

Abstract
Background and Purpose
Current therapies for head and neck cancer frequently are not curative, necessitating novel therapeutic strategies. Thus, we studied whether inhibition of poly (ADP-Ribose) polymerase (PARP), a key DNA repair enzyme, could improve efficacy of radiotherapy in human head and neck cancer.
Material and Methods
UM-SCC1, UM-SCC5, UM-SCC6, and FaDu human head and neck cancer cellular susceptibility to the PARP inhibitor (PARPi) ABT-888 and/or radiation (IR) was assessed using colony formation assays. DNA damage was evaluated using the alkaline comet assay and immunostaining for γ-H2AX foci. Non-homologous end-joining (NHEJ) mediated repair was measured using phospho-DNA-Pk foci. Epidermal growth factor receptor (EGFR) location was assessed by immunostaining. Poly ADP-Ribose polymerization (PAR) levels were assessed using immunoblotting.
Results
Human head and neck cancer cells exhibited enhanced cytotoxicity with IR and ABT-888 compared to either agent alone. This increased susceptibility correlated with reduced nuclear EGFR, attenuation of NHEJ, and persistence of DNA damage following IR. Interestingly, a subset of head and neck cancer cells which had elevated basal PAR levels was susceptible to PARPi alone.
Conclusion
Combining radiotherapy and PARP inhibition may improve outcomes and quality of life for head and neck cancer patients treated with radiotherapy. Furthermore, this novel strategy may also be feasible in other tumor types. Moreover, PAR levels should be investigated as a potential biomarker for tumor susceptibility to PARP inhibition.
Keywords: head and neck cancer, PARP, ABT-888, EGFR, biomarker, H2AX, DNA damage
Introduction
Agents which target cancers that are deficient in homologous recombination (HR)-mediated DNA double strand break (DSB) repair, such as poly (ADP-ribose) polymerase (PARP) inhibitors (PARPi), have gained recent attention due to their highly selective killing of BRCA-associated, DNA repair defective tumors while maintaining minimal toxicity in normal tissues [1–3]. Additionally, PARPi has been reported to enhance cytotoxicity in sporadic tumors when combined with other DNA damaging agents, such as with platinum and cyclophosphamide in breast cancer and with temozolomide in glioblastoma [4]. Thus, much effort has been undertaken to expand the utility of PARPi beyond the realm of BRCA-associated tumors by combining with agents that alter the DNA damage/repair pathways.
Even when head and neck cancers have not metastasized, loco regional therapy is frequently not successful. This may, in part, be due to over expression of the epidermal growth factor receptor (EGFR), which has been implicated in tumorigenesis and disease progression through modulation of proliferation, differentiation, and DNA damage response. Treatment of head and neck cancer typically involves a combination of surgery, chemotherapy, and radiation therapy. It has been reported that, in response to radiotherapy, EGFR is transported to the nucleus and plays a role in radio resistance, poor prognosis, and treatment failures [5–7]. Although targeted chemotherapy and radiation fractionation have had favorable impact, outcomes still remain poor, necessitating novel treatment strategies [8–12].
Thus, in this current study, we hypothesized that enhanced cytotoxicity of head and neck cancer may be achieved by combining radiation (IR), an integral part of therapy that induces cellular DNA damage, and inhibition of PARP, which would theoretically inhibit repair of DNA damage and result in enhanced tumor cytotoxicity.
Consistent with our hypothesis, significantly enhanced cytotoxicity was observed with combination IR and the PARPi ABT-888. This correlated with attenuation of DNA-Pk dependent NHEJ and subsequent persistence of DNA damage. Further dissection of the mechanism of inhibited NHEJ revealed that PARPi attenuated nuclear EGFR levels following IR. Interestingly, a subset of head and neck cancer cells were susceptible to PARPi alone and correlated with an elevated basal poly ADP-Ribose polymerization (PAR) level.
These data support the use of the PARPi ABT-888 in combination with radiotherapy as an innovative treatment strategy to potentially improve outcomes in head and neck cancer patients. Moreover, elevated PAR levels may be used to profile and stratify head and neck cancer patients who may subsequently demonstrate superior response to PARP inhibition. Similarly, γ-H2AX levels may be quantified in patient tumor samples to evaluate patient’s response to therapy. Furthermore, this strategy may also be feasible in other tumors such as breast, brain and lung.
Materials and Methods
Cell culture
The human head and neck squamous carcinoma cell lines (HNSCC) UM-SCC1, UM-SCC5, and UM-SCC6 were obtained courtesy of Dr. Thomas E Carey. FaDu (HTB-43) was obtained from ATCC (Manassas, VA). The PARP inhibitor ABT-888 (Enzo Life Sciences) was utilized in our study. Please refer to SI Materials and Methods for cell culture growth conditions.
Immunofluorescence
Head and neck cell lines were cultured and seeded on sterile cover slips, exposed to 10uM PARPi for 2 hours, and subsequently treated with mock or 3 Gy γ-IR using an X-ray irradiator at 1.225 Gy/min (Kimtron Inc., Woodbury, CT). Immunohistochemistry was performed as previously described [13, 14] with slight modification. Please refer to SI Materials and Methods for details, including antibody used.
Alkaline comet assay
Cells were exposed to ABT-888 and 3 Gy or mock IR and incubated for various times, after which they were prepared and subjected to alkaline comet assay according to the manufacturer’s instructions (catalog # 4250-050-K; Trevigen). Please refer to SI Materials and Methods for details.
Clonogenic survival assay
Cell survival was evaluated by the colony formation assay in the HNSCC cell lines following 3 Gy IR and various doses of ABT-888 (1uM-10μM) as previously described [15]. Briefly, cells were seeded and treated with the indicated doses of ABT 888 (or vehicle) for 2 hours, followed by mock or 3 Gy IR following which the plates were left undisturbed. Please refer to SI Materials and Methods for further details.
Immunoblotting
Cell lysates were prepared using standard protocol. β-Actin levels were analyzed as loading control. Please refer to SI Materials and methods for further details.
Statistical analysis
The data were analyzed via analysis of variance (ANOVA) followed by a Bonferroni post test using GraphPad Prism version 4.02 (GraphPad Software, San Diego, CA). Data presented as average +/− standard error of mean.
Results
PARPi augments tumor response to radiotherapy
UM-SCC1, UM-SCC5, UM-SCC6, and FaDu cells, which are well characterized and representative squamous cell carcinoma of the head and neck, were utilized in this study [16–19]. PARPi has been shown to be an effective strategy to treat DNA repair deficient cancer. Additionally, PARPi has been combined with other DNA damaging agents with improved outcomes in breast cancer patients [20]. Thus, we hypothesized that the PARPi ABT-888 may increase cytotoxicity of head and neck cancer cells following IR, an integral part of standard therapy for head and neck tumors.
To test this hypothesis, we performed colony forming assays with various doses (1–10uM) of ABT-888 in combination with IR (3 Gy). These doses of ABT-888 chosen have been previously reported to be within physiologic range [2, 21]. IR alone reduced colony forming ability of cells by 30–40% (UM-SCC1: 32%, UM-SCC5: 35%, UM-SCC6: 40%, and FaDu: 30%). As shown in Fig. 1 and consistent with our hypothesis, the addition of IR to PARPi significantly reduced the colony forming ability in a dose-dependent manner (70%–95% reduction in cell viability). Interestingly, differential susceptibility to ABT-888 alone was observed. In particular, UM-SCC1 and UM-SCC5 (Fig. 1A–B) cells were exquisitely sensitive to ABT-888 alone (50–75% reduction in cell viability), while UM-SCC6 and FaDu (Fig 1C–D) were not. These results suggest that the combination of radiotherapy and the PARPi ABT-888 may augment cytotoxicity in head and neck tumors. Additionally, a subset of head and neck cancers may be susceptible to PARPi alone.
FIGURE 1. IR augments head and neck tumor susceptibility to the PARP inhibitor ABT-888.
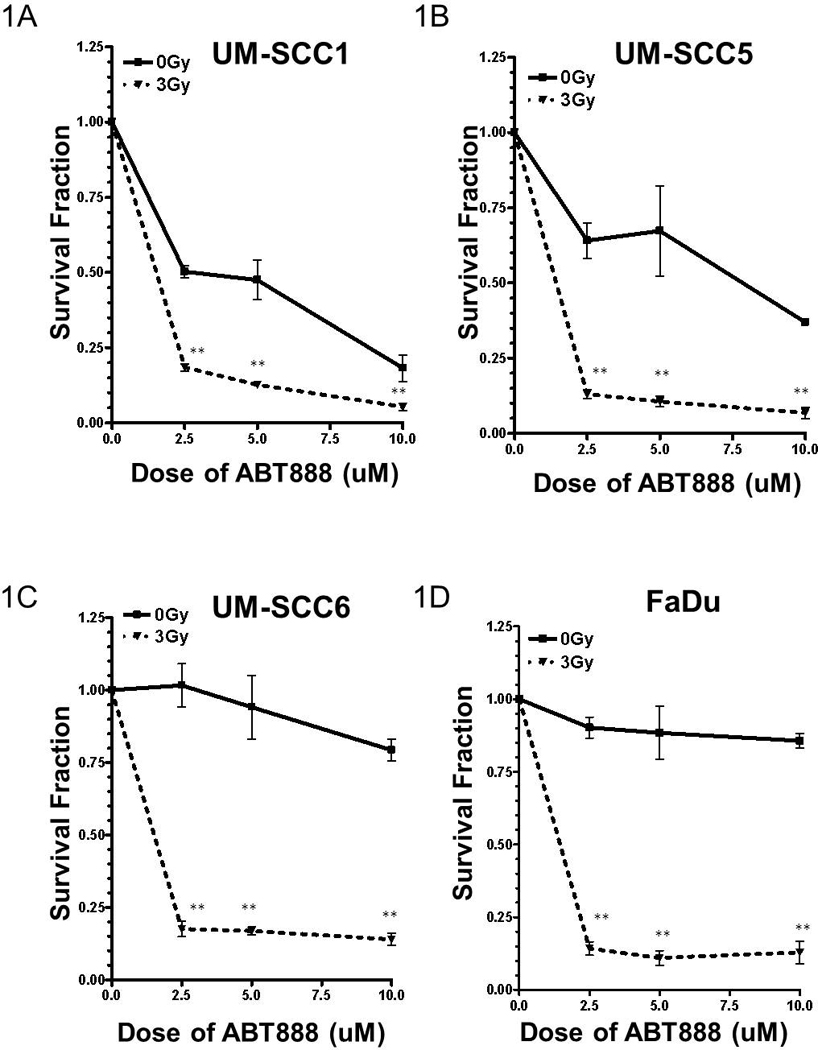
Combination of IR and ABT-888 reduces the viability of (A) UM-SCC1, (B) UM-SCC5, (C) UM-SCC6, and (D) FaDu head and neck cancer cells. Shown is the representative data of at least 3 independent experiments of the cell viability following various treatments as measured by colony formation assay, corrected for the survival fraction following RT (mean +/− SEM, *p<0.01, **p<0.001). Interestingly, UM-SCC1 and UM-SCC5 demonstrate susceptibility to ABT-888 alone.
Persistent DNA damage with PARPi in head and neck cancer cells
Since IR induces SSB and DSB DNA damage, and because PARP is integral in the repair of DNA single strand breaks (SSBs), we hypothesized that the enhanced cytotoxicity with PARPi and IR may be due to inability of cells to repair DNA damage. To assess this notion, total cellular DNA damage (SSBs and DSBs) following IR and/or ABT-888 was evaluated using the alkaline comet assay. Due to the differential effects of ABT-888 and IR observed (Fig. 1), UM-SCC1 (susceptible to PARPi alone) and UM-SCC6 (not susceptible to PARPi alone) were chosen as representative cell lines for further studies.
As shown in Figure 2, as expected, the addition of PARPi alone resulted in increased DNA damage, likely due to inhibition of SSB repair. Additionally, similar levels of DNA damage, as evidenced by the mean tail moment, were observed in both PARPi- and vehicle-treated UM-SCC1 and UM-SCC6 (Fig. 2B–C) cells at 15 minutes after IR. However, at longer time points, increased levels of total DNA damage were observed in irradiated cells treated with PARPi compared with vehicle. Interestingly, at 24 and 48hrs following treatment, levels of DNA damage are further increased by the addition of PARPi.
FIGURE 2. Persistent DNA damage is observed in head and neck cancer cells treated with ABT-888 and radiation.
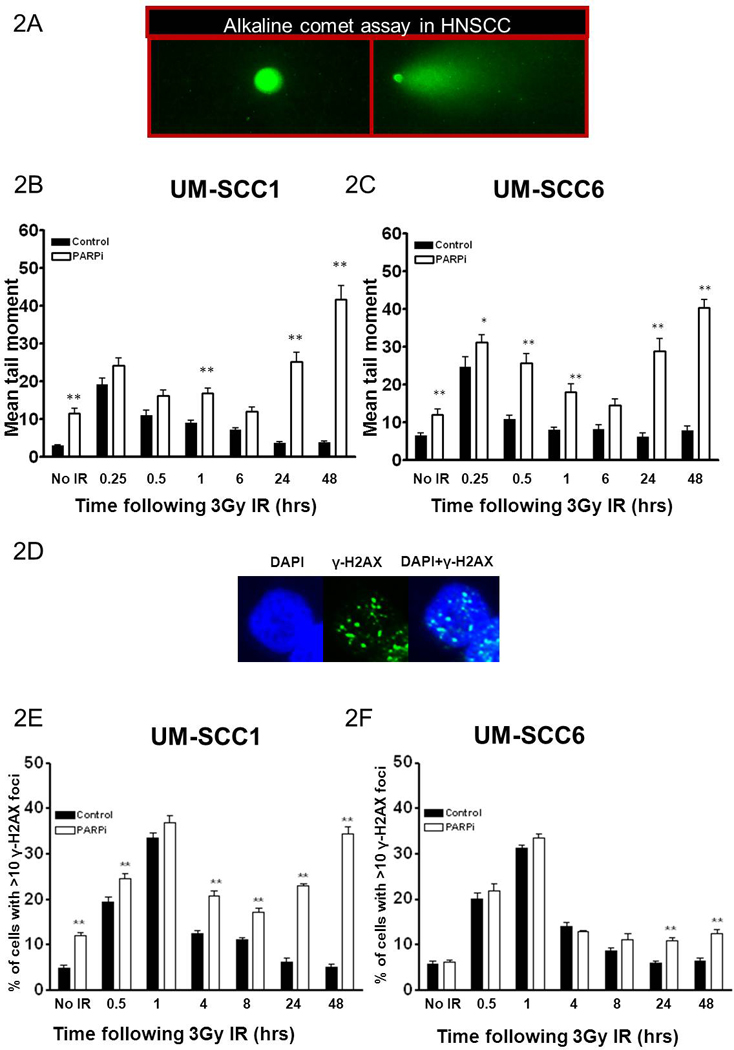
The inset in (A) is a representative image of UM-SCC1 cells exhibiting comet tail following IR. IR enhances the mean tail moment, indicative of total DNA damage (SSB and DSB) in ABT-888 treated (B) UM-SCC1 and (C) UM-SCC6 cell lines, as measured by the alkaline comet assay. Addition of ABT-888 inhibits resolution of IR-induced DNA damage. The inset in (D) is a representative image of UM-SCC1 cells exhibiting γ-H2AX foci, a commonly used marker for DSBs, following IR. The comet data correlates with the number of cells with DSBs in (E) UM-SCC1 and (F) UM-SCC6 cells as evidenced by persistent γ-H2AX foci. Shown is the representative data of 3 independent experiments the % of cells (mean +/− SEM) with >10 foci (*p<0.05, **p<0.01).
Since the critical cellular DNA lesion in relation to cytotoxic effects are DSBs, we next assessed the effect of PARPi on total DSB damage in head and neck cancer cell lines with and without IR. γ-H2AX foci, well established markers of DNA DSBs [22], were evaluated. As shown in Fig. 2 (E–F), all cell lines exhibited robust increase in DNA DSBs following IR as demonstrated by increased percentage of cells with γ-H2AX foci. Interestingly the addition of PARPi to IR resulted in increased unresolved DSBs, which have been implicated in initiation of cell death or senescence. These results support a potentiation of DNA damage by PARPi.
Susceptibility to ABT-888 is associated with inhibition of non-homologous end joining (NHEJ) dependent DNA repair
Since the majority of IR-induced DSBs are repaired via the DNA Pk dependent NHEJ pathway [13, 14], and mounting evidence suggests an interplay between PARP signaling and NHEJ, we hypothesized that the ABT-888-induced radio sensitization may be due to alteration of NHEJ-mediated DSB repair [23–25].
To test this hypothesis, the induction of phospho-Threonine 2609 (Thr2609) DNA-Pk foci, well established markers NHEJ-mediated DSB repair [26, 27], was assessed 1hr following 3 Gy IR. As shown in Fig. 3, IR significantly increased the number of cells with phospho-Thr2609-DNA-Pk-foci. Interestingly, the addition of PARPi significantly attenuated this response by more than 30% in all cell lines examined. Furthermore, there are no cell cycle effects at the time points studied (SI Fig 1). Thus, these results suggest that the enhanced cytotoxicity with PARPi and IR may be due to PARPi-mediated attenuation of NHEJ.
FIGURE 3. ABT-888 attenuates IR-induced non-homologous end joining (NHEJ) in head and neck cancer cells.

The inset in (A) is a representative image of UM-SCC1 cells exhibiting DNA Pk T2609 foci, well characterized markers of NHEJ-mediated DNA DSB repair, following IR. ABT-888 attenuates IR-induced phosphorylated DNA Pk foci in (B) UM-SCC1, (C) UM-SCC5, (D) UM-SCC6, and (E) FaDu cells. Shown is the representative data of 3 independent experiments the % of cells (mean +/− SEM) with >10 foci (*p<0.05, **p<0.01).
PARPi attenuates IR induced nuclear translocation of EGFR
The EGFR has been implicated in a number of cellular processes, including cell proliferation and survival, angiogenesis, and DNA damage response and repair. Specifically, with regards to DNA damage response, IR has been shown to induce EGFR translocation to the nucleus and interaction with DNA-Pk to activate NHEJ [9, 28–34]. To determine whether inhibition of NHEJ by PARPi is due to alterations in EGFR location following IR, we next examined changes in the location of EGFR following PARPi and IR. As shown in Fig. 4, IR induced EGFR nuclear translocation. Interestingly, PARPi inhibited this EGFR nuclear translocation as evidenced by retention of EGFR in the cytoplasm. These data imply that the reduced NHEJ capacity following PARPi and IR are associated with attenuation of IR-induced import of EGFR to the nucleus.
FIGURE 4. ABT-888 reduces nuclear epidermal growth factor receptor (EGFR) following radiation.

The inset in (A) is a representative image of UM-SCC6 cells exhibiting nuclear (N), both nuclear and cytosolic (NC), and cytosolic (C) EGFR. EGFR location was analyzed following IR and cells were assessed as having predominantly nuclear staining, predominantly cytoplasmic staining, or mixed nuclear/cytoplasmic staining. ABT-888 blocks nuclear translocation of EGFR following IR in (B) UM-SCC1, (C) UM-SCC5, (D) UM-SCC6, and (E) FaDu cells. Shown is the representative data of three independent experiments the % of cells (mean ± SEM) with strictly nuclear (N), strictly cytosolic (C), or mixed nuclear/cytosolic (NC) EGFR staining (*p < 0.05, **p < 0.01).
PARPi generates persistent DNA damage in a population of head and neck cancer cells
PARPi inhibits the base excision repair (BER) pathway responsible for resolving SSBs. However, if SSBs persist, they are ultimately converted to DSBs and repaired by HR-mediated repair. Given that UM-SCC1 and UM-SCC5 exhibit susceptibility to PARPi alone, we hypothesized that one explanation for single agent activity may be due to PARPi-induced persistent DNA damage in these cells. Indeed, compared to vehicle control, PARPi induced persistent DNA damage, as assessed using alkaline comet assay, in UM-SCC1 (SI Fig. 2A) and UM-SCC5 (SI Fig. 3A) but not in UM-SCC6 (SI Fig. 2B) and FaDu (SI Fig. 3B) head and neck cancer cells. Moreover, this data correlated with increased γ-H2AX induction (SI Fig. 2C–D, SI Fig 3C–D).
Possible explanations for the persistent DNA damage in these cells may include an inherent HR deficiency and/or inhibition of HR by PARPi [35]. To address this issue, we analyzed IR-induced Rad51 foci, which are well established marker for HR, with and without ABT-888. Surprisingly, all cell lines examined were competent in inducing Rad51 foci, and the addition of ABT-888 did not alter this response (SI Fig.4). These results suggest that cytotoxicity from PARPi may be due to the inability of treated cells to resolve DNA DSBs independent of inhibition of HR or other basal DNA repair deficiencies.
Susceptibility to PARPi correlates with elevated basal PAR levels
Since the head and neck cancer cell lines did not appear to possess an inherent DSB repair deficiency (Fig. 3 and SI Figure 4), we probed for other markers to explain the increased susceptibility to PARPi. As susceptibility to PARPi and chemotherapy has been attributed to elevated PARP activity [20], we assessed PARP activity in head and neck cancer cells by measuring PAR levels, an established surrogate for PARP activity [36]. As shown in Fig. 5, all cell lines exhibited various levels of PAR. Following 2 hours of exposure to PARPi, PAR levels decrease significantly, validating that the dose chosen was suitable. Interestingly, the cell lines that were susceptible to PARPi, UM-SCC1 and UM-SCC5, exhibited significantly elevated basal PAR levels.
FIGURE 5. Basal PAR level correlates with susceptibility of head and neck cancer cells to ABT-888.

Basal PAR levels in head and neck cells were analyzed following ABT-888 treatment and correlated with susceptibility to ABT-888. Shown is a representative western blot of at least 3 independent experiments.
Discussion
In this study, we report that the combination of IR and the PARPi ABT-888 can be an innovative treatment strategy to potentially improve outcomes in head and neck cancer patients. Interestingly, the UM-SCC1 and UM-SCC5 head and neck cancer cells, which exhibit susceptibility to PARPi alone, do not appear to be inherently DSB repair deficient. However, PARPi induces persistent γ-H2AX foci, suggestive of the presence of persistent DSBs. It is intriguing to postulate that there could be other aspects of DNA damage/repair that have not been investigated. In particular, potential defects in other HR repair proteins, recruitment to the DSB, or other upstream/downstream DNA damage response signaling events could explain this susceptibility to PARPi alone.
PARP and DNA Pk have been reported to be critical for DNA repair. Interestingly, both DNA Pk and PARP promote the accelerated repair of IR-induced DSBs and function in the same molecular pathway [23]. ATM, a vital DNA damage response protein, is also activated following PARP inhibition [37]. PAR functions as a docking polymer for a variety of transcription factors, chromatin, DNA replication and repair proteins, e.g. NFκB, PAX 6, AP 2, his tones, XRCC1, Ku, DNA polymerase, DNA Pk, ligases, condensins, and DNA topoisomerases thus modulating gene regulation, chromatin structure, DNA replication and repair [38]. PA Rylation acts as an early DNA damage response, post-translational modification, that recruits repair proteins to the DNA damage site [39]. Thus, a connection between PARP and DSB repair pathways besides HR, such as DNA Pk dependent repair, is one interesting novel mechanism by which PARP inhibition alters cellular viability following DNA damage.
Our study supports the notion that PARPi attenuates DNA Pk dependent NHEJ and may be one potential mechanism of enhanced head and neck tumor susceptibility to radiotherapy. Interestingly, we found that a 6–10% reduction in the percentage of cells with DNA-Pk foci (evident for NHEJ) correlated with a dramatic reduction (70–95%) in colony forming ability of treated cells. This discrepancy may be explained by the notion that contrary to analysis of foci, which demonstrates a snap shot in time, the colony formation assay reflects multiple mechanisms of cell death over a period of 3 weeks. As multiple signaling pathways are involved in regulation and determination of the fate of cell death or survival, our data suggests that DNA-Pk may be one part of the complicated cell signaling/ DNA damage repair network, and may contribute only partly to the overall effect of cell susceptibility to DNA damage.
We also observed via the alkaline comet assay, which detects both SSBs and DSBs, that DNA damage was increased after 24 to 48 hrs compared to the initial damage at 15 min after treatment with PARPi. Since PARP is a SSB DNA repair enzyme, treatment with the PARPi ABT-888 is expected to inhibit SSB repair and thus increase basal levels of SSBs. Addition of IR induces further DNA damage. The increased DNA damage observed at longer time points may be due to persistent DSBs or the result of additional DNA-cuts as a consequence of conversion of SSBs to DSBs during attempted DNA repair or collapsed replication forks. This is supported by the increased % of cells with γ-H2AX foci at later time points. Alternatively, activation of cell death processes such as apoptosis could also induce markers of DNA damage.
EGFR and DNA Pk have been reported to interact as well [28]. As initially reported by Rodemann and colleagues, EGFR translocates to the nucleus following DNA damage and induces DNA repair [40]. In this study we report, for the first time, that the cytotoxicity due to the combination of a PARP inhibitor and IR is mediated by attenuation of IR-induced EGFR nuclear import. This finding advances the intriguing hypothesis that PARP plays a critical role in the nuclear import of EGFR following IR-induced DNA damage. This may be the result of direct interaction between EGFR and PARP, or through a nuclear import/export mediator such as CRM1. Our group is currently pursuing this line of investigation. Nonetheless, targeted therapy using PARPi may be an innovative approach in EGFR over expressing cancers.
There has been active research and effort invested in the discovery of biomarkers to predict tumor response to therapy. Biomarkers are invaluable tools for cancer detection, diagnosis, prognosis, patient stratification, and treatment selection. We propose the use of γ-H2AX foci levels and basal PAR levels as viable protein markers to predict response to PARPi. We do acknowledge that our in vitro work is preliminary and that further validation of these techniques on clinical samples is required prior to implementation in clinic. Our group is currently pursuing this line of investigation.
Our study demonstrates that IR dramatically augments tumor response to PARPi via reduction of NHEJ and attenuation of EGFR nuclear import. These results warrant future studies to compare efficacy versus traditional chemotherapy. More importantly, since maintaining quality of life has become an area of emphasis in head and neck therapy, the use of targeted agents such as ABT-888 may further improve the therapeutic ratio. Lastly, this strategy may also be feasible in other tumors like brain, breast and lung cancers.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
This work was supported by the IMPACT Award from the Department of Radiation Oncology, University of Alabama-Birmingham Comprehensive Cancer Center, the Center for Clinical and Translational Science (CCTS) and the Council of Center Directors (COCD) Translational Research Intramural Pilot Grant Program, and University of Alabama-Birmingham School of Medicine (to E.S.Y.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Presented at the 12th International Wolfsberg Meeting, 2011
Conflict of interest statement
The authors declare no conflict of interest.
References
- 1.Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 2.Fong PC, Boss DS, Yap TA, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361:123–134. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- 3.Rottenberg S, Jaspers JE, Kersbergen A, et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proceedings of the National Academy of Sciences. 2008;105:17079–17084. doi: 10.1073/pnas.0806092105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Donawho CK, Luo Y, Luo Y, et al. ABT-888, an Orally Active Poly(ADP-Ribose) Polymerase Inhibitor that Potentiates DNA-Damaging Agents in Preclinical Tumor Models. Clinical Cancer Research. 2007;13:2728–2737. doi: 10.1158/1078-0432.CCR-06-3039. [DOI] [PubMed] [Google Scholar]
- 5.Peter RU, Beetz A, Ried C, Michel G, van Beuningen D, Ruzicka T. Increased expression of the epidermal growth factor receptor in human epidermal keratinocytes after exposure to ionizing radiation. Radiat Res. 1993;136:65–70. [PubMed] [Google Scholar]
- 6.Rodemann HP, Dittmann K, Toulany M. Radiation-induced EGFR-signaling and control of DNA-damage repair. Int J Radiat Biol. 2007;83:781–791. doi: 10.1080/09553000701769970. [DOI] [PubMed] [Google Scholar]
- 7.Dittmann K, Mayer C, Kehlbach R, Rodemann HP. Radiation-induced caveolin-1 associated EGFR internalization is linked with nuclear EGFR transport and activation of DNA-PK. Molecular Cancer. 2008;7:69. doi: 10.1186/1476-4598-7-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jutten B, Dubois L, Li Y, et al. Binding of cetuximab to the EGFRvIII deletion mutant and its biological consequences in malignant glioma cells. Radiother Oncol. 2009;92:393–398. doi: 10.1016/j.radonc.2009.06.021. [DOI] [PubMed] [Google Scholar]
- 9.Li C, Iida M, Dunn EF, Wheeler DL. Dasatinib blocks cetuximab- and radiation-induced nuclear translocation of the epidermal growth factor receptor in head and neck squamous cell carcinoma. Radiother Oncol. 2010;97:330–337. doi: 10.1016/j.radonc.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meyn RE, Munshi A, Haymach JV, Milas L, Ang KK. Receptor signaling as a regulatory mechanism of DNA repair. Radiother Oncol. 2009;92:316–322. doi: 10.1016/j.radonc.2009.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Santiago A, Eicheler W, Bussink J, et al. Effect of cetuximab and fractionated irradiation on tumour micro-environment. Radiother Oncol. 2010;97:322–329. doi: 10.1016/j.radonc.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 12.Theys J, Jutten B, Dubois L, et al. The deletion mutant EGFRvIII significantly contributes to stress resistance typical for the tumour microenvironment. Radiother Oncol. 2009;92:399–404. doi: 10.1016/j.radonc.2009.06.017. [DOI] [PubMed] [Google Scholar]
- 13.Yang ES, Nowsheen S, Wang T, Thotala DK, Xia F. Glycogen synthase kinase 3{beta} inhibition enhances repair of DNA double-strand breaks in irradiated hippocampal neurons. Neuro Oncol. 2011 doi: 10.1093/neuonc/nor016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang ES, Wang H, Jiang G, et al. Lithium-mediated protection of hippocampal cells involves enhancement of DNA-PKcs dependent repair in mice. The Journal of Clinical Investigation. 2009;119:1124–1135. doi: 10.1172/JCI34051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang H, Yang ES, Jiang J, Nowsheen S, Xia F. DNA Damage Induced Cytotoxicity Is Dissociated from BRCA1's DNA Repair Function but Is Dependent on Its Cytosolic Accumulation. Cancer Research. 2010;70:6258–6267. doi: 10.1158/0008-5472.CAN-09-4713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baker SR. An in vivo model for squamous cell carcinoma of the head and neck. Laryngoscope. 1985;95:43–56. doi: 10.1288/00005537-198501000-00012. [DOI] [PubMed] [Google Scholar]
- 17.Bonner JA, Raisch KP, Trummell HQ, et al. Enhanced Apoptosis With Combination C225/Radiation Treatment Serves as the Impetus for Clinical Investigation in Head and Neck Cancers. Journal of Clinical Oncology. 2000;18:47–53. [PubMed] [Google Scholar]
- 18.Brenner JC, Graham MP, Kumar B, et al. Genotyping of 73 UM-SCC head and neck squamous cell carcinoma cell lines. Head & Neck. 2010;32:417–426. doi: 10.1002/hed.21198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rangan SR. A new human cell line (FaDu) from a hypopharyngeal carcinoma. Cancer. 1972;29:117–121. doi: 10.1002/1097-0142(197201)29:1<117::aid-cncr2820290119>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 20.O'Shaughnessy J, Osborne C, Pippen JE, et al. Iniparib plus chemotherapy in metastatic triple-negative breast cancer. N Engl J Med. 2011;364:205–214. doi: 10.1056/NEJMoa1011418. [DOI] [PubMed] [Google Scholar]
- 21.Kummar S, Kinders R, Gutierrez ME, et al. Phase 0 Clinical Trial of the Poly (ADP-Ribose) Polymerase Inhibitor ABT-888 in Patients With Advanced Malignancies. Journal of Clinical Oncology. 2009;27:2705–2711. doi: 10.1200/JCO.2008.19.7681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bonner WM, Redon CE, Dickey JS, et al. [gamma]H2AX and cancer. Nat Rev Cancer. 2008;8:957–967. doi: 10.1038/nrc2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mitchell J, Smith GCM, Curtin NJ. Poly(ADP-Ribose) Polymerase-1 and DNA-Dependent Protein Kinase Have Equivalent Roles in Double Strand Break Repair Following Ionizing Radiation. International journal of radiation oncology, biology, physics. 2009;75:1520–1527. doi: 10.1016/j.ijrobp.2009.07.1722. [DOI] [PubMed] [Google Scholar]
- 24.Carrozza MJ, Stefanick DF, Horton JK, Kedar PS, Wilson SH. PARP inhibition during alkylation-induced genotoxic stress signals a cell cycle checkpoint response mediated by ATM. DNA Repair (Amst) 2009;8:1264–1272. doi: 10.1016/j.dnarep.2009.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Paddock MN, Bauman AT, Higdon R, Kolker E, Takeda S, Scharenberg AM. Competition between PARP-1 and Ku70 control the decision between high-fidelity and mutagenic DNA repair. DNA Repair (Amst) 2011;10:338–343. doi: 10.1016/j.dnarep.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chan DW, Chen BP, Prithivirajsingh S, et al. Autophosphorylation of the DNA-dependent protein kinase catalytic subunit is required for rejoining of DNA double-strand breaks. Genes Dev. 2002;16:2333–2338. doi: 10.1101/gad.1015202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lieber MR, Ma Y, Pannicke U, Schwarz K. Mechanism and regulation of human non-homologous DNA end-joining. Nat Rev Mol Cell Biol. 2003;4:712–720. doi: 10.1038/nrm1202. [DOI] [PubMed] [Google Scholar]
- 28.Dittmann K, Mayer C, Fehrenbacher B, et al. Radiation-induced Epidermal Growth Factor Receptor Nuclear Import Is Linked to Activation of DNA-dependent Protein Kinase. J. Biol. Chem. 2005;280:31182–31189. doi: 10.1074/jbc.M506591200. [DOI] [PubMed] [Google Scholar]
- 29.Dittmann K, Mayer C, Fehrenbacher B, Schaller M, Kehlbach R, Rodemann HP. Nuclear EGFR shuttling induced by ionizing radiation is regulated by phosphorylation at residue Thr654. FEBS Lett. 2010;584:3878–3884. doi: 10.1016/j.febslet.2010.08.005. [DOI] [PubMed] [Google Scholar]
- 30.Dittmann K, Mayer C, Kehlbach R, Rodemann HP. Radiation-induced caveolin-1 associated EGFR internalization is linked with nuclear EGFR transport and activation of DNA-PK. Mol Cancer. 2008;7:69. doi: 10.1186/1476-4598-7-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wanner G, Mayer C, Kehlbach R, Rodemann HP, Dittmann K. Activation of protein kinase Cepsilon stimulates DNA-repair via epidermal growth factor receptor nuclear accumulation. Radiother Oncol. 2008;86:383–390. doi: 10.1016/j.radonc.2007.10.041. [DOI] [PubMed] [Google Scholar]
- 32.Baumann M, Krause M. Targeting the epidermal growth factor receptor in radiotherapy: radiobiological mechanisms, preclinical and clinical results. Radiother Oncol. 2004;72:257–266. doi: 10.1016/j.radonc.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 33.Skvortsova I, Skvortsov S, Raju U, et al. Epithelial-to-mesenchymal transition and c-myc expression are the determinants of cetuximab-induced enhancement of squamous cell carcinoma radioresponse. Radiother Oncol. 2010;96:108–115. doi: 10.1016/j.radonc.2010.04.017. [DOI] [PubMed] [Google Scholar]
- 34.Zhang N, Erjala K, Kulmala J, et al. Concurrent cetuximab, cisplatin, and radiation for squamous cell carcinoma of the head and neck in vitro. Radiother Oncol. 2009;92:388–392. doi: 10.1016/j.radonc.2009.04.019. [DOI] [PubMed] [Google Scholar]
- 35.Hegan DC, Lu Y, Stachelek GC, Crosby ME, Bindra RS, Glazer PM. Inhibition of poly(ADP-ribose) polymerase down-regulates BRCA1 and RAD51 in a pathway mediated by E2F4 and p130. Proceedings of the National Academy of Sciences. 2010;107:2201–2206. doi: 10.1073/pnas.0904783107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Redon CE, Nakamura AJ, Zhang Y-W, et al. Histone H2AX and Poly(ADP-Ribose) as Clinical Pharmacodynamic Biomarkers. Clinical Cancer Research. 2010;16:4532–4542. doi: 10.1158/1078-0432.CCR-10-0523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bryant HE, Helleday T. Inhibition of poly (ADP-ribose) polymerase activates ATM which is required for subsequent homologous recombination repair. Nucleic Acids Research. 2006;34:1685–1691. doi: 10.1093/nar/gkl108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.D'Amours D, Desnoyers S, D'Silva I, Poirier GG. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem. J. 1999;342:249–268. [PMC free article] [PubMed] [Google Scholar]
- 39.Schreiber V, Dantzer Fo, Ame J-C, de Murcia G. Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol. 2006;7:517–528. doi: 10.1038/nrm1963. [DOI] [PubMed] [Google Scholar]
- 40.Rodemann HP, Dittmann K, Toulany M. Radiation-induced EGFR-signaling and control of DNA-damage repair. International Journal of Radiation Biology. 2007;83:781–791. doi: 10.1080/09553000701769970. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.