

Abstract
The accumulation of β-amyloid protein (Aβ) in the brain is thought to be a primary etiologic event in Alzheimer's disease (AD). Fibrillar Aβ plaques, a hallmark of AD abnormality, are closely associated with activated microglia. Activated microglia have contradictory roles in the pathogenesis of AD, being either neuroprotective (by clearing harmful Aβ and repairing damaged tissues) or neurotoxic (by producing proinflammatory cytokines and reactive oxygen species). Aβ aggregates can activate microglia by interacting with multiple toll-like receptors (TLRs), the pattern-recognition receptors of the innate immune system. Because the adapter protein MyD88 is essential for the downstream signaling of all TLRs, except TLR3, we investigated the effects of MyD88 deficiency (MyD88−/−) on Aβ accumulation and microglial activation in an AD mouse model. MyD88 deficiency decreased Aβ load and microglial activation in the brain. The decrease in Aβ load in an MyD88−/− AD mouse model was associated with increased and decreased protein expression of apolipoprotein E (apoE) and CX3CR1, respectively, compared with that in an MyD88 wild-type AD mouse model. These results suggest that MyD88 deficiency may reduce Aβ load by enhancing the phagocytic capability of microglia through fractalkine (the ligand of CX3CR1) signaling and by promoting apoE-mediated clearance of Aβ from the brain. These findings also suggest that chronic inflammatory responses induced by Aβ accumulation via the MyD88–dependent signaling pathway exacerbate β-amyloidosis in AD.
Accumulation of aggregated β-amyloid protein (Aβ) in the brain is postulated to be a causal event in the etiology of Alzheimer's disease (AD).1 Fibrillar Aβ deposits in the brain are accompanied by activated microglia.2 Increasing lines of evidence support the notion that activated microglia play pivotal dual roles in AD progression: either clearing Aβ deposits by phagocytosis and promoting neuronal survival and plasticity or releasing cytotoxic molecules and proinflammatory cytokines, exacerbating Aβ load and neurodegeneration.3–5 Fibrillar Aβ can activate microglia through interaction with cell surface receptor complexes, resulting in its phagocytosis and inflammatory responses. Some toll-like receptors (TLRs), including TLR2, TLR4, and TLR6, have been shown to be essential components of the receptor complexes for microglial activation by Aβ.6–8 TLRs are a class of pattern-recognition receptors in the innate immune system. One of the important roles of TLRs is to activate phagocytes/microglia in response to insults, including pathogens and damaged host cells, and to clear pathogens, damaged tissues, and accumulated wastes. Stimulation of TLR4 by lipopolysaccharide, a TLR4 ligand, has been repeatedly demonstrated to activate microglia in the brain.4 Hippocampal or intraperitoneal injection of lipopolysaccharide in old AD mouse models decreased diffuse Aβ plaques but not fibrillar Aβ plaques.9–12 Injection of CpG oligodeoxynucleotide, a TLR9 ligand, reduced Aβ load in the brain and restored cognitive deficits in an AD mouse model.13,14 Furthermore, a functionally inactive mutation in the TLR4 gene caused an increase in Aβ load in the brain of an AD mouse model.15 These results suggest that activation of TLRs can be a therapeutic option for AD. On the other hand, repeated injections of lipopolysaccharide induced premature fibrillar Aβ deposits in young AD mouse models16 and caused brain Aβ accumulation and cognitive impairments in mice.17 The latter experiments indicate that activation of TLR4 signaling is detrimental to cognitive functions. At least 10 TLRs in humans and 12 in mice have been reported.18 Except for TLR3, all TLRs use myeloid differentiation primary response protein 88 (MyD88) as an adaptor, which is essential for the downstream signaling culminating in activation of transcription factors. To investigate the roles of TLRs, particularly MyD88-dependent signaling in the pathogenesis of AD, we investigated β-amyloidosis and microglial activation in an AD mouse model deficient for MyD88 (MyD88−/−).
Materials and Methods
Experimental Animals
A congenic C57BL/6 line of AD model mice, B6.Cg-Tg(APPswe, PSEN1dE9) 85Dbo/J mice (APP mice), was purchased from The Jackson Laboratory (Bar Harbor, ME) and propagated by crossing transgenic males with C57BL/6 females. APP mice express chimeric mouse/human amyloid precursor protein (APP) with double mutations (K670N and M671L) and human presenilin 1 with a deletion of exon 9 found in patients with familial AD and develop Aβ deposits.19 Genotyping for the APPswe/PS1dE9 transgenes was performed using the PCR-based method provided by The Jackson Laboratory. MyD88-deficient (MyD88−/−) mice were obtained from Dr. Alan Aderem, Institute for Systems Biology (Seattle, WA), and were backcrossed to C57BL/6 mice more than 10 generations, and the genotyping for the MyD88 gene was performed by PCR.20 All the mice used in this study were C57BL/6 or congenic C57BL/6 mice. First, we produced APP MyD88+/− (hemizygous) mice by mating APP mice with MyD88−/− mice. Next, we mated APP MyD88+/− with MyD88−/− mice. This mating had a small litter size (≤5 pups), and 4 of 150 live pups were APP MyD88−/− mice. Thus, the birthrate of APP MyD88−/− mice of 2.7% was far lower than the expected rate of 25%. All the surviving APP MyD88−/− mice, however, seemed to be normal by appearance and were fertile. When APP MyD88−/− mice were mated with MyD88−/− mice, the birthrate of APP MyD88−/− mice was approximately 25% (half of the expected birthrate). This mating strategy was used to produce APP MyD88−/− and MyD88−/− mice. In this study, four experimental groups of 10-month-old mice that differed in the MyD88 genotype and the APPswe/PS1dE9 transgenes were used: i) APP MyD88−/−, ii) APP, iii) MyD88−/−, and iv) control C57BL/6 (WT) mice. The number of mice used in this study is given in Table 1 for sex, genotype group, and specific analysis. The mice were housed in group cages with bedding and were maintained on a 12-hour light/12-hour dark cycle (on at 6 am) under a specific pathogen-free condition with access to autoclaved food (NIH-31 formula) and water ad libitum. All the animal protocols used for this study were prospectively reviewed and approved by the Institutional Animal Care and Use Committee of the University of Illinois College of Medicine at Peoria.
Table 1.
Body Weight and Number of Mice Used for Each Analysis
| APP MyD88−/− mice |
APP mice |
MyD88−/− mice |
WT mice |
|||||
|---|---|---|---|---|---|---|---|---|
| Male | Female | Male | Female | Male | Female | Male | Female | |
| Body weight, mean ± SEM (g) | 32.6 ± 2.3 | 33.0 ± 2.4 | 32.4 ± 2.4 | 31.9 ± 1.4 | ||||
| Morphometry | 0 | 6 | 0 | 6 | 0 | 2⁎ | 0 | 2⁎ |
| Aβ ELISA | 6 | 0 | 6 | 0 | 2† | 0 | 2† | 0 |
| Immunoblot | 6 | 0 | 6 | 0 | 0 | 0 | 0 | 0 |
| qPCR (5 months old) | 3 | 2 | 1 | 2 | ||||
| qPCR (9 months old) | 4 | 1 | 2 | 2 | ||||
qPCR, quantitative PCR.
Unremarkable.
Indiscernible.
Brain Tissue Lysate Preparation
The mice were sacrificed by lethal injection of pentobarbital, and the brains were quickly removed and processed for enzyme-linked immunosorbent assay (ELISA) and immunoblot analyses. The brain tissues were lysed using the Bio-Plex cell lysis kit (Bio-Rad, Hercules, CA). Briefly, the tissue was rinsed with the cell wash buffer and was cut into 3 × 3-mm pieces. The sample was transferred to a dounce homogenizer on ice and was homogenized in the lysis buffer containing proteinase inhibitors. Then, the sample was sonicated on ice for 30 seconds and was centrifuged at 10,000 × g for 30 minutes at 4°C. The supernatant was collected for ELISA and immunoblotting as buffer-extractable proteins. Protein concentrations of the samples were determined by protein assay (Bio-Rad). The pellets were further dounce homogenized in guanidine hydrochloride (final concentration, 5 mol/L) and then were rock-shaken for 3 to 4 hours at room temperature. The solubilized pellets were used for determination of buffer-unextractable Aβ concentration by ELISA.
Aβ ELISA
After solubilization of brain tissues as described previously herein, tissue samples were further diluted with PBS at various concentrations. Levels of buffer-extractable and buffer-unextractable Aβ in the cerebrum were determined by using the Aβ42 and Aβ40 ELISA kits (Invitrogen, Carlsbad, CA) according to the manufacturer's protocol. Levels of buffer-extractable Aβ are expressed as mean ± SE picograms per milligram of total protein in tissue lysate. Levels of buffer-unextractable Aβ are expressed as mean ± SE nanograms per milligram of wet tissue weight.
Immunoblotting and Densitometric Analysis
Immunoblot analysis was used to quantify levels of APP, the 99–amino acid C-terminal fragment of APP (C99), β-site APP-cleaving enzyme (BACE1), apolipoprotein E (apoE), insulin-degrading enzyme (IDE), neprilysin (NEP), and chemokine (C-X3-C motif) receptor 1 (CX3CR1) in buffer-extractable proteins prepared from the cerebrum as described previously herein. Thirty micrograms of total protein from each sample were applied to 10% to 20% Tris-HCl gradient SDS-PAGE and were electrotransferred to polyvinylidine difluoride membranes (Millipore, Bedford, MA). The membranes were blocked by PBS containing 5% nonfat dried milk (w/v), 0.02% sodium azide, and 0.02% Tween 20 for 1 hour at room temperature; were incubated at 4°C overnight with the primary antibodies 6E10 (Signet Laboratories, Dedham, MA) for APP and C99, BACE1 (Clone 137612; R&D Systems, Minneapolis, MN), anti-NEP (Millipore, Billerica, MA), anti-IDE (PC730; EMD Biosciences, Gibbstown, NJ), apoE antibody (Meridian Life Science, Saco, ME), and CX3CR1 (Santa Cruz Biotechnology, Santa Cruz, CA); and were visualized by the Western Lighting chemiluminescence reagent plus (PerkinElmer, Boston, MA) according to the manufacturer's protocol. The membranes were reprobed with monoclonal antibody against glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Chemicon, Temecula, CA). The ODs of protein bands from the membranes were determined by densitometric scanning using an HP Scanjet G3010 photo scanner (Hewlett-Packard Co., Palo Alto, CA) and ImageJ V1.40 (National Institutes of Health, Bethesda, MD). The OD of each protein band was divided by that of the GAPDH band on the same lane from the same membrane for normalization.
Neuropathologic Assessment
The mice were deeply anesthetized and perfused transcardinally with PBS followed by 4% paraformaldehyde. The brains were removed and postfixed in the perfusate for 16 hours. The brains were then stored overnight in 30% sucrose in 0.1 mol/L PBS and frozen in Tissue-Tek (Sakura Finetek, Torrance, CA) optimal cutting temperature compound. Coronal sections (35 μm thick) of the brains were cut on a freezing-stage cryotome and were kept in 0.1 mol/L PBS with 0.05% sodium azide at 4°C. Sections were subjected to immunohistochemical and histochemical staining. Free-floating immunohistochemical staining was performed using the avidin-biotin immunoperoxidase method (Vector Laboratories, Burlingame, CA). Endogenous peroxidase was eliminated by treatment with 1% H2O2/10% methanol Tris-buffered saline (TBS) for 60 minutes at room temperature. After washing with 0.1 mol/L Tris buffer (pH 7.5) and 0.1 mol/L TBS (pH 7.4), sections were blocked with 5% normal serum (from the same animal species in which the secondary antibody was made) in 0.1 mol/L TBS with 0.5% Triton X-100 (Roche Diagnostics GmbH, Mannheim, Germany) for 60 minutes at room temperature to prevent nonspecific protein binding. The sections were then incubated with primary antibody 6E10 (1:2000), glial fibrillary acidic protein (GFAP; 1:1000), CD11b (1:200) or CD45 (1:100) in 2% bovine serum albumin, and 2% serum in TBS with 0.5% Triton X-100 for 18 to 48 hours at 4°C. The sections were rinsed in 0.1 mol/L TBS and were incubated with biotinylated secondary antibody anti-mouse IgG (1:1000) for 6E10, anti-rat IgG (1:300) for CD11b, anti-rat IgG (1:100) for CD45, or anti-rabbit IgG (1:1000) for GFAP in 2% serum TBS with 0.5% Triton X-100 for 2 hours at room temperature. Finally, the avidin-biotin peroxidase method using 3,3′-diaminobenzidine as a substrate (Vector Laboratories) was performed according to the manufacturer's protocol. For the negative control, slides were processed without primary antibody. Some sections were counterstained with hematoxylin. For thioflavin S staining, tissue sections were stained in 1% thioflavin S (Sigma-Aldrich, St. Louis, MO) and were rinsed with 70% ethanol. After washing with H2O, the sections were mounted in 75% glycerol in H2O.
Morphometry
Histomorphometry for quantification of amyloid deposits, activated microglia, and reactive astrocytes was performed using an Olympus BX61 automated microscope, the Olympus FluoView system (both from Olympus, Center Valley, PA), and Image-Pro Plus v4 image analysis software (Media Cybernetics, Bethesda, MD) capable of color segmentation and automation via programmable macros. Four coronal brain sections from each mouse were analyzed, each section separated by an approximately 500-μm interval, starting 1.3 mm posterior to the bregma to caudal. The neocortex and the hippocampus were found in all the brain sections and were analyzed separately. The stained area was expressed as a percentage of the total area.
BV-2 Cell Culture and Aβ Uptake and Degradation Assay
A mouse cell line of microglia, BV-2 (a gift from Dr. Michael McKinney), was maintained in Dulbecco's minimal essential medium supplemented with 10% FBS, 2 mmol/L l-glutamine, 100 U/mL of penicillin, and 100 μg/mL of streptomycin in 5% CO2 atmosphere at 37°C. BV-2 cells were inoculated at a concentration of 1.1 × 105 cells/1.5 mL/well into Falcon multiwell 6-well plates (Becton Dickinson, Franklin Lakes, NJ). The BV-2 cultures were treated with 0.5 μmol/L monomeric Aβ42 (EZBiolab, Carmel, IN) in the presence or absence of anti-apoE antibody (6 nmol/L) (Novus Biologicals, Littleton, CO) for 1 and 3 hours. As a control, an isotype control antibody (Sigma-Aldrich) was also used. Aβ42 levels in the cell lysates and media were determined by immunoblotting by 6E10 antibody. Briefly, cells were washed with PBS and were incubated in PBS containing 10 μg/mL trypsin on ice for 20 minutes. Trypsin was inactivated by the addition of 100 μg/mL of soybean trypsin inhibitor. After washing with PBS twice, the cells were lysed in 2× Laemmli buffer with protease inhibitors. Protein concentrations of cell lysates were determined by Bio-Rad protein assay. The cell lysates and media were subjected to immunoblotting followed by densitometric analysis as described previously herein except that the OD of each Aβ band from the medium was normalized by the total protein amount in the culture. Three independent experiments were performed.
Quantification of MyD88 mRNA by Real-Time PCR
The cerebra were isolated and soaked in RNAlater tissue collection: RNA stabilization solution (Ambion, Austin, TX) at 4°C overnight and then were stored at −80°C. These tissues were homogenized in TRIzol reagent (Invitrogen) for isolation of RNA. RNA samples were treated with RNase-free DNase (Qiagen, Valencia, CA) for 15 minutes at room temperature, and total RNA was purified using Qiagen RNeasy columns. cDNA was generated from 2 μg of total RNA in a total volume of 20 μL using a SuperScript III first-strand synthesis kit (Invitrogen) according to the manufacturer's protocol. MyD88 mRNA levels were determined by real-time PCR using an iCycler thermal cycler (Bio-Rad). cDNA was amplified using FastStart SYBR Green master mix (Roche Applied Science, Indianapolis, IN) with forward primer (5′-AAGTCTAGGAAGGCCCCAAA-3′) and reverse primer (5′-CTGGGGAGAAAACAGCTGAG-3′). The PCR amplifications were performed as follows: 10 minutes of preincubation at 95°C to activate the FastStart Taq DNA polymerase (Roche Applied Science), 40 cycles of denaturation at 95°C for 15 seconds, and primer annealing and extension for 1 minute at 60°C. PCR product melting curves were examined to confirm the homogeneity of PCR products. MyD88 mRNA levels were normalized by subtracting CT values obtained with GAPDH mRNA using forward primer (5′-CCCACTCTTCCACCTTCGAT-3′) and reverse primer (5′-CCACCACCCTGTTGCTGTAG-3′) and expressed as 2−ΔCT [ΔCT = CT (cytokine or chemokine) – CT (GAPDH)].
Statistics
Data are expressed as mean ± SE. Intergroup differences were assessed by analysis of variance and two-tailed Student's t-tests. P < 0.05 was considered significant.
Results
MyD88 Deficiency Decreases Aβ Deposits and Buffer-Extractable Aβ in the Cerebrum of an AD Mouse Model
To determine whether MyD88 deficiency affects Aβ abnormalities in AD mouse models, we evaluated Aβ deposits in APP MyD88−/− and APP mice at 10 months of age. Diffuse and fibrillar Aβ deposits in the brain were detected by immunohistochemical staining using 6E10 antibody (Figure 1, A, C, and E). Aβ loads are indicated by mean percentages of areas showing Aβ immunoreactivity in the brain. The mean ± SEM Aβ load in the neocortex (0.44% ± 0.07%) and hippocampus (0.30 ± 0.05%) of APP MyD88−/− mice (n = 6) was less than that in the neocortex (0.85% ± 0.07%; P < 0.01) and hippocampus (0.54% ± 0.04%; P < 0.001) of APP mice (n = 6). Fibrillar Aβ deposits were visualized by thioflavin S fluorescence (Figure 1, B, D, and F). The mean ± SEM Aβ load in APP MyD88−/− mice (0.19% ± 0.03% for the neocortex and 0.15% ± 0.02% for the hippocampus; n = 6) was less than that in APP mice (0.30% ± 0.04% for the neocortex and 0.23% ± 0.03% for the hippocampus; P < 0.05 for both; n = 6). Thus, levels of diffuse and fibrillar Aβ deposits decreased in APP MyD88−/− mice. Soluble oligomeric Aβ species have been identified as synaptotoxic and neurotoxic forms of Aβ rather than insoluble amyloid fibrils.21–23 Therefore, we determined the amounts of buffer-extractable and buffer-unextractable Aβ in the cerebrum by the Aβ40- and Aβ42-specific ELISA (Figure 1, G and H). On average, the buffer extractable Aβ40 and Aβ42 content in APP MyD88−/− mice (mean ± SEM: 9.9 ± 1.21 and 15.90 ± 3.42 pg/mg protein, respectively) reduced approximately 40% to 50% of those in APP mice (mean ± SEM: 15.42 ± 2.12 and 28.14 ± 4.20 pg/mg protein, respectively; n = 6; P < 0.05 for both measures). The mean ± SEM cerebral buffer-unextractable Aβ40 and Aβ42 levels in APP MyD88−/− mice (2.69 ± 0.50 and 8.40 ± 0.37 ng/mg wet tissue, respectively; n = 6) were less than those in APP mice (4.55 ± 0.67 and 10.75 ± 0.88 ng/mg wet tissue, respectively; n = 6; P < 0.05 for both measures).
Figure 1.

Detection of diffuse and fibrillar Aβ deposits by anti-Aβ antibody and thioflavin S and quantification of buffer-extractable and buffer-unextractable Aβ by ELISA in APP MyD88−/− and APP mice. Aβ deposits in the brain are visualized by immunohistochemical staining using 6E10 antibody in APP MyD88−/− (A) and APP (C) mice. Percentages of areas showing Aβ immunoreactivity measured by morphometry in the hippocampus and neocortex (E) are shown. Fibrillar Aβ deposits in the brain are visualized by thioflavin S fluorescence in APP MyD88−/− (B) and APP (D) mice. Percentages of areas showing fluorescence measured by morphometry in the hippocampus and neocortex (F) are shown. In the neocortex and hippocampus, the Aβ load in APP MyD88−/− mice is significantly less than that in APP mice. Quantification of buffer-extractable (G) and buffer-unextractable (H) Aβ in the cerebrum of APP MyD88−/− and APP mice at 10 months of age. Levels of buffer-extractable and buffer-unextractable Aβ40 and Aβ42 were determined by ELISA. The data shown are the mean ± SEM of n = 6 for each group (E–H). **P < 0.05, ***P < 0.01, ****P < 0.001. Scale bars = 1 mm.
MyD88 Deficiency Does Not Influence Steady State Levels of APP, a Proteolytic C-Terminal Fragment of APP, BACE1, NEP, or IDE in an AD Mouse Model
To determine whether MyD88 deficiency alters the expression levels of APP and its proteolysis, cerebral homogenates of APP MyD88−/− and APP mice were subjected to immunoblotting followed by densitometric analysis. No significant differences were found in the steady state levels of APP and the β-secretase cleavage product C99 in the brain between the two groups (Figure 2, A and B). Because decreased levels of Aβ deposits in the brain of an AD mouse model deficient for the tumor necrosis factor type 1 death receptor or interferon-γ receptor type I were associated with decreased expression levels of BACE1,24,25 and because MyD88 deficiency may alter expression levels of these cytokines, BACE1 levels were also determined by immunoblotting (Figure 2A). No significant difference was observed in the levels of BACE1 between the two groups (Figure 2B). IDE and NEP have been described as Aβ-degrading enzymes in the brain and are thought to be involved in clearing Aβ from the brain.26,27 To investigate whether the decreased levels of Aβ load in APP MyD88−/− mice are associated with increased levels of IDE and NEP expression, steady state levels of these enzymes were determined by immunoblot analysis (Figure 2, C and E). There were no differences in the expression levels of IDE and NEP in the cerebrum between APP MyD88−/− and APP mice (Figure 2, D and F).
Figure 2.
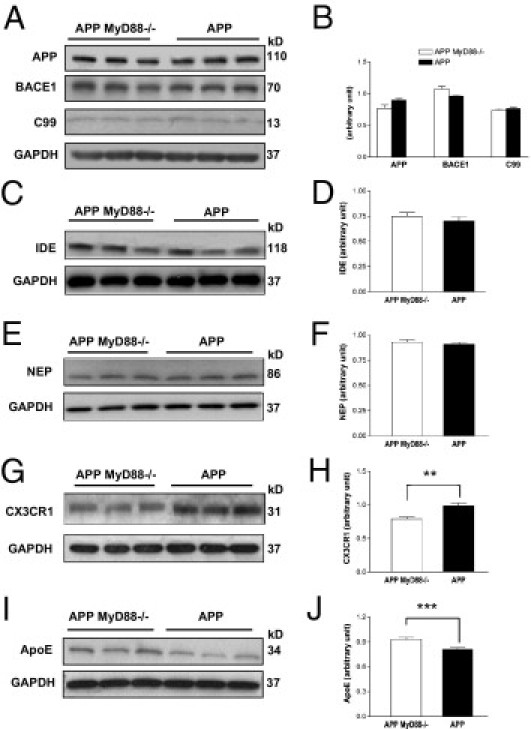
Steady state levels of APP, C99, BACE1, IDE, NEP, CX3CR1, and apoE in APP MyD88−/− and APP mice. A: Representative immunoblots of APP, C99, BACE1, and GAPDH in buffer-extractable tissue lysates prepared from the cerebrum of APP MyD88−/− and APP mice determined by 6E10 antibody for APP and C99, BASE1 antibody, and GAPDH antibody, respectively. Representative immunoblots of IDE and GAPDH (C), NEP and GAPDH (E), CX3CR1 and GAPDH (G), and apoE and GAPDH (I) in the buffer-extractable tissue lysates. The molecular weights of proteins are indicated on the right in each immunoblot. The bar graphs represent densitometric quantification of each protein indicated after normalization with GAPDH (B, D, F, H, and J). There are no differences in APP, BACE1, C99, IDE, and NEP levels between APP MyD88−/− (n = 6) and APP (n = 6) mice. Decreased CX3CR1 expression and increased apoE expression in the brains of APP MyD88−/− mice (n = 6) are shown compared with in APP mice (n = 6). The data shown are the mean ± SE. **P < 0.003, ***P < 0.01.
MyD88 Deficiency Decreases CX3CR1 and Increases ApoE Expression in the Brain of an AD Mouse Model
The chemokine receptor for fractalkine (CX3CL1), CX3CR1, is expressed in microglia and plays an important role in the recruitment of microglia to sites of inflammation and in microglia-mediated neurotoxicity. Recently, CX3CR1 deficiency has been shown to be associated with increased phagocytic ability of microglia and decreased Aβ load in the brains of AD mouse models.28,29 Therefore, we determined the expression levels of CX3CR1 in the cerebrum of APP MyD88−/− and APP mice by immunoblot analysis (Figure 2G). There was decreased expression of CX3CR1 in APP MyD88−/− mice compared with in APP mice (P < 0.003; n = 6 for each group) (Figure 2H).
ApoE has been shown to facilitate clearance of soluble Aβ from the brain.30,31 Therefore, steady state levels of apoE were determined by immunoblot analysis (Figure 2I). Expression levels of apoE were higher in APP MyD88−/− mice than in APP mice (P < 0.01; n = 6 for each group) (Figure 2J).
Decreased Aβ Load in APP MyD88−/− Mice Is Accompanied by Decreased Levels of Markers for Astrocytes and Microglia/Macrophages
Activated microglia and reactive astrocytes are closely associated with fibrillar Aβ deposits in AD mouse models and patients with AD.2,32 GFAP and CD11b (Mac-1) are widely used as markers for reactive astrocytes and activated microglia, respectively, and a common leukocyte antigen, CD45, is a marker to detect activated microglia and monocytes/macrophages that potentially invade the brain. These markers were used to study the effect of MyD88 deficiency on activation of astrocytes and microglia by amyloid plaques in an AD mouse model. Reactive astrocytes in the neocortex and hippocampus of APP MyD88−/− and APP mice were detected by immunohistochemical staining using anti-GFAP antibody (Figure 3, A and D). Morphometric analysis indicated that the hippocampus and neocortex had a lesser degree of astrocytosis in APP MyD88−/− mice (mean ± SEM: 8.0% ± 1.2% and 12.0% ± 1.3%, respectively) than in APP mice (mean ± SEM: 14.0% ± 1.3%, P < 0.05 and 16.4% ± 1.5%, P < 0.05, respectively) (Figure 3G). Brain sections from APP MyD88−/− and APP mice were immunostained with anti-CD45 antibody (Figure 3, B and E). The mean ± SEM CD45 immunoreactivity of the hippocampus and neocortex in APP MyD88−/− mice (0.41% ± 0.07% and 0.69% ± 0.11%, respectively) was less than that in APP mice (0.62% ± 0.02%, P = 0.02 and 1.2% ± 0.17%, P = 0.03, respectively) (Figure 3H). Morphometric analysis of the CD11b-stained brain sections (Figure 3, C and F) revealed that the hippocampus (mean ± SEM: 0.15% ± 0.02%) of APP MyD88−/− mice had less microglial immunoreactivity than did that of APP mice (mean ± SEM: 0.306% ± 0.05%; P = 0.02) (Figure 3I). Mean ± SEM microglial immunoreactivity for CD11b in the neocortex of APP MyD88−/− mice (0.19% ± 0.03%) was reduced compared with that in APP mice (0.28% ± 0.07%), but this did not reach statistical significance (Figure 3I).
Figure 3.

Reduced expression of microglial markers and reactive astrocytes in APP MyD88−/− mice. The frozen sections of cerebral cortices from APP MyD88−/− (A–C) and APP (D–F) mice were stained with anti-GFAP (A and D), anti-CD45 (B and E), and anti-CD11b antibody (C and F). Scale bars = 200 μm. G–I: Mean ± SEM percentages of areas showing fluorescence measured by morphometry in the hippocampus and neocortex. The GFAP (G) and CD45 (H) immunoreactivity of the hippocampus and neocortex in APP MyD88−/− mice (n = 6) is less than that in APP mice (n = 6). The hippocampus of APP MyD88−/− mice (n = 6) had significantly less CD11b immunoreactivity than that of APP mice (n = 6) (I), but the difference in CD11b immunoreactivity in the neocortex did not reach statistical significance. *P < 0.03, **P < 0.05.
Anti-ApoE Antibody Inhibits Aβ Degradation but Not Aβ Uptake by BV-2 Microglial Cells
Because decreased Aβ load in APP MyD88−/− mice was accompanied by increased apoE levels and because apoE has been shown to enhance Aβ clearance by microglia,33 we investigated the role of apoE in Aβ uptake and degradation by a microglial cell line, BV-2. BV-2 cells were incubated with monomeric Aβ42 (0.5 μmol/L) in the presence and absence of anti-apoE antibody (6 nmol/L). As a control, BV-2 cells were treated with an isotype control antibody also. One and 3 hours after incubation, Aβ uptake and degradation by BV-2 cells were quantified by immunoblot analysis of the culture media and cell lysates. There were no differences in the Aβ contents in the culture media among the three groups after 1 and 3 hours of incubation (see Supplemental Figure S1A at http://ajp.amjpathol.org). The Aβ contents in the apoE antibody-treated cell lysates were greater than those in the control lysates after 3 hours of incubation (P < 0.01) (see Supplemental Figure S1B at http://ajp.amjpathol.org). These results suggest that deprivation of apoE by its antibody inhibits degradation of Aβ but does not have any effect on Aβ uptake by microglia.
MyD88 Expression Decreases during Aging
Elderly humans and aged mice are more susceptible to pneumonia, which can be attributable to decreases in TLR1, TLR2, and TLR4 levels in the lung.34 Alterations in expression levels of multiple TLRs in the mouse brain during aging have also been reported.35 Changes in MyD88 expression levels during aging may contribute to Aβ load in the brain. Therefore, we determined expression levels of cerebral MyD88 in 5- and 9-month-old APP (n = 5 for both ages) and WT (n = 3 and 4, respectively) mice. MyD88 mRNA levels at 9 months were lower than those at 5 months in APP mice (P = 0.050). On average, MyD88 mRNA levels at 9 months were lower than those at 5 months in WT mice, but the difference was not significant (P = 0.067) probably owing to the small sample sizes or large variations. Thus, expression levels of cerebral MyD88 seemed to decrease during aging in mice.
Discussion
The present study presents new insights into the role of MyD88 signaling pathway in the pathogenesis of AD. We demonstrate that MyD88 deficiency decreased Aβ load and microglial activation in the brain and that the decrease in Aβ load was accompanied by decreased CX3CR1 expression and increased apoE expression. These results suggest that recognition of Aβ deposits by TLRs induces inflammatory responses through the MyD88 signaling pathway, contributing to exacerbation of β-amyloidosis, and that a blockade of MyD88 signaling slows the progression of AD abnormalities.
The birthrate of APP MyD88−/− mice was less than half of the expected rate. Thus, embryonic lethality seemed to increase in APP MyD88−/− mice. The reasons for the decreased birthrate are not clear. Aberrant activation of TLR signaling during embryonic stages can produce imbalances in fetal brain cytokines, resulting in abnormal brain development,36 and soluble Aβ oligomers can induce inflammatory responses in the brain.37 Such imbalances in cytokines may cause developmental abnormalities in the brains of APP MyD88−/− mice, leading to the decreased birthrate. In addition, the presenilin 1 exon 9 deletion associated with AD sensitizes neuroblastoma cells to hyperosmotic stress–induced apoptosis, and MyD88−/− mice show enhanced epithelial cell apoptosis after lung injury.33,38 Such a combination may also contribute to the increased embryonic lethality. Although APP and MyD88−/− mice were backcrossed to C57BL/6 mice more than 10 generations, it is possible that subtle differences in the genetic background may have an effect on their birthrate. Although an increase in premature sudden death has been reported in APP (APPswe/PS1dE9) mice and is proposed to be caused by epileptic seizures,39 there was no difference in the survival rate between APP and APP MyD88−/− mice after birth. We performed pathologic, bacteriologic, serologic, and parasitologic examinations on APP MyD88−/− mice at 6 weeks of age. Gross observations and microscopic diagnoses found that brain, liver, lung, heart, kidneys, stomach, small intestine, large intestine, reproductive organs, and urinary bladder findings were within normal limits, and no infection was found by these examinations (data not shown). We, however, cannot exclude the possibility that subtle developmental differences between the two groups contributed to a decrease in Aβ load in APP MyD88−/− mice. Adult hippocampal neurogenesis is increased in MyD88−/− mice and decreased in APP mice.40,41 Such a difference in neurogenesis may also affect Aβ load in the brain.
The molecular mechanism by which MyD88 deficiency reduces Aβ load in an AD mouse model is not clear. During the preparation of this article, Hao et al42 reported that bone marrow cells derived from MyD88−/− mice had an enhancement of Aβ phagocytosis and that transplantation of MyD88−/− bone marrow cells into an AD mouse model decreased Aβ load in the brain. Therefore, Aβ clearance by microglia/macrophages may be enhanced in APP MyD88−/− mice, resulting in a decrease in the cerebral Aβ load. In line with this view, CX3CR1 deficiency was shown to enhance the phagocytic capacity of microglia and give rise to a gene dose-dependent reduction in Aβ deposition in AD mouse models.28,29 Thus, the decreased protein levels of CX3CR1 in APP MyD88−/− mice may accelerate Aβ removal by microglia.
ApoE has been identified as the most prominent genetic risk factor for the development of late-onset AD.43 Although the precise mechanism by which the ε4 allele of apoE increases the risk of AD remains to be elucidated, lipidated apoE enhanced Aβ degradation by IDE and NEP and reduced brain Aβ load in an AD mouse model.31 We also showed that Aβ degradation by BV-2 microglial cells was inhibited by deprivation of apoE by its antibody. Therefore, the increased levels of apoE in APP MyD88−/− mice may contribute to a decrease in Aβ load.
We previously demonstrated that B6C3 APPswe/PS1dE9 mice with nonfunctional TLR4 due to the mutation in the TLR4 gene (TLR4M) had increases in buffer-extractable and buffer-unextractable Aβ and amyloid deposits in the brain at 14 to 16 months of age compared with B6C3 APPswe/PS1dE9 mice with functional TLR4.15 In addition, we and others found that activation of TLR2, TLR4, or TLR9 increased uptake of Aβ by cultured microglial cells.15,44,45 Activation of TLR9 by CpG oligodeoxynucleotides reduced soluble Aβ oligomer levels and amyloid deposits in the brain of an AD mouse model.13,14 Therefore, we expected an increase in Aβ load in APP MyD88−/− mice. Contrary to our prediction, APP MyD88−/− mice had less Aβ load than did APP mice at 10 months of age. Richard et al45 also found that APPswe/PS1dE9 mice deficient for TLR2 (TLR2−/−) had decreased levels of Aβ deposits at 3 and 6 months of age compared with TLR2 wild-type APPswe/PS1dE9 mice, but the difference in Aβ deposits was not discernible at 9 months of age. To explain the increase in Aβ load in TLR4M APPswe/PS1dE9 mice and the decrease in TLR2−/− APPswe/PS1dE9 mice, Reed-Geaghan et al8 proposed that during the early stages of AD, TLR-independent Aβ clearance mechanisms were effective in removing Aβ from the brain, and, potentially, the absence of TLR-mediated inflammation made these processes more efficient. According to their hypothesis, decreased Aβ load in the brains of APP MyD88−/− mice can be due to a lack of MyD88 signaling throughout life. They also argued that over time, the rate of Aβ production overwhelmed these processes and the lack of TLRs enhanced Aβ deposition. In line with this concept, MyD88 expression seemed to decrease during aging. However, there are important discrepancies between our APP MyD88−/− and TLR2−/− APPswe/PS1dE9 mice. Levels of Aβ-immunoreactive deposits were similar between TLR2−/− and TLR2+/+ APPswe/PS1dE9 mice at 9 months of age, whereas levels of Aβ-immunoreactive deposits decreased in APP MyD88−/− mice at 10 months compared with in APP mice. Furthermore, TLR2−/− APPswe/PS1dE9 mice had increased levels of soluble Aβ42 compared with TLR2+/+ APPswe/PS1dE9 mice, whereas APP MyD88−/− mice seemed to have decreased levels of buffer-extractable Aβ40 and Aβ42 at 10 months. We find difficulty in explaining these discrepancies solely by the hypothesis set by Reed-Geaghan et al.8 Activation of most TLRs by TLR ligands leads to transcriptional expression of inflammatory mediators, such as cytokines and reactive oxygen species. MyD88 is essential for the downstream signaling of all TLRs, except TLR3. However, depending on the TLRs and cell types, distinct genes are up-regulated, resulting in differences in gene expression profiles. In addition, microorganisms and tissue injuries activate multiple TLRs and other pattern-recognition receptors, further diversifying the expression profiles.18 Therefore, activation and inactivation of innate immune responses by targeting a single TLR is most likely to induce inflammatory responses different from targeting multiple TLRs, resulting in distinctive phenotypic manifestations in AD mouse models. The findings herein signify the importance of comprehensive approaches in delineating the network regulation of inflammatory responses involved in slowing and accelerating the progression of AD.
Acknowledgments
We thank Linda Walter and Pradeep Singanallur for their assistance in preparation of the manuscript.
Footnotes
Supported in part by grants from the National Institutes of Health (AG030399, AG031979, AG029818, and EY018478) and the Alzheimer's Association (IIRG-07-59494).
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.05.045.
Supplementary data
Anti-apoE antibody inhibits Aβ degradation but not Aβ uptake by BV-2 microglial cells (A and B). BV-2 microglial cells were incubated with monomeric Aβ42 (0.5 μmol/L) in the presence or absence of anti-apoE antibody (6 nmol/L). As a control, isotype control antibody (mouse IgG) was also used. One and 3 hours after incubation, Aβ levels in the media (A) and cell lysates (B) were determined by immunoblot analysis using the anti-Aβ antibody 6E10. There were no differences in the Aβ contents in the culture media among the three groups (A). The lane indicated by “no cells” represents Aβ (0.5 μmol/L) in the medium from a 6-well plate without inoculation of BV-2 cells. The Aβ contents in the anti-apoE antibody-treated cell lysates are greater than those in the control groups (B). The bar graphs represents densitometric quantification of each Aβ band after normalization with GAPDH (cell lysate) or total protein amount in the culture (medium). Data shown are the mean ± SE. Expression levels of cerebral MyD88 at 9 months of age tend to decrease compared with those at 5 months in APP and WT mice (C). mRNA levels of cerebral MyD88 in 5- and 9-month-old APP and WT mice were determined by real-time PCR using cDNAs prepared by reverse transcription of mRNA. mRNA levels of MyD88 were normalized by subtracting CT values obtained with GAPDH mRNA and are shown as 2−ΔCt [ΔCT = CT (MyD88) – CT (GAPDH)] (mean ± SEM).
References
- 1.Hardy J., Selkoe D.J. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 2.Akiyama H., Barger S., Barnum S. Inflammation and Alzheimer's disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wyss-Coray T. Inflammation in Alzheimer disease: driving force, bystander or beneficial response? Nat Med. 2006;12:1005–1015. doi: 10.1038/nm1484. [DOI] [PubMed] [Google Scholar]
- 4.Rivest S. Regulation of innate immune responses in the brain. Nat Rev Immunol. 2009;9:429–439. doi: 10.1038/nri2565. [DOI] [PubMed] [Google Scholar]
- 5.Mandrekar-Colucci S., Landreth G.E. Microglia and inflammation in Alzheimer's disease. CNS Neurol Disord Drug Targets. 2010;9:156–167. doi: 10.2174/187152710791012071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu Y., Walter S., Stagi M., Cherny D., Letiembre M., Schulz-Schaeffer W., Heine H., Penke B., Neumann H., Fassbender K. LPS receptor (CD14): a receptor for phagocytosis of Alzheimer's amyloid peptide. Brain. 2005;128:1778–1789. doi: 10.1093/brain/awh531. [DOI] [PubMed] [Google Scholar]
- 7.Stewart C.R., Stuart L.M., Wilkinson K., van Gils J.M., Deng J., Halle A., Rayner K.J., Boyer L., Zhong R., Frazier W.A., Lacy-Hulbert A., El K.J., Golenbock D.T., Moore K.J. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol. 2010;11:155–161. doi: 10.1038/ni.1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reed-Geaghan E.G., Savage J.C., Hise A.G., Landreth G.E. CD14 and toll-like receptors 2 and 4 are required for fibrillar Aβ-stimulated microglial activation. J Neurosci. 2009;29:11982–11992. doi: 10.1523/JNEUROSCI.3158-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DiCarlo G., Wilcock D., Henderson D., Gordon M., Morgan D. Intrahippocampal LPS injections reduce Aβ load in APP+PS1 transgenic mice. Neurobiol Aging. 2001;22:1007–1012. doi: 10.1016/s0197-4580(01)00292-5. [DOI] [PubMed] [Google Scholar]
- 10.Herber D.L., Roth L.M., Wilson D., Wilson N., Mason J.E., Morgan D., Gordon M.N. Time-dependent reduction in Aβ levels after intracranial LPS administration in APP transgenic mice. Exp Neurol. 2004;190:245–253. doi: 10.1016/j.expneurol.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 11.Malm T.M., Koistinaho M., Parepalo M., Vatanen T., Ooka A., Karlsson S., Koistinaho J. Bone-marrow-derived cells contribute to the recruitment of microglial cells in response to β-amyloid deposition in APP/PS1 double transgenic Alzheimer mice. Neurobiol Dis. 2005;18:134–142. doi: 10.1016/j.nbd.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 12.Quinn J., Montine T., Morrow J., Woodward W.R., Kulhanek D., Eckenstein F. Inflammation and cerebral amyloidosis are disconnected in an animal model of Alzheimer's disease. J Neuroimmunol. 2003;137:32–41. doi: 10.1016/s0165-5728(03)00037-7. [DOI] [PubMed] [Google Scholar]
- 13.Scholtzova H., Kascsak R.J., Bates K.A., Boutajangout A., Kerr D.J., Meeker H.C., Mehta P.D., Spinner D.S., Wisniewski T. Induction of toll-like receptor 9 signaling as a method for ameliorating Alzheimer's disease-related pathology. J Neurosci. 2009;29:1846–1854. doi: 10.1523/JNEUROSCI.5715-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Doi Y., Mizuno T., Maki Y., Jin S., Mizoguchi H., Ikeyama M., Doi M., Michikawa M., Takeuchi H., Suzumura A. Microglia activated with the toll-like receptor 9 ligand CpG attenuate oligomeric amyloid β neurotoxicity in in vitro and in vivo models of Alzheimer's disease. Am J Pathol. 2009;175:2121–2132. doi: 10.2353/ajpath.2009.090418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tahara K., Kim H.D., Jin J.J., Maxwell J.A., Li L., Fukuchi K.I. Role of toll-like receptor signalling in Aβ uptake and clearance. Brain. 2006;129:3006–3019. doi: 10.1093/brain/awl249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sheng J.G., Bora S.H., Xu G., Borchelt D.R., Price D.L., Koliatsos V.E. Lipopolysaccharide-induced-neuroinflammation increases intracellular accumulation of amyloid precursor protein and amyloid β peptide in APPswe transgenic mice. Neurobiol Dis. 2003;14:133–145. doi: 10.1016/s0969-9961(03)00069-x. [DOI] [PubMed] [Google Scholar]
- 17.Lee J.W., Lee Y.K., Yuk D.Y., Choi D.Y., Ban S.B., Oh K.W., Hong J.T. Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of β-amyloid generation. J Neuroinflammation. 2008;5:37. doi: 10.1186/1742-2094-5-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takeuchi O., Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 19.Jankowsky J.L., Fadale D.J., Anderson J., Xu G.M., Gonzales V., Jenkins N.A., Copeland N.G., Lee M.K., Younkin L.H., Wagner S.L., Younkin S.G., Borchelt D.R. Mutant presenilins specifically elevate the levels of the 42 residue β-amyloid peptide in vivo: evidence for augmentation of a 42-specific γ secretase. Hum Mol Genet. 2004;13:159–170. doi: 10.1093/hmg/ddh019. [DOI] [PubMed] [Google Scholar]
- 20.Adachi O., Kawai T., Takeda K., Matsumoto M., Tsutsui H., Sakagami M., Nakanishi K., Akira S. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity. 1998;9:143–150. doi: 10.1016/s1074-7613(00)80596-8. [DOI] [PubMed] [Google Scholar]
- 21.Lambert M.P., Barlow A.K., Chromy B.A., Edwards C., Freed R., Liosatos M., Morgan T.E., Rozovsky I., Trommer B., Viola K.L., Wals P., Zhang C., Finch C.E., Krafft G.A., Klein W.L. Diffusible, nonfibrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Walsh D.M., Klyubin I., Fadeeva J.V., Cullen W.K., Anwyl R., Wolfe M.S., Rowan M.J., Selkoe D.J. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 23.Lesne S., Koh M.T., Kotilinek L., Kayed R., Glabe C.G., Yang A., Gallagher M., Ashe K.H. A specific amyloid-β protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- 24.He P., Zhong Z., Lindholm K., Berning L., Lee W., Lemere C., Staufenbiel M., Li R., Shen Y. Deletion of tumor necrosis factor death receptor inhibits amyloid β generation and prevents learning and memory deficits in Alzheimer's mice. J Cell Biol. 2007;178:829–841. doi: 10.1083/jcb.200705042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yamamoto M., Kiyota T., Horiba M., Buescher J.L., Walsh S.M., Gendelman H.E., Ikezu T. Interferon-γ and tumor necrosis factor-α regulate amyloid-β plaque deposition and β-secretase expression in Swedish mutant APP transgenic mice. Am J Pathol. 2007;170:680–692. doi: 10.2353/ajpath.2007.060378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qiu W.Q., Walsh D.M., Ye Z., Vekrellis K., Zhang J., Podlisny M.B., Rosner M.R., Safavi A., Hersh L.B., Selkoe D.J. Insulin-degrading enzyme regulates extracellular levels of amyloid β-protein by degradation. J Biol Chem. 1998;273:32730–32738. doi: 10.1074/jbc.273.49.32730. [DOI] [PubMed] [Google Scholar]
- 27.Iwata N., Tsubuki S., Takaki Y., Shirotani K., Lu B., Gerard N.P., Gerard C., Hama E., Lee H.J., Saido T.C. Metabolic regulation of brain Aβ by neprilysin. Science. 2001;292:1550–1552. doi: 10.1126/science.1059946. [DOI] [PubMed] [Google Scholar]
- 28.Lee S., Varvel N.H., Konerth M.E., Xu G., Cardona A.E., Ransohoff R.M., Lamb B.T. CX3CR1 deficiency alters microglial activation and reduces β-amyloid deposition in two Alzheimer's disease mouse models. Am J Pathol. 2010;177:2549–2562. doi: 10.2353/ajpath.2010.100265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu Z., Condello C., Schain A., Harb R., Grutzendler J. CX3CR1 in microglia regulates brain amyloid deposition through selective protofibrillar amyloid-β phagocytosis. J Neurosci. 2010;30:17091–17101. doi: 10.1523/JNEUROSCI.4403-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shibata M., Yamada S., Kumar S.R., Calero M., Bading J., Frangione B., Holtzman D.M., Miller C.A., Strickland D.K., Ghiso J., Zlokovic B.V. Clearance of Alzheimer's amyloid-ss(1–40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Invest. 2000;106:1489–1499. doi: 10.1172/JCI10498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang Q., Lee C.Y., Mandrekar S., Wilkinson B., Cramer P., Zelcer N., Mann K., Lamb B., Willson T.M., Collins J.L., Richardson J.C., Smith J.D., Comery T.A., Riddell D., Holtzman D.M., Tontonoz P., Landreth G.E. ApoE promotes the proteolytic degradation of Aβ. Neuron. 2008;58:681–693. doi: 10.1016/j.neuron.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morgan D., Gordon M.N., Tan J., Wilcock D., Rojiani A.M. Dynamic complexity of the microglial activation response in transgenic models of amyloid deposition: implications for Alzheimer therapeutics. J Neuropathol Exp Neurol. 2005;64:743–753. doi: 10.1097/01.jnen.0000178444.33972.e0. [DOI] [PubMed] [Google Scholar]
- 33.Jiang D., Liang J., Fan J., Yu S., Chen S., Luo Y., Prestwich G.D., Mascarenhas M.M., Garg H.G., Quinn D.A., Homer R.J., Goldstein D.R., Bucala R., Lee P.J., Medzhitov R., Noble P.W. Regulation of lung injury and repair by toll-like receptors and hyaluronan. Nat Med. 2005;11:1173–1179. doi: 10.1038/nm1315. [DOI] [PubMed] [Google Scholar]
- 34.Hinojosa E., Boyd A.R., Orihuela C.J. Age-associated inflammation and toll-like receptor dysfunction prime the lungs for pneumococcal pneumonia. J Infect Dis. 2009;200:546–554. doi: 10.1086/600870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Letiembre M., Hao W., Liu Y., Walter S., Mihaljevic I., Rivest S., Hartmann T., Fassbender K. Innate immune receptor expression in normal brain aging. Neuroscience. 2007;146:248–254. doi: 10.1016/j.neuroscience.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 36.Meyer U., Feldon J., Fatemi S.H. In-vivo rodent models for the experimental investigation of prenatal immune activation effects in neurodevelopmental brain disorders. Neurosci Biobehav Rev. 2009;33:1061–1079. doi: 10.1016/j.neubiorev.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 37.Ferretti M.T., Cuello A.C. Does a pro-inflammatory process precede Alzheimer's disease and mild cognitive impairment? Curr Alzheimer Res. 2011;8:164–174. doi: 10.2174/156720511795255982. [DOI] [PubMed] [Google Scholar]
- 38.Tanii H., Ankarcrona M., Flood F., Nilsberth C., Mehta N.D., Perez-Tur J., Winblad B., Benedikz E., Cowburn R.F. Alzheimer's disease presenilin-1 exon 9 deletion and L250S mutations sensitize SH-SY5Y neuroblastoma cells to hyperosmotic stress-induced apoptosis. Neuroscience. 2000;95:593–601. doi: 10.1016/s0306-4522(99)00432-7. [DOI] [PubMed] [Google Scholar]
- 39.Minkeviciene R., Rheims S., Dobszay M.B., Zilberter M., Hartikainen J., Fulop L., Penke B., Zilberter Y., Harkany T., Pitkanen A., Tanila H. Amyloid β-induced neuronal hyperexcitability triggers progressive epilepsy. J Neurosci. 2009;29:3453–3462. doi: 10.1523/JNEUROSCI.5215-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rolls A., Shechter R., London A., Ziv Y., Ronen A., Levy R., Schwartz M. Toll-like receptors modulate adult hippocampal neurogenesis. Nat Cell Biol. 2007;9:1081–1088. doi: 10.1038/ncb1629. [DOI] [PubMed] [Google Scholar]
- 41.Crews L., Rockenstein E., Masliah E. APP transgenic modeling of Alzheimer's disease: mechanisms of neurodegeneration and aberrant neurogenesis. Brain Struct Funct. 2010;214:111–126. doi: 10.1007/s00429-009-0232-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hao W., Liu Y., Liu S., Walter S., Grimm M.O., Kiliaan A.J., Penke B., Hartmann T., Rube C.E., Menger M.D., Fassbender K. Myeloid differentiation factor 88-deficient bone marrow cells improve Alzheimer's disease-related symptoms and pathology. Brain. 2011;134:278–292. doi: 10.1093/brain/awq325. [DOI] [PubMed] [Google Scholar]
- 43.Strittmatter W.J., Weisgraber K.H., Huang D.Y., Dong L.M., Salvesen G.S., Pericak-Vance M., Schmechel D., Saunders A.M., Goldgaber D., Roses A.D. Binding of human apolipoprotein E to synthetic amyloid β peptide: isoform-specific effects and implications for late-onset Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90:8098–8102. doi: 10.1073/pnas.90.17.8098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen K., Iribarren P., Hu J., Chen J., Gong W., Cho E.H., Lockett S., Dunlop N.M., Wang J.M. Activation of toll-like receptor 2 on microglia promotes cell up-take of Alzheimer's disease-associated amyloid β peptide. J Biol Chem. 2006;281:3651–3659. doi: 10.1074/jbc.M508125200. [DOI] [PubMed] [Google Scholar]
- 45.Richard K.L., Filali M., Prefontaine P., Rivest S. Toll-like receptor 2 acts as a natural innate immune receptor to clear amyloid β 1–42 and delay the cognitive decline in a mouse model of Alzheimer's disease. J Neurosci. 2008;28:5784–5793. doi: 10.1523/JNEUROSCI.1146-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Anti-apoE antibody inhibits Aβ degradation but not Aβ uptake by BV-2 microglial cells (A and B). BV-2 microglial cells were incubated with monomeric Aβ42 (0.5 μmol/L) in the presence or absence of anti-apoE antibody (6 nmol/L). As a control, isotype control antibody (mouse IgG) was also used. One and 3 hours after incubation, Aβ levels in the media (A) and cell lysates (B) were determined by immunoblot analysis using the anti-Aβ antibody 6E10. There were no differences in the Aβ contents in the culture media among the three groups (A). The lane indicated by “no cells” represents Aβ (0.5 μmol/L) in the medium from a 6-well plate without inoculation of BV-2 cells. The Aβ contents in the anti-apoE antibody-treated cell lysates are greater than those in the control groups (B). The bar graphs represents densitometric quantification of each Aβ band after normalization with GAPDH (cell lysate) or total protein amount in the culture (medium). Data shown are the mean ± SE. Expression levels of cerebral MyD88 at 9 months of age tend to decrease compared with those at 5 months in APP and WT mice (C). mRNA levels of cerebral MyD88 in 5- and 9-month-old APP and WT mice were determined by real-time PCR using cDNAs prepared by reverse transcription of mRNA. mRNA levels of MyD88 were normalized by subtracting CT values obtained with GAPDH mRNA and are shown as 2−ΔCt [ΔCT = CT (MyD88) – CT (GAPDH)] (mean ± SEM).