

Abstract
The α-thalassemias are a group of hereditary disorders caused by reduced synthesis of the α-chain of hemoglobin. We have developed and tested an α-thalassemia assay that uses both multiplex ligation-dependent probe amplification (MLPA) with Luminex-based detection and deletion PCR technologies. The MLPA assay consisted of 20 probes, 15 of which hybridized to the α-globin gene cluster and 5 that served as control probes. A PCR assay was developed to confirm the presence of heterozygous/homozygous 3.7-kb and 4.2-kb deletions. MLPA and PCR results were compared to Southern blot (SB) results from 758 and 133 specimens, respectively. Lastly, MLPA and PCR results were reviewed and summarized from 5386 clinically tested specimens. SB and MLPA results were concordant in 678/687 (99%) specimens. PCR detected all deletions detected by SB with no false positives. No deletions or duplications were identified in 2630 (49%) clinically tested specimens. Extra α-globin copies were identified in 76 patients. A deletion of one or two α-globin genes was identified in 1251 (23%) and 1349 (25%) specimens, respectively, including 15 different genotypes. A deletion of three (hemoglobin H) and four α-globin genes (Hb Bart's) was observed in 65 or 3 specimens, respectively. Six patients had a deletion within the α-globin regulatory region MCS-R2. Thus, MLPA plus deletion PCR identify multiple α-globin gene deletions/duplications in patients being tested for α-thalassemia.
The α-thalassemias are a group of recessively inherited disorders caused by reduced or absent synthesis of the α-chain of hemoglobin (Hb). Approximately 5% of the world's population is thought to be affected by α-thalassemia, making it one of the most common genetic disorders.1 Because of its selective advantage, the carrier rate of α-thalassemia is extremely high (>90%) in tropical and subtropical regions with a history of endemic malaria.2 The phenotype of patients with α-thalassemia is variable depending on the number of dysfunctional or absent α-globin genes. Patients with one dysfunctional α-globin gene (silent carrier) are often asymptomatic and unaware they have the genetic disorder unless they have a complete blood count examined. Similarly, patients with a deletion of two of the four α-globin genes (α-thalassemia trait) may be asymptomatic and require no specific treatment. Hemoglobin H (three α-globin genes deleted/dysfunctional) is often more serious and is characterized by microcytic hypochromic hemolytic anemia, splenomegaly, and jaundice. All four of the α-globin alleles are dysfunctional in patients with Hb Bart's, the most severe and often fatal form of α-thalassemia.2,3
The two genes responsible for synthesis of the α-chain of Hb, HBA1 and HBA2, are located in the telomeric region on chromosome 16 (16p13.3) and encode α1-globin and α2-globin, respectively.2 These two genes are embedded within two highly homologous 4-kb duplication units that can be subdivided into separate homology blocks, written X, Y, and Z.2 Misalignment and recombination within these different homologous regions result in a variety of deletions and duplications that alter the number of α-globin genes, and represent the primary mechanism for α-thalassemia.4,5 There are currently more than 125 classified molecular defects that decrease the production of α-globin chains, including over 50 different deletions and 75 unique mutations.2,3
Numerous testing strategies have been used to detect deletions and mutations within the α-globin gene cluster, including Southern blot (SB), PCR, multiplex ligation-dependent probe amplification (MLPA), and sequence analysis. SB is not as commonly used today because it is time consuming, labor intensive, and requires the use of radioactive materials. The Sanger method of sequencing promoters, exons, and exon–intron boundaries is sensitive for detecting mutations in the α-globin gene cluster, but is currently expensive, time consuming, and unable to detect large deletions. Since its first description in 2002, MLPA has become a favorable technique to assess for deletions or duplications within the α-globin gene region.6 Our institution recently implemented a combined laboratory–developed Luminex-based MLPA and deletion PCR assay (Luminex Corporation, Austin, TX) that can identify a majority of the deletions or duplications that occur in the α-globin gene cluster. This report reviews the development and implementation of this combined assay, and reviews the results from the first 5386 clinically tested specimens.
Materials and Methods
Development of MLPA Assay
A laboratory-developed MLPA assay was used for detecting deletions and duplications within the α-thalassemia gene cluster using both Luminex FlexMAP tag/anti-tag system7,8 and MLPA technologies.6 Twenty MLPA probes were prepared by Integrated DNA Technologies (Coralville, IA). The 20 probes included 4 control probes targeting chromosomal regions 1p22 (probe 1), 3p22 (probe 2), 12p13 (probe 3), and 22q11 (probe 4), 1 gender control probe targeting Xp21 (probe 5), and 15 probes (probes 6 to 20) that spanned from the 5′ regulatory region to the 3′ hypervariable region of the α-globin gene cluster. All MLPA probes were designed using universal primers and an anti-tag sequence complimentary to sequences on Luminex FlexMap beads.
To assess the performance characteristics of this MLPA assay, we performed testing on 678 specimens whose DNA had been extracted using standard techniques and clinically tested by SB for α-thalassemia. MLPA testing began by denaturing 5 μL of patient DNA (diluted to 80 ng/μL) at 98°C for 5 minutes and cooling it to 25°C for 1 minute. The denatured DNA is then mixed with 3 μL of probe mix, which contains equal volumes of the MLPA probe mix and MLPA buffer, and then hybridized at 95°C for 1 minute, followed by incubation at 60°C for 16 to 24 hours. All ligation reagents for MLPA testing were purchased from MRC-Holland (Amsterdam, the Netherlands). Ligation was performed by adding 32 μL of Ligase-65 master mix to the specimen DNA, which was incubated at 54°C for 15 minutes, followed by 95°C for 5 minutes, and a 4°C hold. PCR amplification was then performed using 40 μL of a PCR master mix [26-μL water, 5-μL 10× buffer, 4-μL dNTP (10 mmol/L), 3-μL MgCl2 (25 mmol/L), 0.5 μL of forward and reverse primers, and 1 μL of Platinum Taq polymerase (Life Technologies)] and a 10-μL aliquot of the ligated product.
PCR conditions were as follows: 95°C for 5 minutes, 23 cycles of 95°C for 30 seconds, 60°C for 30 seconds, and 72°C for 60 seconds, followed by a 20-minute final extension at 72°C and a 4°C hold.
After amplification, beads were hybridized by mixing 40 μL of FlexMap bead mix with 10 μL of PCR product in a 96-well plate. The plate was heated to 96°C for 2 minutes followed by incubation at 37°C for 60 minutes. Immediately following bead hybridization, 25 μL of streptavidin phycoerythrin/tetramethylammonium chloride reporter solution [1 μL of streptavidin, R-Phycoerythrin conjugate (1 mg/mL) per 150 μL of 1× TMAC solution] was added to each reaction well. The number of FlexMap beads that successfully hybridized was then enumerated with a Luminex LX100. A median fluorescence intensity value was generated for each of the 20 probe sets for each sample by the Luminex LX100 and saved as a .csv file.
For quantitative data analysis, data from the Luminex LX100 were evaluated using a customized GeneMarker Software program (SoftGenetics, State College, PA). The median fluorescence intensity that was generated for each sample was normalized and compared to control probes (expressed as peak ratio) to determine gene dosage. Probes with a ratio greater than 1.2 (relative to the control probes) were considered gained, whereas probes with ratios below 0.80 were interpreted as deleted for that specific probe region. The thresholds were selected from previous work at our institution where we evaluated six different control probes on 102 specimens and selected threshold ratio values (0.8 and 1.2 for loss and gain, respectively) that exceeded the mean normalized ratio ± 2 SD for each of the control probes.
Multiplex PCR
A multiplex PCR assay was also developed along with MLPA in our laboratory to assess for heterozygous and homozygous 3.7-kb and 4.2-kb deletions within the α-globin gene cluster. The PCR multiplex, first described by Tan et al,9 was prepared by combining 1.2 mmol/L of the forward and reverse 3.7 primers and 5.0 mmol/L of the forward and reverse 4.2 primers. We also added 3.8 mmol/L of the forward and reverse control primers targeting exon 1 of the NPC1 gene (18q11.2) to the PCR multiplex. Three microliters (20 mmol/L) of the PCR primer multiplex, along with 1 μL of patient DNA, was then added to the PCR master mix containing 7.13-μL water, 3.125-μL Platinum Taq buffer, 1.5-μL MgCl2 (25 mmol/L), 2.0-μL dNTP (10 mmol/L), 1.25-μL DMSO, 1.0-μL Platinum Taq polymerase (5 U/μL), and 5-μL betaine. PCR amplification was carried out for 30 cycles (45 seconds at 98°C, 90 seconds at 63.4°C, and 135 seconds at 72°C). Twenty-five microliters of the PCR product, along with appropriate assay controls, were then run on a 2% agarose gel for 2 hours at 110 V and captured using an AlphaImager imaging system (Alpha Innotech, San Leandro, CA).
Clinical Testing Results
The Luminex-based MLPA and deletion PCR α-thalassemia test was clinically implemented at our institution in June 2007. A retrospective review of results obtained from patients undergoing α-thalassemia clinical testing between June 2007 and April 2010 was collected from our reporting system for analysis. Specimens analyzed during this timeframe represented both Mayo Clinic patient specimens and specimens tested through Mayo Medical Laboratories. This study was approved by the Mayo Clinic institutional review board.
Results
MLPA Test Development
The probes for MPLA (Figure 1) were used on residual DNA from 758 patients being tested for α-thalassemia (Table 1). All specimens underwent prior SB analysis as part of routine clinical testing. Interpretable MLPA results were obtained in 687 (91%) of the 758 tested specimens, with 71 (9%) being classified as uninterpretable due to poor sample quality or low sample volume. SB and MLPA results were concordant in 678 (99%) of the 687 interpretable specimens (Table 2). Among the nine discordant cases, five had an αααanti-3.7/-α3.7 SB result, whereas the MLPA findings suggested αα/αα (although αααanti-3.7/-α3.7 could not be ruled out without the deletion PCR). Two other discordant specimens had an SB αα/-α3.7 and MLPA -α3.7/-α3.7 result. Further investigation of both specimens revealed that the incorrect result was reported by SB and that the MLPA results (-α3.7/-α3.7) were accurate. MLPA identified a deletion in the MSC-R2 (HS-40) regulatory region from another discordant case. The SB result was normal on this specimen because the deletion was outside the detection range of the SB assay. The last discordant specimen was interpreted as an αα/αααanti-3.7 mosaic by SB, whereas the MLPA result was αα/αα. A re-review of the MLPA result hinted at a mosaic finding similar to that observed on the SB, but would have been too ambiguous to report in a clinical setting.
Figure 1.
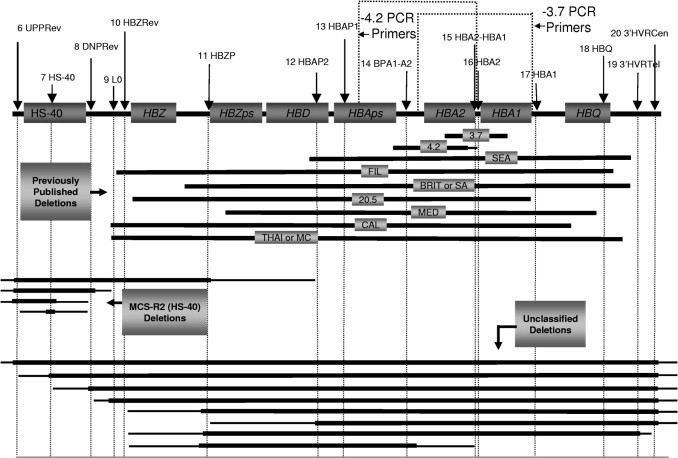
Location of MLPA probes (probes 6 to 20) and corresponding different-sized deletions that were observed during clinical testing within our laboratory. Probes 1 to 5 are excluded from this figure because they represent control/gender probes specific for other chromosomal regions that are not located within the α-globin gene cluster.
Table 1.
Primer and Probe Sequences
| Deletion PCR primers | ||
|---|---|---|
| Primer ID | Probe⁎ | Sequence |
| 3.7 | Forward | 5′-CCCCTCGCCAAGTCCACCC-3′ |
| Reverse | 5′-AAAGCACTCTAGGGTCCAGCG-3′ | |
| 4.2 | Forward | 5′-GGTTTACCCATGTGGTGCCTC-3′ |
| Reverse | 5′-CCCGTTGGATCTTCTCATTTCCC-3′ | |
| Control 18q11.2 | NPC1 forward | 5′-CCCCAGAACAGCACTAC-3′ |
| NPC1 reverse | 5′-CATCGCCAGACCAACTTC-3′ | |
| Multiplex-ligation probe amplification probes⁎ | ||
|---|---|---|
| Probe ID | Probe⁎ | Sequence |
| Control 1 1p22.1 | BCAR3-P1 | 5′-GTTCCTCAAGGTTCCCTCGTCTC-3′ |
| BCAR3-P2 | 5′-CCTCTGCCTGGCTCAACTCAGAGGCCAACTACTGTGAACTGAACCAATCTACACTAACAATTTCATAAC-3′ | |
| Control 2 3p22.1 | CTNNB-P1 | 5′-GTATGCAATGACTCGAGCTCAGAG-3′ |
| CTNNB-P2 | 5′-GGTACGAGCTGCTATGTTCCCTGAGACATTAGATGAGGGCATGCAGTACACTTTATCAAATCTTACAATC-3′ | |
| Control 3 12p13.3 | TNFRSF7-P1 | 5′-GCAACTGCACCATCACTGCCAATG-3′ |
| TNFRSF7-P2 | 5′-CTGAGTGTGCCTGTCGCAATGGCTGGCAGTGCAGGGACAAGGAGTGCTTCAATCATTCAAATCTCAACTTT-3′ | |
| Control 4 12p13.3 | HIRA-P1 | 5′-GCATTCACCAGTCCACCTATG-3′ |
| HIRA-P2 | 5′-GCAAGAGCCTAGCCATCATGACCGAGGCCCAGCTCTCCACAGCAATCCTTTTTACTCAATTCAATCA-3′ | |
| Gender Probe Xp21.1 | DMD-P1 | 5′-TTTGTTTTTCCATGCTAGCTACCCTGAGGCA-3′ |
| DMD-P2 | 5′-TTCCCATCTTGAATTTAGGAGATTCATCTGCTCTTGTACCTATAAACATATTACATTCACATC-3′ | |
| 6 | UPPrev-P1 | 5′-AGCAGGGGAATCAGCACCATTGAGCAG-3′ |
| UPPrev-P2 | 5′-GGTGTTTTCTGATCTGTGTGCATGGTTAGTTACCTTTATACCTTTCTTTTTAC-3′ | |
| 7 | HS40-P1 | 5′-GTGAATGGTACTGCTGATTACAACCTCTGGTGCTGCCTCCCCCTC-3′ |
| HS40-P2 | 5′-CTGTTTATCTGAGAGGGAAGGCCATGCCCAAAGTGCTTTTCATCTTTTCATCTTTCAAT-3′ | |
| 8 | DNPrev-P1 | 5′-ATCAGCATGGTCGCCCGTGTTGCTG-3′ |
| DNPrev-P2 | 5′-GCAGCGTATCTGCTACGCGGCTTACCTTTCAATTACAATACTCATTACA-3′ | |
| 9 | L0-P1 | 5′-GTGACCAGGGAGGGCCAGTTCATCTCGGTCTGAAAGAAGC-3′ |
| L0-P2 | 5′-CCCAGATGAGCAAAGGATACACTGGCCTCCTGTACACTTTCTTTCTTTCTTTCTTT-3′ | |
| 10 | HBZrev-P1 | 5′-CTGGATTCCTGGGTTTCCTCCAAGAAAGCAAGG-3′ |
| HBZrev-P2 | 5′-GTTAGGTCCACCCAGCGCTGAGGGGAGATGTACTGGATCTGCTTTAATCCTTTATCACTTTATCA-3′ | |
| 11 | HBZP-P1 | 5′-CTCTAGGTCACCCTGTCATCACAGGGACAGGGAG-3′ |
| HBZP-P2 | 5′-GTCAAGGACAGTCACTCCTGAGGCCACTTTAATCTCAATCAATACAAATC-3′ | |
| 12 | HBAP2rev-P1 | 5′-TTTTCGGTCAGCACCACGGCCACACCAGTCAG-3′ |
| HBAP2rev-P2 | 5′-GAACTTGTCCCACGCCGCTTGCATTTGCACATCATACATACATACAAATCTACA-3′ | |
| 13 | HBAP1-P1 | 5′-GACTCAGAAATAAGCTGCCGTGGTGCTGTCTC-3′ |
| HBAP1-P2 | 5′-CTGAGGACAAGGCTAACACCAAGGCGGTCTGGGAGATCAAAATCTCAAATACTCAAATCA-3′ | |
| 14 | BPA1-A2-P1 | 5′-CATCCCATGCTGAGGGAACACAG-3′ |
| BPA1-A2-P2 | 5′-CTACATCTACAACTACTGCCACAGGCTCTCTCTACAAACAAACAAACATTATCAA-3′ | |
| 15 | HBA2-HBA1-P1 | 5′-GTGCCAGAACATTTCTCTCATTCC-3′ |
| HBA2-HBA1-P2 | 5′-CACCCCTTCCTGCCAGAGGGTAGGTGTACACAATCTTTTCATTACATCAT-3′ | |
| 16 | HBA2-P1 | 5′-GAAGATCCAACGGGGGAAGCATTG-3′ |
| HBA2-P2 | 5′-CTAAGCTGGTCGGAGCTACTTCCTTCTCATTCATATACATACCAATTCAT-3′ | |
| 17 | HBA1-P1 | 5′-GTGCCAGAACATTTCTCTCATTCC-3′ |
| HBA1-P2 | 5′-CACCCCTTCCTGCCAGAGGGTAGGTGTACACAATCTTTTCATTACATCAT-3′ | |
| 18 | HBQ-P1 | 5′-CTGGACAAGTTCCTGAGCCAC-3′ |
| HBQ-P2 | 5′-GTTATCTCGGCGCTGGTTTCCGAGTAATCATACTCAACTAATCATTCAA-3′ | |
| 19 | 3′HVRTRrev-P1 | 5′-GAGTGTGAGTTTGCACCTGGGTTTCCCTGG-3′ |
| 3′HVRTRrev-P2 | 5′-GCCTTAAGCAGACAGCGTCACCCTCAGAGCCATCCTACATATTCAAATTACTACTTAC-3′ | |
| 20 | 3′HVRcen-P1 | 5′-CTCAAGGGTGGCATGTGTACC-3′ |
| 3′HVRcen-P2 | 5′-CCTGCAGAAACAGAGCGGATGAGGACAATTTACTCATATACATCACTTT-3′ | |
This is the genomic-specific portion of the MLPA probe. Each probe will also have a Universal Primer Sequence included. The P2 probes also contain FlexMap AntiTag sequences.
Table 2.
Comparison of MLPA and Southern Blot Analysis
| Southern blot | Concordant | Not concordant | MLPA failed | Total |
|---|---|---|---|---|
| No apparent deletions (αα/αα) | 317 | 1⁎ | 38 | 356 |
| No apparent deletions with extra α copies | 16 | 1 | 1 | 18 |
| αα/αααanti-3.7 | 11 | 0 | 1 | 12 |
| αα/αααanti-3.7(mosaic) | 0 | 1† | 0 | 1 |
| αα/αααanti-4.2 | 1 | 0 | 0 | 1 |
| αα/ααααanti-3.7 | 1 | 0 | 0 | 1 |
| αα/αααααanti-3.7 | 3 | 0 | 0 | 3 |
| Deletion and extra α copies (4 copies total) | 0 | 5 | 0 | 5 |
| αααanti-3.7“/-α3.7 | 0 | 5† | 0 | 5 |
| Silent (3 α-globin genes present) | 143 | 2 | 17 | 162 |
| αα/-α3.7 | 138 | 2‡ | 17 | 157 |
| αα/-α4.2 | 5 | 0 | 0 | 5 |
| Trait (2 α-globin genes present) | 190 | 0 | 15 | 205 |
| -α3.7/-α3.7 | 130 | 0 | 11 | 141 |
| -α3.7/-α4.2 | 4 | 0 | 0 | 4 |
| αα/–Other deletion | 9 | 0 | 0 | 9 |
| αα/–SEA | 1 | 0 | 0 | 1 |
| αα/–Other deletion | 1 | 0 | 0 | 1 |
| αα/–20.5 | 1 | 0 | 0 | 1 |
| αα/—Brit | 1 | 0 | 0 | 1 |
| αα/–Fil | 4 | 0 | 0 | 4 |
| αα/–SEA | 39 | 0 | 4 | 43 |
| Hemoglobin H (1 α-globin gene present) | 11 | 0 | 0 | 11 |
| -α3.7/–Other deletion | 1 | 0 | 0 | 1 |
| -α3.7/–SEA | 6 | 0 | 0 | 6 |
| -α3.7/–Thai | 1 | 0 | 0 | 1 |
| -α4.2/–Other deletion | 2 | 0 | 0 | 2 |
| -α4.2/–SEA | 1 | 0 | 0 | 1 |
| Bart (–/–) (no α-globin genes present) | 1 | 0 | 0 | 1 |
| Total | 678 (89%) | 9 (1%) | 71 (9%) | 758 |
Deletion identified in MCS-R2 (HS-40) regulatory region by MLPA (outside range of SB).
MLPA interpreted as αα/αα.
MLPA result (-α3.7/-α3.7) was correct (SB result was misinterpreted).
PCR Deletion Test Development
Deletion PCR was performed on 133 residual DNA specimens with the following SB results: heterozygous 3.7-kb deletion (n = 26; 20%), homozygous 3.7-kb deletion (n = 27; 20%), heterozygous 4.2-kb deletion (n = 7; 5%), homozygous 4.2-kb deletions (n = 1; 1%), both heterozygous 3.7-kb and 4.2-kb deletions (n = 2; 2%), and specimens without a 3.7-kb or 4.2-b deletion (n = 70; 53%). The 70 tested patients without a 3.7-kb or 4.2-kb deletion by SB included those with an: αα/αα (n = 46); αα/αααanti-3.7 (n = 5); αα/ααααanti-3.7 (n = 1); αα/αααααanti-3.7 (n = 3); αα/ααααlarge duplication (n = 1); αα/–SEA (n = 9); and αα/–Fil (n = 5) result. The overall concordance between deletion PCR and SB was 100%, with PCR detecting all 63 specimens with a 3.7-kb and/or 4.2-kb deletion but not detecting the targeted deletions in the 70 remaining specimens.
Clinical Testing
A total of 5386 specimens was clinically tested using the MLPA and PCR assay between June 2007 and April 2010. The results of these findings (Table 3, Figures 2 and 3) are summarized. Overall, 50% (n = 2711) of individuals had at least four α-globin genes. This included 2630 patients with no identifiable deletions (αα/αα; Figure 2, A and B), 76 patients with no deletions and five or more α-globin genes (Figure 2G), and 5 patients with a 3.7-kb deletion on one allele and three or four α-globin genes on the other allele (Figure 2E). The five patients with the 3.7-kb deletion and extra copies of the α-globin gene have a sufficient number (four or five) of α-globin genes for their own protein production; however, these patients are at risk for having a child with hemoglobin H if their partner has an αα/– genotype. A large proportion (91%) of the 76 individuals with extra copies of the α-globin gene and no deletions had αα/αααanti-3.7 (n = 57) and αα/αααanti-4.2 (n = 12) genotypes. Five (7%) individuals had a large duplication of the α-globin gene cluster ranging in size from ≥20.9 kb to ≤87.0 kb, with the largest duplication spanning beyond the detectable range of our MLPA assay. In one patient, we were unable to determine whether the patient had a genotype of αα/αα, αα/-αother deletion, or αααanti-3.7/-α20.5. This patient had a deletion that encompassed the targets of MLPA probes 11 to 14 (Figure 3H), suggesting that the deletion could range from 8.2 to 21.7 kb and, depending on the size of the deletion, might delete the α2 gene. It is also possible that the genotype of this individual is αααanti-3.7/-α20.5, suggesting a larger deletion (probes 11 to 16) that would not be evident by MLPA if an extra anti-3.7 α-globin gene was present.
Table 3.
Summary of Clinical Testing Results
| Result | Patients (N) | Total population (%) |
|---|---|---|
| No deletions (αα/αα) | 2630 | 49 |
| No deletions with extra α copies | 76 | 1 |
| αα/αααanti-3.7 | 57 | 1 |
| αα/αααanti-4.2 | 12 | <1 |
| αα/ααααLarge duplication | 4 | <1 |
| αα/ααααanti-3.7or αααanti-3.7/αααanti-3.7 | 2 | <1 |
| αα/αααLarge duplication | 1 | <1 |
| Deletion and extra α copies (total of ≥4 α copies) | 5 | <1 |
| αααanti-3.7/-α3.7 | 4 | <1 |
| ααααanti-3.7/-α3.7 | 1 | <1 |
| Normal or Silent (αα/αα, αα/-αother deletion or αααanti-3.7/-α20.5) | 1 | <1 |
| Silent (3 α-globin genes present) | 1251 | 23 |
| αα/-α3.7 | 1216 | 23 |
| αα/-α4.2 | 31 | 1 |
| αααanti-3.7/–SEA | 3 | <1 |
| αααanti-4.2/–SEA | 1 | <1 |
| Trait (2 α-globin genes present) | 1349 | 25 |
| -α3.7/-α3.7 | 911 | 17 |
| αα/–SEA | 337 | 6 |
| αα/–FIL | 46 | 1 |
| -α3.7/-α4.2 | 19 | <1 |
| αα/—BRIT or SA | 10 | <1 |
| αα/–Other deletion | 10 | <1 |
| αα/–MED | 7 | <1 |
| -α4.2/-α4.2 | 4 | <1 |
| αα/–CAL | 3 | <1 |
| αα/–THAI or MC | 1 | <1 |
| αα/–20.5 | 1 | <1 |
| MCS-R2 (HS-40) deletion (4 α-globin genes present) | 5 | <1 |
| Trait or HbH (HS-40 deletion with αα/-α3.7) | 1 | <1 |
| Hemoglobin H (1 α-globin gene present) | 65 | 1 |
| -α3.7/–SEA | 42 | 1 |
| -α3.7/–FIL | 8 | <1 |
| -α3.7/–BRIT or SA | 6 | <1 |
| -α4.2/–SEA | 3 | <1 |
| -α3.7/–Other deletion | 3 | <1 |
| -α3.7/–MED | 2 | <1 |
| -α3.7/–20.5 | 1 | <1 |
| Bart (–/–) (no α-globin genes present) | 3 | <1 |
| Total | 5386 | 100 |
Figure 2.
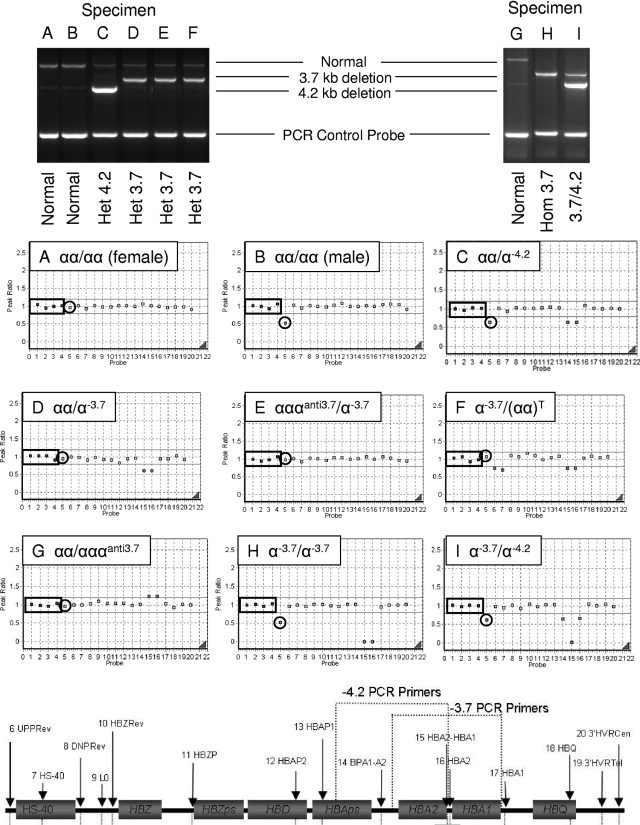
Matched PCR (top) and MLPA results (middle) from nine specimens labeled A–I. Probes 1 to 4 (in small boxes, in panels A–I) represent control probes targeting chromosomes 1p22, 3p22, 12p13, and 22q11, respectively. Probe 5 (circled) represents the gender probe targeting Xp21. The remaining 15 probes (probes 6 to 20) target the α-globin gene cluster. MLPA probes 6 to 20 and 3.7-kb/4.2-kb PCR primer targets are shown at the bottom of the figure. Het, heterozygous; Hom, homozygous.
Figure 3.

MLPA results representing eight unique deletions. Probes 1 to 4 (in small boxes, in panels A–H) represent control probes targeting chromosomes 1p22, 3p22, 12p13, and 22q11, respectively. Probe 5 (circled) represents the gender probe targeting Xp21. The remaining 15 probes (probes 6 to 20) target the α-globin gene cluster as shown at the bottom of the figure. *αα/(αα)T = deletion in MCS-R2 regulatory region.
A deletion of one α-globin gene (silent carrier) was identified in 23% (n = 1251) of tested individuals representing four different genotypes. The four genotypes included the αα/-α3.7 (n = 1216; Figure 2D), αα/-α4.2 (n = 31; Figure 2C), αααanti-3.7/–SEA (n = 3), and αααanti-4.2/–SEA (n = 1). Eleven different-sized deletions were identified in 1349 patients with α-thalassemia trait (Table 3). Of those, the most common deletions included -α3.7/-α3.7 (n = 911; Figure 2H), αα/–SEA (n = 337; Figure 3B), αα/–Fil (n = 46; Figure 3E), and -α3.7/-α4.2 (n = 19; Figure 2I). Ten patients had a genotype of αα/–Brit or SA (Figure 2D). Although the αα/–Brit genotype is more common than the αα/–SA (South African) genotype, the size (∼23 and 26 kb for the –SA and –Brit deletions, respectively) and location of the deletions are similar between the two and give the same MLPA result (reduction in probes 11 to 18; Figure 3D).
Five patients had a deletion within MSC-R2 (HS-40; Figures 1 and 3A) regulatory region, suggesting that these patients would also have a phenotype of α-thalassemia trait. Interestingly, we also had a patient with a deletion within the MSC-R2 region in conjunction with a -α3.7 deletion (Figure 2F), suggesting either αα/(-α3.7)T (trait), or -α3.7/(αα)T (HbH) depending on whether the deletions are in cis or trans. A diagnosis of Hemoglobin H was assigned to 65 patients with genotypes -α3.7/–SEA (n = 42), -α3.7/–Fil (n = 8), -α3.7/–Brit or SA (n = 6), -α4.2/–SEA (n = 3), -α3.7/–Other deletion (n = 3; Figure 3G), -α3.7/–Med (n = 2), and -α3.7/–20.5 (n = 1). Lastly, the most severe and lethal form of α-thalassemia, Hb Bart's (−/−), was diagnosed in 3 of the 5386 specimens tested.
Discussion
In this study, we describe the development and implementation of a laboratory-developed Luminex-based MLPA and deletion PCR assay used to screen for deletions and duplications within the α-globin gene cluster. The first report describing the MLPA technique was published by Schouten et al6 in 2002. Three years later, Harteveld and colleagues10 designed multiple MLPA probe sets targeting novel deletions in and around both the α- and β-globin gene clusters. Since then, multiple investigators have used MLPA to assess for a variety of deletions and mutations within the α-globin gene cluster and flanking genes.11–15 A majority of these publications use MLPA primarily as a tool for detecting and classifying novel deletions that remain uncharacterized by prior testing methods such as SB, sequence analysis, and gap-PCR. To date, there are no known studies other than this study describing the utility of MLPA for detecting α-thalassemia in a routine clinical laboratory setting.
Before implementing this test clinically, our laboratory-developed MLPA assay was validated by comparing our MLPA testing results to clinically validated SB results. As summarized in Table 2, the overall concordance between interpretable MLPA and SB results was 99%, suggesting that MLPA is an accurate technique for diagnosing α-thalassemia. There were, however, nine discordant cases that required further investigation. On further review, MLPA was actually correct in three of the nine specimens due to errors of SB interpretation and due to a MCS-R2 deletion that was not detectable using our SB method. Nonetheless, MLPA failed to produce the correct genotype in the remaining six discordant cases. Because MLPA is a technique that assesses total gene dosage, this assay is only able to determine the total number of HBA1 and HBA2 genes, not the number of genes on each allele. As a result, there were five specimens that had a 3.7-kb duplication on one allele and a 3.7-kb deletion on the other allele (αααanti-3.7/-α3.7) that were incorrectly interpreted as αα/αα by MLPA because there were four α-globin genes present (Figure 2E). It was apparent from these findings that a deletion assay targeting the common 3.7-kb and 4.2-kb deletions was needed in conjunction with MLPA to differentiate αα/αα from the αααanti-3.7/-α3.7 and αααanti-4.2/-α4.2 genotypes, respectively.
In 2001, Tan en al9 described the development of a PCR assay that could reliably detect seven common deletions (-α3.7, -α4.2,-α20.5,–SEA, –FIL, –MED, and –THAI) known to be associated with α-thalassemia. On the basis of this information, we tested the accuracy of this PCR deletion assay using -α3.7 and -α4.2 primers (Table 1). In our PCR validation cohort, PCR detected all of the -α3.7 and -α4.2 deletions that were identified by SB. In addition, none of the patients without the -α3.7 and -α4.2 deletions were positive using these primers. On the basis of the results from both our MLPA and PCR development data, we implemented the combined MLPA/deletion PCR assay in our laboratory in June 2007.
Since introduction of this combined test clinically, we have observed 21 different-sized deletions (Figures 1–3). Eight of these deletions are unclassifiable because they have not been described elsewhere (n = 2; Figure 1) or because they extend beyond the limits of our MLPA assay (n = 6; Figures 1 and 3F). There are also some deletions that we cannot differentiate because they are similar in size and location. For example, our MLPA assay is not able to differentiate between the –Brit and –SA deletions (Figure 3D) because the 5′ end of these deletions are only 3 kb apart and the deletions are similar in size (∼23 and 26 kb for the –SA and –Brit deletions, respectively) (A Database of Human Hemoglobin Variants and Thalassemias, http://globin.bx.psu.edu/hbvar/menu.html, last accessed June 22, 2010). Similarly, we are unable to distinguish between the –MC and –TAI because both encompass probes 9 to 18 in our MLPA assay; however, determining whether a deletion is –Brit, –SA, –MC, or –TAI is of limited clinical value because all of these deletions remove both α-globin genes on that allele. As a result, it is common practice for us to simply report these findings as a deletion of two α-globin genes in cis without reporting the actual size of the deletion.
One exception to not reporting the size of deletions when both α-globin genes are deleted is when the deletion extends beyond the range of our MLPA assay (Figure 3F). In these patients, an unusual form of α-thalassemia called ATR-16 syndrome (OMIM: 141750) must be considered. ATR-16 is a contiguous gene syndrome causing α-thalassemia, developmental abnormalities, and mental retardation.2,12 This syndrome is thought to arise from large chromosomal rearrangements (translocations, inversions/deletions, and subtelomeric truncations) that delete many genes on the short arm of chromosome 16.4 The phenotype of patents with ATR-16 is variable depending on the size of the deletion, which can range from ∼800 to 2000 kb.4,12 Patients with deletions larger than 2000 kb often have more severe mental retardation, tuberous sclerosis, and polycystic kidney disease.2,4 Further testing (eg, conventional cytogenetics, MLPA using additional probes, array comparative genomic hybridization, fluorescence in situ hybridization) may be warranted in individuals who harbor large deletions by MLPA to rule out ATR-16 syndrome, especially if the patient has evidence of developmental delay and/or mental retardation.
Not all types of α-thalassemia are caused by deletions involving the α-globin genes. Deletions in the regulatory region of these genes are also associated with α-thalassemia.2 Human α-globin genes appear to be regulated by one or more regulatory multispecies conserved sequences (MSC-R1 to R4), which correspond to erythroid-specific DNAse1 hypersensitive sites HS-48, HS-40, HS-33, and HS-10, respectively.3,16,17 These regulatory elements are located 10 to 48 kb upstream of the α-globin gene cluster. Deletion of MCS-R2, a 1.1-kb fragment containing HS-40, results in down-regulation of human α-globin gene expression to < 5% of normal.16,17 The exact mechanism of how MSC-R2 regulates gene expression is still unclear. Recent data suggest that intrachromosomal looping allows interaction between protein complexes at the MCS-R2 region and α-globin gene promoters.16,17 Other mechanisms, including tracking, facilitated tracking, and linking, have been proposed by which the regulatory region and promoters interact.16 In our study, there were six patients with a deletion of the MSC-R2 region. Five of these patients had a deletion within the MSC-R2 region without another identifiable deletion [written as αα/(αα)T; Figure 3A].3 However, one patient had a 3.7-kb deletion in addition to the HS-40 deletion suggesting either αα/(-α3.7)T or -α3.7/(αα)T, depending on whether the deletions were in cis or trans. Unfortunately, our current MLPA/PCR assay is unable to determine whether noncontiguous deletions are on the same allele. In rare cases where MLPA identifies noncontiguous deletions, SB analysis, follow-up molecular studies of parents, and/or other clinical/laboratory findings may be necessary to help genotype the individual.
Since the first report on nondeletion α-thalassemia in 1977,18 approximately 70 disease-causing mutations have been reported within the α-thalassemia critical region, nearly two-thirds occurring in the HBA2 gene (written αα/αTα).2 These pathogenic mutations affect mRNA processing (cryptic splicing and altered polyadenylation site sequences), mRNA translation (nonsense mutations and mutations in initiation or termination codons), or α-globin protein stability.2,3 As a group, nondeletion types of α-thalassemia appear to have a more severe clinical phenotype as compared to patients with a simple deletion. It is for this reason that some MLPA assays include probes that detect some of the common mutations associated with α-thalassemia.13 Because of the increasing number of mutations being identified, it may be beneficial to incorporate additional MLPA probes into our current assay, especially the frequent mutations. A better solution may be to sequence the α-globin gene region in patients where α-thalassemia is still suspected after negative α-globin gene deletion testing. Unfortunately, there is currently no one test that detects all of the known causes of α-thalassemia.
In conclusion, this report is the largest report to date describing the use of a combined laboratory–developed Luminex-based MLPA and deletion PCR assay for the detection of α-thalassemia. In the last 4 years, we have identified 21 different-sized deletions representing 23 different genotypes. Some of the identified deletions would not be detectable using commonly used PCR or SB methods. The findings of this study are similar to previous reports suggesting that both MLPA and PCR are reliable techniques for detecting a variety of pathogenic deletions within the α-globin gene cluster. When used together, however, MLPA plus a deletion PCR assay can more accurately identify the wide variety of α-globin gene deletions or duplications that occur in patients undergoing α-thalassemia testing.
References
- 1.Vichinsky E.P. Alpha thalassemia major—new mutations, intrauterine management, and outcomes. Hematology Am Soc Hematol Educ Program. 2009:35–41. doi: 10.1182/asheducation-2009.1.35. [DOI] [PubMed] [Google Scholar]
- 2.Higgs D.R. Disorders of Hemoglobin. In: Steinberg M.H., Forget B.G., Higgs D.R., Weatherall D.J., editors. Cambridge University Press; New York, NY: 2009. pp. 239–320. [Google Scholar]
- 3.Harteveld C.L., Higgs D.R. Alpha-thalassaemia. Orphanet J Rare Dis. 2010;5:21. doi: 10.1186/1750-1172-5-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Higgs D.R., Weatherall D.J. The alpha thalassaemias. Cell Mol Life Sci. 2009;66:1154–1162. doi: 10.1007/s00018-008-8529-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lam K.W., Jeffreys A.J. Processes of copy-number change in human DNA: the dynamics of {alpha}-globin gene deletion. Proc Natl Acad Sci U S A. 2006;103:8921–8927. doi: 10.1073/pnas.0602690103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schouten J.P., McElgunn C.J., Waaijer R., Zwijnenburg D., Diepvens F., Pals G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002;30:e57. doi: 10.1093/nar/gnf056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bortolin S., Black M., Modi H., Boszko I., Kobler D., Fieldhouse D., Lopes E., Lacroix J.M., Grimwood R., Wells P., Janeczko R., Zastawny R. Analytical validation of the tag-it high-throughput microsphere-based universal array genotyping platform: application to the multiplex detection of a panel of thrombophilia-associated single-nucleotide polymorphisms. Clin Chem. 2004;50:2028–2036. doi: 10.1373/clinchem.2004.035071. [DOI] [PubMed] [Google Scholar]
- 8.Monico C.G., Rossetti S., Schwanz H.A., Olson J.B., Lundquist P.A., Dawson D.B., Harris P.C., Milliner D.S. Comprehensive mutation screening in 55 probands with type 1 primary hyperoxaluria shows feasibility of a gene-based diagnosis. J Am Soc Nephrol. 2007;18:1905–1914. doi: 10.1681/ASN.2006111230. [DOI] [PubMed] [Google Scholar]
- 9.Tan A.S., Quah T.C., Low P.S., Chong S.S. A rapid and reliable 7-deletion multiplex polymerase chain reaction assay for alpha-thalassemia. Blood. 2001;98:250–251. doi: 10.1182/blood.v98.1.250. [DOI] [PubMed] [Google Scholar]
- 10.Harteveld C.L., Voskamp A., Phylipsen M., Akkermans N., den Dunnen J.T., White S.J., Giordano P.C. Nine unknown rearrangements in 16p13.3 and 11p15.4 causing alpha- and beta-thalassaemia characterised by high resolution multiplex ligation-dependent probe amplification. J Med Genet. 2005;42:922–931. doi: 10.1136/jmg.2005.033597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coelho A., Picanco I., Seuanes F., Seixas M.T., Faustino P. Novel large deletions in the human alpha-globin gene cluster: clarifying the HS-40 long-range regulatory role in the native chromosome environment. Blood Cells Mol Dis. 2010;45:147–153. doi: 10.1016/j.bcmd.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 12.Harteveld C.L., Kriek M., Bijlsma E.K., Erjavec Z., Balak D., Phylipsen M., Voskamp A., di Capua E., White S.J., Giordano P.C. Refinement of the genetic cause of ATR-16. Hum Genet. 2007;122:283–292. doi: 10.1007/s00439-007-0399-y. [DOI] [PubMed] [Google Scholar]
- 13.Liu J.Z., Han H., Schouten J.P., Wang L.R., Fan X.P., Duarte H.B., Zhu C.J., Cai R., Xiao B., Wang Q.T. Detection of alpha-thalassemia in China by using multiplex ligation-dependent probe amplification. Hemoglobin. 2008;32:561–571. doi: 10.1080/03630260802508111. [DOI] [PubMed] [Google Scholar]
- 14.Phylipsen M., Prior J.F., Lim E., Lingam N., Vogelaar I.P., Giordano P.C., Finlayson J., Harteveld C.L. Thalassemia in Western Australia: 11 novel deletions characterized by Multiplex Ligation-dependent Probe Amplification. Blood Cells Mol Dis. 2010;44:146–151. doi: 10.1016/j.bcmd.2009.12.011. [DOI] [PubMed] [Google Scholar]
- 15.Phylipsen M., Vogelaar I.P., Schaap R.A., Arkesteijn S.G., Boxma G.L., van Helden W.C., Wildschut I.C., de Bruin-Roest A.C., Giordano P.C., Harteveld C.L. A new alpha(0)-thalassemia deletion found in a Dutch family (–(AW)) Blood Cells Mol Dis. 2010;45:133–135. doi: 10.1016/j.bcmd.2010.05.004. [DOI] [PubMed] [Google Scholar]
- 16.Higgs D.R., Wood W.G. Long-range regulation of alpha globin gene expression during erythropoiesis. Curr Opin Hematol. 2008;15:176–183. doi: 10.1097/MOH.0b013e3282f734c4. [DOI] [PubMed] [Google Scholar]
- 17.Vernimmen D., Marques-Kranc F., Sharpe J.A., Sloane-Stanley J.A., Wood W.G., Wallace H.A., Smith A.J., Higgs D.R. Chromosome looping at the human alpha-globin locus is mediated via the major upstream regulatory element (HS-40) Blood. 2009;114:4253–4260. doi: 10.1182/blood-2009-03-213439. [DOI] [PubMed] [Google Scholar]
- 18.Kan Y.W., Dozy A.M., Trecartin R., Todd D. Identification of a nondeletion defect in alpha-thalassemia. N Engl J Med. 1977;297:1081–1084. doi: 10.1056/NEJM197711172972002. [DOI] [PubMed] [Google Scholar]