

Abstract
Gene-targeted mice deficient in the complement mannose-binding lectin-associated serine protease-1 and -3 (MASP1/3−/−) express only the zymogen of factor D (pro-factor D [pro-Df]), a necessary component of the alternative pathway (AP). We used the murine collagen Ab-induced arthritis (CAIA) model, in which the AP is unique among complement pathways in being both necessary and sufficient for disease induction, to determine whether MASP-1/3 are required in vivo for the development of tissue injury. Disease activity scores, complement C3 tissue deposition in the joint, and histopathologic injury scores were markedly decreased in MASP1/3−/− as compared with wild-type (WT) mice. MASP-1 protein was immunochemically localized to synovial cells of knees of WT mice with arthritis. Pro-Df was present in both synovial cells and chondrocytes of knees of WT and MASP1/3−/− mice without arthritis, with increased amounts present in synovial cells of WT mice with CAIA. No conversion of pro-Df to mature Df was detectable in the serum of MASP1/3−/− mice during the evolution of CAIA. C3 activation and deposition as well as C5a generation induced in vitro by adherent anti-type II collagen mAbs were absent using sera from MASP1/3−/− mice under conditions in which only the AP was active. The addition of human Df fully reconstituted in vitro C3 activation and C5a generation using sera from MASP1/3−/− mice. Our studies demonstrate for the first time, to our knowledge, the absolute requirement for the activity of MASP-1 protein in autoimmune-associated inflammatory tissue injury in vivo through activation of the AP of complement by cleavage of pro-Df to mature Df.
The complement system is a component of innate immunity that has evolved to play important roles in host resistance to pathogens, recognition and clearance of apoptotic and injured self-tissues and modulation of adaptive immunity (1-4). Complement activation in the fluid phase as well as in tissues and on the surface of cells takes place by three initiation pathways: the classical pathway (CP), alternative pathway (AP), and lectin pathway (LP). Each of the three initiation pathways has the capability to activate C3, the most abundant serum complement protein, through sequential proteolysis and generation of multiprotein enzyme complexes designated C3 convertases. Following C3 activation, C5 convertases are formed that lead to the cleavage of C5, liberation of C5a, and the generation of C5b, which itself catalyzes the formation of the porelike membrane attack complex.
Although the CP is typically initiated by C1 following engagement of C1q with IgM or IgG Abs (reviewed in Ref. 5) and the LP by the binding of mannose-binding lectin (MBL) and ficolins to carbohydrates or acetylated molecules (reviewed in Refs. 6-9), initiation of the AP is thought to occur by a process termed tickover. Tickover involves the spontaneous hydrolysis of C3 at a rate of ~0.5–1.0% per hour, the interaction of this conformationally altered C3 molecule with factor B and the subsequent cleavage of factor B by the protease factor D (Df). Cleavage of C3 results in the generation of C3b and the covalent attachment of C3b to target surfaces (reviewed in Ref. 10). The AP can also be engaged as an amplification loop when C3b generated from any of the three pathways binds factor B, resulting again in cleavage by Df (10). Notably, recent human genetic studies and the analysis of murine models of human diseases have supported the concept that the AP is the most important of the three pathways in the generation of pathogenic complement activation in vivo, either through direct initiation of the pathway or the magnifying effects of the amplification loop (11, 12).
One important feature of the AP is that, unlike the CP or LP, where the proteins are activated by sequential proteolysis, Df, a key component of the pathway, has been shown to exist in the circulation as a protease that does not require further posttranslational modification prior to binding its substrate factor B (13). Previous analyses have led to the conclusion that a proenzyme form of the molecule, designated pro-factor D (pro-Df), is converted to mature Df by the release of a 5 aa-containing peptide (QPRGR) from the N terminus coincident with its biosynthesis and release from adipocytes (14, 15). However, the precise mechanism of generation of mature Df in vivo has remained unclear. Pro-Df has been shown to be cleaved in vitro by trypsin and in the process releases the N-terminal peptide to generate mature Df. In addition, members of the clotting pathway including thrombin, kallikrein, and plasmin can also cleave pro-Df (14). Although readily demonstrable in vitro, the physiological relevance of these mechanisms to generate Df in vivo has been uncertain. Lastly, mature Df in circulation is inactive and undergoes conformational changes after binding to its substrate, C3bB, to become active.
In the LP, MBL and ficolins are physically associated with four proteins designated MBL-associated serum proteases (MASPs) (16, 17). There are four different types of MASPs that circulate: MASP-1 (18), MASP-2 (19), MASP-3 (20), and MBL-associated plasma protein of 19 kDa, a truncated form of MASP-2 (21, 22). Once MBL is bound to its target, MASPs are activated, resulting in the cleavage of C4 and C2 by MASP-2. MASP-1 can directly cleave C4 but not C2, and MASP-1 can weakly cleave C3 directly, although the biological relevance of this activity is uncertain (23). The MASP1/3 gene encodes both MASP-1 and MASP-3 via alternative splicing (21, 24). Despite their structural similarities, MASP-1 and MASP-3 bind to MBL independently, function independently, and regulate complement independently. For example, MASP-3 may negatively regulate the LP by inhibiting the activation of C4 and MASP-2 (20).
Recently, Takahashi et al. (25) have generated gene-targeted mice that are deficient in MASP-1 and MASP-3, designated MASP1/3−/−. The sera from these mice demonstrated decreased deposition of both C4 and C3 on mannan-coated plates, in comparison with sera from wild-type (WT) mice, and this ability was restored by the addition of rMASP-1K. Furthermore, rMASP-1K was shown to directly activate MASP-2, indicating an important role for MASP-1 in activation of the LP. Sera from MASP1/3−/− mice also demonstrated an inability to activate the AP in a hemolytic assay with rabbit erythrocytes or using zymosan-coated microwells with an associated lack of conversion of serum pro-Df to mature Df (26). MASP-3 may also cleave pro-Df to generate mature Df (26). Thus, MASP-1 and/or MASP-3 appear to be essential for activation of the AP.
In this study, we have examined whether the expression of MASP-1/3 is essential for the development of AP-mediated joint damage in vivo. We have previously demonstrated that the AP is both necessary and sufficient to induce disease in murine collagen Ab-induced arthritis (CAIA) (27, 28). This conclusion was confirmed in recent studies that showed an 88% reduction in clinical disease activity (disease activity score [DAS]) in Df−/− mice at day 10 in comparison with WT mice (DAS in WT mice 10 ± 1.4, mean ± SEM, n = 5, versus Df−/− mice 1.2 ± 0.5, n = 5; p < 0.001) (29). By examining CAIA in MASP1/3−/− mice, we can determine the in vivo importance of MASP-1/3 on activation of the AP through the conversion of pro-Df to mature Df.
Materials and Methods
Mice
Ten-week-old C57BL/6 MASP1/3−/− male and female mice were obtained by breeding of MASP1/3+/− mice (25, 26). MASP-1 and MASP-3 mRNA and protein are absent in MASP1/3−/− mice. Homozygous Df−/− mice on a C57BL/6 background (30) were bred for immunohistochemical studies and to obtain sera for in vitro studies. C4−/−, C3−/−, Bf−/−, C1q−/−Df−/−, and Df−/− mice were all bred onto the C57BL/6 strain at the University of Colorado School of Medicine (Aurora, CO); all genetically deficient mice were bred for at least seven generations. Sera from MBL−/−/FCN A−/− mice (31) were contributed by Dr. K. Takahashi (Massachusetts General Hospital for Children, Boston, MA). For MASP-2 immunohistochemical staining, formalin-fixed knee joints from MASP2−/− mice on a C57BL/6 background were obtained from Drs. T. Fujita and M. Takahashi (Fukushima Medical University School of Medicine, Fukushima, Japan). Ten-week-old C57BL/6 male and female age-matched mice obtained from The Jackson Laboratory (Bar Harbor, ME) were used as controls. Mice were maintained five to a cage in filter-top cages in a barrier animal facility with a climate-controlled environment with 12-h light/dark cycles. All mice were fed breeder’s chow provided by the Center for Laboratory Animal Care, University of Colorado School of Medicine.
Genotyping MASP1/3 gene-targeted mice
Genomic DNA was extracted from the tails of 4-wk-old mice, and PCR was performed using MASP-1 primers as previously described (25). Confirmation of the correct size (539 bp) of the WT MASP1/3 band was performed by separating PCR-amplified DNA using a 2% agarose gel. The presence of MASP-2 DNA was confirmed by PCR in the tails of MASP1/3+/+, MASP1/3+/−, and MASP1/3−/− mice by using the forward primer 5′-GGCGGCTACTATTGCTCCTG-3′ and the reverse primer 5′-AAAGGGCTGAGCACGTCTG-3′.
Induction of CAIA
CAIA was induced in MASP1/3−/− and WT mice by using a mixture of 4 mAb to bovine type II collagn (CII; Arthrogen-CIA, Chondrex, Redmond, WA) suspended in sterile Dulbecco’s PBS (27). Age- and sex-matched WT C57BL/6 mice were used as controls for these studies. All four mAb (three IgG2a and one IgG2b) in this mixture recognize conserved epitopes within the CB11 fragment, for which recognition sequences are shared by CII in many species. All mice received i.p. injections of 8 mg/mouse Arthrogen on day 0, and 50 ug/mouse LPS from Escherichia coli strain 0111B4 was administered i.p. on day 3 to synchronize the development of arthritis. All mice were sacrificed at day 10.
Examination for clinical disease activity
The prevalence of disease and severity of clinical disease activity in all groups of MASP1/3−/− and WT mice was determined every day by two trained laboratory personnel acting independently and blinded to the experimental treatment group. The DAS is based on a three-point scale per paw: 0, normal joint; 1, slight inflammation and redness; 2, severe erythema and swelling affecting the entire paw with inhibition of use; and 3, deformed paw or joint with ankylosis, joint rigidity, and loss of function. The total DAS was based on all four paws with a maximum score of 12 for each mouse.
Histopathology of knee joints
At day 10, both forepaws and the entire right hind limb, including the paw, ankle, and knee, were surgically removed from all mice with and without disease, fixed immediately in 10% neutral buffered formalin (Biochemical Sciences, Swedesboro, NJ), decalcified, paraffin embedded, and stained with toluidine blue. The preparation of tissue samples and histological analyses were performed as previously described (27, 28). All sections were read by a trained observer who was also blinded to the treatment and to the DAS of each mouse. A total of five joints from each mouse [e.g., both front paws with digits and wrist (right and left)] and one hind paw including knee joint, ankle, and paw were scored separately. Each joint was analyzed by a blinded observer for histopathological changes in inflammation, pannus, cartilage, and bone damage on a scale of 0–5. The data were expressed as all joint mean (AJM) score, representing the mean ± SEM for the scores in each individual joint with a maximum score of 5.
Immunohistochemistry for C3, MASP-1, MASP-2, and pro-Df
Knee joints from WT, MASP1/3−/−, and Df−/− mice with and without disease were fixed in 10% neutral buffered formalin to examine for deposition of C3, MASP-1, and MASP-2 proteins. C3 was localized with a primary polyclonal goat anti-mouse C3 antiserum (dilution 1:10,000) (ICN Pharmaceuticals, Costa Mesa, CA) and detected by Goat HRP polymer kit (Biocare Medical, Concord, CA). All nonspecific staining was blocked using a commercial protein block serum-free solution (DakoCytomation, Carpinteria, CA). C3 deposition from all five joints was scored according to our published criteria (27-29). Knee joints from C3−/− mice without disease were used as a negative control. MASP-1 protein was localized using a primary polyclonal rabbit anti-mouse MASP-1 Ab (dilution 1:200) (26) and detected by using EnVision rabbit HRP-conjugated reagent (DakoCytomation). Knee joints from MASP1/3−/− mice without disease were used as a negative control. MASP-2 localization was examined with a specific primary polyclonal goat Ab (dilution 1:200) (Santa Cruz Biotechnology, Santa Cruz, CA) and detected by using a goat HRP polymer kit (Biocare Medical). Knee joints from MASP2−/− mice without disease were used as a negative control. Pro-Df localization was examined with a primary polyclonal rabbit Ab (dilution 1:1000) that recognizes only the five-residue propiece unique to pro-Df (generated by Dr. Minoru Takahashi, Fukushima Medical University School of Medicine), and it was detected by using EnVision rabbit HRP-conjugated reagent (DakoCytomation). Development for all three proteins was carried out by using DAB plus solution substrate (DakoCytomation). This substrate reacts with HRP and produces a brown color for positive staining. We could not localize Df in the knee joints due to the lack of availability of an appropriate Ab to mouse Df that was functional for tissue staining. All slides were observed and scored in a blinded fashion by light microscopy. To quantify the percentages of cells stained positively for MASP-1 protein in the synovium and as well as in the cartilage, boundaries of synovium and cartilage were drawn manually using a scanner (Scan Scope XT, Aperio Technologies, Vista, CA). Although the areas of the synovium varied due to inflammation, the entire synovium was included in assessing the percentages of positive cells. The quantitative data for MASP-1 and pro-Df proteins were obtained from five different sections by using the program Positive Pixel Count. These data were calculated by using the formula total area/positively stained area × 100. The data were expressed as mean ± SEM based on n = 5 separate sections from one joint.
Western blot analysis of sera for detecting MASP-1, Df, and pro-Df
Mannan-agarose beads were used to concentrate MASP-1/3 proteins from sera of WT, MASP1/3+/−, and MASP1/3−/− mice. Briefly, 500 μl mannan-agarose beads were washed and reconstituted in 500 μl 1× TBS buffer (with 5 mM calcium/5 mM magnesium/0.05% Tween 20). Twenty microliters mannan-agarose beads and 20 μl sera were added to 180 μl 1× TBS buffer and incubated for 1 h at 4°C. After washing three times, 20 μl 2× SDS-PAGE buffer was added to each sample, and the samples were then heated at 80°C for 10 min. Samples were electrophoresed in a 10% SDS nonreducing PAGE gel using 1× HEPES/Tris/SDS running buffer. After transfer and blocking in 5% milk, the blots were developed using the rabbit anti-mouse MASP-1 Ab (dilution 1:200) and HRP-conjugated anti-rabbit Ab (dilution 1:5000) as described above. A single band of ~81 kDa MASP-1 protein was detected. Up to three faint separate bands including a possible dimer (~160 kDa) of MASP-1 were occasionally observed, as previously reported (26).
To examine for the presence of mature Df and pro-Df, sera from WT, MASP1/3−/−, and C1q−/−/Df−/− mice were electrophoresed in a 10% NuPAGE Bis-Tris gel under reducing conditions with 1× MOPS buffer. Serum samples were diluted 1:24 in 1× SDS buffer. Posttransfer, the polyvinylidene difluoride membrane was blocked in 5% milk in 1× PBS 0.5% Tween 20 for 2 h. The blots were then incubated for 24 h at 4°C with goat anti-mouse Df Ab (dilution 1:200) (Santa Cruz Biotechnology). This Ab reacts with both pro-Df and mature Df. Rabbit anti-goat HRP was used as the secondary Ab (dilution 1:2000) (Cappel, Costa Mesa, CA), as described above. The blots were washed three times for 10 min each in 1× PBS 0.5% Tween 20. The blots were developed for 3 min by using a 1:1 mixture of SuperSignal West Pico chemiluminescent substrate (Thermo Fisher Scientific, Waltham, MA). To detect pro-Df, a primary Ab (1:5000) was used that reacts only with the five-residue N-terminal peptide (QPRGR) that is present on pro-Df and absent from mature murine Df. The secondary Ab was HRP-conjugated goat anti-rabbit IgG (1:2000; Hycult Biotechnology, Uden, The Netherlands). The blot was developed as described above.
Reconstitution of C3 deposition and C5a generation in vitro by the addition of purified human Df to sera of MASP1/3−/− mice
We examined whether sera from MASP1/3−/− mice could be reconstituted in vitro with human Df (huDf) to restore AP activity as measured by C3 deposition and C5a generation. Sera from MASP1/3−/−, C4−/−, Bf−/−, and Df−/− mice were used in this experiment. To assure specific activation of only the AP, sera were diluted 1:10 in calcium-deficient buffer with or without huDf (Quidel, San Diego, CA). Serum samples were incubated with increasing amounts of huDf (0. 0.5, 1, 2, or 4 ng/10 μl serum) for 30 min at 4°C. These sera were then added to 96-well Costar ELISA plates (Corning, Lowell, MA) precoated with anti-CII mAb (Arthrogen, Chondrex; 2.5 μg/well) and incubated at 37°C for 1 h. C3 deposition and C5a generation were measured by ELISA (28). The reaction was stopped with 2 N H2SO4, and absorbance read at 450 nm, correcting for background at 550 nm.
Levels of complement components in the sera from complement-deficient and WT mice
Absolute serum levels of C1q, C4, C3 factor B, and Df proteins were measured using standard ELISA protocols according to our published methods (28, 29). The absolute levels of MASP-1 protein in sera from WT and MASP-1/3−/− mice were measured by using minor modifications of previously published methods (25).
Cytokine mRNA levels in the knee joints of mice
Cytokine mRNA levels were measured in the knee joints of WT and MASP1/3−/− mice at day 10 in the CAIA experiments. Total RNA was extracted by pulverizing the frozen individual knee joints (synovium, bones, and some adjacent muscles and skin) using TRI Reagent (Sigma-Aldrich, St. Louis, MO) following the manufacturer’s instructions. The mRNA levels of TNF-α, IL-1α, IL-1β, and IL-10 were measured by quantitative RT-PCR, as previously published (27).
Statistical analyses
The p values were calculated by using the parametric Student t test. Before applying this test, the Gaussion distribution of the data was determined using the Shapiro-Wilk normality test; this test has competitive power and performance in determining univariate normality. Based on W and p values using the Shapiro-Wilk test, we decided the type of test to be used for further analyses. All data from the DAS, histopathology, and C3 deposition studies were normally distributed. However, some of the data related to absolute levels of complement shown in Table I were not normally distributed; therefore, the Mann–Whitney U test was used to obtain p values. All significant values were also confirmed by using a nonparametric Mann-Whitney U test and a parametric, two-way ANOVA test. The Pearson test for correlation was used to determine the correlations between DAS and various histopathology scores. The data in all graphs, histograms, and tables have been shown as the mean ± SEM with p < 0.05 considered significant. All graphs and histograms were plotted by using a GraphPad Prism version 4.0 program (GraphPad, San Diego, CA).
Table I. Levels of complement components in sera from WT and MASP1−/− mice.
| C1q | C3 | C4 | Factor B | Dfa | MASP-1 | |
|---|---|---|---|---|---|---|
| WT mice | 0.42 ± 0.03 | 1.81 ± 0.18 | 0.83 ± 0.09 | 0.79 ± 0.05 | 1.07 ± 0.06 | 0.37 ± 0.02 |
| n | 13 | 13 | 13 | 8 | 9 | 9 |
| MASP1−/− mice | 0.43 ± 0.06 | 2.35 ± 0.24 | 0.67 ± 0.21 | 0.78 ± 0.07 | 0.85 ± 0.02 | 0.00 ± 0.00 |
| n | 8 | 7 | 7 | 7 | 4 | 7 |
| p Value | 0.866 | 0.059 | 0.429 | 0.882 | 0.097 |
Data are expressed as OD units with mean ± SEM based on the indicated number of sera (n). To show the specificity of each ELISA, sera from at least three different C1q−/−, C3−/−, C4−/−, fB−/−, and Df−/− mice were used as negative controls for the respective ELISAs; all of the values using these sera were equal to the background levels.
The anti-Df Ab used to measure the absolute levels of Df recognizes both mature Df and pro-Df.
Results
Essential role of MASP-1 and/or MASP-3 proteins in CAIA
CAIA was induced in WT mice and in MASP1/3−/− mice by injecting a mixture of mAb to bovine CII (8 mg/mouse) followed by LPS on day 3. The disease prevalence was 100% in WT mice at days 4 and 10, 0% in MASP1/3−/− mice at day 4, and 50% in MASP1/3−/− mice at day 10 (Fig. 1A). WT mice developed severe disease on day 4, and the DAS was 11.0 ± 0.7 (mean ± SEM; n = 4) at day 10 (Fig. 1B). In contrast, MASP1/3−/− mice developed minimal arthritis when evaluated daily through day 10, with a 93% reduction in the DAS in MASP1/3−/− mice (0.75 ± 0.3, mean ± SEM; n = 8) as compared with WT mice. These results demonstrate that the MASP-1 and/or MASP-3 proteins play an essential role in the development of clinically apparent inflammation and arthritis in this model.
FIGURE 1.

CAIA is reduced in prevalence and disease activity in MASP1/3−/− mice. A mixture of four mAb to CII were injected i.p. on day 0 followed by an i.p. injection of LPS on day 3. Prevalence of disease and DAS were determined daily by an observer blinded to the genotype of each mouse. A, Prevalence (%) of arthritis versus days after mAb injection. B, Clinical disease activity score in OD units versus days after mAb injection. The data represent the mean ± SEM based on: WT, n = 4; and MASP1/3−/−, n = 8. *p < 0.001 for DAS from day 4 to day 10 in MASP1/3−/− mice in comparison with WT mice.
Histopathological evaluation and C3 deposition in mice with CAIA
Histopathological analyses were performed on joints from WT and MASP1/3−/− mice with CAIA at day 10 (Fig. 2A). The histopathological scores paralleled the differences in DAS scores in the WT as compared with MASP1/3−/− mice. There were significant decreases in the individual scores for inflammation, pannus, cartilage, and bone damage at day 10 in MASP1/3−/− mice as compared with WT mice (p < 0.0001) (Fig. 2A), and there was a 59% decrease in the total joint score at day 10 (p < 0.0001) (data not shown). Similarly, the levels of C3 specifically deposited in the synovium and on cartilage, as well as the total joint score for C3 deposition, were significantly reduced in MASP1/3−/− mice as compared with WT mice, with decreases of 79, 82, and 81%, respectively (p < 0.001) (Fig. 2B). These results demonstrate a marked decrease in complement-dependent inflammation and injury in the joint and strongly suggest that there is no effective means in vivo to locally activate C3 in the absence of MASP-1/3.
FIGURE 2.
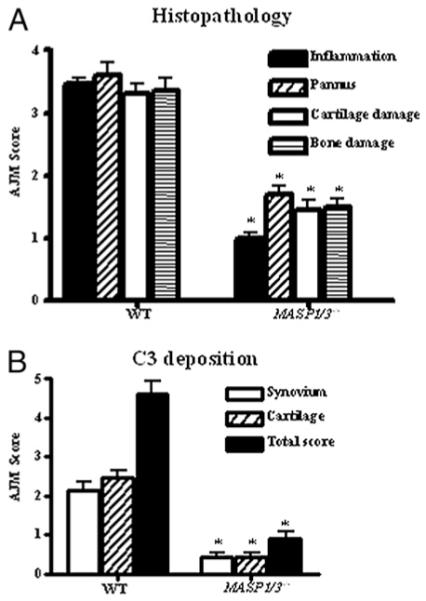
Histopathology scores and immunohistochemical staining for C3 in WT and MASP1/3−/− mice (n = 4 each) with CAIA. All mice were sacrificed at day 10 for these studies. A, Histopathological scores for inflammation, pannus, cartilage damage, and bone damage. Five joints (front right paw, front left paw, hind limb knee joint, hind limb ankle, and hind limb paw) were used. The thickness of histology sections were consistently kept at 7 μm. All sections were cut in duplicate and stained with 0.04% toludine blue dye. AJM ± SEM is shown in this histogram expressed on a scale of 0–5. B, C3 deposition in the knee joints. C3 deposition scores are based on a scale of 0–3 for synovium or cartilage and on a scale of 0–6 for total score. The results are expressed as AJM ± SEM. *p < 0.001 for MASP1/3−/− mice in comparison with WT mice.
MASP-1 and MASP-2 proteins are present in the synovium of mice with CAIA
To extend the analysis, we wanted to determine whether MASP-1 was present and could potentially act locally in the joint. To evaluate this question, immunohistochemical analysis for MASP-1 was performed in the knee joints of WT, MASP1/3−/−, and Df−/− mice with and without disease at day 10. MASP-1 was present in very few synovial cells in WT mice with no disease (data not shown), was absent from the synovium of MASP1/3−/− and Df−/− mice without CAIA (data not shown), and was absent in MASP1/3−/− mice with CAIA (Fig. 3B). In contrast, MASP-1 protein was abundantly present in the synovium of WT and Df−/− mice with CAIA at day 10 (Fig. 3A, 3C). Quantitative analysis of MASP-1 staining in joints from WT mice at day 10 showed an increase from 1.2 ± 0.2% (mean ± SEM; n = 5) positive cells in synovium without disease to 24.6 ± 2.1% (mean ± SEM; n = 5) positive cells in synovium with CAIA (p < 0.00001). Although MASP-1 staining was present in synovial cells in this experiment, there was little staining of chondrocytes. In comparison, MASP-2 was equally present in the synovium, meniscus, and cartilage of WT and MASP1/3−/− mice without CAIA, with an increase in staining intensity in WT mice with disease induction (Fig. 4).
FIGURE 3.
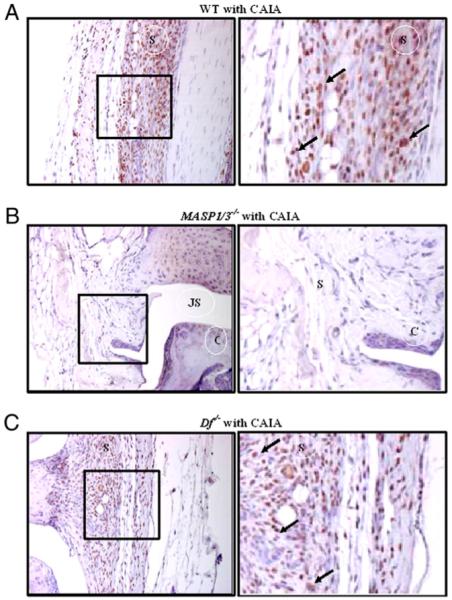
Immunohistochemical localization of MASP-1 protein in knee joints from WT, MASP1/3−/−, and Df−/− mice with CAIA. All mice were sacrificed at day 10 for these studies. In the left panels, the maximum area of the knee joints including cartilage and synovium is shown. The area in the black box is enhanced in the right panels to show specific staining pattern in the synovium and on the surface of cartilage. The presence of MASP-1 protein (brown color) is indicated by black arrows. A, WT mice with CAIA. B, MASP1/3−/− mice with CAIA. C, Df−/− mice with CAIA. No MASP-1 staining was detected in MASP1/3−/− and Df−/− mice without CAIA; some synovial cells were positive for MASP-1 in WT mice (data not shown). Mice having the highest DAS were selected for WT, MASP1/3−/−, and Df−/−. Original magnification for the left panels is ×20. C, cartilage; JS, joint space; S, synovium.
FIGURE 4.
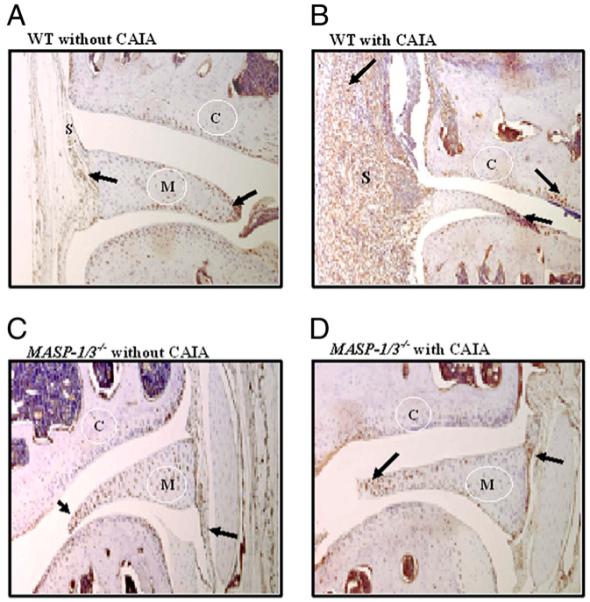
Immunohistochemical localization of MASP-2 protein in the knee joints of WT and MASP1/3−/− mice. The presence of brown stain depicting MASP-2 protein is indicated by the arrows. A, WT mice without CAIA. B, WT mice with CAIA. C, MASP1/3−/− mice without CAIA. D, MASP1/3−/− mice with CAIA. Original magnification ×10. C, cartilage; M, meniscus; S, synovium.
Pro-Df is present in the knee joints of WT and MASP1/3−/− mice
We next examined for the presence of pro-Df locally in the knee joints of WT and MASP1/3−/− mice either without or with CAIA using a mAb for immunohistochemical staining that recognized only the 5 aa propiece unique to pro-Df. Little pro-Df was present in the synovium and on the chondrocytes in the knee joints of WT mice without CAIA (Fig. 5A). However, in WT mice with CAIA pro-Df was abundantly present on multiple cells in the synovium and on a few cells in the cartilage (Fig. 5B). Surprisingly, pro-Df was only weakly present in the synovium as well as on the chondrocytes in the knee joints of MASP1/3−/− mice without CAIA (Fig. 5C) or with CAIA (Fig. 5D). No staining was observed with an isotype IgG control (data not shown).
FIGURE 5.
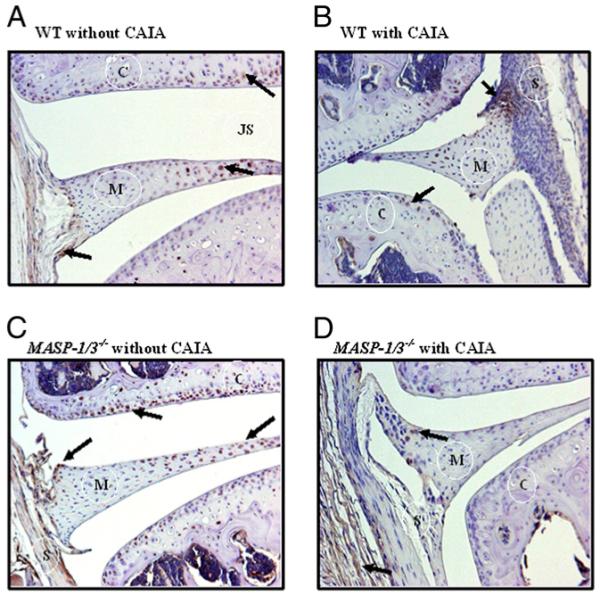
Immunohistochemical localization of pro-Df protein in the synovium and cartilage of knee joints from WT and MASP1/3−/− mice without CAIA and with CAIA. All mice were age and sex matched. The presence of pro-Df protein (brown color) is indicated by black arrows. A, WT mice without CAIA. B, WT mice with CAIA. C, MASP1/3−/− mice without CAIA. D, MASP1/3−/− mice with CAIA. Pro-Df staining in adipose tissue cells was used as a positive control (data not shown). As negative controls, no pro-Df staining was detected in the knee joints from WT and MASP1/3−/− mice using the matched isotope IgG (data not shown). Original magnification ×10. C, cartilage; JS, joint space; M, meniscus; S, synovium.
Cytokine levels in the knee joint
Mice protected from the development of CAIA due to inactivation of the AP typically do not demonstrate significant changes in local cytokine mRNA or protein expression levels in the joint (27, 28). Similarly, no significant differences were seen in the mRNA levels of the proinflammatory cytokines TNF-α, IL-1α, and IL-1β in the knee joints of WT and MASP1/3−/− mice with CAIA (data not shown). mRNA levels of the anti-inflammatory cytokine IL-10 also were not different (data not shown).
Levels of complement proteins in mouse sera
To assure a lack of confounding of the results by unexpected changes in other essential complement activation pathway proteins, the levels of C1q, C3, C4, factor B, Df (both pro-Df and mature forms), and MASP-1 were measured by ELISA in sera from WT and MASP1/3−/− mice (Table I). There were no significant differences in the absolute levels of these complement components between the two strains.
MASP1/3−/− mice have only pro-Df in their serum
Takahashi et al. (26) have shown that pro-Df is the only circulating form of Df present in the serum of MASP1/3−/− mice. We examined sera from MASP1/3−/−, C1q−/−/Df−/−, and WT mice for the presence of pro-Df and mature Df using a Western blot analysis with an Ab that recognizes both forms. The larger three bands in this gel (~95 kDa) represent a complex of glycosylated Df with an unknown binding protein, present in sera from WT mice but not in sera from MASP1/3−/− mice (Fig. 6A) (26, 32).
FIGURE 6.
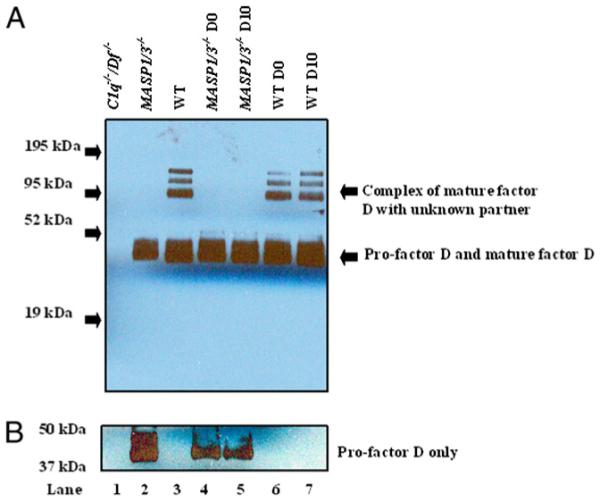
Western blot analysis of pro-Df and active Df before and after CAIA in the sera of C1q−/−/Df−/−, MASP1/3−/−, and WT mice; one representative serum sample was used from each genotype; 10% NuPAGE Bis-Tris gel was used under reducing conditions, as described in Materials and Methods. A, The primary Ab used was goat anti-mouse Df; this Ab detects three bands of Df (~95 kDa) in complex with unknown proteins in sera from WT mice (lanes 3, 6, and 7) and overlapping bands of pro-Df and active Df (~40–44 kDa) in sera from WT and MASP1/3−/− mice (lanes 2–7). Serum from C1q−/−/Df−/− mouse with no CAIA was used as a negative control, and no band was detected (lane 1). Serum from MASP1/3−/− mice with no CAIA contains only one combined band of pro-Df (lane 2), for which the identity has been confirmed by using mass spectrometry (26). Sera from MASP1/3−/− mice at day 0 without CAIA (lane 4) and at day 10 with CAIA (lane 5) still contain only one band of pro-Df (~40–44 kDa) without converstion to active Df. Sera from WT mice at day 0 without CAIA (lane 6) and at day 10 with CAIA (lane 7) still contain a combined band of pro-Df/active Df as well as an active Df along with an unknown partner. B, The primary Ab used was specific for pro-Df and which was present only in sera from MASP1/3−/− mice (lanes 2, 4, and 5).
As expected, sera from C1q−/−/Df−/− mice showed an absence of both forms of Df (Fig. 6A). Sera from WT mice contained both glycosylated pro-Df and mature Df as a broad band of 40–44 kDa; however, this band in sera from MASP1/3−/− mice contained only pro-Df, as determined by experiments using mass spectrometry (26). Furthermore, using an Ab specific for the peptide unique to murine pro-Df, and not found in mature Df, only pro-Df was present in the sera of MASP1/3−/− mice (Fig. 6B). Importantly, no detectable conversion of pro-Df to mature Df was present in the circulation of MASP1/3−/− mice during the evolution of CAIA (Fig. 6A, lane 5). This result suggests that other proteases (trypsin, plasmin, etc.) capable of cleaving pro-Df in vitro are not operative under these inflammatory conditions.
The combination of MASP1/3−/− and Df−/− sera restores full AP activity
We carried out in vitro studies with adherent mAb to CII to initiate the complement system in the presence of calcium-deficient buffer so that only the AP was active (27-29, 33). Neither MASP1/3−/− nor Df−/− sera alone demonstrated any AP activity in this assay (Fig. 7A). However, a significant increase in C3 deposition induced by adherent anti-CII mAb was seen when the sera from MASP1/3−/− and Df−/− mice were mixed (Fig. 7A). A parallel significant increase in C5a levels was also seen when these two sera were mixed (Fig. 7D). The results suggest that MASP-1 from Df−/− serum was able to cleave pro-Df present in MASP1/3−/− serum, consistent with previously reported experiments using WT and MASP1/3−/− sera (26). To confirm that the mixed sera were activating the AP under these assay conditions, a specific inhibitory anti-factor B mAb was used (34). A significant inhibition of C3 deposition and C5a generation was observed in the presence of this mAb (Fig. 7B, 7E). As a positive control for the inhibitory effects of the anti-factor B mAb, sera from C4−/− mice, for which only the AP is active in this assay (28), also showed complete inhibition of C3 deposition and C5a generation by this anti-factor B mAb (Fig. 7C, 7F).
FIGURE 7.

Mixing sera from MASP1/3−/− and Df−/− mice restored full activity of the AP of complement, which was blocked specifically by an inhibitory mouse mAb to anti-factor B. All sera for this experiment were diluted in a calcium-deficient buffer. Levels of C3 deposition and C5a generation were measured by ELISA postinduction by adherent mAb to CII. A, C3 deposition using sera from MASP1/3−/− and Df−/− mice alone and mixed. B, Inhibition of C3 deposition by a mAb to factor B added to the mixture of MASP1/3−/− and Df−/− sera. C, Inhibition of C3 deposition using sera from C4−/− mice. D, C5a generation using sera from MASP1/3−/− and Df−/− mice alone and mixed. E, Inhibition of C5a generation by a mAb to factor B added to the mixture of MASP1/3−/− and Df−/− sera. F, Inhibition of C5a generation using sera from C4−/− mice. The data represent the mean ± SEM based on n = 3 for each sera. *p < 0.001; **p < 0.01 in comparison with control.
Purified human Df restores the AP in the sera of MASP1/3−/− and Df−/− mice
To confirm that Df can restore the defective AP, sera from MASP1/3−/− and Df−/− mice were pretreated for 30 min on ice with different concentrations of purified huDf (0, 0.5, 1, 2, and 4 ng/10 μl sera). There were significant increases in both C3 deposition and C5a generation induced by adherent mAb to CII in the presence of as little as 0.5 ng/μl sera of huDf (Fig. 8A, 8B). Sera from C4−/− mice with an intact AP and sera from Bf−/− mice, which lack the AP, were used as positive and negative controls, respectively. The levels of C5a generation were higher using sera from MASP1/3−/− and Df−/− mice than with sera from C4−/− mice in the presence of 4 ng/10 μl added huDf (Fig. 8B), possibly because the level of Df was increased above normal. Sera from MBL−/−/Df−/− mice also lacked the ability to activate complement in this assay, and it was restored by the addition of huDf (data not shown).
FIGURE 8.

Effect of reconstitution with purified huDf on C3 deposition and C5a generation in vitro. ELISA plates precoated with 4 mAb to CII were incubated with 1:10 dilutions of sera from C4−/−, MASP1/3−/−, Df−/−, and Bf−/− mice for 20 min at 4°C. Different concentrations of huDf were added to the serum prior to incubation on the plate. C3 deposition (A) and C5a generation (B) were measured and expressed in OD units (OD value). The baseline levels of C5a in the sera before incubation on the mAb to CII were subtracted from the measured total C5a at the end of the experiment. The data shown represent the mean ± SEM based on n = 14 for each mouse serum.
Discussion
In this study, we have shown that MASP1/3−/− mice are highly resistant to CAIA as evidenced by a significant decrease in the histological scores and clinical DAS as compared with WT mice. Moreover, consistent with a complement-dependent effect, there was an ~80% decrease in C3 deposition in the synovium and on the surface of cartilage in MASP1/3−/− mice as compared with WT mice. Based on our analyses as well as the original description of these mice (25, 26), it is likely that the primary defect that underlies the lack of robust arthritis in this model is the inability of MASP1/3−/− mice to develop a systemically active complement AP because of an absence of processing of pro-Df to mature Df. These data using MASP1/3−/− mice also indicate that there are no bypass pathways that can substitute for MASP-1/3 in this inflammatory process. Furthermore, there are no pathologically relevant mechanisms to convert pro-Df to mature Df other than through one or both proteins encoded by the MASP-1/3 gene.
We believe that the CAIA model is the ideal one in which to evaluate this question. CAIA is a well-accepted model of immune complex-induced joint injury as may occur in human rheumatoid arthritis (RA) (35). In addition, it is the only in vivo model of arthritis known to date in which the AP has been experimentally shown to demonstrate the unique characteristic of being both necessary and sufficient for the development of tissue injury. To support this conclusion, informative murine strains have been created that are singly deficient in complement pathway components; when these single deficiencies are bred together, a series of strains with combined deficiencies have been generated. Using these unique reagents, we, along with our collaborators who created the strains, have been able to demonstrate that mice singly deficient in the CP or LP develop robust CAIA, whereas in the absence of the AP, there is a significant lack of disease phenotype (27-29). Conversely, mice with combined deficiencies of the CP and LP and only a functional AP are fully susceptible to CAIA, whereas strains possessing only the CP or LP are unable to develop robust arthritis (27-29). We have also shown that adherent anti-CII immune complexes, as occur in the joints of patients with RA, are fully capable of activating the AP (28).
This unique characteristic of being solely dependent upon the AP is particularly important in the analysis of MASP1/3−/− mice. MASP-1/3 DNA and proteins are absent from these mice (25), and normal levels of MASP-2 protein are found in the sera (data not shown). The sera from MASP1/3−/− mice, however, demonstrate impaired MASP-2–induced deposition of C4 and C3 on mannancoated plates, a finding that is indicative of a deficient LP and that is consistent with the role of MASP-1 in activating MASP-2 (25). Thus, it is possible that MASP1/3−/− mice would also demonstrate impaired tissue injury in murine models that are in part dependent upon the LP, but that remains to be determined. In addition, because the AP is also involved in the amplification of tissue injury in CP- and LP-dependent murine disease models (reviewed in Ref. 12), it might not be possible to establish the exact causal relationships between MASP-1/3 deficiency and CP- or LP-dependent pathways of complement activation in induction of changes in phenotype.
The mice we have used are doubly deficient in both MASP-1 and MASP-3, and it could be asked which one of the two proteins underlies the impairment in CAIA in MASP1/3−/− mice. Previous studies have shown that both MASP-1 and MASP-3 can convert pro-Df into active Df in vitro (26); nevertheless, formal proof of that role in vivo will require additional evaluation. MASP-3 has recently been shown to have a regulatory role through binding to ficolin-3 in human sera and inhibiting ficolin-3–mediated complement activation through the LP (36).
Importantly, our study strongly supports the hypothesis that MASP-1 is the only relevant mechanism for cleaving pro-Df both locally and possibly in the circulation during inflammation and that other proposed mechanisms cannot bypass the requirement for MASP-1 in this process. Thus, although trypsin, thrombin, kallikrein, and plasmin can cleave pro-Df in vitro (14), MASP-1/3 remain uniquely required to generate mature Df in vivo. However, it remains uncertain exactly where the conversion of pro-Df to mature Df occurs in vivo. MASP-1/3 is not expressed in adipocytes, the primary site of pro-Df synthesis (26), and where conversion was previously thought to occur during biosynthesis (14). Conversion of pro-Df into mature Df may occur outside of the adipose tissue and possibly in the systemic circulation. This hypothesis is supported by the finding that mixing serum lacking MASP-1/3, but containing pro-Df, with serum containing MASP-1/3, but lacking Df, results in the ability to activate the AP (Fig. 7). In addition, the expression of MASP-1 in synovial cells of WT mice undergoing CAIA (Fig. 3), and the expression of pro-Df in synovial cells and chondrocytes in knee joints of WT and MASP1/3−/− mice without CAIA (Fig. 5), suggests that conversion may occur locally in inflammatory tissues. Pro-Df staining is markedly increased in the inflamed synovium of WT mice with CAIA, suggesting an induction by inflammation. However, we have not examined whether MASP-1 and pro-Df are actually synthesized in the joint and which cells may synthesize them or whether these proteins are secondarily bound to the surface. Defining the exact site(s) of pro-Df conversion to mature Df will require additional studies.
Another relevant question is whether MASP1/3−/− mice are protected from the development of CAIA solely because of lacking pro-Df conversion to mature Df or whether there are other potential injurious effects of MASP-1 or MASP-3 in vivo. MASP-1 demonstrates trypsin-like activity in vitro cleaving fibrinogen and factor XIII (37, 38) engaging protease-activated receptor-4 on endothelial cells (39) and exhibiting structural characteristics of a more promiscuous protease (40). The results of recent studies indicate that MBL and the thrombin-like activity of MASP-1/3 are involved in hemostasis in vivo in mice, possibly mediated through cleavage of protease-activated receptor-4 on endothelial cells (41). Our studies do not formally address the possibility of other functions of MASP-1/3 in vivo, as in this study, we only provide a strong association between the lack of AP activity and the absence of CAIA. Reconstitution of Df in vivo using purified human Df has not been feasible because of a lack of sufficient amounts of material and a short t1/2 postinjection (N. Banda, unpublished observations). Additional methods such as the in vivo use of recombinant murine Df as a slow release depot form or, alternatively, the expression of active murine Df as a transgene in MASP1/3−/− mice, will be necessary to formally address this question.
Another important point relates to the observation that, although it has generally been accepted that MASP proteins present in circulation are associated with MBL and ficolins, the functional requirements for these associations are uncertain (7, 8). Notably, MASP-1 does not require the association in vitro with these recognition molecules to cleave pro-Df (26). Initial studies of sera from MBL−/−/FCN A−/− mice, which lack both classes of LP target-recognition molecules, demonstrate normal in vitro activation of AP induced by anti-CII mAb in the presence of both pro-Df and mature Df by Western blot analysis (data not shown). Additional studies to characterize MASP-1 not bound to MBL or ficolins and its role in the induction of CAIA are necessary to further explore this aspect of the mechanism of priming and activation of the AP in vivo.
In summary, our results show that MASP1/3−/− mice are highly resistant to the development of CAIA. There is no apparent ability in vivo to bypass the effects of this deficient state by converting pro-Df to mature Df by other mechanisms. In the absence of MASP-1/3, no in vitro activation of C3 or C5 induced by adherent mAb CII and mediated by the AP is possible. Whether the absence of other functions proposed for MASP-1 or MASP-3 account for a portion of the protective effect of MASP-1/3 deficiency in vivo cannot be ruled out. However, the lack of AP activity due to a failure to convert pro-Df into mature Df could account for all of the observed effects in MASP1/3−/− mice, as they closely parallel those found in Df−/− mice undergoing CAIA (29). Our results of serum mixing and other experiments suggest that inhibition of the AP initiation and amplification pathways could in principle be accomplished indirectly by systemically blocking MASP-1 activity and impairing conversion of pro-Df to mature Df. Thus, our findings also have potential clinical implications with regard to the development of new therapeutics for the treatment of RA and other forms of inflammatory arthritis in humans.
Acknowledgments
We thank Stephanie Hyatt for measuring absolute levels of various complement components using sera from complement-deficient mice and Umarani Pugazhenthi, PCR Core, University of Colorado School of Medicine, for performing quantitative RT-PCR from the knee joints of mice used in this study.
This work was supported by National Institutes of Health Grant AR051749 to V.M.H.
Abbreviations used in this paper
- AJM
all joint mean
- AP
alternative pathway
- C
cartilage
- CAIA
collagen Ab-induced arthritis
- CII
type II collagen
- CP
classical pathway
- DAS
disease activity score
- Df
factor D
- huDf
human factor D
- JS
joint space
- LP
lectin pathway
- M
meniscus
- MASP
mannose-binding lectin-associated serine protease
- MBL
mannose-binding lectin
- pro-Df
pro-factor D
- RA
rheumatoid arthritis
- S
synovium
- WT
wild-type
Footnotes
Disclosures
V.M.H. owns equity and receives income from Taligen Therapeutics. All other authors have no financial conflicts of interest.
References
- 1.Carroll MC. The role of complement in B cell activation and tolerance. Adv. Immunol. 2000;74:61–88. doi: 10.1016/s0065-2776(08)60908-6. [DOI] [PubMed] [Google Scholar]
- 2.Carroll MC, Holers VM. Innate autoimmunity. Adv. Immunol. 2005;86:137–157. doi: 10.1016/S0065-2776(04)86004-8. [DOI] [PubMed] [Google Scholar]
- 3.Walport MJ. Complement. First of two parts. N. Engl. J. Med. 2001;344:1058–1066. doi: 10.1056/NEJM200104053441406. [DOI] [PubMed] [Google Scholar]
- 4.Walport MJ. Complement. Second of two parts. N. Engl. J. Med. 2001;344:1140–1144. doi: 10.1056/NEJM200104123441506. [DOI] [PubMed] [Google Scholar]
- 5.Lachmann PJ, Hughes-Jones NC. Initiation of complement activation. Springer Semin. Immunopathol. 1984;7:143–162. doi: 10.1007/BF01893018. [DOI] [PubMed] [Google Scholar]
- 6.Takahashi K, Ip WE, Michelow IC, Ezekowitz RA. The mannose-binding lectin: a prototypic pattern recognition molecule. Curr. Opin. Immunol. 2006;18:16–23. doi: 10.1016/j.coi.2005.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wallis R. Interactions between mannose-binding lectin and MASPs during complement activation by the lectin pathway. Immunobiology. 2007;212:289–299. doi: 10.1016/j.imbio.2006.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thiel S. Complement activating soluble pattern recognition molecules with collagen-like regions, mannan-binding lectin, ficolins and associated proteins. Mol. Immunol. 2007;44:3875–3888. doi: 10.1016/j.molimm.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 9.Fujita T, Matsushita M, Endo Y. The lectin-complement pathway—its role in innate immunity and evolution. Immunol. Rev. 2004;198:185–202. doi: 10.1111/j.0105-2896.2004.0123.x. [DOI] [PubMed] [Google Scholar]
- 10.Müller-Eberhard HJ. Molecular organization and function of the complement system. Annu. Rev. Biochem. 1988;57:321–347. doi: 10.1146/annurev.bi.57.070188.001541. [DOI] [PubMed] [Google Scholar]
- 11.Harboe M, Mollnes TE. The alternative complement pathway revisited. J. Cell. Mol. Med. 2008;12:1074–1084. doi: 10.1111/j.1582-4934.2008.00350.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Holers VM. The spectrum of complement alternative pathway-mediated diseases. Immunol. Rev. 2008;223:300–316. doi: 10.1111/j.1600-065X.2008.00641.x. [DOI] [PubMed] [Google Scholar]
- 13.Volanakis JE, Narayana SVL. Complement factor D, a novel serine protease. Protein Sci. 1996;5:553–564. doi: 10.1002/pro.5560050401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yamauchi Y, Stevens JW, Macon KJ, Volanakis JE. Recombinant and native zymogen forms of human complement factor D. J. Immunol. 1994;152:3645–3653. [PubMed] [Google Scholar]
- 15.Lesavre PH, Müller-Eberhard HJ. Mechanism of action of factor D of the alternative complement pathway. J. Exp. Med. 1978;148:1498–1509. doi: 10.1084/jem.148.6.1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wong NK, Kojima M, Dobó J, Ambrus G, Sim RB. Activities of the MBL-associated serine proteases (MASPs) and their regulation by natural inhibitors. Mol. Immunol. 1999;36:853–861. doi: 10.1016/s0161-5890(99)00106-6. [DOI] [PubMed] [Google Scholar]
- 17.Turner MW, Hamvas RM. Mannose-binding lectin: structure, function, genetics and disease associations. Rev. Immunogenet. 2000;2:303–322. [PubMed] [Google Scholar]
- 18.Matsushita M, Fujita T. Activation of the classical complement pathway by mannose-binding protein in association with a novel C1s-like serine protease. J. Exp. Med. 1992;176:1497–1502. doi: 10.1084/jem.176.6.1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thiel S, Vorup-Jensen T, Stover CM, Schwaeble W, Laursen SB, Poulsen K, Willis AC, Eggleton P, Hansen S, Holmskov U, et al. A second serine protease associated with mannan-binding lectin that activates complement. Nature. 1997;386:506–510. doi: 10.1038/386506a0. [DOI] [PubMed] [Google Scholar]
- 20.Dahl MR, Thiel S, Matsushita M, Fujita T, Willis AC, Christensen T, Vorup-Jensen T, Jensenius JC. MASP-3 and its association with distinct complexes of the mannan-binding lectin complement activation pathway. Immunity. 2001;15:127–135. doi: 10.1016/s1074-7613(01)00161-3. [DOI] [PubMed] [Google Scholar]
- 21.Stover CM, Thiel S, Thelen M, Lynch NJ, Vorup-Jensen T, Jensenius JC, Schwaeble WJ. Two constituents of the initiation complex of the mannan-binding lectin activation pathway of complement are encoded by a single structural gene. J. Immunol. 1999;162:3481–3490. [PubMed] [Google Scholar]
- 22.Takahashi M, Endo Y, Fujita T, Matsushita M. A truncated form of mannose-binding lectin-associated serine protease (MASP)-2 expressed by alternative polyadenylation is a component of the lectin complement pathway. Int. Immunol. 1999;11:859–863. doi: 10.1093/intimm/11.5.859. [DOI] [PubMed] [Google Scholar]
- 23.Takahashi M, Mori S, Shigeta S, Fujita T. Role of MBL-associated serine protease (MASP) on activation of the lectin complement pathway. Adv. Exp. Med. Biol. 2007;598:93–104. doi: 10.1007/978-0-387-71767-8_8. [DOI] [PubMed] [Google Scholar]
- 24.Stover CM, Lynch NJ, Dahl MR, Hanson S, Takahashi M, Frankenberger M, Ziegler-Heitbrock L, Eperon I, Thiel S, Schwaeble WJ. Murine serine proteases MASP-1 and MASP-3, components of the lectin pathway activation complex of complement, are encoded by a single structural gene. Genes Immun. 2003;4:374–384. doi: 10.1038/sj.gene.6363970. [DOI] [PubMed] [Google Scholar]
- 25.Takahashi M, Iwaki D, Kanno K, Ishida Y, Xiong J, Matsushita M, Endo Y, Miura S, Ishii N, Sugamura K, Fujita T. Mannose-binding lectin (MBL)-associated serine protease (MASP)-1 contributes to activation of the lectin complement pathway. J. Immunol. 2008;180:6132–6138. doi: 10.4049/jimmunol.180.9.6132. [DOI] [PubMed] [Google Scholar]
- 26.Takahashi M, Ishida Y, Iwaki D, Kanno K, Suzuki T, Endo Y, Homma Y, Fujita T. Essential role of mannose-binding lectin-associated serine protease-1 in activation of the complement factor D. J. Exp. Med. 2010;207:29–37. doi: 10.1084/jem.20090633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Banda NK, Thurman JM, Kraus D, Wood A, Carroll MC, Arend WP, Holers VM. Alternative complement pathway activation is essential for inflammation and joint destruction in the passive transfer model of collagen-induced arthritis. J. Immunol. 2006;177:1904–1912. doi: 10.4049/jimmunol.177.3.1904. [DOI] [PubMed] [Google Scholar]
- 28.Banda NK, Takahashi K, Wood AK, Holers VM, Arend WP. Pathogenic complement activation in collagen antibody-induced arthritis in mice requires amplification by the alternative pathway. J. Immunol. 2007;179:4101–4109. doi: 10.4049/jimmunol.179.6.4101. [DOI] [PubMed] [Google Scholar]
- 29.Banda NK, Levitt B, Wood AK, Takahashi K, Stahl GL, Holers VM, Arend WP. Complement activation pathways in murine immune complex-induced arthritis and in C3a and C5a generation in vitro. Clin. Exp. Immunol. 2010;159:100–108. doi: 10.1111/j.1365-2249.2009.04035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu Y, Ma M, Ippolito GC, Schroeder HW, Jr., Carroll MC, Volanakis JE. Complement activation in factor D-deficient mice. Proc. Natl. Acad. Sci. USA. 2001;98:14577–14582. doi: 10.1073/pnas.261428398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Endo Y, Nakazawa N, Liu Y, Iwaki D, Takahashi M, Fujita T, Nakata M, Matsushita M. Carbohydrate-binding specificities of mouse ficolin A, a splicing variant of ficolin A and ficolin B and their complex formation with MASP-2 and sMAP. Immunogenetics. 2005;57:837–844. doi: 10.1007/s00251-005-0058-1. [DOI] [PubMed] [Google Scholar]
- 32.Rosen BS, Cook KS, Yaglom J, Groves DL, Volanakis JE, Damm D, White T, Spiegelman BM. Adipsin and complement factor D activity: an immune-related defect in obesity. Science. 1989;244:1483–1487. doi: 10.1126/science.2734615. [DOI] [PubMed] [Google Scholar]
- 33.Banda NK, Wood AK, Takahashi K, Levitt B, Rudd PM, Royle L, Abrahams JL, Stahl GL, Holers VM, Arend WP. Initiation of the alternative pathway of murine complement by immune complexes is dependent on N-glycans in IgG antibodies. Arthritis Rheum. 2008;58:3081–3089. doi: 10.1002/art.23865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thurman JM, Kraus DM, Girardi G, Hourcade DE, Kang H-J, Royer PA, Mitchell LM, Giclas PC, Salmon JE, Gilkeson G, Holers VM. A novel inhibitor of the alternative complement pathway prevents antiphospholipid antibody-induced pregnancy loss in mice. Mol. Immunol. 2005;42:87–97. doi: 10.1016/j.molimm.2004.07.043. [DOI] [PubMed] [Google Scholar]
- 35.Monach PA, Benoist C, Mathis D. The role of antibodies in mouse models of rheumatoid arthritis, and relevance to human disease. Adv. Immunol. 2004;82:217–248. doi: 10.1016/S0065-2776(04)82005-4. [DOI] [PubMed] [Google Scholar]
- 36.Skjoedt M-O, Palarasah Y, Munthe-Fog L, Ma YJ, Weiss G, Skjodt K, Koch C, Garred P. MBL-associated serine protease-3 circulates in high serum concentrations predominantly in complex with ficolin-3 and regulates ficolin-3 mediated complement activation. Immunobiology. 2009 doi: 10.1016/j.imbio.2009.10.006. In press. [DOI] [PubMed] [Google Scholar]
- 37.Krarup A, Gulla KC, Gál P, Hajela K, Sim RB. The action of MBL-associated serine protease 1 (MASP1) on factor XIII and fibrinogen. Biochim. Biophys. Acta. 2008;1784:1294–1300. doi: 10.1016/j.bbapap.2008.03.020. [DOI] [PubMed] [Google Scholar]
- 38.Hajela K, Kojima M, Ambrus G, Wong KH, Moffatt BE, Ferluga J, Hajela S, Gál P, Sim RB. The biological functions of MBL-associated serine proteases (MASPs) Immunobiology. 2002;205:467–475. doi: 10.1078/0171-2985-00147. [DOI] [PubMed] [Google Scholar]
- 39.Megyeri M, V Makó, Beinrohr L, Doleschall Z, Prohászka Z, Cervenak L, Závodszky P, Gál P. Complement protease MASP-1 activates human endothelial cells: PAR4 activation is a link between complement and endothelial function. J. Immunol. 2009;183:3409–3416. doi: 10.4049/jimmunol.0900879. [DOI] [PubMed] [Google Scholar]
- 40.Dobó J, Harmat V, Beinrohr L, Sebestyén E, Závodszky P, Gál P. MASP-1, a promiscuous complement protease: structure of its catalytic region reveals the basis of its broad specificity. J. Immunol. 2009;183:1207–1214. doi: 10.4049/jimmunol.0901141. [DOI] [PubMed] [Google Scholar]
- 41.Takahashi K, Chang W-C, Takahashi M, Pavlov V, Ishida Y, La Bonte L, Shi L, Fujita T, Stahl GL, Van Cott EM. Mannose-binding lectin and its associated proteases (MASPs) mediate coagulation and its deficiency is a risk factor in developing complications from infection, including disseminated intravascular coagulation. Immunobiology. 2010 doi: 10.1016/j.imbio.2010.02.005. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]