

Abstract
DNA decoys have been developed for the inhibition of the transcriptional regulation of gene expression. However, the present methodology lacks the spatial and temporal control of gene expression that is commonly found in nature. Here, we report the application of photo-removable protecting groups on nucleobases of NF-κB DNA decoys to regulate NF-κB driven transcription of secreted alkaline phosphatase using light as an external control element. The NF-κB family of proteins is comprised of important eukaryotic transcription factors that regulate a wide range of cellular processes and are involved in immune response, development, cellular growth, and cell death. Several diseases, including cancer, arthritis, chronic inflammation, asthma, neurodegenerative diseases, and heart disease have been linked to constitutively active NF-κB. Through the direct incorporation of caging groups into an NF-κB decoy we were able to disrupt DNA:DNA hybridization and inhibit the binding of the transcription factor to the DNA decoy until UV irradiation removes the caging groups and restores the activity of the oligonucleotide. Excellent light-switching behavior of transcriptional regulation was observed. This is the first example of a caged DNA decoy for the photochemical regulation of gene expression in mammalian cells and represents an important addition to the toolbox of light-controlled gene regulatory agents.
Introduction
Deoxyribonucleic acid decoys have been extensively used to regulate transcription in eukaryotic systems.1–2 Bielinska et al. were the among the first to show that double stranded (ds) phosphorothioate DNA can act as a decoy for sequestering transcription factors through binding to dsDNA.3 A DNA decoy encodes a short consensus binding sequence for a transcription factor and is designed to out-compete the natural, genomic DNA target. Thus, if sufficient quantities of the DNA decoy are present within a cell, the transcription factor will bind preferentially to the decoy and not to its natural binding site, leading to an inhibition of transcription.4–5 Various decoys have been designed for individual transcription factors in order to inhibit gene function with high specificity, including the Stat6 decoy for inhibition of TH2-lymphocyte activity,6 the androgen-responsive element decoy for the regulation of androgen-activated androgen receptors in prostate cancer cells,7 the STAT3 decoy for the induction of apoptosis in A549 cancer cells,8 the est-1 decoy for the inhibition of cell growth of gastric cancer cells,9 and the E2F decoy for the regulation of mesangial cell proliferation.10
One of the first developed DNA decoy was the Nuclear Factor (NF)-κB decoy (Scheme 1).11 NF-κB is an important transcription factor that activates and regulates numerous genes.2,12–13 NF-κB is misregulated in a variety diseases, including diseases associated with inflammatory and oxidative stress response, cardiovascular disease, and cancer. Thus, inhibiting NF-κB has substantial therapeutic potential.13–14 Recent studies have shown that NF-κB DNA decoys inhibit myocardial infarction,5 induce apoptosis in UV damaged skin cells,15 reduce the progression of joint destruction in rheumatoid arthritis,16 and control pulmonary allergy.17
Scheme 1.

NF-κB regulated transcription in the presence of DNA decoys. A) Upon stimulation, NF-κB translocates into the nucleus and binds to its DNA binding site activating transcription. B) In the presence of DNA decoys, NF-κB binds to the DNA decoy over its genomic binding site, leading to an inhibition of transcription.
One method to control NF-κB-mediated in gene expression, is through modified oligonucleotides. Hairpin and dumbbell decoys have been developed to improve the stability and lower the toxicity of phosphorothioate decoys.18–19 Although these modifications have improved certain aspects of oligonucleotide therapy, a methodology to regulate NF-κB function with spatial and temporal resolution has not been reported.
Recently, “caging” technologies have provided an approach to photochemically regulate gene expression in both a spatial and a temporal manner.20–26 By placing a photo-responsive protecting group (a so called “caging group”) directly onto the base of a nucleotide, we (and others) have been able to disrupt DNA:DNA, DNA:RNA and RNA:RNA hybridization, thus rendering the oligonucleotide inactive.27–32 After a brief UV irradiation, the caging group is removed, restoring the activity of the oligonucleotide. This approach has been successfully applied to the photochemical regulation of gene translation.30,33–34 Here, we are presenting the first examples of photochemical control of gene transcription through the application of caged oligonucleotides. We hypothesized that caged, light-activated hairpin and dumbbell DNA decoys allow for the photochemical regulation of NF-κB activation of gene expression (Scheme 2). The caged oligonucleotides will not be able to form the double-stranded DNA required for transcription factor binding due to the blocking of Watson-Crick base pairing at nucleobase-caged thymidines. Removal of the caging groups through a brief UV irradiation restores duplex formation and thus activates decoy function leading to sequestering of the transcription factor.
Scheme 2.

Photochemical control over NF-κB activation of gene expression. The caging groups disrupt Watson-Crick base-pairing of DNA and thus hairpin formation, rendering the decoy inactive. Therefore, NF-κB binds to its native genomic binding site and initiates transcription. After a brief irradiation with UV light, the caging groups are removed, the hairpin decoy forms, and out-competes the natural NF-κB binding site, leading to the inhibition of gene expression.
Results and Discussion
Non-caged and caged deoxynucleotides containing NPOM (6-nitropiperonyloxymethyl)-caged thymidine groups were synthesized under standard DNA synthesis conditions to provide NF-κB DNA decoys that can be regulated photochemically. The required NPOM-caged thymidine phosphoramidite was synthesized as previously reported.35–36 In order to confirm that DNA decoy formation can be disrupted through the installation of NPOM caging groups on the bases of selected nucleotides, the melting temperature of the DNA decoy hairpins and dumbbells were determined (Table 1 and Supporting Information Figure S1). As expected, the hairpin decoy D2 had a significantly higher Tm than the simple double-stranded DNA decoys D0 & D1. With the introduction of 3 caging groups into the hairpin decoy D3, the melting temperature decreased approximately 30 °C. After addition of a fourth caging group (in D4), no Tm and thus no hairpin formation could be detected. However, upon UV irradiation (365 nm, 10 min, 25 W), the caging groups are removed and hairpin formation occurs as demonstrated by observing melting temperatures comparable to the non-caged DNA hairpin decoy D2. The dumbbell decoy D5 consists of two hairpin structures at the 5′ and 3′ termini of the DNA in order to protect the decoy from exonuclease degradation. The introduction of 3 and 4 caging groups at selected thymidine residues fully abolished the formation of the dumbbell structure, as indicated by the absence of a sigmoidal melting curve for D6 & D7. Decaging through UV irradiation regenerated the dumbbell DNA D2 (as shown by a melting temperature of 74 °C) and thus led to the light-induced formation of a DNA decoy. Dumbbell precursors containing less than 3 caging groups were not investigated because previous studies have shown that complete disruption of DNA hybridization requires a caging group every 4–6 bases throughout a DNA sequence,30–31,36 and because the presence of only 3 caging groups on the decoy D3 (which non-caged version D2 has a similar Tm as the non-caged dumbbell D5) was not sufficient to fully inhibit DNA hairpin formation.
Table 1.
Sequences and melting temperatures of synthesized NF-κB DNA decoys.
| DNA | Sequence | Tm/°C | |
|---|---|---|---|
| −UV | +UV | ||
| D0 | 5′ T* T* G* C* C* G* T* A* C* C* T* G* A* C* T A* A* C* G* G* C* A* T* G* G* A* C* T* G* A |
52.1 ± 0.5 | - |
|
| |||
| D1 | 5′ T* T* G* G* G* G* A* C* T* T* T* C* C* A* G A* A* C* C* C* C* T* G* A* A* A* G* G* T* C |
45.8 ± 1.5 | - |
|
| |||
| D2 |
|
84.4 ± 0.0 | - |
|
| |||
| D3 |
|
54.4 ± 1.7 | 81.8±0.6 |
|
| |||
| D4 |
|
NA | 83.1 ± 0.6 |
|
| |||
| D5 |
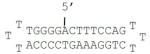
|
74.8 ± 0.6 | - |
|
| |||
| D6 |
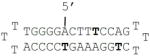
|
NA | 73.8 ± 1.5 |
|
| |||
| D7 |
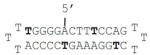
|
NA | 74.4 ± 0.9 |
Indicates a phosphorothioate bond; a bold T denotes a caged thymidine nucleotide; NA indicates that no melting temperature could be measured and thus no formation of dsDNA was observed.
The melting temperature measurements confirmed that we could photochemically control DNA hybridization of the hairpin and dumbbell decoys through the site-specific introduction of caging groups.33,37 Next, we wanted to test if the caged and non-caged DNA decoys would bind to NF-κB. Nuclear extracts were isolated from NF-κB/SEAP HEK 293 cells (IMGENEX) that were induced with TNFα and a gel shift assay was performed. Similar gel shift assays have previously been performed to determine the specificity of DNA decoys for the NF-κB protein complex.38 The nuclear extracts were incubated with excess radiolabeled DNA decoys and analyzed by gel electrophoresis (Figure 1). With the addition of nuclear extracts, the negative control scrambled phosphorothioate decoy D0 showed no binding affinity to NF-κB, as expected (lanes 1–2). The phosphorothioate decoy D1 binds to NF-κB as shown by the band shift induced in the presence of nuclear extracts (lane 3–4), confirming its ability to sequester the transcription factor. The two bands shown in the band shift correspond to the NF-κB heterodimer.15,39 The hairpin decoy D2 and the dumbbell decoy D5 also bind to NF-κB protein as indicated by the band shift (lanes 5–6 and 7–8). The varying intensities of the bands from the different decoys, demonstrate different binding affinities of the decoy for the NF-κB complex. For example, D5 (lane 8) has a higher band intensity than D1 and D2 (lanes 4 and 6), leading to the conclusion that D5 presumably has as higher binding affinity to the NF-κB complex than the phosphorothioate decoy D1 and the hairpin decoy D2.
Figure 1.
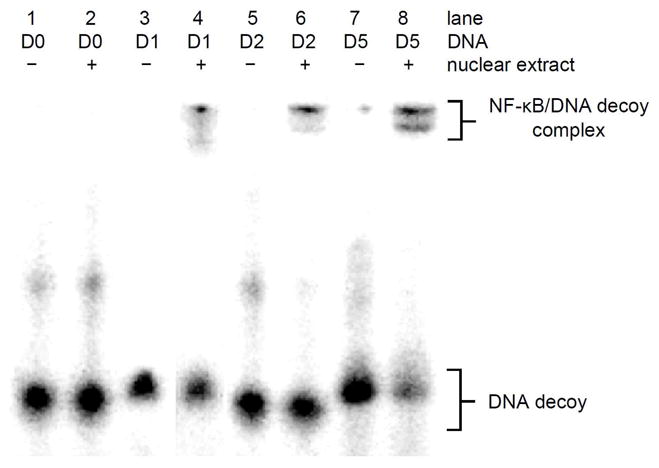
In vitro binding of NF-κB DNA decoys to the NF-κB protein complex. Nuclear extracts were isolated from NF-κB/SEAP HEK 293 cells and incubated with radiolabelled NF-κB DNA decoys at room temperature for 20 min. Samples were analyzed on a 16% native PAGE gel and imaged with a Typhoon 7000 phosphorimager.
The same gel shift assay was then used to investigate the light-activation of the caged DNA decoys in vitro. First an irradiation time course with D4 was performed to optimize the decaging conditions (Supporting Information Figure S2). The caged decoys were either activated through irradiation for 1 min (365 nm, 25 W) or kept in the dark. As we have previously seen in Figure 1, the D2 decoy showed a band shift in the presence of nuclear extract (Figure 2, lanes 1–2). However, despite the observed meting point and thus partial decoy formation for the caged oligonucleotide D3, the presence of the 3 caging groups on the hairpin sufficiently perturbed protein binding to an extent that the presence of NF-κB does not induce a gel shift (Figure 2, lane 4); gratifyingly, upon irradiation, the caging groups are removed and the NF-κB protein binds to the DNA (Figure 2, lane 5). The presence of 4 caging groups completely inhibited DNA hairpin formation (Table 1), and as expected the NF-κB protein does not undergo binding to D4 (Figure 2, lane 7) until the caging groups are removed through UV irradiation (Figure 2, lane 8).
Figure 2.

Light-activation of NF-κB DNA decoy binding. Nuclear extracts were isolated from NF-κB/SEAP HEK 293 cells and caged decoys were irradiated for 1 min (365 nm, 25 W) and incubated with nuclear extracts at room temperature for 20 min. Samples were analyzed on a 16 % Native PAGE gel and imaged with a Typhoon 7000 phosphorimager.
Similar to the hairpin decoys, DNA duplex formation was completely inhibited in the presence of 3–4 caging groups in case of the dumbbell decoys D6 and D7, and thus no NF-κB binding affinity and gel shifts were observed (Figure 2, lanes 11–12 and 14–15). The formation of the dumbbell decoys through UV decaging led to NF-κB complex formation and the expected gel shifts (Figure 2, lanes 13 and 16) matching the gel shift of the non-caged positive control decoy D5 (Figure 2, lanes 9–10). In summary, these experiments demonstrate that the introduction of 3–4 caged thymidine residues enables the photochemical regulation of DNA hairpin and dumbbell decoy formation and thus the light-regulation of NF-κB binding.
In order to investigate the photochemical control of DNA decoy activity in mammalian tissue culture, a NF-κB/SEAP HEK 293 stable cell line,40 that contains a NF-κB activated secreted alkaline phosphatase (SEAP) reporter gene was used. NF-κB decoy concentration and irradiation conditions were first optimized (see Supporting Information Figures S3 and S4). In addition, just NF-κB decoys were tested as well in order to ensure the inhibition of induced transcription of NF-κB activated gene expression rather than simply the basal levels of transcription (see Supporting Information Figure S5). Then NF-κB/SEAP HEK 293 cells were transfected with the non-caged or caged DNA decoys. After a brief UV irradiation (365 nm, 2 min, 25 W), NF-κB phoshorylation and activation was induced through addition of TNFα to the media. The NF-κB driven SEAP activity was assayed using the Phopha Light System kit (Applied Biosystems), which was normalized to a Cell Titer Glo viability assay (Promega) to account for differences in cell viability. The phosphorothioate DNA decoy D1 had very little effect on NF-κB driven SEAP expression with only 19% reduction in reporter gene activity in comparison to the scrambled phosphorothioate control D0 (Figure 3). Conversely, the hairpin DNA decoy D2 down-regulated NF-κB-mediated gene expression by 60%. The caged hairpin D3 (despite the presence of three caging groups and its complete inactivity in the gel shift assay shown in Figure 2) still showed a substantial, 37% inhibition of SEAP activity presumably due to partial hairpin formation (see Table 1) over the extended time course of the cell based experiment. However, after decaging the decoy D3 very effectively sequestered NF-κB complexes, inhibiting transcription of SEAP to the same extent of D2. In contrast to D3, the caged hairpin D4 (containing four caging groups) was rendered completely inactive and showed no inhibition of SEAP expression and thus no background activity in its caged form. Most importantly, the caged dumbbell decoys D6 and D7 are also fully inactive. As in the case of D4, UV irradiation of 365 nm efficiently removes the caging groups and induces dumbbell formation, resulting in the effective sequestering of NF-κB and the inhibition SEAP expression. Thus, caged dumbbell decoys, D6 and D7, exhibit excellent on/off light-switching behavior as shown by their complete inactivity before irradiation (100% SEAP activity, identical with the negative control oligonucleotide D0, and no background activity) and the complete restoration of DNA decoy activity after irradiation (5% SEAP activity, identical with the positive control D5). Overall, the (caged and non-caged) dumbbell decoys D5–D7 are more efficient inhibitors of NF-κB than the phosphorothioate and hairpin decoys, presumably due to an enhanced cellular stability41,42 and a higher binding affinity of the dumbbell decoy to the NF-κB complex as observed in Figures 1–2. In addition, the dumbbell decoy activity can be completely abolished with as little as three caging groups while the hairpin decoy requires four caging groups to completely inhibit hybridization as shown by their melting temperatures (Table 1) and reporter assay activity (Figure 3).
Figure 3.
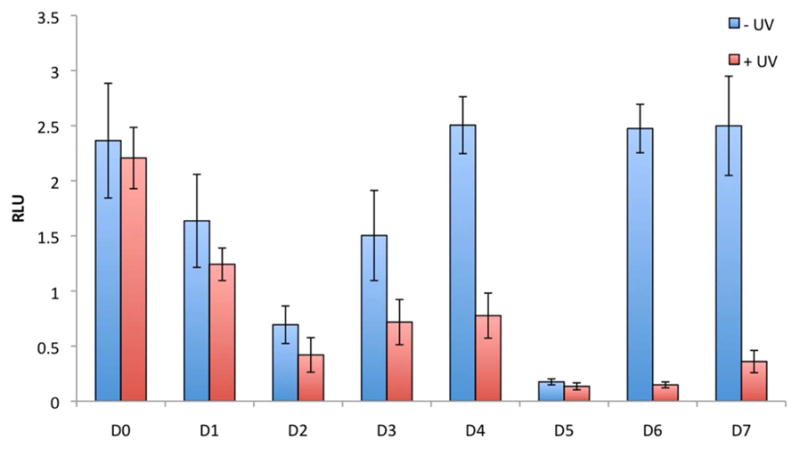
Photochemical activation of NF-κB induced SEAP expression. NF-κB/SEAP HEK293 cells were transfected with caged and non-caged DNA decoys using X-tremeGENE. Cells were either irradiated for 2 min (365 nm, 25 W) or kept in the dark. TNFα was added after 4 hours, and a SEAP assay was conducted after 24 hours using a Phospha Light Systems kit (Applied Biosystems). Cell viability was assayed using a Cell Titer Glo assay (Promega), and the SEAP signal was normalized to the Cell Titer Glo signal. All experiments were performed in triplicate and error bars represent standard deviations.
The developed light-regulation methodology was then applied to temporal control over NF-κB induced gene expression. Thus, a time course assay was performed in which the scrambled (D0), the dumbbell (D5), and the caged dumbbell (D6) decoys were transfected into NF-kB/SEAP HEK 293 stable cells. The cells were irradiated (365 nm, 2 min, 25 W) 12 hrs post induction of SEAP expression by TNFα and aliquots of the supernatant were taken at 1, 8, 12, 24, 36, and 48 hrs (Figure 4). SEAP expression was quantified and normalized as described above. The scrambled decoy D0 served as an inactive control and showed steadily increasing SEAP activity over the entire experiment. As previously seen in Figure 3, the dumbbell decoy D5 completely inhibits SEAP expression and only a slight increase can be detected over time. Importantly, the caged dumbbell D6 displays complete inactivity and shows virtually identical SEAP expression levels as D0 for the first 12 hrs of the experiment. After irradiation at 12 hrs, SEAP expression still continues to increase for D6, reaching its maximum expression level at 24 hrs. After 24 hrs, the reporter gene expression level is constant for another 12 hrs then it starts to slowly decrease, in contrast to the control D0 that shows a constant linear increase in SEAP expression. Although light-induced decoy formation and thus NF-κB sequestering is most likely instantaneous after UV decaging, the presence of already transcribed SEAP mRNA may lead to the further increase in activity until 24 hrs into the time course. Once the dumbbell decoy is formed, further SEAP transcription is inhibited and the concentration of the reporter protein remains constant due to the SEAP protein’s stability of 2–3 days.43 These experiments demonstrate that precise temporal control over gene transcription can be achieved with light-activated DNA decoys.
Figure 4.
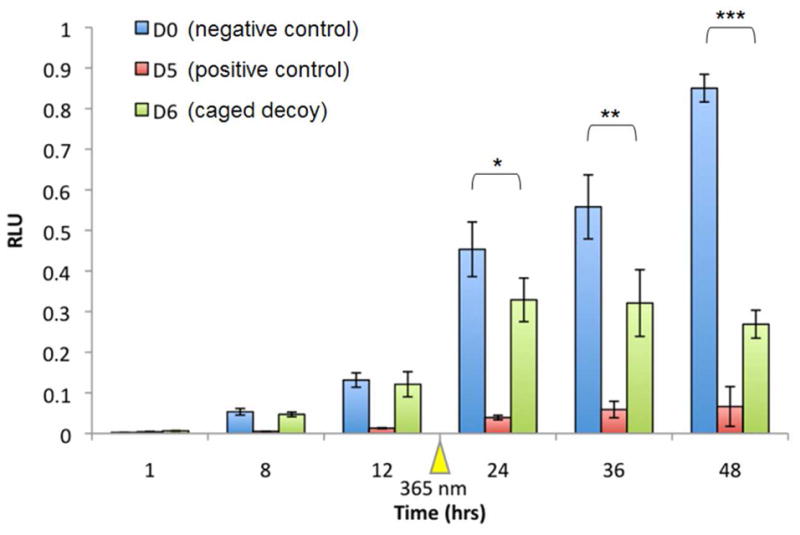
Temporally controlled deactivation of SEAP expression with caged NF-κB decoys. NF-κB/SEAPorter HEK293 cells were transfected with inactive (D0), non-caged (D5), and caged (D6) DNA decoys using X-tremeGENE, followed by TNFα addition 4 hrs post-transfection. Aliquots of the supernatant were taken at 1, 8, 12, 24 and 48 hrs after induction with TNFα followed by SEAP quantification. Cells were irradiated for 2 min (365 nm, 25 W) at 12 hrs after induction. The SEAP signal (Phospha Light Systems kit, Applied Biosystems) was normalized to cell viability (Cell Titer Glo, Promega). All experiments were performed in triplicate and error bars represent standard deviations. Asterisks indicate t-test results: * p < 0.5, ** p < 0.05, and *** p < 0.0005.
Conclusion
In summary, we have developed light-activated NF-κB DNA decoys through the site-specific installation of caging groups on select nucleobases of oligonucleotides. These decoys were used to photochemically regulate gene function in mammalian cells. The presented approach enables the precise activation of genes that are regulated by the NF-κB transcription factor, but is easily adaptable to any DNA decoy. Melting temperatures and gel shift assays verified that the NPOM-caging groups are sufficient to disrupt DNA:DNA hybridization and thus the affinity of the caged decoy to the targeted transcription factor (NF-κB). Irradiation with UV light led to decoy activation and complete restoration of transcriptional inhibition. Decoy activation and gene downregulation with UV light was furthermore demonstrated in a time-resolved fashion. DNA decoys have great potential as therapeutic agents for the treatment of various diseases, including atherosclerosis,44 tumorgensis,45 and inflammation,46 and are currently being evaluated in multi-cellular model organism. The developed caged DNA decoys will enable the study of gene expression and gene function in biological pathways with unprecedented spatial and temporal resolution.
Experimental Section
DNA synthesis protocol
DNA synthesis was performed using an Applied Biosystems (Foster City, CA) Model 394 automated DNA/RNA Synthesizer using standard β-cyanoethyl phosphoramidite chemistry. The caged DNA decoys were synthesized using 40 nmole scale, low volume solid phase supports obtained from Glenn Research (Sterling, VA). Reagents for automated DNA synthesis were also obtained from Glenn Research. Standard synthesis cycles provided by Applied Biosystems were used for all normal bases using 2 minute coupling times. The coupling time was increased to 10 minutes for the positions at which the caged thymidine phosphoramidites36 were incorporated. Each synthesis cycle was monitored by following the release of dimethoxy trityl (DMT) cations after each deprotection step. No significant loss of DMT was noted following the addition of the caged-T to the DNA, thus 10 minutes was sufficient to allow maximal coupling of the caged thymidine. Yields of caged decoys were close to theoretical values routinely obtained.
Melting Temperatures
The melting temperature (Tm) of each NF-κB DNA decoy was measured using a Cary 100 Bio UV/Vis spectrometer with a temperature controller (Varian). NF-κB DNA Decoys (1 μM) were incubated in 0.15 M NaCl, 0.05 M NaH2PO4, pH 7.2 buffer. The samples were protected from light or irradiated at 365 nm with a UV transilluminator for 10 min, heated to 100 °C for 2 min, and then cooled to 20 °C at a rate of 2 °C/min, held at 20 °C for 5 min, then heated to 100 °C at a rate of 2 °C/min. Absorbance was recorded at 260 nm every 1 °C. The Tm was determined by the maximum of the first derivative of the absorbance vs. temperature plot. Standard deviations were calculated form three individual experiments.
Nuclear Extract Gel Shift Assay
NF-κB/SEAP Human embryonic kidney (HEK) 293 cells were induced with TNFα (20 ng/mL) for 24 hrs then the nuclear extracts were isolated using the NE-PER Nuclear and Cytoplasmic Extraction kit (Pierce Biotechnology). The nuclear extract gel shift was performed as described previously with the following modification.38 Radiolabeled NF-κB Decoys (5,000 cpm) were incubated with 20 μg nuclear extracts at room temperature for 20 min. Samples were resolved on a 16% native PAGE gel and visualized on a Typhoon 7000 phosphorimager.
Cell Culture
NF-κB/SEAP HEK 293 cells were grown at 37 °C and 5% CO2 in Dulbecco’s modified Eagle’s medium (Hyclone), supplemented with 10% fetal bovine serum (Hyclone), 10% streptomycin/penicillin (MP Biomedicals), 1 mM sodium pyruvate (Alfa Aesar), and 500 μg/mL G418 (Sigma Aldrich). Cells were passaged into a 96-well plate (200 μL per well, ~104 cells per well) and grown to ~70% confluence within 24 h. The medium was changed to Optimem (Invitrogen), and the cells were transfected with 150 ng DNA decoy using X-tremeGENE (3:2 reagent/DNA ratio, Roche). All transfections were performed in triplicate. Cells were incubated at 37 °C for 4 hrs, and the transfection medium was removed, replaced with standard growth medium, and NF-κB expression was induced with TNFα (20 ng/mL). After a 24 hr incubation, gene expression was assayed using the Phospha Light systems assay (Applied Biosystems) according to the manufacturers protocol. A Cell Titer Glo viability assay (Promega) was performed and the Phospha Light assay was normalized to the cell viability assay. For each of the triplicates, the data were averaged and standard deviations were calculated.
Temporal Control of DNA Decoy Activation
NF-κB/SEAP HEK 293 cells were passaged into a 96-well plate (200 μL per well, ~104 cells per well) and grown to ~ 70 % confluence within 24 hrs. The medium was changed to Optimem (Invitrogen), and the cells were transfected with 150 ng DNA decoy using X-tremeGENE (3:2 reagent/DNA ratio, Roche). All transfections were performed in triplicate. Cells were incubated at 37 °C for 4 hrs, and the transfection medium was removed, replaced with standard growth medium, and NF-κB expression was induced with TNFα (20 ng/mL). After 1, 8, 12, 24, 36, and 48 hrs of induction with TNFα, media samples (10 μL) were taken and stored at 4 °C. At 12 hrs post induction, cells were irradiated for 2 min at 365 nm on a UV transilluminator. After 48 hrs, the medium was assayed using the Phospha Light systems assay (Applied Biosystems) according to the manufacturers protocol. A Cell Titer Glo viability assay (Promega) was performed and the Phospha Light assay was normalized to the cell viability assay. For each of the triplicates, the data were averaged, and standard deviations were calculated.
Supplementary Material
Acknowledgments
This research was supported in part by the National Institutes of Health (R01GM079114), the Beckman Foundation (Beckman Young Investigator Award for AD), and the Research Corporation (Cottrell Scholar Award for AD). We would like to thank Hrvoje Lusic for the synthesis of the caged thymidine phosphoramidite.
Footnotes
Supporting Information Available: Melt curves of DNA decoy hybridization, in vitro irradiation time course, and optimization and validation of DNA decoy experiments in mammalian cells. This information is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Gambari R. Curr Drug Targets. 2004;5:419. doi: 10.2174/1389450043345416. [DOI] [PubMed] [Google Scholar]
- 2.Isomura I, Morita A. Microbiol Immunol. 2006;50:559. doi: 10.1111/j.1348-0421.2006.tb03827.x. [DOI] [PubMed] [Google Scholar]
- 3.Bielinska A, Shivdasani RA, Zhang LQ, Nabel GJ. Science. 1990;250:997. doi: 10.1126/science.2237444. [DOI] [PubMed] [Google Scholar]
- 4.De Stefano D, De Rosa G, Carnuccio R. Curr Opin Mol Ther. 2010;12:203. [PubMed] [Google Scholar]
- 5.Morishita R, Sugimoto T, Aoki M, Tomita N, Moriguchi A, Kida I, Maeda K, Sawa Y, Kaneda Y, Higaki J, Ogihara T. Circulation. 1996;94:1313. doi: 10.1038/nm0897-894. [DOI] [PubMed] [Google Scholar]
- 6.Wang LH, Yang XY, Kirken RA, Resau JH, Farrar WL. Blood. 2000;95:1249. [PubMed] [Google Scholar]
- 7.Zhang P, Zhang J, Young CY, Kao PC, Chen W, Jiang A, Zhang L, Guo Q. Ann Clin Lab Sci. 2005;35:278. [PubMed] [Google Scholar]
- 8.Zhang X, Zhang J, Wang L, Wei H, Tian Z. BMC Cancer. 2007;7:149. doi: 10.1186/1471-2407-7-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Taniguchi H, Fujiwara Y, Doki Y, Sugita Y, Sohma I, Miyata H, Takiguchi S, Yasuda T, Tomita N, Morishita R, Monden M. Int J Cancer. 2007;121:1609. doi: 10.1002/ijc.22870. [DOI] [PubMed] [Google Scholar]
- 10.Tomita N, Horiuchi M, Tomita S, Gibbons GH, Kim JY, Baran D, Dzau VJ. Am J Physiol. 1998;275:F278. doi: 10.1152/ajprenal.1998.275.2.F278. [DOI] [PubMed] [Google Scholar]
- 11.Mann MJ, Dzau VJ. J Clin Invest. 2000;106:1071. doi: 10.1172/JCI11459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mankan AK, Lawless MW, Gray SG, Kelleher D, McManus R. J Cell Mol Med. 2009;13:631. doi: 10.1111/j.1582-4934.2009.00632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harari OA, Liao JK. Ann N Y Acad Sci. 2010;1207:32. doi: 10.1111/j.1749-6632.2010.05735.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin Y, Bai L, Chen W, Xu S. Expert Opin Ther Targets. 2010;14:45. doi: 10.1517/14728220903431069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yokoyama S, Nakano H, Yamazaki T, Tamai K, Hanada K, Takahashi GBr. J Dermatol. 2005;153:47. doi: 10.1111/j.1365-2133.2005.06969.x. [DOI] [PubMed] [Google Scholar]
- 16.Tomita T, Takano H, Tomita H, Morishita R, Kaneko M, Shi K, Takahi K, Nakase T, Kaneda Y, Yoshikawa H, Ochi T. Rheumatology. 2000;39:749. doi: 10.1093/rheumatology/39.7.749. [DOI] [PubMed] [Google Scholar]
- 17.Edwards MR, Bartlett NW, Clarke D, Birrell M, Belvisi M, Johnston SL. Pharmacol Ther. 2009;121:1. doi: 10.1016/j.pharmthera.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim KH, Lee ES, Cha SH, Park JH, Park JS, Chang YC, Park KK. Exp Mol Pathol. 2009;86:114. doi: 10.1016/j.yexmp.2008.11.011. [DOI] [PubMed] [Google Scholar]
- 19.Hosoya T, Takeuchi H, Kanesaka Y, Yamakawa H, Miyano-Kurosaki N, Takai K, Yamamoto N, Takaku H. FEBS Lett. 1999;461:136. doi: 10.1016/s0014-5793(99)01450-7. [DOI] [PubMed] [Google Scholar]
- 20.Riggsbee CW, Deiters A. Trends Biotechnol. 2010;28:468. doi: 10.1016/j.tibtech.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deiters A. Chembiochem. 2010;11:47. doi: 10.1002/cbic.200900529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deiters A. Curr Opin Chem Biol. 2009;13:678. doi: 10.1016/j.cbpa.2009.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meng XM, Chen XY, Fu Y, Guo QX. Prog Chem. 2008;20:2034. [Google Scholar]
- 24.Dmochowski IJ, Tang XJ. Biotechniques. 2007;43:161. doi: 10.2144/000112519. [DOI] [PubMed] [Google Scholar]
- 25.Young DD, Deiters A. Org Biomol Chem. 2007;5:999. doi: 10.1039/b616410m. [DOI] [PubMed] [Google Scholar]
- 26.Mayer G, Heckel A. Angew Chem, Int Ed. 2006;45:4900. doi: 10.1002/anie.200600387. [DOI] [PubMed] [Google Scholar]
- 27.Deiters A, Garner RA, Lusic H, Govan JM, Dush M, Nascone-Yoder NM, Yoder JA. J Am Chem Soc. 2010;132:15644. doi: 10.1021/ja1053863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Richards JL, Seward GK, Wang YH, Dmochowski IJ. Chembiochem. 2010;11:320. doi: 10.1002/cbic.200900702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Casey JP, Blidner RA, Monroe WT. Mol Pharm. 2009;6:669. doi: 10.1021/mp900082q. [DOI] [PubMed] [Google Scholar]
- 30.Young DD, Lusic H, Lively MO, Yoder JA, Deiters A. Chembiochem. 2008;9:2937. doi: 10.1002/cbic.200800627. [DOI] [PubMed] [Google Scholar]
- 31.Young DD, Edwards WF, Lusic H, Lively MO, Deiters A. Chem Commun. 2008:462. doi: 10.1039/b715152g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mikat V, Heckel A. RNA. 2007;13:2341. doi: 10.1261/rna.753407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Young D, Lively M, Deiters A. J Am Chem Soc. 2010;132:6183. doi: 10.1021/ja100710j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tang X, Swaminathan J, Gewirtz AM, Dmochowski IJ. Nucleic Acids Res. 2008;36:559. doi: 10.1093/nar/gkm1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lusic H, Deiters A. Synthesis-Stuttgart. 2006:2147. [Google Scholar]
- 36.Lusic H, Young DD, Lively MO, Deiters A. Org Lett. 2007;9:1903. doi: 10.1021/ol070455u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kröck L, Heckel A. Angew Chem Int Ed Engl. 2005;44:471. doi: 10.1002/anie.200461779. [DOI] [PubMed] [Google Scholar]
- 38.Shibuya T, Takei Y, Hirose M, Ikejima K, Enomoto N, Maruyama A, Sato N. Biochem Biophys Res Commun. 2002;298:10. doi: 10.1016/s0006-291x(02)02369-0. [DOI] [PubMed] [Google Scholar]
- 39.Fisher L, Samuelsson M, Jiang Y, Ramberg V, Figueroa R, Hallberg E, Langel U, Iverfeldt K. J Mol Neurosci. 2007;31:209. doi: 10.1385/jmn:31:03:209. [DOI] [PubMed] [Google Scholar]
- 40.Feng D, Sangster-Guity N, Stone R, Korczeniewska J, Mancl ME, Fitzgerald-Bocarsly P, Barnes BJ. J Immunol. 2010;185:6003. doi: 10.4049/jimmunol.1000482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Osako MK, Tomita N, Nakagami H, Kunugiza Y, Yoshino M, Yuyama K, Tomita T, Yoshikawa H, Ogihara T, Morishita R. J Gene Med. 2007;9:812. doi: 10.1002/jgm.1077. [DOI] [PubMed] [Google Scholar]
- 42.Lee IK, Ahn JD, Kim HS, Park JY, Lee KU. Curr Drug Targets. 2003;4:619. doi: 10.2174/1389450033490821. [DOI] [PubMed] [Google Scholar]
- 43.Yang TT, Sinai P, Kitts PA, Kain SR. Biotechniques. 1997;23:1110. doi: 10.2144/97236pf01. [DOI] [PubMed] [Google Scholar]
- 44.Kim SJ, Park JH, Kim KH, Lee WR, Lee S, Kwon OC, Kim KS, Park KK. Basic Clin Pharmacol Toxicol. 2010;107:925. doi: 10.1111/j.1742-7843.2010.00617.x. [DOI] [PubMed] [Google Scholar]
- 45.Wang T, Li QH, Hao GP, Zhai J. Asian Pac J Cancer Prev. 2010;11:193. [PubMed] [Google Scholar]
- 46.De Stefano D, De Rosa G, Maiuri MC, Ungaro F, Quaglia F, Iuvone T, Cinelli MP, La Rotonda MI, Carnuccio R. Pharmacol Res. 2009;60:33. doi: 10.1016/j.phrs.2009.03.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.