

Abstract
Febrile seizures, or febrile convulsions (FEB), represent the most common form of childhood seizures and are believed to be influenced by variations in several susceptibility genes. Most of the associated loci, however, remain ‘orphan’, i.e. the susceptibility genes they contain still remain to be identified. Further orphan loci have been mapped for a related disorder, genetic (generalized) epilepsy with febrile seizures plus (GEFS+).
We show that both spatially mapped and ‘traditional’ gene expression data from the human brain can be successfully employed to predict the most promising candidate genes for FEB and GEFS+, apply our prediction method to the remaining orphan loci and discuss the validity of the predictions. For several of the orphan FEB/GEFS+ loci we propose excellent, and not always obvious, candidates for mutation screening in order to aid in gaining a better understanding of the genetic origin of the susceptibility to seizures.
Introduction
Febrile seizures, or febrile convulsions (FEB), are acute symptomatic seizures that occur in response to fever and represent the most common form of childhood seizures, affecting worldwide between 2 and 14% of infants before five years of age (reviewed in [1]–[4]). Clinically, febrile seizures are often divided in a mostly benign ‘simple’ type (about 60–70% of incidences), consisting of single generalized tonic-clonic seizures of less than 10–15 minutes without focal neurological features, and a ‘complex’ type of prolonged seizures (15 minutes or more) with possible long-term consequences, having focal neurological features or recurrence within the same febrile illness [5]–[7]. FEB is a complex and heterogeneous disorder in which environmental factors play an important role, the most obvious being an immature brain (suggested by the age-specificity) and fever. However, both experimental data on animal models and known familial cases with Mendelian inheritance demonstrate the significant role that genetic factors may play in the etiology of the disease [2], [3]. Several loci have been associated with FEB predisposition and/or genetic (generalized) epilepsy with febrile seizures plus (GEFS+) [8], [9], often with an autosomal dominant mode of inheritance with incomplete penetrance, suggesting a possibly simultaneous involvement of multiple genes [1], [10]. However, so far only few susceptibility genes have been found [11].
Although FEB is considered to be a distinct syndrome with a generally very good prognosis, and not a true epileptic disease, some evidence points towards a higher risk of patients with a FEB history to develop epileptic disorders later in life [1]–[4], [7], [12]. While most prospective studies have found no evidence for this hypothesis, retrospective studies indicate that a high fraction of patients that require surgery for treatment-resistant temporal lobe epilepsy (TLE) have a history of FEB, and large cohort studies have linked a history of FEB to a greater than five-fold increase in epilepsy later in life, although the overall probability of the latter event remains low.
Moreover, mutations in the genes encoding for the voltage-gated sodium channel  -1,
-1,  -2 and
-2 and  -9 subunits ( SCN1A, SCN2A and SCN9A) and the GABA
-9 subunits ( SCN1A, SCN2A and SCN9A) and the GABA receptor
receptor  -2 subunit gene ( GABRG2)–four out of five known FEB susceptibility genes according to the OMIM database (see Table 1)–have been identified in families with GEFS+. In many of the reported cases of familial FEB, at least some of the affected members developed also epileptic seizures [1], suggesting that many FEB loci actually represent genetic loci for GEFS+ [11]. Only the FEB2, FEB5 and FEB8 loci have been reported for pure febrile convulsions [13]–[15] (although the gene for FEB8, GABRG2, has also been found mutated in GEFS+ [16]). This may, of course, reflect only the appearance of febrile seizures in GEFS+, as a continuum between febrile and afebrile seizures is typical of GEFS+ [1]. On the other hand, it may also hint at a more profound relation between FEB and true epileptic disorders.
-2 subunit gene ( GABRG2)–four out of five known FEB susceptibility genes according to the OMIM database (see Table 1)–have been identified in families with GEFS+. In many of the reported cases of familial FEB, at least some of the affected members developed also epileptic seizures [1], suggesting that many FEB loci actually represent genetic loci for GEFS+ [11]. Only the FEB2, FEB5 and FEB8 loci have been reported for pure febrile convulsions [13]–[15] (although the gene for FEB8, GABRG2, has also been found mutated in GEFS+ [16]). This may, of course, reflect only the appearance of febrile seizures in GEFS+, as a continuum between febrile and afebrile seizures is typical of GEFS+ [1]. On the other hand, it may also hint at a more profound relation between FEB and true epileptic disorders.
Table 1. Genes known to be involved in FEB and/or GEFS+.
| Gene | Entrez ID | Phenotype(s) | OMIM ID(s) | Locus | References |
| GABRD | 2563 | GEFS+5 | #604233 | 1p36.3 | [77] |
| GABRG2 | 2566 | FEB8, GEFS+3 | #611277, #604233 | 5q34 | [13], [16], [78], [79] |
| SCN1A | 6323 | FEB3A, GEFS+2 | #604403, #604233 | 2q24.3 | [80]–[83] |
| SCN1B | 6324 | GEFS+1 | #604233 | 19q13.1 | [84] |
| SCN2A | 6326 | FEB/GEFS+ | (–)† | 2q24.3 | [85] |
| SCN9A | 6335 | FEB3B, GEFS+7 | #604403, #604233 | 2q24 | [86] |
Although the association of FEB with an increased risk of adult epileptic disorders such as TLE remains controversial [1], [17], febrile seizures can lead to epilepsy in some animal models, probably due to an imbalance of excitation and inhibition in the limbic system [4]. Due to the obvious differences between animal models and FEB in humans (see [2], [4] for a discussion), however, the interpretation of the obtained results requires some caution.
On the other hand, direct human studies of the mechanisms underlying FEB have obvious limitations. In this report, we try to aid in elucidating the genetic factors that influence FEB by applying computational disease gene prioritization to evaluate the ‘positional’ candidate genes in previously mapped ‘orphan’ FEB and GEFS+ loci (listed in Table 2), for which the involved susceptibility genes have not yet been determined. We first show that both spatially mapped and ‘traditional’ gene expression data from the human brain can be successfully used to prioritize candidate genes for Mendelian disorders related to the central nervous system (CNS), and that the results obtained for the two data sources are highly complementary. We then demonstrate that this works particularly well for ‘re-discovering’ known FEB/GEFS+ susceptibility genes, suggesting that a prioritization of the candidate genes included in orphan loci may actually yield biologically meaningful predictions. Finally, we apply the method to all orphan FEB and GEFS+ loci and present the most promising candidates. We hope that our predictions may eventually help in gaining a better understanding of the genes that influence the susceptibility to seizures.
Table 2. Orphan FEB and GEFS+ loci.
| Phenotype | OMIM ID | Locus | Coordinates | Candidates | Ref. | ||
| total | HBA | GEO | |||||
| FEB1 | %121210 | 8q13-q21 | chr8:66,932,638–72,988,347 | 31 | 19 | 28 | [29] |
| FEB2 | %602477 | 19p13.3 | chr19:3,075,844–7,583,551 | 112 | 76 | 105 | [14] |
| FEB4† | #604352 | 5q14-q15 | chr5:89,166,516–110,036,710 | 50 | 39 | 49 | [40] |
| FEB4 | #604352 | 5q14.3-q23.1 | chr5:88,728,695–122,125,234 | 93 | 70 | 88 | [28] |
| (Deprez et al.) ‡ | |||||||
| FEB5 | %609255 | 6q22-q24 | chr6:129,947,710–133,546,636 | 29 | 12 | 27 | [15] |
| FEB6 | %609253 | 18p11.2 | chr18:10,133,108–12,597,248 | 12 | 11 | 12 | [52] |
| FEB7 | %611515 | 21q22 | chr21:32,533,246–39,359,893 | 59 | 43 | 54 | [58] |
| FEB9 | %611634 | 3p24.2-p23 | chr3:24,924,673–33,687,557 | 32 | 26 | 29 | [53] |
| FEB10 | %612637 | 3q26.2-q26.33 | chr3:170,902,825–181,419,957 | 28 | 24 | 28 | [63] |
| GEFS+4 | %609800 | 2p24 | chr2:17,491,922–20,329,508 | 13 | 8 | 11 | [67] |
| GEFS+6 | %612279 | 8p23-p21 | chr8:6,940,747–15,649,945 | 56 | 20 | 36 | [72] |
| GEFS+6 | %612279 | 8p23.1-p21.3 | chr8:6,940,747–20,367,401 | 79 | 35 | 59 | [72] |
| (family 15173)* | |||||||
| GEFS+N | – | 6q16.3-q22.31 | chr6:101,352,134–119,553,944 | 89 | 60 | 85 | [73] |
Mapped FEB and GEFS+ loci for which the susceptibility or disease-causing gene has not yet been identified. Coordinates correspond to the regions flanked by genetic markers identified in the respective studies. The number of candidate genes in the mapped regions is indicated as a total number and the subset of genes for which HBA and GEO expression profiles are available, respectively (see Table S4 for the complete lists of candidates). Note: “GEFS+N” is not an official symbol (N = novel).
The implication of MASS1 in FEB4 [25] is controverse; Deprez et al [28] describe a case with an overlapping linkage interval (‡) without evidence for MASS1 mutations (see text).
*Baulac et al [72] describe also a family with a larger linkage interval; since it is not clear whether the genetic cause is the same, we consider this interval as a distinct orphan locus.
In our previous proof-of-concept study [18] we have shown that spatially mapped, i.e. 3D, high-resolution gene expression data covering the entire adult brain of the C57BL/6J mouse strain–the anatomically comprehensive Allen Mouse Brain Atlas (MBA) [19]–can be successfully exploited for the prioritization of positional candidates for both mouse phenotypes and human CNS-related hereditary disorders. This particular type of gene expression data is distinct from ‘traditional’ heterogeneous datasets containing samples from multiple tissues and cell types, due to its three-dimensional geometry and tissue-specificity. We have used this dataset to suggest some promising novel candidates for X-linked mental retardation [18].
However, the important differences between human FEB and animal models of the disorder [2], [4], and the fact that differences between mouse models and human brains can yield important insight also for other neurological disorders [20], suggest that an application of our method to the Allen Institute’s recently published Human Brain Atlas (HBA) [21] may be more appropriate, even if the HBA is still in a preliminary version.
In contrast to the more complete MBA, the HBA is not derived from in situ hybridization (ISH) images of many different brains, but instead uses a microarray technology on samples from a single individual. It does not yet provide expression data for all consecutive positions of the entire organ since the project is scheduled to be completed in 2013. Nonetheless, the current preliminary version already includes samples for most of the anatomical structures of the human brain.
Results
Evaluation for all CNS-related disorders
We evaluated the possibility to predict gene–phenotype associations based on 13,204 HBA-derived 3D gene expression profiles from the human brain using a large-scale leave-one-out cross validation (LOOCV; see Methods) over all CNS-related Mendelian disorders from the Online Mendelian Inheritance in Man (OMIM) database [22] (see Text S1) and compared the results to those previously obtained for the MBA [18].
As in our previous study, we prioritized a list of candidate genes taken from “artificial” orphan loci of various sizes centered around the true phenotype-causing gene 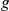 and verified how often
and verified how often 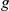 was ranked first (
was ranked first (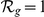 ), among the top ten (
), among the top ten (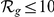 ), and among the upper 10% (
), and among the upper 10% (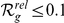 ) of the candidates. For this purpose, the 3D gene expression profiles of the candidates were compared to those of a set of disease-related reference genes (see Methods).
) of the candidates. For this purpose, the 3D gene expression profiles of the candidates were compared to those of a set of disease-related reference genes (see Methods).
As can be seen in Table S2, the procedure yielded for all sizes of artificial loci a significantly higher number of positive results than expected by chance, suggesting that spatial gene expression data from the HBA can be an important source of information to predict novel gene–phenotype associations. The HBA expression profiles perform similarly to those of the MBA, in some cases even better. This may seem obvious, given that we consider human disorders, but it is less clear when taking into account that the mouse 3D expression profiles cover the entire brain and have a far higher-resolution than the preliminary human data and that the two brain atlases are based on completely different technologies (ISH vs. microarray).
Better results are obtained when taking the known molecular basis of the disorders as reference (simulating phenotypes with partially known molecular basis), instead of relying on reference genes from similar OMIM phenotypes (to simulate phenotypes with unknown molecular basis; see Text S1). Nonetheless, the results obtained when simulating an unknown molecular basis are highly significant and suggest that the method can be used also for phenotypes to which so far no disease genes have been associated.
Complementarity of HBA and GEO data
Since the HBA is based on microarray technology, in contrast to the MBA that uses ISH, we asked how the disease gene prioritization method performs using ‘traditional’ microarray data from multiple individuals. For this purpose we compiled a dataset of 453 normal brain samples for 19,946 genes from the Gene Expression Omnibus (GEO) [23].
The GEO dataset performs better than the HBA data (see Table S2). This is likely due to the greater number of genes for which expression profiles are available. Nonetheless, given the different nature of the two datasets the positive predictions derived from both have only a limited overlap (no overlap for 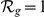 and less than 30% for both
and less than 30% for both 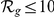 and
and 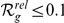 ; see Table S3). This suggests that the 3D gene expression data–spatially distributed samples from a single individual–provides information that is complementary to that provided by ‘traditional’ expression data–arbitrarily distributed samples from many individuals. Also the fact that different microarray platforms have been used for the two datasets is likely to play a role [24].
; see Table S3). This suggests that the 3D gene expression data–spatially distributed samples from a single individual–provides information that is complementary to that provided by ‘traditional’ expression data–arbitrarily distributed samples from many individuals. Also the fact that different microarray platforms have been used for the two datasets is likely to play a role [24].
Consequently, some cases can be successfully predicted when relying on spatial expression information while others using ‘traditional’ expression data. We therefore suggest that while the best candidates are those derived from an intersection of the prioritizations from the two data sources, those candidates obtained from only one of the datasets should still be taken into consideration. Although the GEO dataset performs generally better, it cannot replace the HBA predictions.
Evaluation for FEB and GEFS+
The effectiveness of our approach for CNS-related disease phenotypes in general does not necessarily imply that it can be successfully used for FEB and GEFS+ in particular. Febrile seizures are age-dependent and occur in immature brains of infants and young children. It is therefore imperative to specifically evaluate whether in this case expression data from the adult brain, that shows important differences in gene expression patterns, can aid in prioritizing candidate genes.
In order to verify the performance of our approach for the specific case of FEB and GEFS+, we performed an LOOCV taking into account only the reference genes listed in Table 1, found to be associated with FEB and GEFS+ in OMIM and/or the literature. From this list, we excluded MASS1 ( GPR98) that was suggested by Nakayama et al [25] to be the disease gene for the FEB4 locus because the gene had been reported to be involved in audiogenic seizures in the Frings mouse strain [26], [27] and they found a nonsense mutation in a small family with febrile and afebrile seizures. However, the cosegregation of the mutation with the seizure phenotype was not unambiguous due to the small family size. Moreover, another linkage study has mapped FEB/GEFS+ to an overlapping (although larger) locus, but did not find disease-causing mutations in the exons of MASS1 (or VLGR1 that includes most MASS1 exons), suggesting that another gene may be underlying the FEB4 locus [28]. Due to this uncertainty, we chose to consider this linkage interval as an additional orphan locus (see Table 2) and MASS1 as a candidate, rather than a disease gene.
As for the large-scale LOOCV performed over all CNS-related disorders, we constructed artificial loci of various sizes centered around each of the FEB or GEFS+ genes and took the remaining five genes as reference genes for the prioritization.
Table 3 demonstrates that in most of the prioritizations the true FEB/GEFS+ gene is ranked among the top ten and/or the best 10% of the candidates from the artificial locus. Most important, four of the six genes ( GABRD, GABRG2, SCN1B, and SCN2A) rank in at least one of the two datasets among the first three candidates for artificial loci of similar sizes (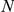 = 50) as the orphan loci (between 12 and 112 genes, see Table 2). Even for far larger loci of up to 401 candidates (
= 50) as the orphan loci (between 12 and 112 genes, see Table 2). Even for far larger loci of up to 401 candidates (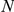 = 200) half of the disease genes have
= 200) half of the disease genes have 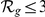 , suggesting that the method may be particularly effective for FEB and GEFS+.
, suggesting that the method may be particularly effective for FEB and GEFS+.
Table 3. Leave-one-out cross-validation of known FEB and GEFS+ genes.
Gene 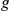
|
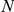
|
HBA | GEO | ||
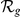
|
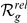
|
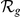
|
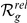
|
||
| GABRD | 50 |
3

|
0.0435 |
2

|
0.0217 |
| 100 |
5

|
0.0467 |
3

|
0.0210 | |
| 200 |
6

|
0.0390 |
3

|
0.0140 | |
| 400 |
10

|
0.0357 |
4

|
0.0101 | |
| GABRG2 | 50 |
3

|
0.0455 |
4

|
0.0421 |
| 100 |
4

|
0.0294 |
4

|
0.0214 | |
| 200 |
6

|
0.0287 |
4

|
0.0134 | |
| 400 |
9

|
0.0257 |
4

|
0.0082 | |
| SCN1A | 50 | 12 | 0.1644 |
4

|
0.0404 |
| 100 | 28 | 0.2137 |
6

|
0.0319 | |
| 200 | 42 | 0.1615 |
6

|
0.0164 | |
| 400 | 72 | 0.1426 |
9

|
0.0126 | |
| SCN1B | 50 |
3

|
0.0448 |
1

|
0.0105 |
| 100 |
8

|
0.0541 |
1

|
0.0054 | |
| 200 | 18 | 0.0629 |
3

|
0.0082 | |
| 400 | 39 | 0.0721 |
5

|
0.0069 | |
| SCN2A | 50 |
7

|
0.1000 |
2

|
0.0202 |
| 100 |
8

|
0.0611 |
3

|
0.0160 | |
| 200 |
10

|
0.0385 |
3

|
0.0082 | |
| 400 | 12 | 0.0237 |
3

|
0.0042 | |
| SCN9A | 50 | 8
|
0.1111 | 65 | 0.6566 |
| 100 | 12 | 0.0923 | 118 | 0.6277 | |
| 200 | 20 | 0.0769 | 230 | 0.6284 | |
| 400 | 27 | 0.0536 | 459 | 0.6411 | |
Absolute (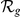 ) and relative (
) and relative (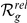 ) rankings of the known FEB and GEFS+ genes
) rankings of the known FEB and GEFS+ genes 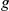 (see Table 1) for LOOCVs using different artificial locus sizes
(see Table 1) for LOOCVs using different artificial locus sizes 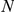 (up to 2
(up to 2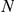 +1 genes). Results are shown for both gene expression datasets, HBA and GEO. Ranks among the best 10% (
+1 genes). Results are shown for both gene expression datasets, HBA and GEO. Ranks among the best 10% (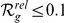 ) are evidenced by bold face font. Ranks among the top 10 (
) are evidenced by bold face font. Ranks among the top 10 (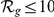 ) are additionally marked by a single star (
) are additionally marked by a single star ( ) and ranks among the top 3 (
) and ranks among the top 3 (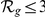 ) by three stars (
) by three stars ( ).
).
Although, as for the large-scale LOOCV, the GEO expression data generally performs better, in some cases the rankings obtained with the HBA data are better. Interestingly, this is the case for SCN9A that does not show a particularly strong relationship with the other reference genes, but whose involvement in FEB/GEFS+ is significantly better recalled when applying spatially mapped expression profiles.
Prediction for orphan FEB and GEFS+ loci
Given the good performance of our prioritization procedure, as demonstrated by the CNS-related and the GEFS+/FEB-specific LOOCVs, we processed all orphan disease loci (see Table 2) to find the most promising candidates for an involvement in FEB or GEFS+ susceptibility using only the known disease genes (Table 1) as reference genes.
Since we found the HBA and GEO expression data to be complementary for the purpose of disease gene prediction for CNS-related disorders, in Table 4 we report for all orphan loci the best ranking candidate for each of the two data sources, as well as the candidate that has the best average relative rank (average of 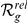 ) over both datasets. For the complete prioritized candidate lists that contain further interesting candidates, please see Table S4.
) over both datasets. For the complete prioritized candidate lists that contain further interesting candidates, please see Table S4.
Table 4. Prediction results for orphan FEB and GEFS+ loci.
| Phenotype / | Candidate | HBA | GEO | average | |||
| Orphan locus | Entrez ID | Symbol |
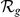
|
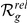
|
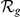
|
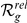
|
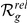
|
| FEB1 | 10565 | ARFGEF1 | 1 | 0.0526 | 19 | 0.6786 | 0.3656 |
| FEB1 | 80124 | VCPIP1 | 3 | 0.1579 | 1 | 0.0357 | 0.0968 |
| FEB2 | 56961 | SHD | 1 | 0.0132 | 8 | 0.0762 | 0.0447 |
| FEB2 | 60680 | BRUNOL5 | 3 | 0.0395 | 1 | 0.0095 | 0.0245 |
| FEB2 | 79187 | FSD1 | 2 | 0.0263 | 2 | 0.0190 | 0.0227 |
| FEB4 | 2745 | GLRX | 1 | 0.0256 | 10 | 0.2041 | 0.1149 |
| FEB4 | 5066 | PAM | 17 | 0.4359 | 1 | 0.0204 | 0.2282 |
| FEB4 | 1946 | EFNA5 | 4 | 0.1026 | 2 | 0.0408 | 0.0717 |
| FEB4 (Deprez et al.) | 9315 | C5orf13 | 1 | 0.0143 | 12 | 0.1364 | 0.0753 |
| FEB4 (Deprez et al.) | 5066 | PAM | 33 | 0.4714 | 1 | 0.0114 | 0.2414 |
| FEB4 (Deprez et al.) | 814 | CAMK4 | 4 | 0.0571 | 2 | 0.0227 | 0.0399 |
| FEB5 | 9465 | AKAP7 | 1 | 0.0833 | 7 | 0.2593 | 0.1713 |
| FEB5 | 114801 | TMEM200A | 3 | 0.2500 | 1 | 0.0370 | 0.1435 |
| FEB5 | 285735 | LOC285735 | - | - | 2 | 0.0741 | 0.0741 |
| FEB6 | 8774 | NAPG | 1 | 0.0909 | 1 | 0.0833 | 0.0871 |
| FEB7 | 3763 | KCNJ6 | 1 | 0.0233 | 2 | 0.0370 | 0.0301 |
| FEB7 | 7074 | TIAM1 | 20 | 0.4651 | 1 | 0.0185 | 0.2418 |
| FEB9 | 7342 | UBP1 | 1 | 0.0385 | 2 | 0.0690 | 0.0537 |
| FEB9 | 25827 | FBXL2 | 10 | 0.3846 | 1 | 0.0345 | 0.2095 |
| FEB10 | 5290 | PIK3CA | 1 | 0.0417 | 3 | 0.1071 | 0.0744 |
| FEB10 | 57552 | AADACL1 | 2 | 0.0833 | 1 | 0.0357 | 0.0595 |
| GEFS+4 | 7447 | VSNL1 | 1 | 0.1250 | 1 | 0.0909 | 0.1080 |
| GEFS+6 | 157627 | LOC157627 | 1 | 0.0500 | 1 | 0.0278 | 0.0389 |
| GEFS+6 (family 15173) | 286097 | EFHA2 | 1 | 0.0286 | 2 | 0.0339 | 0.0312 |
| GEFS+6 (family 15173) | 157627 | LOC157627 | 2 | 0.0571 | 1 | 0.0169 | 0.0370 |
| GEFS+N | 7259 | TSPYL1 | 1 | 0.0167 | 1 | 0.0118 | 0.0142 |
For each orphan FEB or GEFS+ locus (see Table 2) we report the best ranking candidate gene for HBA expression data, for GEO expression data and the candidate gene with the best average 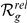 (indicated by bold face font). For the complete prioritized candidate lists, see Table S4.
(indicated by bold face font). For the complete prioritized candidate lists, see Table S4. 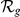 = absolute rank;
= absolute rank; 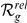 = relative rank (among candidates with expression profiles). Note: “GEFS+N” is not an official symbol (N = novel).
= relative rank (among candidates with expression profiles). Note: “GEFS+N” is not an official symbol (N = novel).
Strikingly, for most of the orphan loci, the results obtained with HBA and GEO data support each other. Indeed, in the large majority of cases, the first ranking candidate of one data source often obtained a high rank also for the other data source and vice versa, so that one of the two first ranking candidates (from HBA or GEO) remains the best candidate also on average. Only for four of the thirteen loci (FEB2, both FEB4 loci, and FEB5) the best average candidate did not rank first for HBA or GEO. Most important, for several of the loci (FEB6, GEFS+4, GEFS+6, and GEFS+N), both spatially mapped and traditional expression data agree upon the best candidate. This result is especially remarkable for the GEFS+N locus, being the third largest orphan locus with a total of 89 candidates (see Table 2).
Discussion
Overall, the good agreement between the prioritizations for FEB/GEFS+ obtained starting from HBA and GEO expression data increases the likelihood that the best candidates are functionally correlated to known FEB and GEFS+ genes and thus potentially disease relevant. Therefore, in the following sections we discuss particularly interesting candidates for the known orphan loci.
FEB1
The first FEB locus was mapped by Wallace et al [29] in a large Australian family spanning three generations. Results supported a hypothesis of autosomal dominant inheritance with incomplete penetrance, i.e. conditional upon the involvement of other modifier loci or environmental factors.
Wallace et al [29] have proposed corticotropin-releasing hormone (CRH) as one of the most interesting candidates of this locus. Indeed, in our prioritizations, CRH ranks 4th for the HBA expression data and 2nd for GEO, as well as 2nd with respect to the average relative rank over both datasets (see Table S4). CRH is known to be involved in an age-dependent manner in seizures in rat models [30], [31] and its activation is thought to be an important mechanism for generating developmentally regulated, triggered seizures [32]. Notably, some studies support a region-specific regulation of CRH gene expression by gamma-aminobutyric acid (GABA). Tran et al [33], for example, showed that the GABA degradation blocker gamma-vinyl-GABA (VGB) that is used for clinical seizure treatment causes a reduction of both CRH gene expression and secretion in the hypothalamus of 9-day-old rats. Finally, an excess in CRH has been hypothesized to underlie West syndrome, in which seizures during infancy play an important role [34], and CRH has been found to play a role in the increased susceptibility to seizures in shigellosis [35].
Taken together, CRH is a likely candidate for this locus, and a mutation that causes an increase in CRH levels or activity would be consistent with an autosomal dominant inheritance. Our two first ranking candidates ARFGEF1 and VCPIP1, instead, have so far not been implicated in seizures or epileptic disorders, but their coexpression with the reference genes in the human brain suggests a potential functional relationship.
FEB2
Johnson et al [14] mapped the FEB2 locus in a large multi-generational family with an autosomal dominant inheritance of febrile convulsions.
A potentially interesting candidate for the largest FEB/GEFS+ orphan locus is the ATCAY gene, which has been found mutated in human Cayman ataxia [36] and in a form of rat dystonia [37]. This gene ranks 5th for the HBA expression data and 3rd for GEO, as well as 4th with respect to the average relative rank over both datasets (see Table S4). ATCAY encodes for BNIP-H or Caytaxin, a brain-specific member of the BNIP-2 family that reduces the steady-state levels of glutamate by inhibiting kidney-type glutaminase (KGA) enzyme activity, affecting glutamate synthesis at synapses during neurotransmission [38]. Loss of function of ATCAY may lead to deregulated glutamatergic activation or even to glutamate excitotoxicity [38]. Hence, we propose that a mutation of the ATCAY gene may potentially lead to an imbalance between excitation and inhibition that increases the susceptibility to seizures.
The coexpression of BRUNOL5, or CELF5, with the reference genes is also intriguing. Indeed, this gene encodes for a brain-specific splicing factor (although its function is likely not limited to the regulation of alternative splicing) [39] that may in theory affect the splicing of other genes related to FEB/GEFS+.
FEB4
We have considered two different linkage intervals for FEB4, since Deprez et al [28] mapped a locus that overlapped the one first identified by Nakayama et al [40], who had suggested MASS1 ( GPR98) as the disease causing gene [25]. The evidence, however, was not conclusive and Deprez et al [28] did not find any disease-causing mutations in the exons of MASS1 (see above), so that we considered it as a regular candidate.
In our prioritization for the original FEB4 locus, GPR98 ranks 16th and 20th for HBA and GEO expression data, respectively. In the larger locus identified by Deprez et al [28] the ranks are even less suggestive (31st for HBA and 39th for GEO, see Table S4). Hence, at least with respect to expression profiles in the human brain, as compared to those of the reference genes, MASS1 does not seem to be a particularly strong candidate. Of course this result does not imply that this gene is not involved, as we do not expect all disease genes to be revealed by a coexpression-based approach. Its involvement in audiogenic seizures in the Frings mouse strain [26], [27] remains a strong argument for a possible implication in FEB/GEFS+.
However, among the other candidates found in these overlapping loci, we think that the peptidylglycine  -amidating monooxygenase ( PAM) gene is a particularly promising alternative. Indeed, although PAM is not a very strong candidate with the HBA dataset, it is the top scoring candidate in both intervals with the GEO dataset. The protein encoded by the PAM gene is a cuproenzyme essential for the synthesis of many neuropeptides [41]. Finally, and quite remarkably, it has recently been shown that mice heterozygous for a null mutation in the PAM gene are more susceptible to chemically induced seizures [42]. Taken together, these evidences point to PAM as a very strong candidate for FEB4.
-amidating monooxygenase ( PAM) gene is a particularly promising alternative. Indeed, although PAM is not a very strong candidate with the HBA dataset, it is the top scoring candidate in both intervals with the GEO dataset. The protein encoded by the PAM gene is a cuproenzyme essential for the synthesis of many neuropeptides [41]. Finally, and quite remarkably, it has recently been shown that mice heterozygous for a null mutation in the PAM gene are more susceptible to chemically induced seizures [42]. Taken together, these evidences point to PAM as a very strong candidate for FEB4.
But also GLRX and C5orf13 are potentially interesting candidates. Glutaredoxin (thioltransferase), or GLRX, regulates the activity of the copper-transporting P-type ATPases ATP7A and ATP7B [43] that are involved in Menkes disease (OMIM: #309400) and Wilson disease (OMIM: #277900), respectively. Since epilepsy is a major feature of Menkes disease [44], [45], and seizures occur at low frequency in patients affected by Wilson disease [46], [47], it is conceivable that reduced function of GLRX may lead to increased neuronal excitability. On the other hand, C5orf13 (also PTZ17 or P311) lies outside of the original FEB4 locus but might be a candidate for the larger locus reported by Deprez et al [28], since it was identified as differentially expressed after inducing seizures in mice [48]. However, no seizures were reported in C5orf13 knockout mice [49].
FEB5
Nabbout et al [15] suggested that the locus mapping to 6q22-q24 was the first locus identified for pure simple febrile seizures, the most frequent form of FEB. They excluded the implication of several candidate genes, such as syntaxin 7 ( STX7), A kinase anchor protein 7 ( AKAP7, also AKAP18), putative neurotransmitter receptor ( PNR), G protein receptor 58 ( GPR58) and G protein receptor 57 ( GPR57) by sequencing their coding exons and exon–intron boundaries.
According to our analysis, AKAP7 is one of the best candidates, ranking 1st for the HBA. AKAP7, that is expressed among other tissues in the cerebral cortex, targets the cAMP-dependent protein kinase (PKA) to the plasma membrane, permitting its functional coupling with L-type calcium channels [50]. These voltage-gated channels control a variety of neuronal functions that are implicated in epileptogenesis [51]. It is in principle possible that the affected family members studied by Nabbout et al [15] are carriers of a mutation that effects regulatory elements or other essential non-coding residues of AKAP7. Hence, we suggest not to exclude the possibility that a mutation in AKAP7 is the true cause for the FEB5 locus.
FEB6
The haplotype analysis performed by Nakayama et al [52] indicated a possible role for the IMPA2 gene, which is located in the FEB6 locus. Moreover, Nabbout et al [53] have found a locus on chromosome 18p to contain a modifier gene who's cosegregation with the FEB9 locus on 3p (see FEB9) is associated with more severe disorders like childhood absence epilepsy (CAE) and TLE, but is likely to also contribute to febrile seizures per se [53], suggesting that the modifier locus could actually be the FEB6 locus. Thus, they sequenced the IMPA2 gene as most likely candidate, but found no disease-causing mutations. In our analysis, IMPA2 has no significant rankings (5th out of 11 for HBA and 8th out of 12 for GEO expression data). However, neither the lack of mutations in [53] nor our negative result represent sufficient evidence to exclude this candidate.
The best candidate identified by our approach is the N-ethylmaleimide-sensitive factor attachment protein gamma ( NAPG) that ranks first with both the HBA and the GEO datasets. The encoded protein belongs to the SNAP family, a group of factors involved in membrane fusion events by allowing NSF proteins to target membranes. Accordingly, the NAPG protein has been shown to mediate platelet exocytosis and to control the membrane fusion events of this process [54]. Although the gene has never been studied in a neuronal context, genetic studies suggest that it could play an important role in neurons, because a possible involvement in bipolar disorders has been detected by several independent reports [55]–[57].
FEB7
The orphan FEB7 locus was mapped by Hedera et al [58] in a five-generation family with autosomal dominant febrile seizures. Of 13 individuals affected by FEB, three showed coexisting afebrile seizures. The authors sequenced all exons of four ion-channel genes–the potassium channel genes KCNE1, KCNE2 and KCNJ6 and the intracellular chloride channel gene CLIC6–but did not identify any disease-causing mutations. Three of these candidates ( KCNE1, KCNE2 and CLIC6) have no HBA expression profiles available, but rankings obtained with the GEO dataset are rather low (37th, 26th and 36th out of 54, respectively). KCNJ6, however, was ranked 1st for the HBA expression data and 2nd for GEO. As for AKAP7 and FEB5, a mutation in a regulatory element or in another essential non-coding region should not be excluded.
On the other hand, even the TIAM1 gene, that ranks first with the GEO dataset, could be an interesting candidate, because studies conducted in rodents have shown that the encoded protein plays a role in axonogenesis [59], in neuronal migration [60] and in the formation of dendritic spines [61].
FEB9
Nabbout et al [53] mapped the FEB9 locus in a large French family with febrile seizures but also CAE and TLE that were largely associated with an additional modifier locus on chromosome 18p (see FEB6). The authors have sequenced the coding exons, exon-intron boundaries and translation start sites of six potential candidates of the FEB9 locus without finding disease-causing mutations. Of these, the SCL4A7 gene encoding the sodium bicarbonate cotransporter NBC3 scored best in our prioritizations (3rd for HBA, 10th for GEO and 5th for its average relative rank).
The two candidates that rank first for HBA and GEO data, respectively, are difficult to link to FEB or GEFS+.
However, GPD1L, which ranks 5th for HBA, 3rd for GEO, and 3rd with respect to its average relative rank, encodes the glycerol phosphate dehydrogenase 1-like protein, an enzyme that may modify heart excitability by regulating the activity of the sodium channel SCN5A through PKC-dependent phosphorylation [62]. SCN5A itself was one of the candidates sequenced by Nabbout et al [53] (although it lies for about 5-Mb outside of the FEB9 locus), but had no causal mutations. It is in principle thinkable that GPD1L instead may be responsible for the phenotype through its capability to regulate the activity of SCN5A or other sodium channels in the brain, some of which have already been implicated in FEB/GEFS+ (see Table 1).
FEB10
Dai et al [63] mapped the FEB10 locus in a four-generation Chinese family with autosomal dominant febrile seizures and epilepsy. By mutational analysis, they have excluded the two potassium channel genes KCNMB2 and KCNMB3, none of which ranks among the top five in our analysis (average relative rank over HBA and GEO).
The strongest candidates identified by our analysis are AADACL1 and PIK3CA that rank very favorably with both datasets. The involvement of the first gene is not supported by other evidences, because AADACL1, also known as NCHE1 (neutral cholesterol ester hydrolase 1), has never been studied in a neuronal context. On the other hand, PIK3CA, the p110 alpha subunit of PI3K, which is known mostly for its role in growth factor-activated signaling and for its oncogenic potential, has recently been shown to negatively regulate neuronal excitability [64]. In addition, levels of phosphorylated (inactive) FOXO, a product of PI3K/Akt activity, are strongly modulated in experimental models of epilepsy and in the hippocampi of epileptic patients [65]. Finally, it has been recently observed that application of leptin, a known PI3K activator, inhibits seizures in rats in a PI3K-dependent manner [66]. Thus, it is conceivable that mutations leading to reduced PI3K activity might be a cause of increased neuronal excitability, possibly leading to febrile seizures.
GEFS+4
Audenaert et al [67] have suggested that KCNS3 (voltage gated potassium channel, subfamily S, member 3) is one of the most attractive candidate genes in the GEFS+4 locus, since it encodes a subunit of the voltage-gated potassium channel of the delayed rectifier type and is abundantly expressed in the brain [68]. We find this gene ranking 3rd for HBA and 6th for GEO (5th with respect to its average relative rank), but given the small locus size, these ranks cannot be deemed as very significant.
According to our prioritization, we propose VSNL1 (also VILIP-1) as a potential alternative candidate. Indeed, besides ranking first for both the HBA and the GEO dataset, this gene may be strongly involved in regulating neuronal excitability. VSNL1 is a member of the visinin/recoverin subfamily of neuronal calcium sensor proteins [69]. Interestingly, it has been recently found that VSNL1 may lead to the upregulation of functional  4
4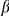 2 nicotinic acetylcholine receptors (nAChR) in hippocampal neurons [70]. This observation could be very important, because a dysfunction of the
2 nicotinic acetylcholine receptors (nAChR) in hippocampal neurons [70]. This observation could be very important, because a dysfunction of the  4
4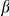 2 nAChR has been previously implicated in frontal lobe epilepsy [71]. Therefore, a mutation that should result in increased function of VSNL1 would be predicted to produce increased neuronal excitability.
2 nAChR has been previously implicated in frontal lobe epilepsy [71]. Therefore, a mutation that should result in increased function of VSNL1 would be predicted to produce increased neuronal excitability.
GEFS+6
The locus for GEFS+6 was mapped by Baulac et al [72] in two independent French families. More precisely, the largest family (family 15173) showed a linkage to a 13-Mb interval between markers D8S1706 and D8S258 (chr8:6,840,747-20,367,401), but since a second family showed a linkage to an overlapping region, the authors assumed that in both families the same gene was affected and hence narrowed to region down to a 7.3-Mb candidate interval (chr8:6,940,747-15,649,945).
However, it is in principle possible that the genetic causes are distinct for the two families and hence we have performed the prioritization for both intervals. The results of these prioritizations are in good agreement, since the top three genes (with respect to average relative rank) for the narrow locus– LOC157627, MTMR9, PRAGMIN–rank 2nd, 4th, and 3rd for family 15173, respectively (see Table S4). LOC157627 is the best ranking gene for GEO in both the narrowed and the larger locus. EFHA2, although lying for about 1.2-Mb outside of the narrowed GEFS+6 locus, has particularly good rankings for the locus of family 15173 (1st for HBA, 2nd for GEO, and 1st regarding the average relative rank) and might thus be taken as an additional interesting candidate.
Baulac et al [72] have sequenced the coding exons of six candidates– MTMR9, MTMR7, CTSB, SGCZ, SG223 ( PRAGMIN), and ATP6V1B–according to their expression in the brain and putative function, but all identified variants were either known polymorphisms or found in 100 matched French controls. Two of these candidates are suggested as interesting by our analysis: MTMR9 and PRAGMIN (see Table S4). Their rankings for the larger locus of family 15173 are less suggestive, but still good. It is in principle possible that mutations in their regulatory elements or other essential non-coding regions are causative for GEFS+. Therefore, we suggest not to exclude them from further studies.
GEFS+N
Not having found an official denomination, we called this locus “GEFS+N” (where N stands for ‘novel’). The locus contains no known genes associated with ion channels or neurotransmitter receptors, therefore Poduri et al [73] suggest that the identification of the responsible gene may lead to novel insight into the mechanisms of febrile seizures and inherited epilepsy. This makes our prediction results for this locus particularly interesting, but also more difficult to interpret, since a coexpression with known disease genes (reference genes) is more likely if a candidate is involved in a similar function.
Of the 16 candidates sequenced and excluded by Poduri et al [73], KPNA5 obtained the highest average relative rank in our prioritization (9th for HBA, 11th for GEO, 7th with respect to the average relative rank).
The TSPYL1 and TSPYL4 genes come out as outstanding candidates from our analysis. Indeed, they are found as the first and second ranking genes, respectively, for both the HBA and GEO dataset; a very strong result if considering that the locus contains a total of 89 candidates. The two genes are closely related members of the Nucleosome Assembly Protein (NAP) family. Both genes have been investigated as candidates for a genetic syndrome characterized by sudden infant death from cardiac and respiratory arrest, associated with dysgenesis of the testes, and null mutations have been found in TSPYL1 [74]. However, the molecular mechanism leading to this phenotype is not understood. Therefore, we think that it would be very interesting to evaluate both genes for mutations in GEFS+N. Moreover, it could be worth addressing whether increased or decreased function of these genes may affect the electrical behavior of excitable cells, such as neurons and cardiomyocytes. These studies could contribute to elucidating the mechanisms underlying both febrile seizures and sudden death syndrome.
Conclusions
In this study, we have shown that the preliminary Human Brain Atlas (HBA), although not yet complete, is already a powerful tool for disease gene prediction and provides results that are complementary to those obtained from a traditional microarray dataset.
Despite the fact that the HBA and GEO gene expression data represent mostly the adult brain and not the immature brain, an application to FEB/GEFS+ seemed particularly promising, given that, in an LOOCV with artificial loci of approximately the same size as the largest FEB or GEFS+ orphan loci, four out of six known disease genes ranked among the first three candidates for at least one of the two human brain gene expression atlases. This allowed us to propose several strong candidates for the true orphan loci and to discuss their possible involvement in FEB/GEFS+. Several of them would be excellent candidates for mutation screenings, however also high ranking candidates that are less obviously related to neuronal excitability should not be excluded from further analysis. In fact, the purpose and strength of computational approaches like the one presented here is to potentially uncover so far unknown and non obvious functional relationships.
Methods
Spatial human brain gene-expression data
We downloaded the spatial gene-expression data of the Human Brain Atlas (HBA) from [21] on 20 July 2010, but used only the most recent experiment for each tissue ID, resulting in a total of 798 tissue samples. After normalization with the “Agi4x44PreProcess” Bioconductor R package, we mapped the probesets of the custom Agilent 8x60 Whole Human Genome array (that is derived from the standard Agilent 4x44 Whole Human Genome array) to Entrez gene IDs via the RefSeq RNA nucleotide accessions reported in the downloaded data files. We considered only unique mappings to Entrez IDs and averaged expression profiles of genes with multiple probesets, obtaining expression profiles for a total of 13,204 genes.
Traditional microarray expression data
For comparison with the spatially mapped gene expression data we compiled a microarray dataset from GEO [23], containing 453 samples of normal human brain tissue (excluding tumor samples or other diseases) performed on a standard Affymetrix HG-U133 Plus 2.0 chip. We chose the Affymetrix platform over the Agilent 4x44 because for the latter only four normal brain samples were available. A list of the GEO accessions of all samples can be found in Table S1. We normalized the dataset with the Affymetrix MAS5.0 algorithm and mapped probeset IDs to Entrez IDs using the official Affymetrix annotation (na30), but accepted only probesets with unique Entrez IDs (19,946 genes). For genes with multiple probesets, we used averaged expression profiles.
Candidate gene prioritization
The evaluation and prioritization of positional candidates was performed as in our previous study. The following is a brief summary of the procedure, for a more detailed description, please see [18].
First, a set of ‘reference genes’ is defined as the genes that are involved in the given phenotype or in similar phenotypes (see Text S1). Then, for each reference gene, all other genes are ranked in a genome-wide coexpression list according to their decreasing Pearson correlation coefficient with the reference gene.
The relative ranks of the positional candidates within these reference coexpression lists are determined (i.e. the ranks divided by the number of genes in the lists) and overall scores for the single candidates are determined as the product of their relative ranks.
Finally, positional candidates are sorted, i.e. prioritized, according to their increasing overall scores, since lower scores indicate a higher correlation with the reference genes and thus a higher probability of being functionally related to (and hence more likely involved in) the given phenotype.
Leave-one-out
We performed large-scale LOOCVs over all CNS-related Mendelian disorders for both the spatially mapped expression data from the HBA and the GEO dataset. For each known gene–disease association, we constructed an artificial locus comprising the disease gene itself and the 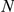 closest genes on both sides of the chromosome (for
closest genes on both sides of the chromosome (for 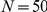 ,
, 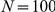 ,
, 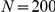 , and
, and 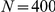 ), hence containing at the most
), hence containing at the most 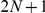 genes. Then, we prioritized the candidates from the artificial loci and verified the absolute rank (
genes. Then, we prioritized the candidates from the artificial loci and verified the absolute rank (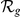 ) and relative rank (
) and relative rank (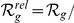 number of candidates) of the true disease gene
number of candidates) of the true disease gene 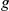 within the prioritized candidate list. For more details, please see Supporting Text S1.
within the prioritized candidate list. For more details, please see Supporting Text S1.
Candidate gene lists
To identify candidate genes (in terms of Entrez gene IDs) residing between or at the markers flanking the mapped linkage intervals, reported in the respective publications (see Table 2), we used the UCSC Genome Browser [75].
Supporting Information
List of GEO accessions of the ‘traditional’ microarray expression data set.
(PDF)
Results of the leave-one-out tests for human CNS-related OMIM phenotypes. Legend: 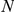 represents the size of the artificial loci having a maximum of 2
represents the size of the artificial loci having a maximum of 2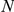 +1 genes. The average numbers of effective candidates with expression profiles and the numbers of evaluated
+1 genes. The average numbers of effective candidates with expression profiles and the numbers of evaluated 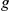 –
–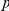 pairs are shown. The observed and expected numbers of
pairs are shown. The observed and expected numbers of 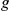 –
–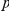 pairs, for which the true phenotype-causing gene
pairs, for which the true phenotype-causing gene 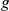 ranks first, among the top ten and within the best 10% of the prioritized list, is reported along with the corresponding
ranks first, among the top ten and within the best 10% of the prioritized list, is reported along with the corresponding 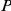 -values (one-tailed Fisher exact test). Significant
-values (one-tailed Fisher exact test). Significant 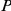 -values are highlighted (
-values are highlighted (
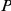
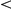 0.05;
0.05; 
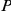
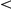 0.01;
0.01; 
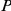
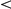 0.001). Reference genes (Ref.) were either taken from similar phenotypes (sim.), or from the OMIM disease phenotype itself (dis.).
0.001). Reference genes (Ref.) were either taken from similar phenotypes (sim.), or from the OMIM disease phenotype itself (dis.).  Results taken from [18]; artificial loci with
Results taken from [18]; artificial loci with 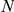 = 400 have not been evaluated in our previous study.
= 400 have not been evaluated in our previous study.
(PDF)
Overlap of correct leave-one-out predictions (over all CNS-related OMIM phenotypes) for reference genes from similar phenotypes. Legend: 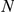 represents the size of the artificial loci, having a maximum of 2
represents the size of the artificial loci, having a maximum of 2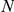 +1 genes. The number of predictions with the true phenotype-causing gene
+1 genes. The number of predictions with the true phenotype-causing gene 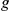 ranking first, among the top ten and within the best 10% of the prioritized list (see also Table S2, is reported for the Human Brain Atlas (HBA) and the GEO microarray dataset (GEO) along with their overlap. Parentheses indicate the corresponding fraction of the maximum possible overlap.
ranking first, among the top ten and within the best 10% of the prioritized list (see also Table S2, is reported for the Human Brain Atlas (HBA) and the GEO microarray dataset (GEO) along with their overlap. Parentheses indicate the corresponding fraction of the maximum possible overlap.
(PDF)
Candidate gene prioritizations/predictions for orphan FEB and GEFS+ loci. Legend: locus = orphan FEB/GEFS+ locus; candidate_id/ symbol = Entrez gene ID and symbol of the positional candidate; rank_HBA/ GEO = rank among the prioritized positional candidates for the HBA/GEO dataset; relRank_HBA/GEO = relative rank among the prioritized positional candidates for the HBA/GEO dataset (rank divided by number of candidates with expression data); avRelRank_HBA+GEO = average relative rank, i.e. 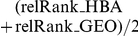 if the candidate has expression data in both datasets, otherwise the average relative rank is taken as either relRank_HBA or relRank_GEO if only one dataset provides expression data, or remains undefined if no expression data is available.
if the candidate has expression data in both datasets, otherwise the average relative rank is taken as either relRank_HBA or relRank_GEO if only one dataset provides expression data, or remains undefined if no expression data is available.
(XLS)
Supplementary Methods. Definition of CNS-related Mendelian disorders; similarity of human disease phenotypes; leave-one-out cross validation.
(PDF)
Acknowledgments
We thank Paolo Provero of the University of Torino for proof-reading and critical discussions, as well as Han Brunner and Martin Oti of the Nijmegen Centre for Molecular Life Sciences, The Netherlands, and Marc van Driel of the Netherlands Bioinformatics Centre for providing us with software and precious help regarding MimMiner [76].
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
Funding: Funding was provided by Regione Piemonte (to F.D.C.); the FIRB-Italbionet program of the Italian Ministry of Education, University and Research (to F.D.C.); the Fondazione San Paolo (to F.D.C.); and Progetto Neuroscienze (to F.D.C.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Baulac S, Gourfinkel-An I, Nabbout R, Huberfeld G, Serratosa J, et al. Fever, genes, and epilepsy. Lancet Neurol. 2004;3:421–430. doi: 10.1016/S1474-4422(04)00808-7. [DOI] [PubMed] [Google Scholar]
- 2.Koyama R, Matsuki N. Novel etiological and therapeutic strategies for neurodiseases: mechanisms and consequences of febrile seizures: lessons from animal models. J Pharmacol Sci. 2010;113:14–22. doi: 10.1254/jphs.09r19fm. [DOI] [PubMed] [Google Scholar]
- 3.Nakayama J, Arinami T. Molecular genetics of febrile seizures. Epilepsy Res. 2006;70S:S190–S198. doi: 10.1016/j.eplepsyres.2005.11.023. [DOI] [PubMed] [Google Scholar]
- 4.Reid AY, Galic MA, Teskey GC, Pittman QJ. Febrile seizures: current views and investigations. Can J Neurol Sci. 2009;36:679–686. doi: 10.1017/s0317167100008246. [DOI] [PubMed] [Google Scholar]
- 5.Annegers JF, Hauser WA, Shirts SB, Kurland LT. Factors prognostic of unprovoked seizures after febrile convulsions. N Engl J Med. 1987;316:493–498. doi: 10.1056/NEJM198702263160901. [DOI] [PubMed] [Google Scholar]
- 6.Berg AT, Shinnar S. Complex febrile seizures. Epilepsia. 1996;37:126–133. doi: 10.1111/j.1528-1157.1996.tb00003.x. [DOI] [PubMed] [Google Scholar]
- 7.Lewis DV. Febrile convulsions and mesial temporal sclerosis. Curr Opin Neurol. 1999;12:197–201. doi: 10.1097/00019052-199904000-00011. [DOI] [PubMed] [Google Scholar]
- 8.Scheffer IE, Berkovic SF. Generalized epilepsy with febrile seizures plus. A genetic disorder with heterogeneous clinical phenotypes. Brain. 1997;120:479–490. doi: 10.1093/brain/120.3.479. [DOI] [PubMed] [Google Scholar]
- 9.Scheffer IE, Berkovic SF. Generalized (genetic) epilepsy with febrile seizures plus. Engel JJ, Pedley T, editors. Epilepsy: a comprehensive textbook, Philadelphia: Lippincott, Williams & Wilkins. 2008;2nd edition:2553–2558. [Google Scholar]
- 10.Ito M, Yamakawa K, Sugawara T. Phenotypes and genotypes in epilepsy with febrile seizures plus. Epilepsy Res. 2006;70:S199–S205. doi: 10.1016/j.eplepsyres.2005.11.028. [DOI] [PubMed] [Google Scholar]
- 11.Nakayama J. Progress in searching for the febrile seizure susceptibility genes. Brain Dev. 2009;31:359–365. doi: 10.1016/j.braindev.2008.11.014. [DOI] [PubMed] [Google Scholar]
- 12.Hesdorffer DC, Hauser WA. Febrile seizures and the risk for epilepsy. In: Baram TZ, Shinnar S, editors. Febrile Seizures, San Diego: Academic Press; 2002. pp. 63–76. [Google Scholar]
- 13.Audenaert D, Schwartz E, Claeys KG, Claes L, Deprez L, et al. A novel GABRG2 mutation associated with febrile seizures. Neurology. 2006;67:687–690. doi: 10.1212/01.wnl.0000230145.73496.a2. [DOI] [PubMed] [Google Scholar]
- 14.Johnson EW, Dubovsky J, Rich SS, O'Donovan CA, Orr HT, et al. Evidence for a novel gene for familial febrile convulsions, FEB2, linked to chromosome 19p in an extended family from the Midwest. Hum Mol Genet. 1998;7:63–67. doi: 10.1093/hmg/7.1.63. [DOI] [PubMed] [Google Scholar]
- 15.Nabbout R, Prud'homme JF, Herman A, Feingold J, Brice A, et al. A locus for simple pure febrile seizures maps to chromosome 6q22-q24. Brain. 2002;125:2668–2680. doi: 10.1093/brain/awf281. [DOI] [PubMed] [Google Scholar]
- 16.Baulac S, Huberfeld G, Gourfinkel-An I, Mitropoulou G, Beranger A, et al. First genetic evidence of GABA(A) receptor dysfunction in epilepsy: a mutation in the gamma2-subunit gene. Nat Genet. 2001;28:46–48. doi: 10.1038/ng0501-46. [DOI] [PubMed] [Google Scholar]
- 17.Dubé CM, Brewster AL, Baram TZ. Febrile seizures: mechanisms and relationship to epilepsy. Brain & Development. 2009;31:366–371. doi: 10.1016/j.braindev.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Piro RM, Molineris I, Ala U, Provero P, Di Cunto F. Candidate gene prioritization based on spatially mapped gene expression: an application to XLMR. Bioinformatics. 2010;26:i618–i624. doi: 10.1093/bioinformatics/btq396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- 20.Miller JA, Horvath S, Geschwind DH. Divergence of human and mouse brain transcriptome highlights Alzheimer disease pathways. Proc Natl Acad Sci U S A. 2010;107:12698–12703. doi: 10.1073/pnas.0914257107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Human Brain Atlas. Available: http://human.brain-map.org/. Accessed 30 Jul 2011.
- 22.Amberger J, Bocchini CA, Scott AF, Hamosh A. McKusick's Online Mendelian Inheritance in Man (OMIM). Nucl Acids Res. 2009;37:D793–D796. doi: 10.1093/nar/gkn665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barrett T, Troup DB, Wilhite SE, Ledoux P, Rudnev D, et al. NCBI GEO: archive for high-throughput functional genomic data. Nucl Acids Res. 2009;37:D885–D890. doi: 10.1093/nar/gkn764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Russ J, Futschik ME. Comparison and consolidation of microarray data sets of human tissue expression. BMC Genomics. 2010;11:305. doi: 10.1186/1471-2164-11-305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakayama J, Fu YH, Clark AM, Nakahara S, Hamano K, et al. A nonsense mutation of the MASS1 gene in a family with febrile and afebrile seizures. Ann Neurol. 2002;52:654–657. doi: 10.1002/ana.10347. [DOI] [PubMed] [Google Scholar]
- 26.Skradski SL, White HS, Ptácek LJ. Genetic mapping of a locus (mass1) causing audiogenic seizures in mice. Genomics. 1998;49:188–192. doi: 10.1006/geno.1998.5229. [DOI] [PubMed] [Google Scholar]
- 27.Skradski SL, Clark AM, Jiang H, White HS, Fu YH, et al. A novel gene causing a mendelian audiogenic mouse epilepsy. Neuron. 2001;31:537–544. doi: 10.1016/s0896-6273(01)00397-x. [DOI] [PubMed] [Google Scholar]
- 28.Deprez L, Claes LR, Claeys KG. Genome-wide linkage of febrile seizures and epilepsy to the FEB4 locus at 5q14.3-q23.1 and no MASS1 mutation. Hum Genet. 2006;118:618–625. doi: 10.1007/s00439-005-0077-x. [DOI] [PubMed] [Google Scholar]
- 29.Wallace RH, Berkovic SF, Howell RA, Sutherland GR, Mulley JC, et al. Suggestion of a major gene for familial febrile convulsions mapping to 8q13-21. J Med Genet. 1996;33:308–312. doi: 10.1136/jmg.33.4.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baram TZ, Schultz L. Corticotropin-releasing hormone is a rapid and potent convulsant in the infant rat. Brain Res Dev Brain Res. 1991;61:97–101. doi: 10.1016/0165-3806(91)90118-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smith BN, Dudek EE. Age-related epileptogenic effects of corticotropin-releasing hormone in the isolated CA1 region of rat hippocampal slices. J Neurophysiol. 1994;72:2328–2333. doi: 10.1152/jn.1994.72.5.2328. [DOI] [PubMed] [Google Scholar]
- 32.Baram TZ, Hatalski CG. Neuropeptide-mediated excitability: a key triggering mechanism for seizure generation in the developing brain. Trends Neurosci. 1998;21:471–476. doi: 10.1016/s0166-2236(98)01275-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tran V, Hatalski CG, Yan XX, Baram TZ. Effects of blocking GABA degradation on corticotropin-releasing hormone gene expression in selected brain regions. Epilepsia. 1999;40:1190–1197. doi: 10.1111/j.1528-1157.1999.tb00847.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brunson KL, Eghbal-Ahmadi M, Baram TZ. How do the many etiologies of West syndrome lead to excitability and seizures? The corticotropin releasing hormone excess hypothesis. Brain Dev. 2001;23:533–538. doi: 10.1016/s0387-7604(01)00312-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yuhas Y, Weizman A, Chrousos GP, Ovadia H, Ashkenazi S. Involvement of the neuropeptide corticotropinreleasing hormone in an animal model of Shigella-related seizures. J Neuroimmunol. 2004;153:36–39. doi: 10.1016/j.jneuroim.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 36.Bomar JM, Benke PJ, Slattery EL, Puttagunta R, Taylor LP, et al. Mutations in a novel gene encoding a CRALTRIO domain cause human Cayman ataxia and ataxia/dystonia in the jittery mouse. Nat Genet. 2003;35:264–269. doi: 10.1038/ng1255. [DOI] [PubMed] [Google Scholar]
- 37.Xiao J, Ledoux MS. Caytaxin deficiency causes generalized dystonia in rats. Brain Res Mol Brain Res. 2005;141:181–192. doi: 10.1016/j.molbrainres.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 38.Buschdorf JP, Chew L, Zhang B, Cao Q, Liang FY, et al. Brain-specific BNIP-2-homology protein Caytaxin relocalises glutaminase to neurite terminals and reduces glutamate levels. J Cell Sci. 2006;119:3337–3350. doi: 10.1242/jcs.03061. [DOI] [PubMed] [Google Scholar]
- 39.Ladd AN, Charlet N, Cooper TA. The CELF family of RNA binding proteins is implicated in cell-specific and developmentally regulated alternative splicing. Mol Cell Biol. 2001;21:1285–1296. doi: 10.1128/MCB.21.4.1285-1296.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nakayama J, Hamano K, Iwasaki N, Nakahara S, Horigome Y, et al. Significant evidence for linkage of febrile seizures to chromosome 5q14–q15. Hum Mol Genet. 2000;9:87–91. doi: 10.1093/hmg/9.1.87. [DOI] [PubMed] [Google Scholar]
- 41.Eipper BA, Bloomquist BT, Husten EJ, Milgram SL, Mains RE. Peptidylglycine alpha-amidating monooxygenase and other processing enzymes in the neurointermediate pituitary. Ann N Y Acad Sci. 1993;680:147–160. doi: 10.1111/j.1749-6632.1993.tb19681.x. [DOI] [PubMed] [Google Scholar]
- 42.Bousquet-Moore D, Prohaska JR, Nillni EA, Czyzyk T, Wetsel WC, et al. Interactions of peptide amidation and copper: novel biomarkers and mechanisms of neural dysfunction. Neurobiol Dis. 2010;37:130–140. doi: 10.1016/j.nbd.2009.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Singleton WC, McInnes KT, Cater MA, Winnall WR, McKirdy R, et al. Role of glutaredoxin1 and glutathione in regulating the activity of the copper-transporting P-type ATPases, ATP7A and ATP7B. J Biol Chem. 2010;285:27111–27121. doi: 10.1074/jbc.M110.154468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bahi-Buisson N, Kaminska A, Nabbout R, Barnerias C, Desguerre I, et al. Epilepsy in Menkes disease: analysis of clinical stages. Epilepsia. 2006;47:380–386. doi: 10.1111/j.1528-1167.2006.00432.x. [DOI] [PubMed] [Google Scholar]
- 45.Kaler SG, Liew CJ, Donsante A, Hicks JD, Sato S, et al. Molecular correlates of epilepsy in early diagnosed and treated Menkes disease. J Inherit Metab Dis. 2010;33:583–589. doi: 10.1007/s10545-010-9118-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pestana Knight EM, Gilman S, Selwa L. Status epilepticus in Wilson's disease. Epileptic Disord. 2009;11:138–143. doi: 10.1684/epd.2009.0254. [DOI] [PubMed] [Google Scholar]
- 47.Prashanth LK, Sinha S, Taly AB, Mahadevan A, Vasudev MK, et al. Spectrum of epilepsy in Wilson's disease with electroencephalographic, MR imaging and pathological correlates. J Neurol Sci. 2010;291:44–51. doi: 10.1016/j.jns.2010.01.007. [DOI] [PubMed] [Google Scholar]
- 48.Kajiwara K, Nagawawa H, Shimizu-Nishikawa S, Ookura T, Kimura M, et al. Molecular characterization of seizure-related genes isolated by differential screening. Biochem Biophys Res Commun. 1996;219:795–799. doi: 10.1006/bbrc.1996.0313. [DOI] [PubMed] [Google Scholar]
- 49.Taylor GA, Rodriguiz RM, Greene RI, Daniell X, Henry SC, et al. Behavioral characterization of P311 knockout mice. Genes Brain Behav. 2008;7:786–795. doi: 10.1111/j.1601-183X.2008.00420.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fraser ID, Tavalin SJ, Lester LB, Langeberg, LK, Westphal AM, et al. A novel lipid-anchored A-kinase anchoring protein facilitates cAMP-responsive membrane events. EMBO J. 1998;17:2261–2272. doi: 10.1093/emboj/17.8.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Empson RM, Jefferys JG. Ca(2+) entry through L-type Ca(2+) channels helps terminate epileptiform activity by activation of a Ca(2+) dependent afterhyperpolarisation in hippocampal CA3. Neuroscience. 2001;102:297–306. doi: 10.1016/s0306-4522(00)00494-2. [DOI] [PubMed] [Google Scholar]
- 52.Nakayama J, Yamamoto N, Hamano K, Iwasaki N, Ohta M, et al. Linkage and association of febrile seizures to the IMPA2 gene on human chromosome 18. Neurology. 2004;63:1803–1807. doi: 10.1212/01.wnl.0000144499.34164.e0. [DOI] [PubMed] [Google Scholar]
- 53.Nabbout R, Baulac S, Desguerre I, Bahi-Buisson N, Chiron C, et al. New locus for febrile seizures with absence epilepsy on 3p and a possible modifier gene on 18p. Neurology. 2007;68:1374–1381. doi: 10.1212/01.wnl.0000260062.02829.e3. [DOI] [PubMed] [Google Scholar]
- 54.Lemons PP, Chen D, Bernstein AM, Bennett MK, Whiteheart SW, et al. Regulated secretion in platelets: identification of elements of the platelet exocytosis machinery. Blood. 1997;90:1490–1500. [PubMed] [Google Scholar]
- 55.Li X, Zhang J, Wang Y, Ji J, Yang F, et al. Association study on the NAPG gene and bipolar disorder in the Chinese Han population. Neurosci Lett. 2009;457:159–162. doi: 10.1016/j.neulet.2009.03.070. [DOI] [PubMed] [Google Scholar]
- 56.Weller AE, Dahl JP, Lohoff FW, Ferraro TN, Berrettini WH. Analysis of variations in the NAPG gene on chromosome 18p11 in bipolar disorder. Psychiatr Genet. 2006;16:3–8. doi: 10.1097/01.ypg.0000180678.88169.b0. [DOI] [PubMed] [Google Scholar]
- 57.Yosifova A, Mushiroda T, Stoianov D, Vazharova R, Dimova I, et al. Case-control association study of 65 candidate genes revealed a possible association of a SNP of HTR5A to be a factor susceptible to bipolar disease in Bulgarian population. J Affect Disord. 2009;117:87–97. doi: 10.1016/j.jad.2008.12.021. [DOI] [PubMed] [Google Scholar]
- 58.Hedera P, Ma S, Blair MA, Taylor KA, Hamati A, et al. Identification of a novel locus for febrile seizures and epilepsy on chromosome 21q22. Epilepsia. 2006;47:1622–1628. doi: 10.1111/j.1528-1167.2006.00637.x. [DOI] [PubMed] [Google Scholar]
- 59.Kunda P, Paglini G, Quiroga S, Kosik K, Caceres A. Evidence for the involvement of Tiam1 in axon formation. J Neurosci. 2001;21:2361–2372. doi: 10.1523/JNEUROSCI.21-07-02361.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kawauchi T, Chihama K, Nabeshima Y, Hoshino M. The in vivo roles of STEF/Tiam1, Rac1 and JNK in cortical neuronal migration. EMBO J. 2003;22:4190–4201. doi: 10.1093/emboj/cdg413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sasaki N, Kurisu J, Kengaku M. Sonic hedgehog signaling regulates actin cytoskeleton via Tiam1-Rac1 cascade during spine formation. Mol Cell Neurosci. 2010;45:335–344. doi: 10.1016/j.mcn.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 62.Valdivia CR, Ueda K, Ackerman MJ, Makielski JC. GPD1L links redox state to cardiac excitability by PKC-dependent phosphorylation of the sodium channel SCN5A. Am J Physiol Heart Circ Physiol. 2009;297:H1446–H1452. doi: 10.1152/ajpheart.00513.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dai XH, Chen WW, Wang X, Zhu QH, Li C, et al. A novel genetic locus for familial febrile seizures and epilepsy on chromosome 3q26.2–q26.33. Hum Genet. 2008;124:423–429. doi: 10.1007/s00439-008-0566-9. [DOI] [PubMed] [Google Scholar]
- 64.Howlett E, Lin CC, Lavery W, Stern M. A PI3-kinase-mediated negative feedback regulates neuronal excitability. PLoS Genet. 2008;4:e1000277. doi: 10.1371/journal.pgen.1000277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shinoda S, Schindler CK, Meller R, So NK, Araki T, et al. Bim regulation may determine hippocampal vulnerability after injurious seizures and in temporal lobe epilepsy. J Clin Invest. 2004;113:1059–1068. doi: 10.1172/JCI19971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xu L, Rensing N, Yang XF, Zhang HX, Thio LL, et al. Leptin inhibits 4-aminopyridine- and pentylenetetrazoleinduced seizures and AMPAR-mediated synaptic transmission in rodents. J Clin Invest. 2008;118:272–280. doi: 10.1172/JCI33009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Audenaert D, Claes L, Claeys KG, Deprez L, Van Dyck T, et al. A novel susceptibility locus at 2p24 for generalized epilepsy with febrile seizures plus. J Med Genet. 2005;42:947–952. doi: 10.1136/jmg.2005.031393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stocker M, Kerschensteiner D. Cloning and tissue distribution of two new potassium channel alpha-subunits from rat brain. Biochem Biophys Res Commun. 1998;248:927–934. doi: 10.1006/bbrc.1998.9072. [DOI] [PubMed] [Google Scholar]
- 69.Braunewell KH, Klein-Szanto AJ. Visinin-like proteins (VSNLs): interaction partners and emerging functions in signal transduction of a subfamily of neuronal Ca2+ -sensor proteins. Cell Tissue Res. 2009;335:301–316. doi: 10.1007/s00441-008-0716-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhao CJ, Noack C, Brackmann M, Gloveli T, Maelicke A, et al. Neuronal Ca2+ sensor VILIP-1 leads to the upregulation of functional alpha4beta2 nicotinic acetylcholine receptors in hippocampal neurons. Mol Cell Neurosci. 2009;40:280–292. doi: 10.1016/j.mcn.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Klaassen A, Glykys J, Maguire J, Labarca C, Mody I, et al. Seizures and enhanced cortical GABAergic inhibition in two mouse models of human autosomal dominant nocturnal frontal lobe epilepsy. Proc Natl Acad Sci U S A. 2006;103:19152–19157. doi: 10.1073/pnas.0608215103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Baulac S, Gourfinkel-An I, Couarch P, Depienne C, Kaminska A, et al. A novel locus for generalized epilepsy with febrile seizures plus in French families. Arch Neurol. 2008;65:943–951. doi: 10.1001/archneur.65.7.943. [DOI] [PubMed] [Google Scholar]
- 73.Poduri A, Wang Y, Gordon D, Barral-Rodriguez S, Barker-Cummings C, et al. Novel susceptibility locus at chromosome 6q16.3-22.31 in a family with GEFS+. Neurology. 2009;73:1264–1272. doi: 10.1212/WNL.0b013e3181bd10d3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Puffenberger EG, Hu-Lince D, Parod JM, Craig DW, Dobrin SE, et al. Mapping of sudden infant death with dysgenesis of the testes syndrome (SIDDT) by a SNP genome scan and identification of TSPYL loss of function. Proc Natl Acad Sci U S A. 2004;101:11689–11694. doi: 10.1073/pnas.0401194101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, et al. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.van Driel MA, Bruggeman J, Vriend G, Brunner HG, Leunissen JA. A text-mining analysis of the human phenome. Eur J Hum Genet. 2006;14:535–542. doi: 10.1038/sj.ejhg.5201585. [DOI] [PubMed] [Google Scholar]
- 77.Dibbens LM, Feng HJ, Richards MC, Harkin LA, Hodgson BL, et al. GABRD encoding a protein for extra- or perisynaptic GABAA receptors is a susceptibility locus for generalized epilepsies. Hum Mol Genet. 2004;13:1315–1319. doi: 10.1093/hmg/ddh146. [DOI] [PubMed] [Google Scholar]
- 78.Kananura C, Haug K, Sander T, Runge U, Gu W, et al. A splice-site mutation in GABRG2 associated with childhood absence epilepsy and febrile convulsions. Arch Neurol. 2002;59:1137–1141. doi: 10.1001/archneur.59.7.1137. [DOI] [PubMed] [Google Scholar]
- 79.Wallace RH, Marini C, Petrou S, Harkin LA, Bowser DN, et al. Mutant GABA(A) receptor gamma2-subunit in childhood absence epilepsy and febrile seizures. Nat Genet. 2001;28:49–52. doi: 10.1038/ng0501-49. [DOI] [PubMed] [Google Scholar]
- 80.Escayg A, MacDonald BT, Meisler MH, Baulac S, Huberfeld G, et al. Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nat Genet. 2000;24:343–345. doi: 10.1038/74159. [DOI] [PubMed] [Google Scholar]
- 81.Mantegazza M, Gambardella A, Rusconi R, Schiavon E, Annesi F, et al. Identification of an Nav1.1 sodium channel (SCN1A) loss-of-function mutation associated with familial simple febrile seizures. Proc Natl Acad Sci U S A. 2005;102:18177–18182. doi: 10.1073/pnas.0506818102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Schlachter K, Gruber-Sedlmayr U, Stogmann E, Lausecker M, Hotzy C, et al. A splice site variant in the sodium channel gene SCN1A confers risk of febrile seizures. Neurology. 2009;72:974–978. doi: 10.1212/01.wnl.0000344401.02915.00. [DOI] [PubMed] [Google Scholar]
- 83.Sijben AE, Sithinamsuwan P, Radhakrishnan A, Badawy RA, Dibbens L, et al. Does a SCN1A gene mutation confer earlier age of onset of febrile seizures in GEFS+? Epilepsia. 2009;50:953–956. doi: 10.1111/j.1528-1167.2009.02023.x. [DOI] [PubMed] [Google Scholar]
- 84.Wallace RH, Wang DW, Singh R, Scheffer IE, George AL, et al. Febrile seizures and generalized epilepsy associated with a mutation in the Na+-channel beta1 subunit gene SCN1B. Nat Genet. 1998;19:366–370. doi: 10.1038/1252. [DOI] [PubMed] [Google Scholar]
- 85.Sugawara T, Tsurubuchi Y, Agarwala KL, Ito M, Fukuma G, et al. A missense mutation of the Na+ channel alpha II subunit gene Na(v)1.2 in a patient with febrile and afebrile seizures causes channel dysfunction. Proc Natl Acad Sci U S A. 2001;98:6384–6389. doi: 10.1073/pnas.111065098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Singh NA, Pappas C, Dahle EJ, Claes LRF, Pruess TH, et al. A role of SCN9A in human epilepsies, as a cause of febrile seizures and as a potential modifier of Dravet syndrome. PLoS Genet. 2009;5:e1000649. doi: 10.1371/journal.pgen.1000649. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of GEO accessions of the ‘traditional’ microarray expression data set.
(PDF)
Results of the leave-one-out tests for human CNS-related OMIM phenotypes. Legend: represents the size of the artificial loci having a maximum of 2+1 genes. The average numbers of effective candidates with expression profiles and the numbers of evaluated – pairs are shown. The observed and expected numbers of – pairs, for which the true phenotype-causing gene ranks first, among the top ten and within the best 10% of the prioritized list, is reported along with the corresponding -values (one-tailed Fisher exact test). Significant -values are highlighted (
0.05;
0.01;
0.001). Reference genes (Ref.) were either taken from similar phenotypes (sim.), or from the OMIM disease phenotype itself (dis.). Results taken from [18]; artificial loci with = 400 have not been evaluated in our previous study.
(PDF)
Overlap of correct leave-one-out predictions (over all CNS-related OMIM phenotypes) for reference genes from similar phenotypes. Legend: represents the size of the artificial loci, having a maximum of 2+1 genes. The number of predictions with the true phenotype-causing gene ranking first, among the top ten and within the best 10% of the prioritized list (see also Table S2, is reported for the Human Brain Atlas (HBA) and the GEO microarray dataset (GEO) along with their overlap. Parentheses indicate the corresponding fraction of the maximum possible overlap.
(PDF)
Candidate gene prioritizations/predictions for orphan FEB and GEFS+ loci. Legend: locus = orphan FEB/GEFS+ locus; candidate_id/ symbol = Entrez gene ID and symbol of the positional candidate; rank_HBA/ GEO = rank among the prioritized positional candidates for the HBA/GEO dataset; relRank_HBA/GEO = relative rank among the prioritized positional candidates for the HBA/GEO dataset (rank divided by number of candidates with expression data); avRelRank_HBA+GEO = average relative rank, i.e. if the candidate has expression data in both datasets, otherwise the average relative rank is taken as either relRank_HBA or relRank_GEO if only one dataset provides expression data, or remains undefined if no expression data is available.
(XLS)
Supplementary Methods. Definition of CNS-related Mendelian disorders; similarity of human disease phenotypes; leave-one-out cross validation.
(PDF)