSup as compared with untreated NecSup. B–D:In vitro U87 GBM cell colony formation. U87 GBM cells suspended in soft agar containing different dilutions of NecSup were layered onto the bottom agar in 24-well plates. Cells were grown in culture medium for 2 weeks (B). Scale bar = 100 μm. The numbers of all colonies (C) and colonies greater than 100 μm in diameter (D) were counted. Data are mean ± SD *P < 0.05, compared with DMEM control group (−). †P < 0.05, compared with untreated NecSup. Experiments were performed two or three times.
We additionally examined the capability of NecSup to promote colony formation by tumor cells in soft agar. After 2 weeks of culture in soft agar, U87 GBM cell colonies were counted. Figure 3, B–D shows that tumor cells in the presence of NecSup formed a higher number of colonies, which grew more rapidly as compared with tumor cells in the absence of NecSup. After removal of Anx A1, the tumor cell colony stimulating activity of NecSup was considerably reduced (Figure 3, B–D). Thus, Anx A1 in the NecSup has potent tumor cell growth–promoting activity.
Decreased Rate of Tumor Progression by Knocking Down Anx A1 in U87 GBM Cells
We then examined the effects of Anx A1 siRNA on the growth of tumors formed by U87 GBM cells in immunocompromised mice. We transiently transfected Anx A1 siRNA into U87 GBM cells (Anx KD). After 48 hours, we detected reduced protein and mRNA levels of Anx A1 in Anx KD cells (data not shown). We next examined the proliferation of the cells containing Anx A1 KD or FPR1 KD constructs. The results showed that knocking down Anx A1 (Anx KD) or FPR1 (FPR1 KD) alone reduced the rate of U87 GBM cell proliferation. Knocking down both Anx A1 and FPR1 (Double KD) further reduced the growth of U87 GBM cells in vitro (Figure 4A). The growth of Anx KD-control cells and FPR1 KD-control cells was similar to that of the parental U87 GBM cells (Figure 4A). U87 GBM and Anx KD cells were subsequently s.c. injected into nude mice. Figure 4B shows high levels of Anx A1 in tumors formed by U87 GBM cells (Figure 4B, left panels). In tumors formed by Anx KD U87 GBM cells, the expression of Anx A1 was considerably reduced as measured by immunofluorescence (Figure 4B, right panels and Figure 4C).
Figure 4.
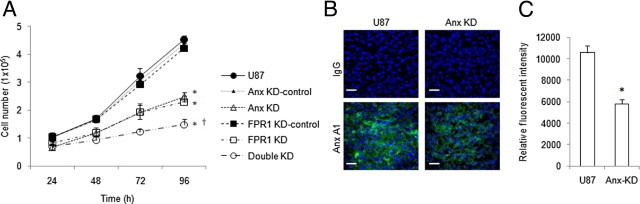
The effect of siRNA knockdown of Anx A1 on U87 GBM cell growth. A: The growth curves of U87 GBM cells containing scrambled siRNAs (Anx KD-control and FPR1 KD-control), siRNAs targeting Anx A1 (Anx KD), FPR1 (FPR1 KD), or both (Double KD) were seeded (4 × 104 cells) in triplicates in 12-well plate. After 24, 48, 72, and 96 hours, cell number was counted. Experiments were performed three times. *P < 0.05, compared with U87 GBM cells. †P < 0.05, compared with FPR1 KD-control cells. B: Anx A1 expression in tumors formed by U87 GBM cells containing Anx A1 siRNA. U87 GBM and Anx KD cells were s.c. injected into nude mice. Tumors were harvested on day 36 and sectioned, and the expression of Anx A1 was detected with immunofluorescence. Bar, 20 μm. Representative images are shown. C: The Anx A1 fluorescence intensity in transplanted tumors calculated by ImageJ.
The progression of tumors formed by Anx KD U87 GBM cells was significantly reduced as compared with tumors formed by U87 GBM cells (Figure 5A), despite the fact that knocking down of Anx A1 was partial as shown in tumor section (Figure 4B, right panels). To investigate the role of FPR1/Anx A1 axis in GBM progression, we transfected Anx A1 siRNA into U87 GBM cells that contain FPR1 siRNA (FPR1 KD) to establish double knocking down cells (Double KD). We found that whereas tumors formed by Anx KD or FRR1 KD cells grew more slowly than those formed by U87 GBM cells, Double KD cells showed a further reduction in tumorigenicity (Figure 5A). Figure 5B shows the survival of mice injected with U87 GBM cells containing Anx A1 and FPR1 siRNAs. All mice injected with U87 GBM tumor cells were dead within 38 days after tumor cell implantation. Mice injected with Anx KD or FPR1 KD tumor cells showed significantly prolonged survival. Interestingly, all mice injected with Double KD cells remained alive up to day 150 after tumor cell implantation, considerably longer than mice injected with GBM cells containing Anx A1 siRNA or FPR1 siRNA alone (Figure 5B). Overall, these results indicate an important role of FPR1/Anx A1 axis in promoting the tumorigenicity of GBM cells.
Figure 5.
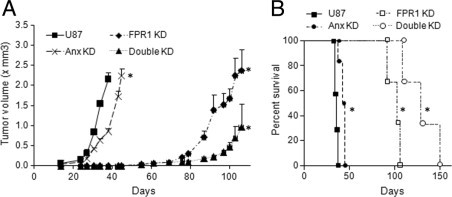
The tumorigenicity of U87 GBM cells after knockdown Anx A1. A: The tumorigenicity of U87 GBM cells containing Anx A1 or FPR1 siRNA. After s.c. injection of U87 GBM, Anx KD, FPR1 KD, and Double KD U87 GBM cells into nude mice, tumor volume was measured at different time points. *P < 0.01, compared with tumors formed by U87 GBM cells. Data are mean ± SEM. B: The survival curves of mice bearing tumors formed by U87 GBM cells containing siRNAs targeting Anx A1, FPR1, and both. *P < 0.01, compared with mice bearing tumors formed by U87 GBM cells. Data (A and B) are representative of results from two independent experiments with three to five mice in each group.
Because FPR1 is also expressed by myeloid cells and Anx A1 induces chemotaxis of these cells in vitro,4 we examined the infiltration of leukocytes in tumors formed by U87 GBM cells in nude mice. Figure 6A shows higher levels of infiltrating CD11b+ mouse myeloid cells in tumors formed by U87 GBM cells as compared with tumors formed by Anx KD cells. There was no significant reduction in myeloid cell infiltration in tumors formed by FPR1 KD cells when the tumors grew to the size similar to those formed by control cells. In tumors formed by FPR1/Anx A1 Double KD cells, the level of myeloid cell infiltration was comparable to tumors formed by Anx KD cells, and reduced as compared with tumors formed by U87 or FPR1 KD cells. We then examined myeloid cell subtypes in transplanted tumors. We found that in tumors formed by U87 GBM cells, the infiltrating myeloid cells were composed mainly of Ly6G+ granulocytes (61%) and F4/80+ macrophages (36%) (Figure 6B). Thus, Ly6G+ granulocytes constitute the major part of the infiltrating cells. Moreover, we found that in tumors formed by Anx A1 siRNA knocking down and Anx A1/FPR1 double knocking down U87 GBM cells, the reduced infiltrating leukocytes were also mainly Ly6G+ granulocytes. These results suggest that Anx A1 expressed by tumor cells may also play a role in recruiting myeloid cell infiltration in the tumor tissue, because myeloid cells express FPR1 and FPR2, and both may recognize Anx A1 produced by tumor cells.
Figure 6.
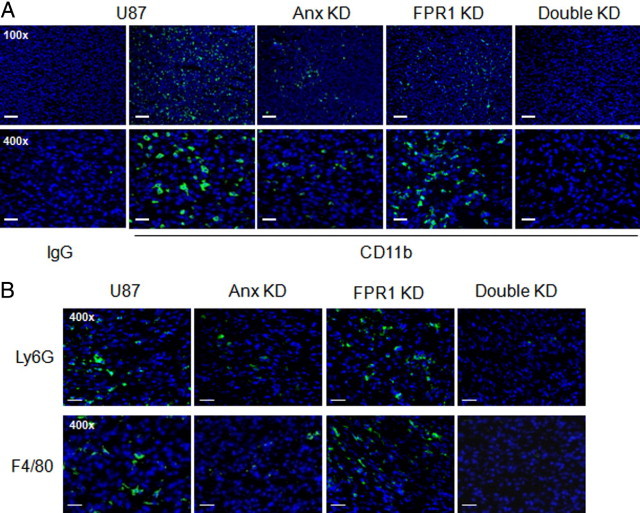
Detection of tumor-infiltrating myeloid cells. Nude mice were s.c. injected with control, Anx KD, FPR1 KD, and Double KD U87 GBM cells. Tumors in different groups were harvested when they reached a comparable size. On day 36 or 93 after s.c. injection, tumors were excised and sectioned. Tumors formed by U87 and Anx KD cells were taken on day 36, whereas tumors formed by FPR1 KD and Double KD cells were harvested on day 93. A: Tumor sections were stained by anti-CD11b antibody. CD11b+ myeloid cells are shown in green fluorescence. Sections were counterstained by DAPI to show nuclei. Scale bar = 100 µm. B: Tumor sections were stained by anti-Ly6G and F4/80 antibodies. Ly6G+ (61% of total myeloid cells) or F4/80+ (36% of total myeloid cells) cells were shown in green fluorescence. Scale bar = 40 μm.
Detection of Anx A1 in Human Primary Gliomas
To determine the clinical relevance of Anx A1 in human gliomas, we examined its expression in human primary tumor specimens. Anx A1 expression was not detected in peri-tumor normal brain tissues, but was increased in tumor tissues (Table 1; see also Supplemental Figure S1 at http://ajp.amjpathol.org). We scored Anx A1expression and found that among 102 total cases tested, 42% of Grade II, 100% of Grade III, and 88% of Grade IV tumors are positive for Anx A1 staining. In addition, the highest level expression of Anx A1 (scores ≥9) was detected in higher Grade (III and IV) gliomas. The results confirmed the expression of Anx A1 by human primary gliomas, in particular by higher-grade tumors. The high level expression of Anx A1 in GBMs in conjunction with the selective expression of the receptor FPR11 suggests an important role for this ligand/receptor axis in exacerbating the malignant behavior of human GBMs.
Table 1.
Expression of Anx A1 in Human Primary Gliomas with Different Grades
| Histological grade | Case no. | Positive (%) | Scores⁎ |
||
|---|---|---|---|---|---|
| 1–4 | 5–8 | ≥9 | |||
| Normal | 8 | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| II | 33 | 14 (42)† | 9 (27) | 5 (15) | 0 (0) |
| III | 11 | 11 (100)† | 7 (64) | 3 (27) | 1 (9) |
| IV | 58 | 51 (88)† | 16 (28) | 24 (41)† | 11 (19)† |
Sections were prepared from peri-tumor normal tissues and tumors of glioma patients receiving surgical therapy. Anx A1 was detected by histochemical staining with an anti–Anx A1 antibody and hematoxylin counterstaining. Anx A1 stianing was scored by area scores and intensity scores as described in Materials and Methods.
Data are case numbers, with percentages in parentheses.
Significantly increased rate of Anx A1 expression and intensity score as compared with normal brain tissue or lower-grade tumors as measured by SPSS 10.0 software.
Discussion
In this study, we reported that Anx A1 in the necrotic glioma cell supernatant is a potent activator of FPR1 expressed by viable tumor cells. Anx A1 is a member of calcium-dependent phospholipid-binding proteins with diverse functions. Anx A1 was originally characterized as an endogenous anti-inflammatory protein. In inflammatory responses, Anx A1 suppresses leukocytes infilatration into the inflamed tissue.13,14 The 2- to 26-amino acid N-terminal region is unique for the annexin family and accounts for the anti-inflammatory activities of the protein. Recently, the inhibitory effect of N-terminal Anx A1 peptides as well as the full-length Anx A1 was reported to be based on interaction with FPR1 as well as FPRL1 (FPR2).15,16 Anx A1 was also reported to chemoattract certain tumor cells and its overexpression in head and neck squamous cell carcinoma,17 pancreatic cancer,18,19 and melanoma20 was associated with increased tumor invasiveness and metastasis. Small interfering RNA–mediated knockdown of Anx A1 expression resulted in significant reduction of SKCO-15 colorectal adenocarcinoma cell invasion.7 The tumor-promoting effects of Anx A1 has been attributed to its interaction with FPR members on tumor cells in a paracrine or autocrine fashion. Recently, Anx A1 has been implicated in tumor angiogenesis, as tumors transplanted into Anx A1 null mice grew more slowly and formed fewer metastasis because of defective formation of tumor vasculature.20
Our previous studies have shown that highly malignant GBM cells express FPR1, which reacts to necrotic tumor cell derived agonist(s) by an increase in tumor cell migration, growth, and the production of angiogenic factors.1,5 However, the identity of the FPR1 agonist released by tumor cells remained unclear. In this study, we have identified Anx A1 as an FPR1 agonist released by necrotic GBM cells, as most of the tumor-promoting activity of the supernatant was reduced after absorption by anti–Anx A1 antibody. Thus, our study supported the hypothesis that the capability of necrotic cell–derived Anx A1 accounted for the FPR1 agonist effects on stimulating GBM cell growth and invasion in a paracrine manner.
Malignant tumors possess the unique capability to detect signals in the microenvironment to favor their survival, growth, invasion, and metastasis.5,21 Some growth factors such as EGF and CSF-1 (MCSF), and the chemokine CXCL12 have been implicated as being present at high local concentrations as microenvironmental tumor stimulants. The antimicrobial peptide LL-37, which is a ligand of the FPR1 variant FPRL1 (FPR2), was capable of stimulating the growth of epithelial tumor cells as well as recruiting mesenchymal stem cells into tumors to facilitate angiogenesis.22–24 Our study revealed that knocking down either Anx A1 or its receptor FPR1 reduced the tumorigenicity of U87 GBM cells. Double knocking down FPR1/Anx A1 in U87 GBM cells further reduced their capacity to form actively growing tumors. Thus, our observations expanded the spectrum of locally available tumor stimulants to include Anx A1 and the receptor FPR1 as playing an important role in promoting the progression of GBM. Because FPR1 is abundantly expressed by myeloid cells, it will be important to elucidate the potential for Anx A1 produced by GBM cells to recruit such cells into tumor tissues, to promote tumor growth, invasion, and dissemination.25 In our initial effort to examine this issue, we found that s.c. tumors formed by control U87 GBM cells contained high levels of infiltrating myeloid cells. In contrast, the myeloid infiltration was markedly reduced in tumors formed by Anx KD cells in association with considerably decreased rate of tumor growth. Moreover, the reduced infiltrating leukocytes were mainly Ly6G+ granulocytes. Thus, Anx A1 may indeed play a role in chemoattracting tumor-promoting myeloid cells into more actively growing tumors. Further studies in human primary gliomas are required to more clearly elucidate the contribution of Anx A1 to immune cell infiltration in tumors.
The staining of human primary gliomas in this study shows that there was an increased Anx A1 expression in higher grade tumor specimens (Grade III and IV), similar to the expression pattern of FPR1.1 These results indicate a clear correlation between the level of Anx A1 expression and the malignancy of gliomas. However, in Grade IV GBM specimens, a small number (12%, 7/58) of cases were negative for Anx A1. Whether this was caused by technical limitations in sampling and staining (ie, to avoid necrotic areas that easily cause nonspecific staining) or by the presence of subtypes of GBM requires further clarification. Also, we did not detect a clear difference in the rate of survival of Grade IV GBM cases in relation to Anx A1 expression, presumably because of the poor prognosis of the cases and short follow-up time. This issue remains to be determined in future studies.
In conclusion, we have shown that Anx A1 accounts for most of the activities of necrotic GBM cell supernatant to induce migration, invasion, proliferation, and colony formation of live tumor cells through activation of FPR1. Because depletion of either FPR11,12 or Anx A1 reduced the growth of transplanted tumors formed by U87 GBM cells, and double knocking down both molecules even further reduced the capacity of GBM cells to form tumors in vivo, the axis of Anx A1 and FPR1 in GBM cells should constitute molecular targets for the development of novel anti-GBM therapy.
Acknowledgments
The authors thank Dr. Joost J. Oppenheim for reviewing the manuscript and Cheryl F. Lamb for secretarial assistance.
Footnotes
Supported in part by federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E, and in part by the Intramural Research Program of the NCI, NIH. J.H. was funded in part by the National Natural Science Foundation of China (#30973446).
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.05.059.
The content of this publication do not necessarily reflect the views or policies of the Department of Health and Human Services, nor does the mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Y.Y. and Y.L. contributed equally to this study
Supplementary data
Anx A1 expression in human GBM specimens. A: Peri-tumor normal brain tissues. B–E: Different scores for Anx A1 expression in GBM tissues. The upper panels are H&E staining images. The lower panels show immunohistochemical staining of different levels of Anx A1 in GBMs with corresponding scores. Scale bar, 20 μm.
References
- 1.Zhou Y., Bian X., Le Y., Gong W., Hu J., Zhang X., Wang L., Iribarren P., Salcedo R., Howard O.M.Z., Farrar W., Wang J.M. Formylpeptide receptor FPR and the rapid growth of malignant human gliomas. J Natl Cancer Inst. 2005;97:823–835. doi: 10.1093/jnci/dji142. [DOI] [PubMed] [Google Scholar]
- 2.Huang J., Chen K., Gong W., Dunlop N.M., Wang J.M. G-protein coupled chemoattractant receptors and cancer. Front Biosci. 2008;13:3352–3363. doi: 10.2741/2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ye R.D., Boulay F., Wang J.M., Dahlgren C., Gerard C., Parmentier M., Serhan C.N., Murphy P.M. International Union of Basic and Clinical Pharmacology: LXXIII Nomenclature for the formyl peptide receptor (FPR) family. Pharmacol Rev. 2009;61:119–161. doi: 10.1124/pr.109.001578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Le Y., Murphy P.M., Wang J.M. Formyl-peptide receptors revisited. Trends Immunol. 2002;23:541–548. doi: 10.1016/s1471-4906(02)02316-5. [DOI] [PubMed] [Google Scholar]
- 5.Huang J., Chen K., Gong W., Zhou Y., Le Y., Bian X., Wang J.M. Receptor “hijacking” by malignant glioma cells: a tactic for tumor progression. Cancer Lett. 2008;267:254–261. doi: 10.1016/j.canlet.2008.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rabiet M.J., Huet E., Boulay F. Human mitochondria-derived N-formylated peptides are novel agonists equally active on FPR and FPRL1, while Listeria monocytogenes-derived peptides preferentially activate FPR. Eur J Immunol. 2005;35:2486–2495. doi: 10.1002/eji.200526338. [DOI] [PubMed] [Google Scholar]
- 7.Babbin B.A., Lee W.Y., Parkos C.A., Winfree L.M., Akyildiz A., Perretti M., Nusrat A. Annexin I regulates SKCO-15 cell invasion by signaling through formyl peptide receptors. J Biol Chem. 2006;281:19588–19599. doi: 10.1074/jbc.M513025200. [DOI] [PubMed] [Google Scholar]
- 8.Sun R., Iribarren P., Zhang N., Zhou Y., Gong W., Cho E.H., Lockett S., Chertov O., Bednar F., Rogers T.J., Oppenheim J.J., Wang J.M. Identification of neutrophil granule protein cathepsin G as a novel chemotactic agonist for the G protein-coupled formyl peptide receptor. J Immunol. 2004;173:428–436. doi: 10.4049/jimmunol.173.1.428. [DOI] [PubMed] [Google Scholar]
- 9.Yao X.H., Ping Y.F., Chen J.H., Xu C.P., Chen D.L., Zhang R., Wang J.M., Bian X.W. Glioblastoma stem cells produce vascular endothelial growth factor by activation of a G-protein coupled formylpeptide receptor FPR. J Pathol. 2008;215:369–376. doi: 10.1002/path.2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang J., Chen K., Huang J., Gong W., Dunlop N.M., Howard O.M.Z., Bian X., Gao Y., Wang J.M. Regulation of the leucocyte chemoattractant receptor FPR in glioblastoma cells by cell differentiation. Carcinogenesis. 2009;30:348–355. doi: 10.1093/carcin/bgn266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Le Y., Iribarren P., Zhou Y., Gong W., Hu J., Zhang X., Wang J.M. Silencing the formylpeptide receptor FPR by short-interfering RNA. Mol Pharmocol. 2004;64:1022–1028. doi: 10.1124/mol.104.001982. [DOI] [PubMed] [Google Scholar]
- 12.Huang J., Hu J., Bian X., Chen K., Gong W., Dunlop N.M., Howard O.M.Z., Wang J.M. Transactivation of the epidermal growth factor receptor by formylpeptide receptor exacerbates the malignant behavior of human glioblastoma cells. Cancer Res. 2007;67:5906–5913. doi: 10.1158/0008-5472.CAN-07-0691. [DOI] [PubMed] [Google Scholar]
- 13.Getting S.J., Flower R.J., Perretti M. Inhibition of neutrophil and monocyte recruitment by endogenous and exogenous lipocortin 1. Br J Pharmacol. 1997;120:1075–1082. doi: 10.1038/sj.bjp.0701029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Flower R.J., Rothwell N.J. Lipocortin-1: cellular mechanisms and clinical relevance. Trends Pharmacol Sci. 1994;15:71–76. doi: 10.1016/0165-6147(94)90281-x. [DOI] [PubMed] [Google Scholar]
- 15.Walther A., Riehemann K., Gerke V. A novel ligand of the formyl peptide receptor: annexin I regulates neutrophil extravasation by interacting with the FPR. Mol Cell. 2000;5:831–840. doi: 10.1016/s1097-2765(00)80323-8. [DOI] [PubMed] [Google Scholar]
- 16.Perretti M., Getting S.J., Solito E., Murphy P.M., Gao J.L. Involvement of the receptor for formylated peptides in the in vivo anti-migratory actions of Anx A1 and its mimetics. Am J Pathol. 2001;158:1969–1973. doi: 10.1016/S0002-9440(10)64667-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu W., Tang X., Hu W., Lotan R., Hong W.K., Mao L. Identification and validation of metastasis-associated proteins in head and neck cancer cell lines by two-dimensional electrophoresis and mass spectrometry. Clin Exp Metastasis. 2002;19:319–326. doi: 10.1023/a:1015515119300. [DOI] [PubMed] [Google Scholar]
- 18.Bai X.F., Ni X., Zhao P., Liu S.M., Wang H.X., Guo B., Zhou L.P., Liu F., Zhang J.S., Wang K., Xie Y.Q., Shao Y.F., Zhao X.H. Overexpression of Anx A1 in pancreatic cancer and its clinical significance. World J Gastroenterol. 2004;10:1466–1470. doi: 10.3748/wjg.v10.i10.1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rondepierre F., Bouchon B., Papon J., Bonnet-Duquennoy M., Kintossou R., Moins N., Maublant J., Madelmont J.C., D'Incan M., Degoul F. Proteomic studies of B16 lines: involvement of annexin A1 in melanoma dissemination. Biochim Biophys Acta. 2009;1794:61–69. doi: 10.1016/j.bbapap.2008.09.014. [DOI] [PubMed] [Google Scholar]
- 20.Yi M., Schnitzer J.E. Impaired tumor growth, metastasis, angiogenesis and wound healing in annexin A1-null mice. Proc Natl Acad Sci USA. 2009;106:17886–17891. doi: 10.1073/pnas.0901324106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liotta L.A., Kohn E.C. The microenvironment of the tumour-host interface. Nature. 2001;411:375–379. doi: 10.1038/35077241. [DOI] [PubMed] [Google Scholar]
- 22.De Y., Chen Q., Schmidt A.P., Anderson G.M., Wang J.M., Wooters J., Oppenheim J.J., Chertov O. LL-37, the neutrophil granule- and epithelial cell-derived cathelicidin, utilizes formyl peptide receptor-like 1 (FPRL1) as a receptor to chemoattract human peripheral blood neutrophils, monocytes, and T cells. J Exp Med. 2000;192:1069–1074. doi: 10.1084/jem.192.7.1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tokumaru S., Sayama K., Shirakata Y., Komatsuzawa H., Ouhara K., Hanakawa Y., Yahata Y., Dai X., Tohyama M., Nagai H., Yang L., Higashiyama S., Yoshimura A., Sugai M., Hashimoto K. Induction of keratinocyte migration via transactivation of the epidermal growth factor receptor by the antimicrobial peptide LL-37. J Immunol. 2005;175:4662–4668. doi: 10.4049/jimmunol.175.7.4662. [DOI] [PubMed] [Google Scholar]
- 24.Coffelt S.B., Marini F.C., Watson K., Zwezdaryk K.J., Dembinski J.L., LaMarca H.L., Tomchuck S.L., Honer zu Bentrup K., Danka E.S., Henkle S.L., Scandurro A.B. The pro-inflammatory peptide LL-37 promotes ovarian tumor progression through recruitment of multipotent mesenchymal stromal cells. Proc Natl Acad Sci USA. 2009;106:3806–3811. doi: 10.1073/pnas.0900244106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Joyce J.A., Pollard J.W. Microenvironmental regulation of metastasis. Nat Rev Cancer. 2009;9:239–252. doi: 10.1038/nrc2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Anx A1 expression in human GBM specimens. A: Peri-tumor normal brain tissues. B–E: Different scores for Anx A1 expression in GBM tissues. The upper panels are H&E staining images. The lower panels show immunohistochemical staining of different levels of Anx A1 in GBMs with corresponding scores. Scale bar, 20 μm.