

Abstract
Hemorrhage under the neural retina (subretinal hemorrhage) can occur in the context of age-related macular degeneration and induce subsequent photoreceptor cell death and permanent vision loss. Current treatments with the objective of removing or displacing the hemorrhage are invasive and of mixed efficacy. We created a mouse model of subretinal hemorrhage to characterize the inflammatory responses and photoreceptor degeneration that occur in the acute aftermath of hemorrhage. It was observed that microglial infiltration into the outer retina commences as early as 6 hours after hemorrhage. Inflammatory cells progressively accumulate in the outer nuclear layer concurrently with photoreceptor degeneration and apoptosis. Administration of minocycline, an inhibitor of microglial activation, decreased microglial expression of chemotactic cytokines in vitro and reduced microglial infiltration and photoreceptor cell loss after subretinal hemorrhage in vivo. Inflammatory responses and photoreceptor atrophy occurred after subretinal hemorrhage, however, the degree of response and atrophy were similar between C3-deficient and C3-sufficient mice, indicating a limited role for complement-mediated processes. Our data indicate a role for inflammatory responses in driving photoreceptor cell loss in subretinal hemorrhage, and it is proposed that microglial inhibition may be beneficial in the treatment of subretinal hemorrhage.
In the industrialized world, age-related macular degeneration (AMD) is the leading cause of legal blindness in persons older than 65 years1,2 and represents one of the largest challenges to visual health worldwide.3 The exudative, or “wet,” form of AMD, the largest cause of AMD-related vision loss, has undergone recent therapeutic advances with the introduction of anti-angiogenic treatments that have significantly improved visual outcomes.4,5 However, choroidal neovascularization that grows and extends into the subretinal space in wet AMD can in some situations produce substantial bleeding, resulting in formation of large hemorrhages in the subretinal space that detach the retina from the supporting retinal pigment epithelial layer. These large subretinal hemorrhages are inadequately addressed with use of current anti-angiogenic therapies alone, with the result that the visual prognosis in these situations remains comparatively uncertain and suboptimal.6–9
Subretinal hemorrhages often result in permanent vision loss because they induce extensive photoreceptor atrophy in the overlying neuroretina.10 The objective of previously investigated therapies was to physically remove or displace these subretinal hemorrhages, either surgically via evacuation of the subretinal clot11–14 or by pneumatic displacement after pharmacologic fibrinolysis.15–18 These approaches, largely explored in uncontrolled clinical trials, have thus far produced mixed results. In the only controlled clinical trial to date that compared surgical hemorrhage evacuation versus no treatment, vision improvement or stabilization resulting from surgery could not be demonstrated.19 In addition, logistical considerations for surgery often entail some time delay between diagnosis and treatment, during which photoreceptor degeneration in response to subretinal hemorrhage can progress substantially. As a result, more broadly effective and readily accessible therapeutic regimens for this clinical entity are desired.
The cellular mechanisms underlying photoreceptor cell death in subretinal hemorrhage are currently incompletely understood but may be revealing of new therapeutic strategies. Pathogenic mechanisms previously proposed include the direct toxic effects of blood components such as hemoglobin20 and iron,21 the mechanical traction effects from fibrinous attachments on photoreceptor outer segments,22,23 and the barrier effects to nutrient and oxygen transport to the retina.7 The role of inflammation as a contributor to photoreceptor toxicity, however, has been incompletely explored. In studies of hemorrhage occurring in other central nervous system locations such as the cerebral cortex and microglia, brain resident immune cells have been noted to migrate rapidly to the site of intracerebral hemorrhage.24–26 Once there, they participate in the phagocytosis of blood products but also induce increased inflammation at the injury site.27–29 The cellular influences induced by this population of activated microglia also have been thought to contribute to neuronal cell death.30 In addition, studies in brain have indicated activation of the complement system in intracerebral hemorrhage,31 which has been proposed to contribute to pathologic neurodegeneration.32 Whether these cellular and molecular mechanisms also contribute to photoreceptor degeneration in the context of subretinal hemorrhage has not been closely explored.
In the present study, to address the nature of inflammatory cellular responses in subretinal hemorrhage and their relationship to photoreceptor degeneration, a murine model for subretinal hemorrhage was created that simulates key clinical features observed in hemorrhagic AMD. This model was used to characterize the nature and time course of the inflammatory processes after subretinal hemorrhage in terms of microglia and macrophage infiltration and activation in the outer retina occurring concurrent with photoreceptor apoptosis, as well as changes in expression of inflammatory cytokines and chemokines. Also evaluated was the effect of administration of minocycline, an inhibitor of microglial activation, in the mouse model. The role of complement activation in photoreceptor cell death was also examined via induction of subretinal hemorrhage in mice deficient in complement protein C3, a central component of the alternative pathway of complement activation.
The results obtained using a murine model of subretinal hemorrhage indicate that microglial involvement in disease was initiated shortly after hemorrhage induction and that microglial infiltration was spatially and temporally associated with photoreceptor apoptosis. Expression levels of inflammatory cytokines, chemotactic chemokines, and adhesion molecules were elevated as a result of the presence of blood in the subretinal space. Microglial inhibition by systemic minocycline decreased microglial activation and infiltration and delayed the progression of photoreceptor apoptosis and degeneration, which suggests a potential therapeutic axis that may be helpful in the treatment of subretinal hemorrhage.
Materials and Methods
Animal Surgery to Create a Model for Subretinal Hemorrhage
Two- to 3-month old C57BL/6 mice (Charles River Laboratories International, Inc., Wilmington, MA) and transgenic mice on a C57BL/6 background, deficient in complement component C3 (C3−/−)33 (B6.129S4-C3tm1Crr; Jackson Laboratory, Bar Harbor, ME) were used in this study. C3−/− transgenic mice lack C3 in serum and in the ocular aqueous and vitreous of the eye. They also demonstrate normal ocular anatomy and intact ocular responses to bacterial infection.34 All animal experiments were conducted according to the Association for Research in Vision and Ophthalmology statement for the use of animals, and were approved by the National Eye Institute Institutional Animal Care and Use Committee. To create the mouse model of subretinal hemorrhage, animals were anesthetized via i.p. injection of a mixture of 90 mg/kg ketamine and 8 mg/kg xylazine. Autologous blood was collected from a tail vein and immediately used for subretinal injections. Subretinal injections were performed under a dissecting microscope (model SZX16; Olympus America, Central Valley, PA). In brief, the scleral surface on the temporal quadrant was exposed, and a tunneled scleral incision was made using a sharp 29-gauge needle at approximately 0.5 mm posterior to the limbus. The tip of a blunt 33-gauge needle that had been connected to a Hamilton microsyringe containing autologous blood was introduced through the incision and advanced toward the posterior pole at a 5-degree angle to the scleral surface. On obtaining access to the subretinal space with the tip of the blunt needle, 1.6 μL autologous blood was slowly injected, forming an elevated retinal bleb. The needle was carefully withdrawn, and topical antibiotic ointment (Bacitracin Ophthalmic Ointment USP; E. Fougera & Co., division of Altana, Inc., Melville, NY) was applied. An anticoagulant was not used together with the injection of autologous blood, and a subretinal blood clot was allowed to form in the subretinal space after injection. In control experiments, instead of autologous blood, 1.6 μL 0.5% sodium hyaluronate (Provisc; Alcon Laboratories, Inc., Fort Worth, TX) was injected in the contralateral eye using the same technique into the subretinal space.
Imaging of Mouse Fundus after Subretinal Blood Injection
Proper localization of the injected autologous blood in the subretinal space was confirmed in vivo at various times after subretinal injection using fundus imaging and optical coherence tomography (OCT). Animals that previously received subretinal blood injections were anesthetized, and their pupils were dilated using eye drops containing 1% tropicamide and 2.5% phenylephrine hydrochloride. Color fundus images were captured using a fundus imaging system (Micron II; Phoenix Research Laboratories, Inc., Pleasanton, CA) to visualize the size and location of retinal blebs resulting from subretinal blood injection. OCT imaging was performed using a device adapted for small-animal and rodent imaging (SDOCT; Bioptigen, Inc., Research Triangle Park, NC). Animals were positioned in a rodent-alignment stage, and the scanning beam was directed at the temporal retinal quadrant over the area of injection. Series of B-scans, each 1.2 mm long and traversing the area of subretinal hemorrhage, were collected.
Histopathologic Analysis
Experimental animals were euthanized via CO2 inhalation at various times (6 hours to 10 days) after subretinal injection. For preparation of plastic-embedded sections, enucleated globes were fixed via immersion in a solution of 2.5% glutaraldehyde and 2% paraformaldehyde in PBS, embedded in methacrylate, serially sectioned through the vertical pupillary–optic nerve plane, and stained using H&E. The plane of sectioning was oriented in a nasal-to-temporal direction to traverse the site of subretinal injection in the temporal quadrant. For immunohistochemical studies, harvested globes were dissected to remove the anterior segments, immersed overnight in a solution of 4% paraformaldehyde in PBS, and washed overnight with PBS. The fixed eye cups were embedded in 7% agarose, after which serial sections 100 μm thick were cut using a vibrating microtome (VT1000S; Leica Microsystems, Inc., Bannockburn, IL) with the cutting plane in the nasal-temporal direction and vertically oriented in the pupillary–optic nerve axis, and were collected in PBS. Sections traversing the geometric center of the area of hemorrhage, where the outer nuclear layer was subject to the greatest attenuation, were identified and collected. The sections were stained with primary antibodies against ionized calcium-binding adaptor molecule-1 (1:500; Wako, Richmond, VA) and CD11b (1:50; catalog No. MCA711; AbD Serotec USA, Raleigh, NC) to label microglia, glial fibrillary acidic protein (GFAP) (1L100; catalog No. 13–0300; Invitrogen Corp., Carlsbad, CA) to label astrocytes and processes of gliotic Müller cells, C3 [1:25; catalog No. HM1045; HyCult Biotechnology (Hycult Biotech), Uden, The Netherlands] to characterize complement deposition, NF-κB p65 subunit (1:50; catalog No. 4764; Cell Signaling Technology, Inc., Beverly, MA) to evaluate NF-κB activation, and tumor necrosis factor-α (TNF-α) (catalog No. ab1793; Abcam, Inc., Cambridge, MA) to detect microglial activation. Secondary antibodies were added at a 1:400 dilution and incubated for 4 hours. Sections were washed in ICC buffer (PBS containing 0.5% bovine serum albumin, 0.2% Tween 20, and 0.05% sodium azide), pH 7.3, labeled with DAPI (catalog No. D1306; Molecular Probes, Inc., Eugene, OR) to label cellular nuclei, and mounted on glass slides with mounting medium (Fluoromount; Sigma-Aldrich Corp., St. Louis, MO). To label apoptotic cells, retinal sections were also labeled using the terminal dUTP-mediated TUNEL technique, performed according to the manufacturer's protocol (In Situ Cell Death Detection Kit; Hoffman-La Roche AG, Basel, Switzerland). In brief, vibratome sections were permeabilized in 300 μL 1% Triton X-100 for 60 minutes, after which 100 μL TUNEL reaction mixture was added and incubated for 1 hour at 37°C.
Confocal Imaging and Morphometric Analysis
Slide-mounted sections after immunohistochemical staining and/or TUNEL labeling were imaged using confocal microscopy (SP2, Leica Microsystems GmbH, Wetzlar, Germany; or FluoView 1000, Olympus Corp., Tokyo, Japan). Sections traversing the area of the subretinal injection were identified by the dome-shaped profile of the detached retina. The section passing through the central portion of the area of subretinal injection was selected, and the imaging field was centered on the central portion of the lesion. Confocal stacks of images of uniform z-depth (21 images per z-stack, 1 μm apart; total z-depth, 20 μm) were collected at ×40 magnification using standardized settings. To avert variability in tissue pathologic features that may exist along the central-to-periphery axis of the lesion, cell counts of the number of TUNEL- and Iba-1–labeled cells in the outer nuclear layer were performed in only one central imaging field in each eye at the location where pathologic changes were maximal. Cell counts were performed with masking of the status of the experimental animal. The limits of outer nuclear layer (ONL) visible in the central retinal section were delineated using a region-of-interest tool within an image-processing computer program (ImageJ; National Institutes of Health, Bethesda, MD), and the area of the delineated section (ONLarea) was measured. The length of the vitreal (ONLL,V) and scleral (ONLL,S) borders of the outer nuclear layer belonging to the region of interest were then separately measured. The outer nuclear layer thickness for that particular section was calculated as the circumscribed area of the outer nuclear layer divided by the mean measured lengths of the scleral and vitreal borders of the outer nuclear layer in that section: [ONLthickness = ONLarea/0.5*(ONLL,V + ONLL,S)]. The density of counted cells in a section was determined by dividing the number of cells in the outer nuclear layer by the measured area of the outer nuclear area (ONLarea).
Measurements of Protein Levels using Enzyme-Linked Immunosorbent Assay
Changes in protein level in the retina after subretinal blood injections were evaluated. At days 1 and 3 after subretinal blood injection, experimental animals were euthanized, and their eyes were enucleated. Retinal tissue overlying the areas of subretinal hemorrhage was dissected free in oxygenated Ringer's solution and immediately transferred into a 1.5-mL Eppendorf tube containing 150 μL lysis buffer (RIPA buffer; Sigma-Aldrich Corp.) containing a protease inhibitor cocktail (Calbiochem, Gibbstown, NJ). Retinal tissue was then homogenized and incubated on ice for 20 minutes. Cell lysates were centrifuged for 20 minutes at 21,000g, and the supernatants were isolated. The overall protein level in retinal lysates was quantitated using a BCA Protein Assay Kit (Pierce Chemical Co., Rockford, IL). The protein levels in cytokines and adhesion molecules were assayed using a commercial chemiluminescence-based enzyme-linked immunosorbent assay (ELISA) (Searchlight; Aushon Biosystems, Inc., Billerica, MA). At least three replicates of each experimental condition were performed.
In Vivo and in Vitro Administration of Minocycline
Experimental animals received subretinal injections of autologous blood, and were immediately administered i.p. injections of minocycline (50 mg/kg b.i.d. dissolved in an injection volume of 0.3 mL PBS; catalog No. M9511-1G; Sigma-Aldrich Corp.). As controls, experimental animals that received subretinal blood injections were treated with an equal volume of PBS on the same schedule. Animals were euthanized at 5 and 10 days after treatment, and their eyes were harvested for analysis.
In vitro effects of minocycline administration on cultured microglial cells were also evaluated. In brief, microglia were isolated as a pure population from mouse retina as previously described.35 Cultured retinal microglia were seeded into a six-well tissue culture plate (Corning, Inc., Corning, NY) at a density of 1 × 106 cells/well, and were allowed to settle overnight. Blood was collected via the orbital sinus from a 2-month-old wild-type C57BL/6 mouse and was centrifuged immediately at 8000g for 3 minutes. The resulting erythrocyte cell pellet was frozen to induce cell lysis, and was resuspended in culture medium at a dilution of 1:100,000. Cultured retinal microglia were divided into three groups and incubated for 24 hours with either: i) culture medium only, ii) culture medium containing freshly lysed blood cells and their hemoglobin-containing contents, or iii) the same contents as in ii) with added 30 μmol/L minocycline. Microglial cells were harvested from the culture wells, and protein level was assessed using ELISA as described above.
Statistical Analysis
Statistical analysis were performed using statistical software (GraphPad Software, Inc., San Diego, CA). One-way analysis of variance using the Dunnett's or Tukey's multiple-comparison posttest was performed to analyze the results of ELISA measurements. Data sets from in vivo experiments evaluating the effect of minocycline and C3 deficiency on outer nuclear layer thickness, TUNEL staining density, and outer nuclear layer microglial density were evaluated using normality tests. Data were analyzed using the unpaired Student's t-test in the case of gaussian distribution or the nonparametric Mann-Whitney U-test if the data did not follow a normal distribution. P < 0.05 was set as the basis for rejection the null hypothesis.
Results
Creation of Mouse Model of Subretinal Hemorrhage
A model for subretinal hemorrhage in the murine eye was established that recapitulated the major pathologic features observed in human disease. A murine model confers the advantages of a small-animal model and provides opportunities to study genetic contributions to disease pathogenesis in genetically manipulated mice. Our murine model was examined for key pathologic phenotypes found in clinical subretinal hemorrhage. The most common cause of subretinal hemorrhage in patients is AMD of the wet, or exudative, form. In these clinical cases, a large subretinal hemorrhage is typically characterized by the acute appearance of a dome-shaped accumulation of blood in the subretinal space (Figure 1, A, B, and D), and is formed as a result of bleeding from choroidal neovascularization, new vessels that grow from the choroid through Bruch's membrane into the retina. The overlying neuroretina is elevated from its normal position and separated from the retinal pigment epithelium. The accumulated subretinal blood gradually resolves over a course of weeks but is accompanied by concurrent outer retinal atrophy that results in substantial and irreversible loss of vision (Figure 1, C and E).
Figure 1.
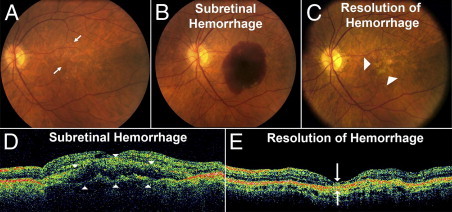
Clinical findings in subretinal hemorrhage in the context of neovascular (wet) AMD. Fundus photographs demonstrate the typical clinical setting in which subretinal hemorrhage develops in the context of AMD. A: Subretinal hemorrhage can occur acutely in an eye at risk such as one containing signs of intermediate AMD in the form of drusen and pigmentary changes (arrows). B: Appearance of an acute macular subretinal hemorrhage as a result of choroidal neovascularization formation. C: Subretinal blood gradually resolves over a period of weeks, with the affected area developing extensive retinal atrophy and loss of central vision (arrowheads). D: Corresponding images obtained using OCT in the same eye demonstrate in vivo cross-sectional views of the subretinal hemorrhage that detaches the retina at the macula (arrowheads) in a dome-shaped configuration. E: With resolution of the hemorrhage, concurrent retinal atrophy and loss of the outer retinal lamina in the affected area is typically observed (arrows).
Subretinal hemorrhage was simulated in our mouse model via injection of a small volume of autologous blood into the temporal subretinal space, creating an elevated dome-shaped retinal detachment (Figure 2A). The anatomical configuration of the subretinal blood accumulation as visualized using in vivo OCT imaging (Figure 2B) and histopathology (Figure 2C) resembled that observed in clinical exudative AMD. The subretinal hemorrhage in the mouse model also underwent gradual resolution within a few weeks after induction. Extensive outer retinal atrophy developed concurrently in the area overlying the hemorrhage (Figure 2, C and D). These results demonstrated that the anatomical configuration and degenerative consequences of subretinal hemorrhage were recapitulated in the mouse model of the disease.
Figure 2.
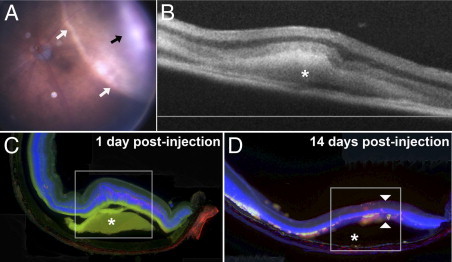
Anatomical and pathologic findings in a murine model of subretinal hemorrhage. A model for subretinal hemorrhage was created in the adult mouse by injecting a small volume (1.6 μL) of autologous blood into the subretinal space via a transscleral route using a small-gauge (33G) needle. A:In vivo photograph of the mouse fundus of the treated eye at 5 hours after subretinal blood injection reveals an area of dome-shaped retinal detachment (white arrows) containing a central blood clot (black arrow). B:In vivo cross-sectional view of the subretinal hemorrhage (asterisk) at 3 days after treatment as visualized using OCT. C: Histologic section of the retina in the area of hemorrhage reveals an area of retinal detachment (box) overlying subretinal hemorrhage (asterisk). Photoreceptor apoptosis in the area overlying the hemorrhage is labeled using TUNEL (red). Nuclear staining was performed using DAPI (blue), and the green channel was used to demonstrate the autofluorescent nature of the subretinal hemorrhage. D: Histologic section of the retina at 2 weeks after subretinal blood injection demonstrates resolution of the hemorrhage (asterisk) and development of outer retinal atrophy over the area of hemorrhage (arrowheads).
Apoptosis and Microglial and Macrophage Infiltration in Subretinal Hemorrhage
With establishment of a mouse model, we evaluated the nature of cellular and tissue changes occurring in the retina in response to subretinal blood. To differentiate the cellular effects resulting from the presence of blood components in the subretinal space from those resulting from mechanical detachment of the retina, an equivalent volume of inert viscoelastic material was injected in the contralateral eye as a control. Histologic analysis in retinal sections obtained at 1, 3, and 5 days after injection of the viscoelastic material revealed moderate attenuation of the outer nuclear layer after detachment in the control eyes (Figure 3). However, in the contralateral eyes, which were injected with autologous blood, there was a considerably greater extent of outer nuclear layer atrophy overlying the area of hemorrhage at all times examined (Figure 3). Outer nuclear layer atrophy was apparent at 1 day after subretinal blood injection, and progressed rapidly with time; at 5 days, the outer nuclear layer was reduced to a single irregular layer of cells. This indicated that outer retinal degeneration after subretinal hemorrhage was in large part contributed to by the presence of blood constituents in the subretinal space rather than detachment from the retinal pigment epithelial layer itself.
Figure 3.
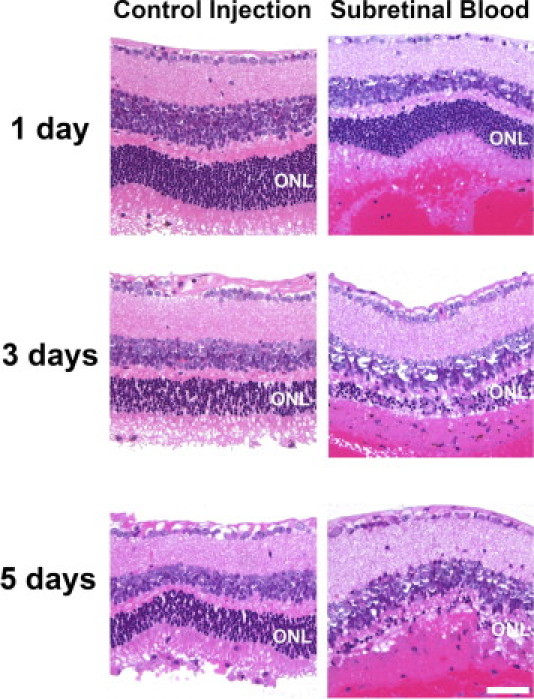
Outer retinal atrophy after subretinal hemorrhage in a mouse model of subretinal hemorrhage. Experimental mice received a subretinal injection of autologous blood (1.6 μL) in the treated eye, and in the contralateral control eye, a subretinal injection of an equal volume of an inert viscoelastic substance (0.5% sodium hyaluronate) to simulate an equivalent retinal detachment without the presence of subretinal blood. Representative photomicrographs of thin plastic retinal sections stained using H&E are shown at 1, 3, and 5 days after injection. Outer nuclear layer thickness (ONL) was reduced in eyes receiving subretinal blood injections (right panels) to a significantly greater extent than in eyes receiving control injections (left panels). Scale bar = 50 μm.
We also evaluated the mechanism of cell death after subretinal hemorrhage in the mouse model. Apoptosis was assessed after subretinal injections of blood and control viscoelastic material by performing TUNEL staining on vibratome-sectioned retinas. At 12 hours after injection, TUNEL-positive cells in the outer nuclear layer were visible in substantial numbers in eyes injected with subretinal blood but not in control eyes injected with viscoelastic material (Figure 4, A and B). At 1, 3, and 5 days after injection, the density of TUNEL-positive cells increased in the outer nuclear layer in both control eyes and those injected with subretinal blood; however, the density was significantly greater in eyes that received subretinal blood injections (Figure 4, C–H). These results indicated that outer nuclear layer cell death was initiated hours after subretinal blood injection, and was likely driven by apoptosis.
Figure 4.
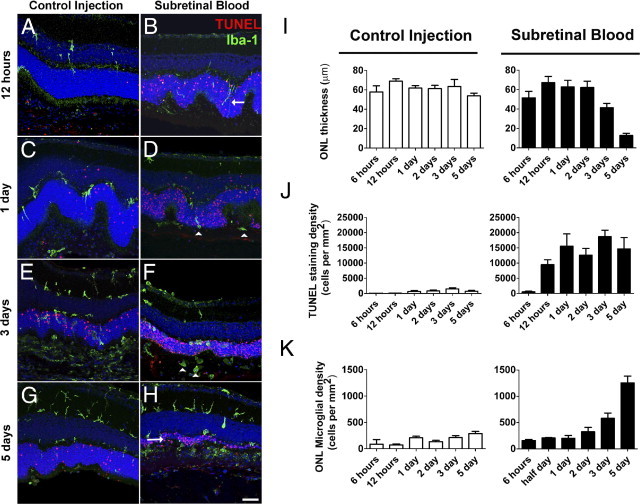
Photoreceptor apoptosis and microglial activation after subretinal hemorrhage. Experimental animals received subretinal injections of either blood or viscoelastic substance (0.5% sodium hyaluronate; control). Eyes were harvested for histopathologic analysis at various times after injection, and were analyzed using TUNEL staining (red) or immunohistochemistry for Iba-1 (to label microglia, green). A and B: At 12 hours after injection, retinal microglia, which are normally confined to the inner retina, could be seen extending vertically oriented processes and migrating into the outer nuclear layer in eyes injected with subretinal blood (B, arrow) but to a smaller extent in control eyes (A). TUNEL positivity is observed in the outer nuclear layer in eye injected with subretinal blood but was largely absent in control eyes. C and D: At 1 day after injection, the number of TUNEL-positive cells increased in both control eyes (C) and eyes injected with subretinal blood eyes (D) but remained significantly higher in eyes injected with subretinal blood. Microglia were also observed to migrate into the outer retina, translocating their somata into the outer nuclear layer and subretinal space (arrowheads). E and F: At 3 days, the thickness of the outer nuclear layer was considerably decreased, to a greater extent in eyes injected with subretinal blood (F) than in control eyes (E). F: Microglia in the outer nuclear layer and subretinal space also acquired a rounded amoeboid architecture in eyes injected with subretinal blood (arrowheads). G and H: At 5 days, the outer nuclear layer in eyes injected with subretinal blood was reduced to only one or two cells thick (H, arrow) but remained substantial in control eyes (G). I: Quantification of outer retinal layer thickness, photoreceptor apoptosis, and outer retinal microglial infiltration after control and subretinal blood injections demonstrated that outer nuclear layer thickness was largely maintained over 5 days after injection in control eyes (left), whereas after subretinal blood injection, it decreased significantly at 3 and 5 days after injection (right). J: Density of TUNEL-positive cells in the outer nuclear layer remained relatively low in control eyes (left), whereas those in eyes injected with subretinal blood increased markedly at 12 hours (right). K: Density of microglial infiltration into the outer nuclear layer increased over time, and was relatively higher at 3 and 5 days after subretinal blood injection compared with control injection. Scale bar = 50 μm.
Also assessed were changes in the distribution of inflammatory cells after injection at immunohistochemical staining for Iba-1, a marker for microglia and macrophages. At 12 hours after injection, we observed in eyes with subretinal blood injections the rapid extension of Iba-1–positive microglial processes into the outer nuclear layer, a region normally devoid of microglia (Figure 4, A and B). Increased numbers of Iba-1–positive cells were also observed to migrate into the outer nuclear layer and the subretinal space after postinjection day 1 (Figure 4C–H). A higher density of Iba-1–positive cells in the outer nuclear layer were observed to accumulate in blood-injected retinas than in control-injected retinas, indicating that that the presence of blood in the subretinal space induced an early and pronounced influx of activated microglia and macrophages into the outer retina concurrent with outer nuclear layer apoptosis. Microglia and macrophages in the outer nuclear layer and subretinal space exhibited rounded amoeboid architecture, indicative of an activated phenotype (Figure 4, D and F).
Quantitation of the changes in outer nuclear layer thickness, density of TUNEL-positive cells in the outer nuclear layer, and density of outer nuclear layer microglia and macrophages (Figure 4, I–K) demonstrated that outer nuclear layer apoptosis, microglial infiltration, and photoreceptor degeneration increased progressively during the first 5 days after subretinal hemorrhage. These effects were significantly more prominent in eyes injected with subretinal blood than in control-injected eyes.
Microglia and Müller Cell Gliosis in Subretinal Hemorrhage
Injected retinas were evaluated for the presence of gliotic processes. Müller cell gliosis, as detected by the presence and increase in GFAP staining in Müller cell processes, was observed at low levels at 1 day after injection in both control- and blood-injected retinas (Figure 5). GFAP positivity in Müller cell processes increased at 5 days after injection, and was more prominent and extensive in blood-injected than in control-injected retinas (Figure 5). The presence of microgliosis was also observed prominently in blood-injected retinas, with the appearance of deramified and amoeboid Iba-1–positive cells in the outer nuclear layer and subretinal space. These cells also stained positively for Mac2, a marker of activated microglia (data not shown).
Figure 5.
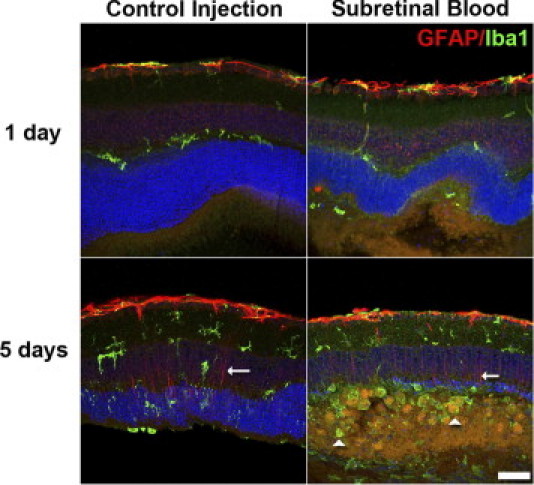
Microglia and Müller cell gliosis in mouse model of subretinal hemorrhage. Microglia gliosis and Müller cell gliosis were followed up at 1 and 5 days after injection with immunohistochemical staining for Iba-1 (green) to label microglia and GFAP (red), astrocytes, and activated Müller cells. At 1 day after injection, weak GFAP immunopositivity in Müller cell processes was observed in both control eyes and eyes injected with subretinal blood. At 5 days after injection, Müller cell processes (arrows) were clearly labeled with GFAP in both control eyes and those that received subretinal injections, indicating an increase in Müller cell gliosis. Large numbers of microglia with prominent, rounded, amoeboid architecture were apparent in the outer nuclear layer and subretinal space (arrowheads), indicating a more extensive degree of microgliosis in eyes injected with subretinal blood compared with control eyes injected with viscoelastic material. Scale bar = 50 μm.
Increased Expression of Inflammatory Cytokines, Chemokines, and Adhesion Molecules in Subretinal Hemorrhage
To investigate the molecular nature of the inflammatory response in the retina after subretinal hemorrhage, retinas were isolated from eyes that received subretinal injections of either viscoelastic material or autologous blood and eyes that received no injections. Retinas were dissected at 3 days after injection, and levels of inflammatory cytokines, chemokines, and adhesion molecules in the retina were measured using ELISA. It was observed that although not all comparisons achieved statistical significance, levels of inflammatory cytokines TNF-α, IL-6, and transforming growth factor-beta were highest in retinas injected with subretinal blood, followed by retinas injected with inert viscoelastic material, and with lowest levels detected in eyes that received no injections (Figure 6, A–C). Similar trends were observed for the levels of chemotactic cytokines CCL2 [chemokine (C-C motif) ligand 2] and SDF-1 (stromal cell–derived factor 1) (Figure 6, D and E) and for vascular cell adhesion molecule-1 and intercellular adhesion molecule-1 (Figure 6, G and H). Levels of CCL5, another chemokine, were, however, relatively low (<1 pg/mg) and remained unchanged (Figure 6F). These consistent changes indicated that although retinal detachment alone, as induced by subretinal viscoelastic, induced increased expression of these molecules, the additional presence of hemorrhage in the subretinal space further increased inflammatory responses in the retina.
Figure 6.
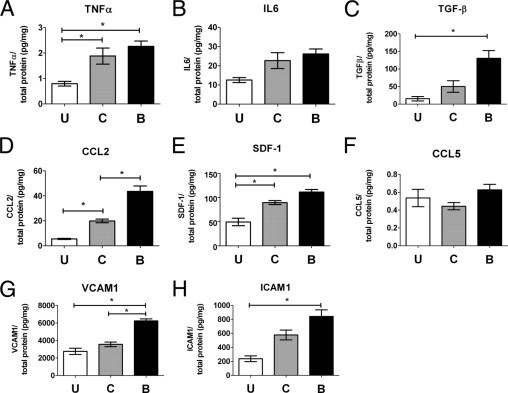
Inflammatory cytokines, chemotactic cytokines, and adhesion molecules are up-regulated after subretinal hemorrhage. Retinal levels of cytokines and adhesion molecules in the retina were assessed using ELISA in uninjected eyes (U) and in eyes injected subretinally 3 days before with either control (C) or autologous blood (B). Inflammatory cytokines TNF-α (A), IL-6 (B), and transforming growth factor-β (C) were generally elevated in injected eyes compared with uninjected eyes, with the highest levels in eyes injected with autologous blood. Similar trends were observed for chemotactic cytokines CCL2 (D) and SDF-1 (E) and adhesion molecules VCAM-1 (G) and ICAM-1 (H). Levels of chemokine CCL5 (F) were low and relatively unchanged. *P < 0.05).
Effect of Minocycline on Photoreceptor Cell Death and Inflammatory Cell Infiltration in Subretinal Hemorrhage
We examined the effect of minocycline, an inhibitor of microglial activation, on retinal changes occurring after subretinal hemorrhage in our murine model. Minocycline administration (i.p. injection, 50 mg/kg b.i.d.) was started immediately after subretinal blood injection, and was continued until the animals were sacrificed at days 5 and 10. Control animals received injections of PBS on the same schedule after subretinal blood injection. It was observed that, compared with control animals, minocycline-treated animals at 5 days after injection of subretinal blood demonstrated a less attenuated outer nuclear layer thickness overlying the area of hemorrhage (Figure 7A, B, and E), with a lower density of TUNEL-positive cells (Figure 7F) and microglial cells (Figure 7G) in the outer nuclear layer relative to control. At 10 days after injection, control animals were observed to have either a completely absent outer nuclear layer or an attenuated layer composed of a single layer of cells, whereas minocycline-treated animals exhibited a more preserved outer nuclear layer (Figure 7, C–E). Owing to the degree of attenuation of the outer nuclear layer in control animals at 10 days, the density of TUNEL-positive cells and outer nuclear layer microglia could not be adequately determined.
Figure 7.
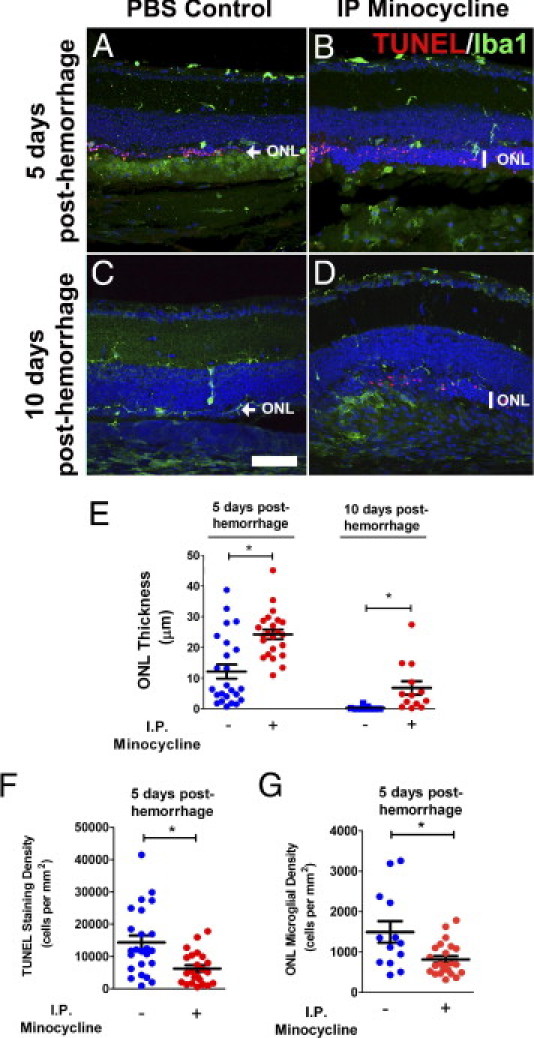
Minocycline treatment reduces outer nuclear layer apoptosis and atrophy after subretinal injection. Experimental animals were treated with minocycline (i.p. injection, 50 mg/kg b.i.d.) beginning immediately after injection of subretinal blood. As control, experimental animals were injected with PBS at the same schedule after subretinal blood injection. Representative retinal sections comparing atrophy in the outer nuclear layer (ONL) in PBS-treated control animals and minocycline-treated animals at 5 days (A and B) and 10 days (C and D) after injection of subretinal blood (TUNEL staining in red, and Iba-1 staining in green). At 5 days after injection, outer nuclear layer thickness was typically greater in minocycline-treated animals than in control-treated animals. Density of TUNEL-positive cells and microglia in the outer retina were correspondingly lower in minocycline-treated animals. At 10 days after injection, outer nuclear layer thickness in control retinas was either completely attenuated or reduced to a layer one cell thick (arrow). Outer nuclear layer thickness was significantly greater in minocycline-treated animals (vertical bar). E: Quantitation of outer nuclear layer thickness in eyes from control (−) and minocycline-treated (+) animals at 5 and 10 days after hemorrhage. F: Density of TUNEL-positive cells in the outer nuclear layer in control (−) and minocycline-treated (+) animals at 5 days after subretinal injection. G: Density of microglial cells in the outer nuclear layer in control (−) and minocycline-treated (+) animals at 5 days after subretinal injection. Data for postinjection day 5: n = 24 eyes from 12 control animals and 23 eyes from 12 treated animals. Data for postinjection day 10: n = 9 eyes from 6 control animals and 13 eyes from 7 treated animals. *P < 0.05). Scale bar = 50 μm.
To explore the mechanisms underlying the inflammatory reaction to subretinal hemorrhage and the effect of minocycline administration, cultured retinal microglia were exposed to lysed blood cells in the presence and absence of minocycline and were evaluated for expression of inflammatory cytokines and chemokines in microglia. In vitro exposure of cultured retinal microglial cells to lysed red blood cells in the absence of minocycline increased the levels of CCL2, CCL5, and TNF-α but not IL-6, or SDF-1 (Figure 8), which suggests that retinal microglia respond to hemorrhage by increasing their activation state and expression of chemotactic cytokines. The addition of minocycline decreased levels of CCL2, CCL5, and TNF-α to close to baseline, whereas levels of IL-6 and SDF-1 were not significantly changed. Levels of TGFβ were also decreased. These results suggest that the activation of retinal microglia induced by hemorrhage may be suppressed by minocycline and may be a mechanism by which minocycline decreases apoptotic cell death and microglial infiltration in the context of subretinal hemorrhage in vivo.
Figure 8.
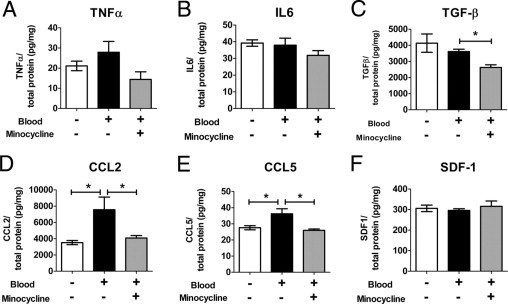
Effect of minocycline on expression of cytokines by retinal microglia after exposure to red blood cells. Levels of cytokines in cultured retinal microglia were assessed using ELISA under baseline conditions (white bars), after exposure to lysed red blood cells (black bars), and after exposure to lysed red blood cells and minocycline together (gray bars). A: TNF-α. B: IL-6. C: Transforming growth factor-β. D: CCL2. E: CCL5. F: SDF-1. *P < 0.05).
The effects of minocycline in vivo on microglial activation were also assessed. NF-κB, a transcription factor involved in regulation of multiple proinflammatory cytokines, is translocated from the cytoplasm to the nucleus during activation.36 Immunohistochemical localization of the p65 subunit of NF-κB was assessed in outer retinal microglia after subretinal hemorrhage. In PBS-treated animals, NF-κB was localized primarily to microglial nuclei, indicative of activation, and in minocycline-treated animals, a more diffuse pattern of NF-κB staining was observed in both nucleus and cytoplasm, suggesting an inhibitory effect of minocycline on NF-κB activation (Figure 9, A and B). Immunopositivity of TNF-α was also reduced in outer retinal microglia in minocycline-treated animals compared with PBS-treated control animals (Figure 9, C and D). Changes in the overall tissue levels of inflammatory cytokines in response to minocycline administration were also assessed. Inasmuch as the inhibitory effects of minocycline on retinal microglia were likely partial and the representation of microglial cells in the overall sampled population of retinal cells was relatively small, ELISA measurements made on retinal tissue may not prominently reflect changes occurring in microglia. Although comparisons did not reach statistical significance, levels of cytokines and adhesion molecules were generally decreased or stable in minocycline-treated retina compared with controls, with decreases most prominent for CCL2 and IL-6 (see Supplemental Figure S1 at http://ajp.amjpathol.org).
Figure 9.
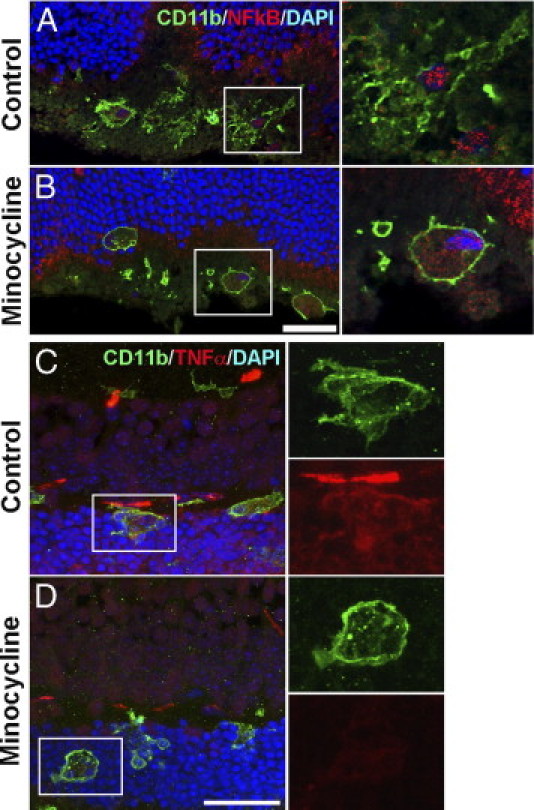
Effect of minocycline on microglial activation in the retina after subretinal hemorrhage. Subretinal hemorrhage was induced in experimental animals, and treated daily with i.p. injection of minocycline or PBS. Retinal tissue was isolated after 3 days of treatment. A and B: Microglia in the outer retina were labeled with CD11b (green), and their nuclei were marked using DAPI staining. Cellular localization of NF-κB (subunit p65) was followed by immunohistochemical staining (red). A: Microglia in PBS-treated control retina demonstrated a pattern of NF-κB staining that was primarily confined to the nucleus, suggestive of NF-κB activation. B: Microglia in minocycline-treated retina demonstrated a more diffuse cytoplasmic pattern of NF-κB staining, indicative of a less-activated state. C and D: CD11b-positive microglia (green) in the outer retina were co-labeled with an antibody to TNF-α (red). Microglia in PBS-treated control retinas (C) demonstrated a higher level of TNF-α immunopositivity than did those in minocycline-treated retinas (D), indicating a suppressive effect on microglial TNF- α expression by minocycline. Representative images shown were captured using confocal microscopy under similar settings to provide a relative comparison of expression levels. Scale bar = 30 μm.
Role of C3 in Photoreceptor Cell Death and Microglial Activation in Subretinal Hemorrhage
The presence of C3 deposition in the subretinal space after subretinal hemorrhage was evaluated using immunohistochemical staining for C3. Deposition of C3 was detected in the subretinal space and along Bruch's membrane in wild-type mice at 5 and 10 days after subretinal blood injection (Figure 10, A and D). C3 immunopositivity was not detected in control experiments (in the absence of the primary antibody to C3) (Figure 10, B and E), in injected transgenic mice deficient in C3 (C3−/−) mice) (Figure 10, C and F), or in mice that received no injections (data not shown). The association of C3 deposition with subretinal hemorrhage raised the possibility that complement activation may have contributed to outer nuclear layer apoptosis after hemorrhage. However, experiments involving subretinal blood injections into transgenic mice deficient in C3, a central component in the alternative complement cascade, demonstrated a similar profile of outer nuclear layer atrophy and thinning at 5 and 10 days after hemorrhage (Figure 10G). In addition, no differences were observed between wild-type C57Bl6 mice and C3−/− transgenic mice insofar as outer nuclear layer thickness (Figure 10H), density of TUNEL-positive cells (Figure 10D), or density of outer nuclear layer microglia (Figure 10I), which indicates that the absence of C3 deposition in the outer retina after subretinal hemorrhage did not significantly alter the progression of retinal disease.
Figure 10.
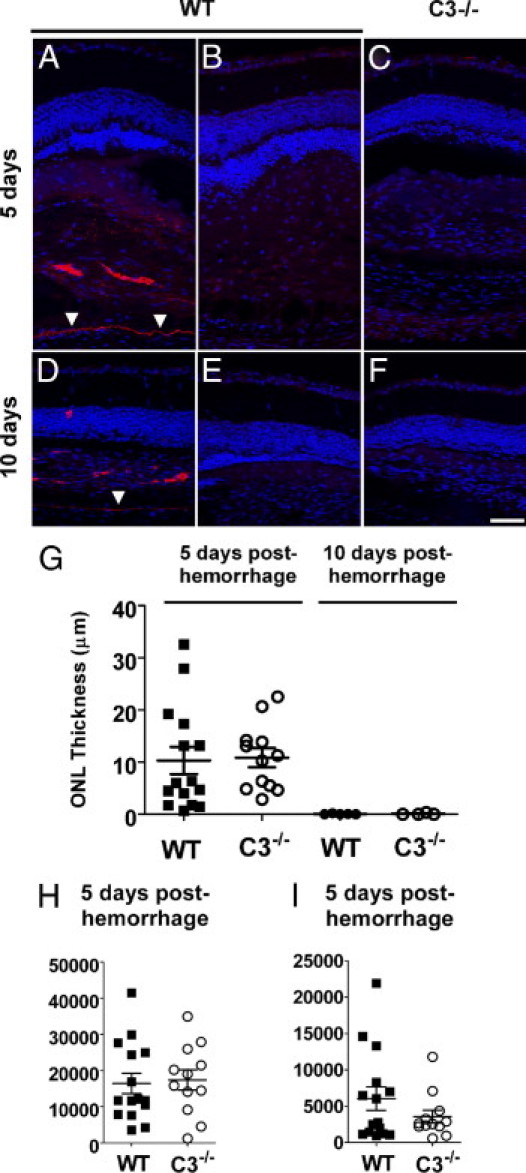
Role of complement in outer nuclear layer atrophy in a mouse model of subretinal blood. Immunohistochemical staining for C3 after subretinal injection demonstrated C3 deposition in the subretinal space and along Bruch's membrane. Representative retinal sections demonstrated C3 deposition in a wild-type C57BL/6 mouse at 5 days (A) and 10 days (D) after injection of subretinal blood. Absence of C3 staining was observed in control experiments in which the primary antibody to C3 was omitted (B and E) and when similar sections from a C3−/− mouse on a C57Bl6 background were tested (C and F). Arrowheads indicate the position of Bruch's membrane. G: Quantitation of outer nuclear layer thickness in eyes from wild-type and C3−/− animals at 5 and 10 days after injection of subretinal blood. H: Density of TUNEL-positive cells in the outer nuclear layer in wild-type and C3−/− animals at 5 days after subretinal injection. I: Density of microglial cells in wild-type and C3−/− animals at 5 days after subretinal injection. No significant differences were detected (P > 0.05) for all comparisons between wild-type and C3−/− animals. Data for postinjection day 5: n = 15 eyes from 8 wild-type animals and 12 eyes from 6 C3−/− animals. Data for postinjection day 10: n = 5 eyes from 3 wild-type animals and 4 eyes from 3 C3−/− animals. Scale bar = 50 μm.
Discussion
In the present study, a model was established for subretinal hemorrhage in the mouse eye that recapitulates the primary pathologic features of clinical subretinal hemorrhage such as that resulting from exudative AMD.37 Previous studies have investigated the use of larger animal models such as monkey,20 rabbit,22 cat,23 and rat,38; however, to our knowledge, no previous reports have described the use of the mouse model to study subretinal hemorrhage. The murine model provides the advantages of a small-animal model and enables access to genetically manipulated models for molecular study of disease mechanisms. We used a combination of in vivo fundus imaging and OCT imaging to guide accurate placement of subretinal blood injections, generating hemorrhages that were consistent in location and size and, thus, provided a reproducible model for experimental study.
Quantitative histologic analyses were performed in our mouse model to explore and describe the nature of cellular reactions after subretinal hemorrhage. It was documented that in the first 5 days after subretinal hemorrhage, progressive thinning and atrophy occur in the outer retina, driven by apoptotic photoreceptor death in the outer nuclear layer. The appearance of the first TUNEL-positive apoptotic photoreceptors was detected as early as 6 hours after hemorrhage, concurrent with initiation of microglial migration from the inner to the outer retina. The subsequent increase in the density of TUNEL-positive cells in the outer nuclear layer over the next 5 days was also accompanied by increased density of microglia and macrophages in the outer retina, many of which appeared activated by architecture (rounded amoeboid cells) and by the expression of activation markers. Although the density of microglia in the outer nuclear layer will increase as that layer thins, separate counts of the number of microglia per unit length of retinal section also demonstrated increasing microglia over time. This process of microgliosis was also accompanied by an increase in Müller cell gliosis, as evidenced by the increase in GFAP immunopositivity in Müller cell processes. While it is possible that activated inflammatory cells in the outer nuclear layer may have also included macrophages infiltrating from the choroidal circulation, the apparently migratory and polarized morphologic features of resident retinal microglia in the inner retina indicate that they likely contribute to the aggregated population. In previous studies, we demonstrated that resident retinal microglia can migrate rapidly through retinal tissue in response to tissue injury39 under the guidance of chemotactic cytokines.40 These experiments are the first, to our knowledge, to describe the inflammatory reaction to subretinal hemorrhage in the form of rapid onset of microglial activation and migration to the outer nuclear layer in the first hours after hemorrhage, and the accumulation of activated microglia and macrophages in the outer retina that forms concurrently with the wave of apoptotic photoreceptor cell death in the outer nuclear layer. These results posit the involvement of activated microglia at the earliest stages of disease pathogenesis and link changes in microglial physiologic features to onset of apoptotic photoreceptor cell death. The dynamics of this response reflect the need for early intervention in the clinical context of subretinal hemorrhage and suggest microglial inhibition as a potential therapeutic target.
Formation of large subretinal hemorrhages induces both physical detachment of photoreceptors from the retinal pigment epithelial layer and the presence of blood components in the subretinal space. Our experiments in the mouse model comparing the effects of subretinal injections of blood to equivolume subretinal injections of inert viscoelastic material demonstrate that although some measure of outer nuclear layer apoptosis may be induced from physical retinal detachment alone, the presence of blood in the subretinal space contributed additionally and significantly to outer retinal atrophy. Red blood cells, which form most of the cellular content of the hemorrhage, likely constitute an important contributing blood constituent because separate subretinal injections of cell-free mouse serum samples produced less outer nuclear layer apoptosis and atrophy (data not shown). Also, the degree of microglial infiltration was significantly greater in subretinal blood versus subretinal viscoelastic injections, underlining the influence of hemorrhage in inducing migration and influx of inflammatory cells into the outer retina.30 ELISA analyses of retinal tissue after subretinal injection demonstrated that although increases in inflammatory cytokines, chemotactic cytokines, and adhesion molecules occurred after control viscoelastic material injections, these factors were all increased to a greater general extent after subretinal blood injections. It is possible that the presence of subretinal blood additionally increases activation of retinal microglia, inducing increased expression of microglial factors that can attract additional microglia from the inner retina (chemotactic cytokines), retain them in the outer retina (adhesion molecules), and contribute to their further activation (inflammatory cytokines). This notion is supported by in vitro experiments that demonstrated the ability of red blood cells to increase TNF-α, CCL2, and CCL5 in cultured retinal microglia. The activation of Müller cells may additionally result in expression of chemokines that recruit and activate inflammatory microglia and macrophages.41
We hypothesize that the increasing density of microglia and macrophages after subretinal hemorrhage transforms the environment of the outer retina from an immune-privileged zone42 normally devoid of retinal microglia43 into a highly proinflammatory region, which in turn potentiates processes of cellular apoptosis and photoreceptor degeneration. Our experimental results involving microglial inhibition with minocycline lend support to this notion. Minocycline is a semi-synthetic tetracycline derivative that, in addition to its antimicrobial action, possesses anti-inflammatory properties including inhibition of central nervous system microglia.44–46 It has a favorable distribution across the blood–central nervous system barrier, has high bioavailability, and is clinically well tolerated,47 all of which have promoted its potential use.48 In in vivo retinal studies, minocycline reduced microglial activation in models of glaucoma49 and retinal degeneration,50 and improved pathologic outcome measures in retinal degeneration,50 retinal detachment,51 and diabetic retinopathy.52 In the present study, we observed that minocycline treatment can significantly decrease microglial infiltration in the outer nuclear layer and reduce TUNEL positivity and atrophy in the outer retina. Our in vitro studies of minocycline in cultured mouse retinal microglial cells indicated that the ameliorative effects may relate to suppression of cytokines TNF-α, CCL2, and CCL5. The role of inflammatory reactions in potentiating photoreceptor cell death is also supported by previous observations that triamcinolone, an immunosuppressive corticosteroid, can reduce photoreceptor cell death in a rabbit model of subretinal hemorrhage.38
We recognize from our results that despite exerting an ameliorative effect on photoreceptor apoptosis, minocycline treatment alone does not completely arrest photoreceptor degeneration in subretinal hemorrhage. Although outer nuclear layer atrophy was slowed with treatment, it nevertheless continued to progress. Minocycline monotherapy may thus be useful only in transiently delaying the progression of photoreceptor degeneration so that an extended window of opportunity may be instituted to enable addition of other surgical interventions. Our results demonstrate that onset of inflammatory reactions and commencement of cellular apoptosis after subretinal hemorrhage occur rapidly, and underscore the need for readily instituted neuroprotective strategies shortly after occurrence of hemorrhage. Future work will investigate other potential microglial inhibitors that may exert more sustained therapeutic effects either alone or in combination with surgical strategies.
In the present study, we also investigated the involvement of the complement system in the inflammatory pathophysiologic features of subretinal hemorrhage. The complement cascade, involved in a variety of inflammatory, opsonization, and cytolytic reactions in many central nervous system disorders, is composed of a number of pathways in which C3 has a central role.32 Involvement of the complement cascade has been implicated in intracerebral hemorrhage31, in which cleavage of C3 releases the C3a anaphylatoxin, which activates resident microglia and attracts circulating leukocytes53 and also forms the C5b-9 membrane attack complex, which induces erythrocyte lysis and also perhaps bystander neuronal apoptosis.54 Induction of intracerebral hemorrhage in C3-deficient mice has also resulted in less brain edema, lower microglial activation, and fewer neurologic deficits than in wild-type control mice.55,56 In our mouse model of subretinal hemorrhage, it was observed that C3 deposition is increased in the subretinal space in the area of the hemorrhage and also more diffusely in Bruch's membrane. Minimal or no C3 deposition was observed in either C3-deficient mice with subretinal hemorrhage or in uninjected wild-type mice (data not shown). Although complement deposition was associated with the presence of subretinal hemorrhage in our model, microglial and macrophage infiltration, outer nuclear layer apoptosis, and photoreceptor atrophy all occurred to similar extents in both wild-type and C3-deficient mice. These results suggest that the absence of the central component of the complement system is not sufficient to ameliorate the inflammatory and degenerative consequences of subretinal hemorrhage and that complement activation may not be required to induce outer retinal atrophy.
In summary, we have established a murine model for subretinal hemorrhage that recapitulates features of the clinical disease and may be useful for examining underlying cellular and molecular pathogenic mechanisms of photoreceptor atrophy and for evaluating potential experimental therapies. We have also characterized the inflammatory component of subretinal hemorrhage as an early and progressive infiltration of microglia and macrophages into the outer retina that occurs concurrent with initiation of photoreceptor apoptosis. This inflammatory reaction likely contributes to photoreceptor atrophy because minocycline, an inhibitor of microglial activation, decreases microglial chemokine production, reduces microglial infiltration in vivo, and exerts a neuroprotective effect on photoreceptor apoptosis and atrophy. Microglial inhibition using agents such as minocycline that have high bioavailability and a favorable adverse effect profile may represent a useful ancillary therapeutic strategy to provide neuroprotection to photoreceptors at risk in the acute aftermath of subretinal hemorrhage, before the institution of surgical and pneumatic displacement approaches, with the final goal of minimizing photoreceptor cell death and permanent visual loss in this challenging clinical scenario.
Acknowledgments
We thank Drs. Mausam Damani and Catherine Cukras for comments and suggestions.
Footnotes
Supported by a grant from Prevention of Blindness Metropolitan Washington and by the National Eye Institute Intramural Research Program.
L.Z. and W.M. conceived and carried out experiments, W.T.W. conceived experiments and analyzed data, and R.N.F. analyzed and interpreted data. All authors were involved in writing the paper and had final approval of the submitted and published versions.
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.05.042.
Supplementary data
Effect of minocycline on expression of cytokines and adhesion molecules in the retina after subretinal hemorrhage. Subretinal hemorrhage was induced in experimental animals and treated daily with i.p. injections of minocycline or PBS. Retinal tissue was isolated after 3 days of treatment. Levels of cytokines and adhesion molecules in retinal tissue were assessed using ELISA in nine control PBS-treated animals (white bars) and in nine minocycline-treated animals (black bars). Compared with PBS-treated controls, levels of assayed proteins were either stable or decreased, although no decreases reached statistical significance. Levels of TNF-α were relatively low and were not consistently above the lower limit of detection of the assay.
References
- 1.Congdon N., O'Colmain B., Klaver C.C., Klein R., Munoz B., Friedman D.S., Kempen J., Taylor H.R., Mitchell P. Causes and prevalence of visual impairment among adults in the United States. Arch Ophthalmol. 2004;122:477–485. doi: 10.1001/archopht.122.4.477. [DOI] [PubMed] [Google Scholar]
- 2.Rein D.B., Wittenborn J.S., Zhang X., Honeycutt A.A., Lesesne S.B., Saaddine J. Forecasting age-related macular degeneration through the year 2050: the potential impact of new treatments. Arch Ophthalmol. 2009;127:533–540. doi: 10.1001/archophthalmol.2009.58. [DOI] [PubMed] [Google Scholar]
- 3.Kawasaki R., Yasuda M., Song S.J., Chen S.J., Jonas J.B., Wang J.J., Mitchell P., Wong T.Y. The prevalence of age-related macular degeneration in Asians: a systematic review and meta-analysis. Ophthalmology. 2010;117:921–927. doi: 10.1016/j.ophtha.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 4.Mousa S.A., Mousa S.S. Current status of vascular endothelial growth factor inhibition in age-related macular degeneration. BioDrugs. 2010;24:183–194. doi: 10.2165/11318550-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 5.Bressler N.M. Antiangiogenic approaches to age-related macular degeneration today. Ophthalmology. 2009;116:S15–S23. doi: 10.1016/j.ophtha.2009.06.048. [DOI] [PubMed] [Google Scholar]
- 6.Avery R.L., Fekrat S., Hawkins B.S., Bressler N.M. Natural history of subfoveal subretinal hemorrhage in age-related macular degeneration. Retina. 1996;16:183–189. doi: 10.1097/00006982-199616030-00001. [DOI] [PubMed] [Google Scholar]
- 7.Bennett S.R., Folk J.C., Blodi C.F., Klugman M. Factors prognostic of visual outcome in patients with subretinal hemorrhage. Am J Ophthalmol. 1990;109:33–37. doi: 10.1016/s0002-9394(14)75575-8. [DOI] [PubMed] [Google Scholar]
- 8.Chang M.A., Do D.V., Bressler S.B., Cassard S.D., Gower E.W., Bressler N.M. Prospective one-year study of ranibizumab for predominantly hemorrhagic choroidal neovascular lesions in age-related macular degeneration. Retina. 2010;30:1171–1176. doi: 10.1097/IAE.0b013e3181dd6d8a. [DOI] [PubMed] [Google Scholar]
- 9.Stifter E., Michels S., Prager F., Georgopoulos M., Polak K., Hirn C., Schmidt-Erfurth U. Intravitreal bevacizumab therapy for neovascular age-related macular degeneration with large submacular hemorrhage. Am J Ophthalmol. 2007;144:886–892. doi: 10.1016/j.ajo.2007.07.034. [DOI] [PubMed] [Google Scholar]
- 10.Hochman M.A., Seery C.M., Zarbin M.A. Pathophysiology and management of subretinal hemorrhage. Surv Ophthalmol. 1997;42:195–213. doi: 10.1016/s0039-6257(97)00089-1. [DOI] [PubMed] [Google Scholar]
- 11.de Juan E., Jr, Machemer R. Vitreous surgery for hemorrhagic and fibrous complications of age-related macular degeneration. Am J Ophthalmol. 1988;105:25–29. doi: 10.1016/0002-9394(88)90116-x. [DOI] [PubMed] [Google Scholar]
- 12.Ibanez H.E., Williams D.F., Thomas M.A., Ruby A.J., Meredith T.A., Boniuk I., Grand M.G. Surgical management of submacular hemorrhage: a series of 47 consecutive cases. Arch Ophthalmol. 1995;113:62–69. doi: 10.1001/archopht.1995.01100010064022. [DOI] [PubMed] [Google Scholar]
- 13.Thompson J.T., Sjaarda R.N. Vitrectomy for the treatment of submacular hemorrhages from macular degeneration: a comparison of submacular hemorrhage/membrane removal and submacular tissue plasminogen activator–assisted pneumatic displacement. Trans Am Ophthalmol Soc. 2005;103:98–107. discussion 107. [PMC free article] [PubMed] [Google Scholar]
- 14.Vander J.F., Federman J.L., Greven C., Slusher M.M., Gabel V.P. Surgical removal of massive subretinal hemorrhage associated with age-related macular degeneration. Ophthalmology. 1991;98:23–27. doi: 10.1016/s0161-6420(91)32348-0. [DOI] [PubMed] [Google Scholar]
- 15.Chen C.Y., Hooper C., Chiu D., Chamberlain M., Karia N., Heriot W.J. Management of submacular hemorrhage with intravitreal injection of tissue plasminogen activator and expansile gas. Retina. 2007;27:321–328. doi: 10.1097/01.iae.0000237586.48231.75. [DOI] [PubMed] [Google Scholar]
- 16.Hassan A.S., Johnson M.W., Schneiderman T.E., Regillo C.D., Tornambe P.E., Poliner L.S., Blodi B.A., Elner S.G. Management of submacular hemorrhage with intravitreous tissue plasminogen activator injection and pneumatic displacement. Ophthalmology. 1999;106:1900–1906. doi: 10.1016/S0161-6420(99)90399-8. [DOI] [PubMed] [Google Scholar]; discussion 1906–1907
- 17.Hesse L., Schmidt J., Kroll P. Management of acute submacular hemorrhage using recombinant tissue plasminogen activator and gas. Graefes Arch Clin Exp Ophthalmol. 1999;237:273–277. doi: 10.1007/s004170050232. [DOI] [PubMed] [Google Scholar]
- 18.Schulze S.D., Hesse L. Tissue plasminogen activator plus gas injection in patients with subretinal hemorrhage caused by age-related macular degeneration: predictive variables for visual outcome. Graefes Arch Clin Exp Ophthalmol. 2002;240:717–720. doi: 10.1007/s00417-002-0516-5. [DOI] [PubMed] [Google Scholar]
- 19.Bressler N.M., Bressler S.B., Childs A.L., Haller J.A., Hawkins B.S., Lewis H., MacCumber M.W., Marsh M.J., Redford M., Sternberg P., Jr, Thomas M.A., Williams G.A. Surgery for hemorrhagic choroidal neovascular lesions of age-related macular degeneration: ophthalmic findings; sST report No. 13. Ophthalmology. 2004;111:1993–2006. doi: 10.1016/j.ophtha.2004.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sanders D., Peyman G.A., Fishman G., Vlchek J., Korey M. The toxicity of intravitreal whole blood and hemoglobin. Albrecht Von Graefes Arch Klin Exp Ophthalmol. 1975;197:255–267. doi: 10.1007/BF00410870. [DOI] [PubMed] [Google Scholar]
- 21.Wong R.W., Richa D.C., Hahn P., Green W.R., Dunaief J.L. Iron toxicity as a potential factor in AMD. Retina. 2007;27:997–1003. doi: 10.1097/IAE.0b013e318074c290. [DOI] [PubMed] [Google Scholar]
- 22.Glatt H., Machemer R. Experimental subretinal hemorrhage in rabbits. Am J Ophthalmol. 1982;94:762–773. doi: 10.1016/0002-9394(82)90301-4. [DOI] [PubMed] [Google Scholar]
- 23.Toth C.A., Morse L.S., Hjelmeland L.M., Landers M.B., III Fibrin directs early retinal damage after experimental subretinal hemorrhage. Arch Ophthalmol. 1991;109:723–729. doi: 10.1001/archopht.1991.01080050139046. [DOI] [PubMed] [Google Scholar]
- 24.Xi G., Keep R.F., Hoff J.T. Mechanisms of brain injury after intracerebral haemorrhage. Lancet Neurol. 2006;5:53–63. doi: 10.1016/S1474-4422(05)70283-0. [DOI] [PubMed] [Google Scholar]
- 25.Xue M., Del Bigio M.R. Intracerebral injection of autologous whole blood in rats: time course of inflammation and cell death. Neurosci Lett. 2000;283:230–232. doi: 10.1016/s0304-3940(00)00971-x. [DOI] [PubMed] [Google Scholar]
- 26.Gong C., Hoff J.T., Keep R.F. Acute inflammatory reaction following experimental intracerebral hemorrhage in rat. Brain Res. 2000;871:57–65. doi: 10.1016/s0006-8993(00)02427-6. [DOI] [PubMed] [Google Scholar]
- 27.Zhao X., Zhang Y., Strong R., Zhang J., Grotta J.C., Aronowski J. Distinct patterns of intracerebral hemorrhage-induced alterations in NF-kappaB subunit, iNOS, and COX-2 expression. J Neurochem. 2007;101:652–663. doi: 10.1111/j.1471-4159.2006.04414.x. [DOI] [PubMed] [Google Scholar]
- 28.Power C., Henry S., Del Bigio M.R., Larsen P.H., Corbett D., Imai Y., Yong V.W., Peeling J. Intracerebral hemorrhage induces macrophage activation and matrix metalloproteinases. Ann Neurol. 2003;53:731–742. doi: 10.1002/ana.10553. [DOI] [PubMed] [Google Scholar]
- 29.Wang J., Dore S. Inflammation after intracerebral hemorrhage. J Cereb Blood Flow Metab. 2007;27:894–908. doi: 10.1038/sj.jcbfm.9600403. [DOI] [PubMed] [Google Scholar]
- 30.Loftspring M.C., Hansen C., Clark J.F. A novel brain injury mechanism after intracerebral hemorrhage: the interaction between heme products and the immune system. Med Hypotheses. 2010;74:63–66. doi: 10.1016/j.mehy.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hua Y., Xi G., Keep R.F., Hoff J.T. Complement activation in the brain after experimental intracerebral hemorrhage. J Neurosurg. 2000;92:1016–1022. doi: 10.3171/jns.2000.92.6.1016. [DOI] [PubMed] [Google Scholar]
- 32.Ducruet A.F., Zacharia B.E., Hickman Z.L., Grobelny B.T., Yeh M.L., Sosunov S.A., Connolly E.S., Jr The complement cascade as a therapeutic target in intracerebral hemorrhage. Exp Neurol. 2009;219:398–403. doi: 10.1016/j.expneurol.2009.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wessels M.R., Butko P., Ma M., Warren H.B., Lage A.L., Carroll M.C. Studies of group B streptococcal infection in mice deficient in complement component C3 or C4 demonstrate an essential role for complement in both innate and acquired immunity. Proc Natl Acad Sci USA. 1995;92:11490–11494. doi: 10.1073/pnas.92.25.11490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Engelbert M., Gilmore M.S. Fas ligand but not complement is critical for control of experimental Staphylococcus aureus endophthalmitis. Invest Ophthalmol Vis Sci. 2005;46:2479–2486. doi: 10.1167/iovs.04-1139. [DOI] [PubMed] [Google Scholar]
- 35.Ma W., Zhao L., Fontainhas A.M., Fariss R.N., Wong W.T. Microglia in the mouse retina alter the structure and function of retinal pigmented epithelial cells: a potential cellular interaction relevant to AMD. PLoS One. 2009;4:e7945. doi: 10.1371/journal.pone.0007945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kawai T., Akira S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol Med. 2007;13:460–469. doi: 10.1016/j.molmed.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 37.Green W.R., Key S.N., III Senile macular degeneration: a histopathologic study. Trans Am Ophthalmol Soc. 1977;75:180–254. [PMC free article] [PubMed] [Google Scholar]
- 38.Bhisitkul R.B., Winn B.J., Lee O.T., Wong J., de S Pereira D., Porco T.C., He X., Hahn P., Dunaief J.L. Neuroprotective effect of intravitreal triamcinolone acetonide against photoreceptor apoptosis in a rabbit model of subretinal hemorrhage. Invest Ophthalmol Vis Sci. 2008;49:4071–4077. doi: 10.1167/iovs.08-1892. [DOI] [PubMed] [Google Scholar]
- 39.Lee J.E., Liang K.J., Fariss R.N., Wong W.T. Ex vivo dynamic imaging of retinal microglia using time-lapse confocal microscopy. Invest Ophthalmol Vis Sci. 2008;49:4169–4176. doi: 10.1167/iovs.08-2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liang K.J., Lee J.E., Wang Y.D., Ma W., Fontainhas A.M., Fariss R.N., Wong W.T. Regulation of dynamic behavior of retinal microglia by CX3CR1 signaling. Invest Ophthalmol Vis Sci. 2009;50:4444–4451. doi: 10.1167/iovs.08-3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nakazawa T., Hisatomi T., Nakazawa C., Noda K., Maruyama K., She H., Matsubara A., Miyahara S., Nakao S., Yin Y., Benowitz L., Hafezi-Moghadam A., Miller J.W. Monocyte chemoattractant protein 1 mediates retinal detachment–induced photoreceptor apoptosis. Proc Natl Acad Sci USA. 2007;104:2425–2430. doi: 10.1073/pnas.0608167104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zamiri P., Masli S., Streilein J.W., Taylor A.W. Pigment epithelial growth factor suppresses inflammation by modulating macrophage activation. Invest Ophthalmol Vis Sci. 2006;47:3912–3918. doi: 10.1167/iovs.05-1267. [DOI] [PubMed] [Google Scholar]
- 43.Santos A.M., Calvente R., Tassi M., Carrasco M.C., Martin-Oliva D., Marin-Teva J.L., Navascues J., Cuadros M.A. Embryonic and postnatal development of microglial cells in the mouse retina. J Comp Neurol. 2008;506:224–239. doi: 10.1002/cne.21538. [DOI] [PubMed] [Google Scholar]
- 44.Amin A.R., Attur M.G., Thakker G.D., Patel P.D., Vyas P.R., Patel R.N., Patel I.R., Abramson S.B. A novel mechanism of action of tetracyclines: effects on nitric oxide synthases. Proc Natl Acad Sci USA. 1996;93:14014–14019. doi: 10.1073/pnas.93.24.14014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wasserman J.K., Schlichter L.C. Neuron death and inflammation in a rat model of intracerebral hemorrhage: effects of delayed minocycline treatment. Brain Res. 2007;1136:208–218. doi: 10.1016/j.brainres.2006.12.035. [DOI] [PubMed] [Google Scholar]
- 46.Yrjanheikki J., Tikka T., Keinanen R., Goldsteins G., Chan P.H., Koistinaho J. A tetracycline derivative, minocycline, reduces inflammation and protects against focal cerebral ischemia with a wide therapeutic window. Proc Natl Acad Sci USA. 1999;96:13496–13500. doi: 10.1073/pnas.96.23.13496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Aronson A.L. Pharmacotherapeutics of the newer tetracyclines. J Am Vet Med Assoc. 1980;176:1061–1068. [PubMed] [Google Scholar]
- 48.Wirostko E., Wirostko W.J., Wirostko B.M. Age-related macular degeneration is an inflammatory disease possibly treatable with minocycline. Acta Ophthalmol Scand. 2004;82:243–244. doi: 10.1111/j.1600-0420.2004.00150d.x. [DOI] [PubMed] [Google Scholar]
- 49.Bosco A., Inman D.M., Steele M.R., Wu G., Soto I., Marsh-Armstrong N., Hubbard W.C., Calkins D.J., Horner P.J., Vetter M.L. Reduced retina microglial activation and improved optic nerve integrity with minocycline treatment in the DBA/2J mouse model of glaucoma. Invest Ophthalmol Vis Sci. 2008;49:1437–1446. doi: 10.1167/iovs.07-1337. [DOI] [PubMed] [Google Scholar]
- 50.Yang L.P., Li Y., Zhu X.A., Tso M.O. Minocycline delayed photoreceptor death in rds mice through iNOS-dependent mechanism. Mol Vis. 2007;13:1073–1082. [PMC free article] [PubMed] [Google Scholar]
- 51.Yang L., Kim J.H., Kovacs K.D., Arroyo J.G., Chen D.F. Minocycline inhibition of photoreceptor degeneration. Arch Ophthalmol. 2009;127:1475–1480. doi: 10.1001/archophthalmol.2009.288. [DOI] [PubMed] [Google Scholar]
- 52.Chen Y.D., Xu X., Xia X., Wu H., Liu K., Zheng Z., Zhu D. MMP9 is involved in glycation end-products induced increase of retinal vascular permeability in rats and the therapeutic effect of minocycline. Curr Eye Res. 2008;33:977–983. doi: 10.1080/02713680802450984. [DOI] [PubMed] [Google Scholar]
- 53.Rynkowski M.A., Kim G.H., Garrett M.C., Zacharia B.E., Otten M.L., Sosunov S.A., Komotar R.J., Hassid B.G., Ducruet A.F., Lambris J.D., Connolly E.S. C3a receptor antagonist attenuates brain injury after intracerebral hemorrhage. J Cereb Blood Flow Metab. 2009;29:98–107. doi: 10.1038/jcbfm.2008.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Park C.C., Shin M.L., Simard J.M. The complement membrane attack complex and the bystander effect in cerebral vasospasm. J Neurosurg. 1997;87:294–300. doi: 10.3171/jns.1997.87.2.0294. [DOI] [PubMed] [Google Scholar]
- 55.Yang S., Nakamura T., Hua Y., Keep R.F., Younger J.G., He Y., Hoff J.T., Xi G. The role of complement C3 in intracerebral hemorrhage–induced brain injury. J Cereb Blood Flow Metab. 2006;26:1490–1495. doi: 10.1038/sj.jcbfm.9600305. [DOI] [PubMed] [Google Scholar]
- 56.Yang S., Nakamura T., Hua Y., Keep R.F., Younger J.G., Hoff J.T., Xi G. Intracerebral hemorrhage in complement C3–deficient mice. Acta Neurochir Suppl. 2006;96:227–231. doi: 10.1007/3-211-30714-1_49. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Effect of minocycline on expression of cytokines and adhesion molecules in the retina after subretinal hemorrhage. Subretinal hemorrhage was induced in experimental animals and treated daily with i.p. injections of minocycline or PBS. Retinal tissue was isolated after 3 days of treatment. Levels of cytokines and adhesion molecules in retinal tissue were assessed using ELISA in nine control PBS-treated animals (white bars) and in nine minocycline-treated animals (black bars). Compared with PBS-treated controls, levels of assayed proteins were either stable or decreased, although no decreases reached statistical significance. Levels of TNF-α were relatively low and were not consistently above the lower limit of detection of the assay.