

The NFκB transcription factor family is associated with a broad spectrum of human cancers and inflammatory diseases. It was shown to be required for viability of leukemic cells and recently suggested as a predictor of relapse in T cell acute lymphoblastic leukemia (T-ALL) (1–3). The NOTCH1 signaling pathway, commonly activated in T-ALL (4), was demonstrated to enhance transcriptional function of NFκB via several mechanisms, including transcriptional activation and nuclear retention of the NFκB subunits, activation of upstream regulators and by a mechanism that prevents termination of NFκB signaling (1,2,5).
Combination therapy is the standard approach used to treat T-ALL (6). Thus inhibition of both NOTCH1 and NFκB was suggested to be used in combination with existing therapeutic agents (1,2). However, the mechanism of action of ionizing radiation and the majority of conventional chemotherapeutic drugs is based on activation of various types of stress such as DNA damage, mitotic perturbations or the unfolded protein response. Frequently, NFκB is activated as a part of the stress response and, depending on yet unknown factors, it either protects cells from the drug or mediates apoptosis (7,8). Here we report that T-ALL cells responded to NFκB inhibition in opposite ways depending on whether they were treated with a stress-inducing chemotherapeutic agent or not. Moreover, NOTCH1 enhanced NFκB pro-apoptotic function in the stressed cells. Our data is in agreement with clinical observations in which T-ALL cases carrying mutations activating NOTCH1 exhibit a better initial response to treatment (9), suggesting that mutant NOTCH1 contributes to chemotherapy-induced cell death. Thus we raise the question: Is it beneficial to inhibit the NOTCH-NFκB pathway in combination with conventional stress-inducing anti-T-ALL therapies?
As examples of stress-inducing therapeutic agents, we used etoposide, bortezomib and vincristine. Etoposide is a topoisomerase II inhibitor that activates a stress response to DNA double-strand breaks and is a key component of reinduction chemotherapies being evaluated to treat relapsed T-ALL patients (10). Bortezomib is a proteasome inhibitor activating the unfolded protein response, which is also being evaluated clinically for relapsed ALL patients (ClinicalTrials.gov NCT00873093). Vincristine is an inhibitor of chromosomal spindle formation that causes stress associated with mitotic perturbations and is a central component of most anti-T-ALL protocols (6). To inhibit NFκB we used BMS-345541 (BMS), a highly selective inhibitor of IκB kinase (IKK), the major upstream regulator of NFκB. BMS was previously reported to have anti-T-ALL activity (1). We performed the experiments with five cell lines derived from T-ALL patient samples: ALL-SIL, K3P, DND-41, HPB-ALL and LOUCY. We measured cell growth with alamarBlue, and apoptosis by annexin-V staining or detection of caspase-3 processing. We correlated the data with the expression levels of selected NFκB target genes and the levels of activation of the IKK complex (by detecting protein levels of the IKK substrate, IκBα, which is degraded following phosphorylation) (7).
We found that partial inhibition of the IKK complex counteracts the cytotoxic effects of etoposide, bortezomib and vincristine. Briefly, Fig. 1A shows growth response as a function of BMS concentration for four of the T-ALL-derived cell lines tested. We observed that BMS treatment alone caused inhibition of growth, consistent with the pro-survival role of NFκB in T-ALL reported by others (1,2). However, when the cells were pre-treated with etoposide, the response to BMS was altered; instead of inhibiting growth, BMS treatment caused an increase in the number of ALL-SIL, K3P and DND-41 cells or had no effect in the case of Loucy (Fig. 1A). HPB-ALL cells were resistant to etoposide under the conditions tested (data not shown). We obtained essentially similar results measuring apoptosis in ALL-SIL cells (Fig. 1B) and using 4 µM BAY 11-7082 (another inhibitor of IκBα phosphorylation) in DND-41 cells (Fig. 1C). Etoposide was previously reported to activate NFκB (11). Consistent with this, we observed that etoposide activated the IKK complex (Fig. 1D), induced RELA nuclear translocation (not shown) and upregulated the NFκB target genes FAS, BCL2L11 (BIM), BCL2L1 (BCLXL) and NFKBIA (Fig. 1E). Treatment with 5 µM BMS partially reversed these effects (Fig. 1D,E). In summary, our data indicated that etoposide-mediated IKK activation contributes to stress-induced T-ALL cell killing; thus combining IKK inhibitors with etoposide might not be therapeutically beneficial.
Figure 1.
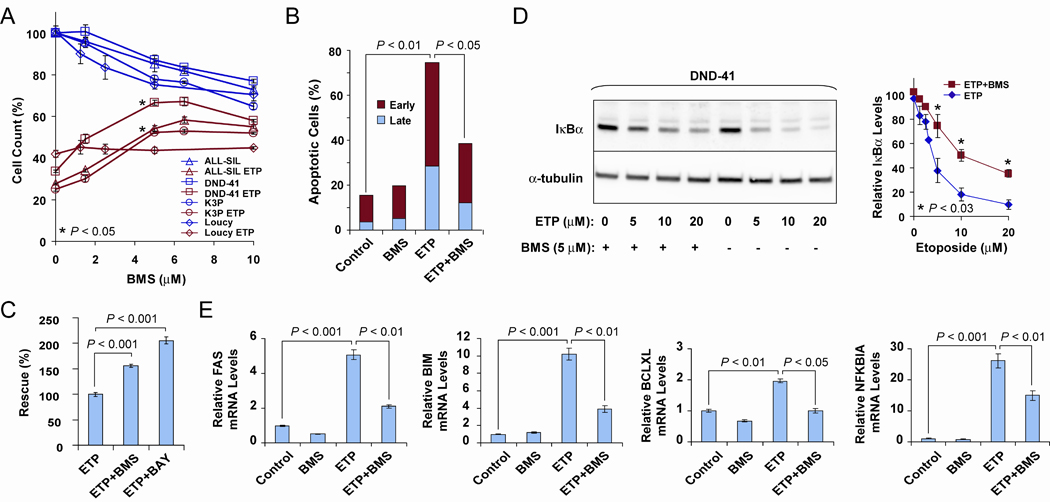
BMS counteracts etoposide effects on cell growth, IKK activation levels and NFκB target expression. A, AlamarBlue cell growth assay (25). ALL-SIL, DND-41, K3P and Loucy T-ALL cells, untreated or treated with 5 µM etoposide (ETP) and BMS at the indicated concentrations for 18 h. P values indicate significant differences in cell counts for etoposide-treated cells in the presence of 5 µM BMS (asterisks) versus etoposide alone. Measurements were performed in triplicate. Similar results were obtained for DND-41 cells (n = 5) and ALL-SIL cells (n = 2) using different times of incubation, order of etoposide and BMS addition and different sources of serum. B, Apoptosis assay (25). ALL-SIL cells were seeded at 2.5 × 105/ml and early and late apoptotic cells were measured by flow cytometry after staining with annexin-PE and 7-AAD. Conditions: Control, 0.06% dimethyl sulfoxide; ETP, 10 µM etoposide; BMS, 10 µM BMS; ETP+BMS, 10 µM etoposide plus 10 µM BMS. Treatment was for 24 h. A representative graph is shown for two biological replicates. Similar results were obtained for K3P cells. C, AlamarBlue cell growth assay. DND-41 cells were treated with 5 µM etoposide and with 8 µM BMS or 4 µM BAY 11-7082 (BAY) for 18 h, as indicated. D, IKK kinase assay based on Western blot detection of IκBα degradation in DND-41 cells after 6 h treatment as indicated: ETP, etoposide. (Left) Shown is a representative blot of three independent experiments. (Right) Quantitative analysis of IκBα levels demonstrating statistically significant partial inhibition of IκBα degradation for etoposide-treated cells in the presence of 5 µM BMS (asterisks) versus etoposide alone (n = 3). E, DND-41 cells were treated as described in D and expression of NFκB targets, FAS, BIM, BCLXL and NFKBIA, was determined by qRT-PCR (19). Experiments were performed in triplicate for two biological replicates of three cell lines, ALL-SIL, K3P, and DND-41, with similar results obtained. Student’s t test for all comparisons.
Since HPB-ALL was resistant to etoposide under the conditions tested, we examined the effects of treating these cells with bortezomib and vincristine. As an additional approach to inhibit NFκB function, we expressed a trans-dominant “super repressor” form of IκBα (IκB-SR) harboring two amino acid substitutions (S32A/S36A) which renders it resistant to phosphorylation and degradation, preventing a signal transfer from IKK to NFκB (12). Treatment with bortezomib and vincristine gave essentially similar results, consistent with activation of an IKK and NFκB pro-apoptotic function; BMS treatment and IκB-SR expression partially reversed the cytotoxic effects of both drugs (Fig. 2A). Interestingly, the effects of BMS and IκB-SR expression were cooperative, since HPB-ALL cells expressing IκB-SR that were also treated with BMS were the most resistant to bortezomib or vincristine (Fig. 2A). These findings suggest that the mechanism targeted by IKK inhibition may have an NFκB-independent component (7). Bortezomib treatment caused induction of the pro-apoptotic genes FAS and BIM and repression of pro-survival BCLXL; these transcriptional responses were attenuated by BMS treatment and IκB-SR expression (Fig. 2B). In support of this data, bortezomib was recently shown to activate the IKK-NFκB pathway in other human malignancies (13). While our observations indicate that etoposide action is mediated by IKK-NFκB transcriptional activation of apoptotic genes, other topoisomerase II poisons (such as doxorubicin) have been demonstrated to induce IKK-NFκB-dependent apoptosis via repression of BCLXL (11) similar to what we observed for bortezomib. Despite differences in the downstream events, IKK activation contributed to apoptosis induced by all of the above therapeutic agents tested; thus inhibition of the IKK pathway in T-ALL may not provide a therapeutic advantage when combined with these drugs.
Figure 2.
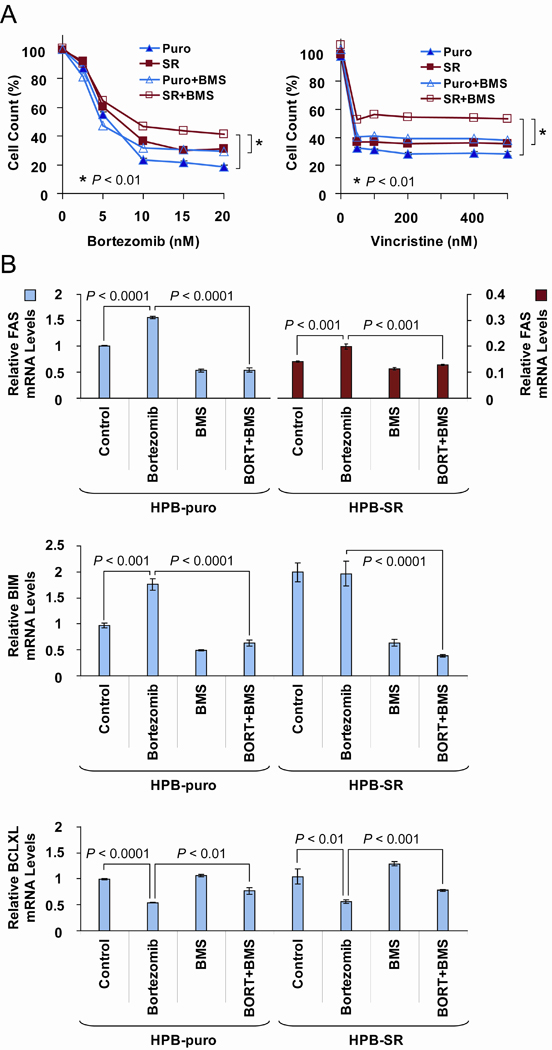
BMS counteracts bortezomib and vincristine effects on cell growth and NFκB target expression in HPB-ALL cells. HPB-SR cells (SR) express a trans-dominant “super repressor” of IκBα (IκB-SR); HPB-puro (Puro) is an empty vector-transduced control. A, AlamarBlue cell growth assay. Cells were treated with 5 µM BMS for 48 h in the presence of bortezomib (Left) or vincristine (Right). P values indicate significant differences in cell counts at the highest concentrations of the drugs (asterisks). Experiments were performed in triplicate; representative graphs are shown. B, Expression of BIM, FAS, and BCLXL was determined by qRT-PCR; same conditions as in A. BORT, bortezomib. Experiments were performed in triplicate for two biological replicates. Student’s t test for all comparisons.
To directly test the role of NOTCH1 in the NFκB pro-apoptotic response, we modulated NOTCH1 signaling by γ-secretase inhibition or by co-culturing T-ALL cells on OP9-DL1 monolayers presenting the NOTCH1 ligand Delta-like 1 (14). Briefly, we treated ALL-SIL, K3P and DND-41 cells with the γ-secretase inhibitor Compound E; after several days of treatment, we observed a decrease in etoposide-induced cytotoxicity concomitant with a reduced ability of BMS to counteract the cytotoxic effects (Fig. 3A). Thus our data indicated that IKK contribution to etoposide-induced T-ALL cell apoptosis is diminished by NOTCH1 inhibition. Conversely, we found that Delta-like 1-mediated activation of NOTCH signaling in OP9-DL1 co-cultures enhanced the response to etoposide; BMS or Compound E treatment partially protected the T-ALL cells from etoposide-induced death under these conditions (Fig. 3B).
Figure 3.
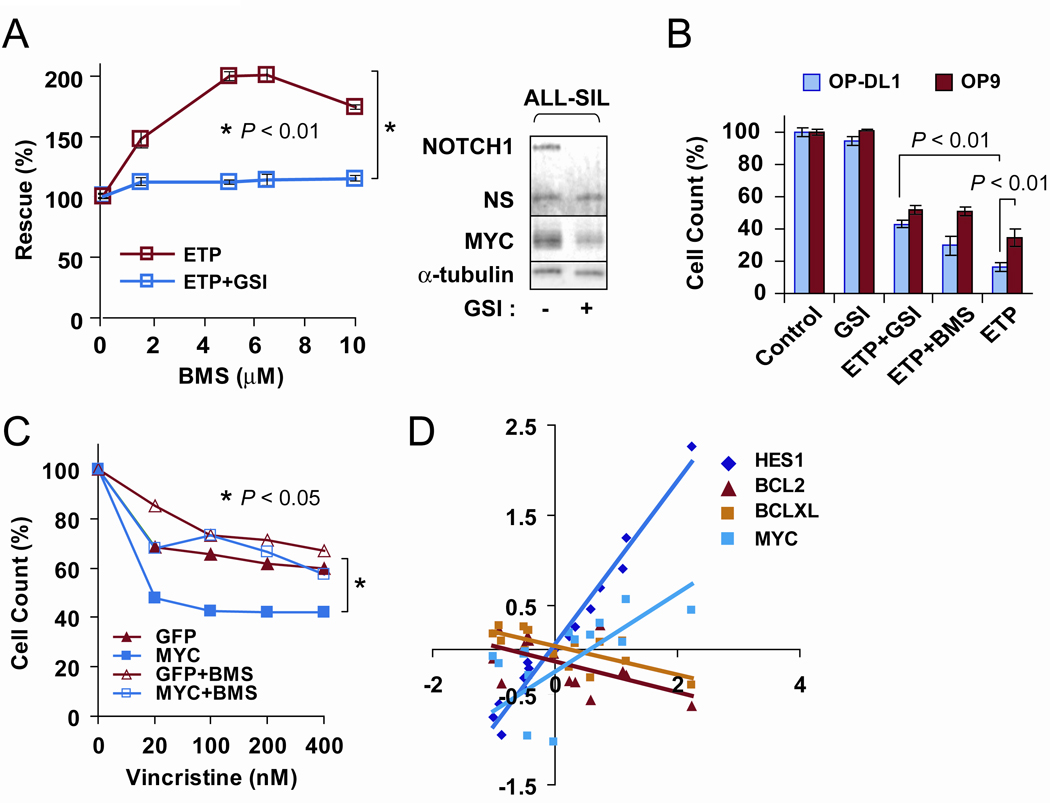
Role of the NOTCH1/MYC-NFκB pathway in T-ALL response to etoposide and vincristine: NOTCH1 inhibition prevents BMS “rescue” effect. A, (Left) After pre-treatment of ALL-SIL cells in suspension cultures with 1 µM of the γ-secretase inhibitor (GSI) Compound E or 0.01% dimethyl sulfoxide for 10 days, cell counts were determined by alamarBlue assay. P value indicates significant difference in the presence of 5 µM BMS (asterisk). (Right) Western blot analysis showing inhibition of activated NOTCH1 and downregulation of MYC protein levels in GSI-treated ALL-SIL cells (19). NS, nonspecific band. Similar results were obtained for K3P and DND-41 cells. B, DND-41 cells were grown on monolayers for 3 days; the protective effect was more prominent in the case of cells cocultured with OP9-DL1 than with OP9; 5 µM etoposide, 5 µM BMS, 1 µM Compound E as indicated for 18 h treatment. The experiment was performed in triplicate for two biological replicates. C, AlamarBlue cell growth assay. Immortalized mouse hematopoietic progenitors overexpressing MYC (18,19) were treated with vincristine in the presence or absence of 5 µM BMS, 18 h treatment. GFP is an empty vector-transduced control. The experiment was performed in triplicate; a representative graph is shown. P value indicates significant differences in cell counts at the highest concentration of the drug (asterisk). Student’s t test for all comparisons. D, Expression of NOTCH1 target genes HES1 and MYC inversely correlates with expression of BCL2 and BCLXL pro-survival genes in T-ALL patient samples (23). Samples were ranked on HES1 levels and average values for groups of 6 were plotted. The correlation coefficients were determined for two independent HES1 probe sets as indicated in Table 1.
MYC is a downstream target of NOTCH1 in T-ALL (15,16). Other studies have shown that enforced expression of MYC in hematopoietic cells enhances sensitivity to etoposide and other stress-inducing therapeutic agents (17). We observed that overexpression of MYC in immortalized mouse hematopoietic progenitors (18,19) also increased sensitivity to vincristine (Fig. 3C). Moreover, the effect was alleviated by BMS, suggesting that the MYC-enhanced response to vincristine is mediated by IKK (Fig. 3C). In this regard, the cytotoxic effects of vincristine on T-ALL cells have previously been shown to be attenuated by γ-secretase inhibitor treatment (20). Although the mechanism remains to be defined, our data indicate that MYC is one of the downstream components of NOTCH signaling that contributes to the NFκB pro-apoptotic response to chemotherapy. Notably, activation of the IKK-NFκB pathway by vincristine was previously implicated in the induction of apoptotic cell death in other human tumor types; moreover, the investigators showed that glucocorticoids, which are known to partially act via inhibition of NFκB, attenuated vincristine-induced death (21). These findings might explain why additional courses of vincristine and dexamethasone combination therapy did not result in improved treatment outcomes in a recent worldwide survey of T-ALL clinical trials (22).
Therefore, our data suggest that NOTCH1 or MYC activation levels are potential candidates to predict the apoptotic role of NFκB in response to stress-inducing chemotherapy. We analyzed publicly available microarray profiling data of 92 T-ALL patients (23). We plotted the expression levels of HES1 as a measure of NOTCH1 pathway activation versus pro-survival BCL2 and BCLXL expression levels and found that they are inversely correlated throughout the range of NOTCH1 activation (Fig. 3D). As expected, HES1 mRNA levels directly correlated with MYC mRNA levels, consistent with findings that NOTCH1 induces MYC expression in T-ALL cells (15,16). These correlations support the notion that when NOTCH1 is activated, the balance of NFκB targets may shift towards an apoptotic outcome.
Despite an overall cure rate of 80% in children, overall survival of adult ALL patients is only around 35% (6). For both patient populations, relapse remains a serious problem in T-ALL. The fate of relapsed patients is dismal. To prevent relapse, T-ALL patients have historically undergone an aggressive combination therapy so intense that toxicity had become a significant cause of unfavorable outcome (24). New more effective therapeutic agents and a better understanding of the compatibility with existing drugs are needed. We found that under the conditions examined chemotherapy-induced stress changes the function of the NOTCH-NFκB signaling pathway in T-ALL cell lines from pro-survival to pro-apoptotic. Thus the data strongly argue for further studies of the circumstances determining NFκB action in T-ALL patients on different treatment protocols and the impact of activating NOTCH1 mutations.
Table 1.
Expression of NOTCH1 target genes HES1 and MYC inversely correlates with expression of BCL2 and BCLXL pro-survival genes in T-ALL patient samples.
| HES1 Affymetrix Probes | Correlation Coefficients | |||
|---|---|---|---|---|
| HES1 | BCL2 | BCLXL | MYC | |
| 203394_s_at | 0.99 | −0.56 | −0.72 | 0.57 |
| 203395_s_at | 1.00 | −0.49 | −0.68 | 0.61 |
Acknowledgments
Grant Support
This work was supported in part by an American Cancer Society Pilot Grant for Junior Faculty from The George Washington University Cancer Institute (IRG-08-091-01) and a Pilot Project Award from the Clinical and Translational Science Institute at Children’s National Medical Center (NIH UL1RR031988) (I.R.), and by NIH Grants R01HL65519 and R01HL66305, an Elaine H. Snyder Cancer Research Award and a King Fahd Endowment from The George Washington University Medical Center (R.G.H.). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health.
Footnotes
Disclosure of Potential Conflicts of Interest
The authors have no potential conflicts of interest.
References
- 1.Vilimas T, Mascarenhas J, Palomero T, Mandal M, Buonamici S, Meng F, et al. Targeting the NF-κB signaling pathway in Notch1-induced T-cell leukemia. Nat Med. 2007;13:70–77. doi: 10.1038/nm1524. [DOI] [PubMed] [Google Scholar]
- 2.Espinosa L, Cathelin S, D'Altri T, Trimarchi T, Statnikov A, Guiu J, et al. The Notch/Hes1 pathway sustains NF-κB activation through CYLD repression in T cell leukemia. Cancer Cell. 2010;18:268–281. doi: 10.1016/j.ccr.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cleaver AL, Beesley AH, Firth MJ, Sturges NC, O'Leary RA, Hunger SP, et al. Gene-based outcome prediction in multiple cohorts of pediatric T-cell acute lymphoblastic leukemia: a Children's Oncology Group study. Mol Cancer. 2010;9:105. doi: 10.1186/1476-4598-9-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weng AP, Ferrando AA, Lee W, Morris JP, Silverman LB, Sanchez-Irizarry C, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306:269–271. doi: 10.1126/science.1102160. [DOI] [PubMed] [Google Scholar]
- 5.Shin HM, Minter LM, Cho OH, Gottipati S, Fauq AH, Golde TE, et al. Notch1 augments NF-κB activity by facilitating its nuclear retention. EMBO J. 2006;25:129–138. doi: 10.1038/sj.emboj.7600902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pui CH, Robison LL, Look AT. Acute lymphoblastic leukaemia. Lancet. 2008;371:1030–1043. doi: 10.1016/S0140-6736(08)60457-2. [DOI] [PubMed] [Google Scholar]
- 7.Perkins ND. Integrating cell-signalling pathways with NF-κB and IKK function. Nat Rev Mol Cell Biol. 2007;8:49–62. doi: 10.1038/nrm2083. [DOI] [PubMed] [Google Scholar]
- 8.Wu ZH, Miyamoto S. Many faces of NF-κB signaling induced by genotoxic stress. J Mol Med. 2007;85:1187–1202. doi: 10.1007/s00109-007-0227-9. [DOI] [PubMed] [Google Scholar]
- 9.Kox C, Zimmermann M, Stanulla M, Leible S, Schrappe M, Ludwig WD, et al. The favorable effect of activating NOTCH1 receptor mutations on long-term outcome in T-ALL patients treated on the ALL-BFM 2000 protocol can be separated from FBXW7 loss of function. Leukemia. 2010;24:2005–2013. doi: 10.1038/leu.2010.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tallen G, Ratei R, Mann G, Kaspers G, Niggli F, Karachunsky A, et al. Long-term outcome in children with relapsed acute lymphoblastic leukemia after time-point and site-of-relapse stratification and intensified short-course multidrug chemotherapy: results of trial ALL-REZ BFM 90. J Clin Oncol. 2010;28:2339–2347. doi: 10.1200/JCO.2009.25.1983. [DOI] [PubMed] [Google Scholar]
- 11.Campbell KJ, O'Shea JM, Perkins ND. Differential regulation of NF-κB activation and function by topoisomerase II inhibitors. BMC Cancer. 2006;6:101. doi: 10.1186/1471-2407-6-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boehm JS, Zhao JJ, Yao J, Kim SY, Firestein R, Dunn IF, et al. Integrative genomic approaches identify IKBKE as a breast cancer oncogene. Cell. 2007;129:1065–1079. doi: 10.1016/j.cell.2007.03.052. [DOI] [PubMed] [Google Scholar]
- 13.Li C, Chen S, Yue P, Deng X, Lonial S, Khuri FR, et al. Proteasome inhibitor PS-341 (bortezomib) induces calpain-dependent IκBα degradation. J Biol Chem. 2010;285:16096–16104. doi: 10.1074/jbc.M109.072694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Pooter R, Zuniga-Pflucker JC. T-cell potential and development in vitro: the OP9-DL1 approach. Curr Opin Immunol. 2007;19:163–168. doi: 10.1016/j.coi.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 15.Palomero T, Lim WK, Odom DT, Sulis ML, Real PJ, Margolin A, et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc Natl Acad Sci U S A. 2006;103:18261–18266. doi: 10.1073/pnas.0606108103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weng AP, Millholland JM, Yashiro-Ohtani Y, Arcangeli ML, Lau A, Wai C, et al. c-Myc is an important direct target of Notch1 in T-cell acute lymphoblastic leukemia/lymphoma. Genes Dev. 2006;20:2096–2109. doi: 10.1101/gad.1450406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nesbit CE, Tersak JM, Grove LE, Drzal A, Choi H, Prochownik EV. Genetic dissection of c-myc apoptotic pathways. Oncogene. 2000;19:3200–3212. doi: 10.1038/sj.onc.1203636. [DOI] [PubMed] [Google Scholar]
- 18.Hawley RG, Fong AZC, Lu M, Hawley TS. The HOX11 homeobox-containing gene of human leukemia immortalizes murine hematopoietic precursors. Oncogene. 1994;9:1–12. [PubMed] [Google Scholar]
- 19.Riz I, Hawley TS, Luu TV, Lee NH, Hawley RG. TLX1 and NOTCH coregulate transcription in T-cell acute lymphoblastic leukemia cells. Mol Cancer. 2010;9:181. doi: 10.1186/1476-4598-9-181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.De Keersmaecker K, Lahortiga I, Mentens N, Folens C, Van NL, Bekaert S, et al. In vitro validation of γ-secretase inhibitors alone or in combination with other anti-cancer drugs for the treatment of T-cell acute lymphoblastic leukemia. Haematologica. 2008;93:533–542. doi: 10.3324/haematol.11894. [DOI] [PubMed] [Google Scholar]
- 21.Huang Y, Fang Y, Wu J, Dziadyk JM, Zhu X, Sui M, et al. Regulation of Vinca alkaloid-induced apoptosis by NF-κB/IκB pathway in human tumor cells. Mol Cancer Ther. 2004;3:271–277. [PubMed] [Google Scholar]
- 22.Eden TO, Pieters R, Richards S. Systematic review of the addition of vincristine plus steroid pulses in maintenance treatment for childhood acute lymphoblastic leukaemia - an individual patient data meta-analysis involving 5,659 children. Br J Haematol. 2010;149:722–733. doi: 10.1111/j.1365-2141.2010.08148.x. [DOI] [PubMed] [Google Scholar]
- 23.Soulier J, Clappier E, Cayuela JM, Regnault A, Garcia-Peydro M, Dombret H, et al. HOXA genes are included in genetic and biologic networks defining human acute T-cell leukemia (T-ALL) Blood. 2005;106:274–286. doi: 10.1182/blood-2004-10-3900. [DOI] [PubMed] [Google Scholar]
- 24.Pui CH, Pei D, Sandlund JT, Ribeiro RC, Rubnitz JE, Raimondi SC, et al. Long-term results of St. Jude Total Therapy Studies 11, 12, 13A, 13B, and 14 for childhood acute lymphoblastic leukemia. Leukemia. 2010;24:371–382. doi: 10.1038/leu.2009.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Riz I, Hawley TS, Johnston H, Hawley RG. Role of TLX1 in T-cell acute lymphoblastic leukaemia pathogenesis. Br J Haematol. 2009;145:140–143. doi: 10.1111/j.1365-2141.2008.07556.x. [DOI] [PMC free article] [PubMed] [Google Scholar]