

Abstract
Endothelial function refers to a multitude of physiological processes that maintain healthy homeostasis of the vascular wall. Exposure of the endothelium to cardiac risk factors results in endothelial dysfunction and is associated with an alteration in the balance of vasoactive substances produced by endothelial cells. These include a reduction in nitric oxide (NO), an increase in generation of potential vasoconstrictor substances and a potential compensatory increase in other mediators of vasodilation. The latter has been surmised from data demonstrating persistent endothelium-dependent vasodilatation despite complete inhibition of NO and prostaglandins. This remaining non-NO, non-prostaglandin mediated endothelium-dependent vasodilator response has been attributed to endothelium-derived hyperpolarizing factor/s (EDHF). Endothelial hyperpolarization is likely due to several factors that appear to be site and species specific. Experimental studies suggest that the contribution of the EDHFs increase as the vessel size decreases, with a predominance of EDHF activity in the resistance vessels, and a compensatory up-regulation of hyperpolarization in states characterized by reduced NO availability. Since endothelial dysfunction is a precursor for atherosclerosis development and its magnitude is a reflection of future risk, then the mechanisms underlying endothelial dysfunction need to be fully understood, so that adequate therapeutic interventions can be designed.
1. Introduction
Endothelial function refers to a multitude of physiological processes of the vascular endothelium that maintain healthy homeostasis of the vascular wall and may be used as a “barometer” of the injury/repair inflicted by multiple environmental and genetic factors [1–3]. Vascular endothelial dysfunction is associated with a reduction in nitric oxide (NO) bioavailability, an increase in generation of potential vasoconstrictor substances such as superoxide anions and endothelin–1, and a potential compensatory increase in other mediators of vasodilation. This remaining non-NO, nonprostaglandin-mediated endothelium-dependent vasodilation has been partly attributed to endothelium-derived hyperpolarizing factor/s (EDHF). Endothelial hyperpolarization is likely due to several factors that are site- and species-specific, ultimately causing vascular smooth muscle hyperpolarization and relaxation. Experimental studies suggest that the contribution of EDHFs increase as the vessel size decreases, with predominant EDHF activity in the resistance vessels and a compensatory upregulation of EDHFs in states characterized by reduced NO availability [4–12]. Whereas prostacyclin and NO bioavailability have been extensively investigated in the human circulation in vivo, little is known about endothelial hyperpolarization.
2. Endothelium-Derived Hyperpolarizing Factor (EDHF)
Potential EDHFs differ by species and vascular bed, but act by increasing potassium (K+) conductance resulting in the subsequent propagation of depolarization of vascular smooth muscle cells and relaxation [13–15] (Figure 1). Acetylcholine causes hyperpolarization of vascular smooth muscle in arteries with an intact endothelium but not in its absence [16–19]. This hyperpolarization is mimicked by certain K+ channel agonists and is unaffected by inhibitors of nitric oxide synthase or cyclooxygenase and has been attributed to release of EDHF/s. EDHFs appear to open smooth muscle cell K+ channels, allowing K+ efflux along its chemical gradient resulting in membrane hyperpolarization. Thus, EDHF activity may be defined functionally as agonist-induced, endothelium-dependent relaxation that is not blocked by inhibitors of NO synthase or cyclooxygenase but can be inhibited, at least in part, by K+ channel blockers.
Figure 1.
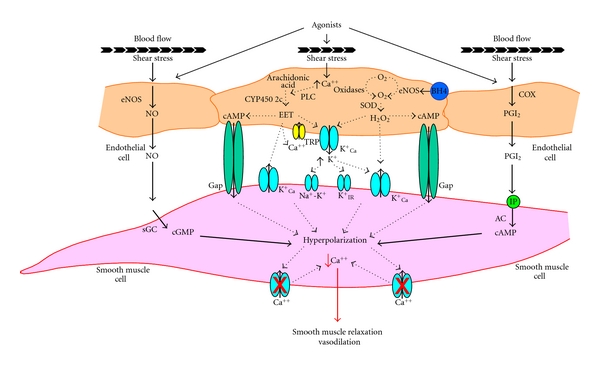
Mechanisms for endothelial cell mediated relaxation. Agonist (bradykinin/acetylcholine/substance P) or shear stress increases the activity of endothelial NO synthase (eNOS) and cyclooxygenase (COX), providing nitric oxide x(NO) and prostacyclin(PGI2)-mediated dilation. There are multiple potential EDHF pathways. Increases in intracellular calcium activates phospholipase A2 (PLC) to produce arachidonic acid. Its metabolism by cytochrome P450 2C (CYP4502c) generates eicosatrienoic acids (EETs) that can stimulate calcium dependent potassium (KCa +) channels in endothelial and smooth muscle cells. EETs may also directly activate gap junctions (Gap). EETs may also act in an autocrine manner on endothelial cells by activating transient receptor potential (TRP) V4 channels, which promote Calcium (Ca++) influx further increasing the calcium concentration and activating KCa + channels to cause hyperpolarization and release of K+ ions into the subendothelial space. The increase in potassium in the interstitium may activate KCa + channels, inwardly rectifying potassium channels (KIR +), or the Na+-K+ pump on smooth muscle cells and cause hyperpolarization. Smooth muscle hyperpolarization in turn results in relaxation by closing voltage-gated channels leading to a fall in Ca++ concentration and subsequent vasodilation. The action of eNOS (with cofactor tetrahydrobiopterin [BH4]) and oxidases on oxygen (O2) produces the reactive oxygen species superoxide (.O2−). Hydrogen peroxide (H2O2) generated by dismutation of superoxide anions by superoxide dismutase (SOD) can also cause hyperpolarization by activating endothelial and smooth muscle KCa + channels or by gap junctions. Adenylyl cyclase: AC; cyclic Adenosine monophosphate: cAMP; cyclic guanosine monophosphate: cGMP; soluble guanylyl cyclase: sGC; prostacyclin receptor, IP.
2.1. Potassium (KCa +) Channel Activation (Figure 1)
2.1.1. Calcium-Activated Potassium (KCa +) Channels
Agonists such as bradykinin stimulate endothelial G protein-coupled receptors provoking an increase in intracellular calcium [Ca2+]i in the endothelial cell [20, 21]. Endothelium-dependent hyperpolarization of smooth muscle cells results from the opening of K+ channels in the smooth muscle plasmalemma and is abolished by K+ concentrations higher than 25 mM [22]. Agonists that produce hyperpolarization also stimulate efflux of K+ [17, 23]. However, there are several smooth muscle K+ channels; endothelium-dependent hyperpolarization is not prevented by glibenclamide, an inhibitor of ATP-dependent K+ channels (KATP +), or inhibitors of inwardly rectifying potassium channels (KIR +) [24]. However, both barium chloride (<100 μmol/L) and ouabain (2.7 nmol/min), inhibitors of KIR + channels, reduced resting flow in healthy subjects, an effect that was lower in obesity, even though these are not endothelium-dependent responses [25].
The hallmark of the EDHF-mediated responses is its abolition by the combination of apamin (a specific inhibitor of KCa + channels of small conductance (SKCa + channels)) plus charybdotoxin (a nonselective inhibitor of large-conductance (BKCa +) and intermediate-conductance (IKCa +) channels), and of some voltage-dependent K+(KV +) channels [26–28]. This toxin combination targets KCa + channels on endothelial cells rather than K+ channels located on smooth muscle cells. Increasing intracellular free calcium in endothelial cells opens KCa + channels allowing efflux and accumulation of K+ into the myoendothelial space. This triggers several processes that explain the EDHF phenomena; including (1) synthesis of cytochrome that P450 (CYP450) metabolites, a family of epoxides, (2) transmission of endothelial cell hyperpolarization to the vascular smooth muscle via gap junctions, and (3) K+ released from the endothelial cells via KCa + channels induces smooth muscle hyperpolarization by activating KCa + channels and/or Na+-K+-ATPase on vascular smooth muscle cells.
Study of gastroepiploic conductance arteries and microvessels revealed that the contribution of EDHF to endothelium-dependent relaxations is significantly larger in human microvessels than in large arteries, that EDHF-mediated relaxations are mediated via activation of K+ channels, and that risk factors, particularly hypercholesterolemia and aging, were associated with impaired EDHF-mediated dilation [4]. Convincing evidence has been presented to suggest that a CYP450-dependent EDHF plays a significant role in the regulation of coronary arteriolar tone by KCa + channel activation and smooth muscle hyperpolarization [8, 29]. Thus, L-NG-nitro arginine/indomethacin-insensitive component of acetylcholine-mediated relaxation was sensitive to 25 mmol/L K+, but not to glibenclamide, a KATP channel inhibitor. Importantly, relaxation in human subcutaneous resistance arteries was abolished by a combination of charybdotoxin and apamin indicating the crucial role for endothelial KCa + channel activation [7].
Human studies have used tetraethylammonium chloride (TEA) to investigate the role of large KCa + channel activation on forearm blood flow and on the vascular effects of bradykinin. TEA selectively blocks KCa + channels in arterial smooth muscle cells at concentrations of >1 mmol/L [29]. At these doses, TEA decreased resting forearm blood flow by 23% and radial artery diameter by 5%, and it also inhibits bradykinin- and substance P-induced, but not acetylcholine-mediated vasodilation after inhibition of NO and prostaglandins in the forearm microcirculation [30–32]. Moreover, resting radial arterial blood flow and diameter were reduced only with combined blockade of NO synthesis and KCa + channels but not with either blockade individually, suggesting an important interaction between these two vasodilator systems [31]. These observations demonstrate the contribution of both NO and KCa + channel activation to resting conductance artery and microvascular tone in the healthy human forearm circulation.
2.2. Epoxyeicosatrienoic Acids (EETS) (Figure 1)
Epoxyeicosatrienoic acids (EETs) are arachidonic acid derived products of cytochrome P450 (CYP450) epoxygenases [33]. CYP450 enzymes are membrane-bound, heme-containing terminal oxidases in a multienzyme system. The arachidonic acid metabolizing CYP450 enzymes with prominent roles in vascular regulation are the epoxygenases of the CYP 2 gene family (e.g., CYP 2B, 2C8, 2C9, 2C10, and 2J2 in humans) that generate a series of region-specific and stereo-specific epoxides (5,6-, 8,9-, 11,12-, and 14,15-EETs), and the arachidonic acid v-hydroxylases belonging to the CYP 4A family which form hydroxyeicosatetraenoic acids (HETEs) which can demonstrate organ specific opposed actions [34].
Evidence in favor of EDHF being a short-lived metabolite of the cytochrome P450 epoxygenase pathway has been obtained using bovine [35, 36], porcine [36, 37], canine [38, 39], murine [40, 41], and human coronary arteries [8, 9, 29, 42–45]. EDHF-mediated responses are blocked by nonspecific CYP450 inhibitors such as miconazole, 17-octadecynoic acid, and more selective epoxygenase inhibitors [6-(2-proparglyloxyphenyl) hexanoic acid and N-methylsulphonyl-6-(2-propargyloxyphenyl) hexanamide] [46–48]. RT-PCR, Western blotting, and immunofluorescence techniques have demonstrated that native coronary endothelial cells express CYP epoxygenases, including CYP 2C8, CYP 2C9, and CYP 2J2 [49–52]. EET-induced activation of KCa + channels appears to be mediated by a cascade of intracellular events involving the ADP ribosylation of cellular proteins including anti-G(S)alpha antibody [53, 54] ultimately hyperpolarizing smooth muscle cells by increasing the open-state probability of KCa + channels [35, 41, 50, 55]. In porcine coronary arteries, a CYP450-derived epoxide, namely 11,12-epoxyeicosatrienoic acid (11,12-EET) has been shown to possess EDHF properties because (1) both 11,12-EET and bradykinin elicit hyperpolarization; (2) endothelial cells, but not smooth muscle cells, expressed mRNA and protein for the epoxygenase enzyme of the CYP2C family and released 11,12-EET; (3) induction of CYP2C8 or CYP2C34 increased epoxygenase expression, which was associated with increased release of 11, 12-EET, and enhanced relaxation and hyperpolarization in response to bradykinin; (4) an antisense oligonucleotide directed at the endothelial epoxygenase reduced both CYP2C mRNA and protein expression and the capacity to generate 11,12-EET, concomitantly with a reduction in the vasorelaxant and hyperpolarizing response to bradykinin [50]. The finding that sulfaphenazole, a selective inhibitor of CYP 2C9 [56, 57], inhibits EDHF-mediated responses [50] and potentiates non-NO-mediated relaxation in the porcine coronary artery [33] suggests that the CYP isoform required for the generation of EDHF is a porcine equivalent of CYP 2C9 [58]. Further, there is evidence in some species that CYP450-derived epoxides stimulate Na+/K+ ATPase [59]. These data strongly suggest that the activation of a cytochrome P450 epoxygenase is a prerequisite for the generation of EDHF-mediated relaxation in certain species.
Other intracellular second messenger roles of EETs may be equally as important in the control of vascular homeostasis. EETs (in particular 11,12- and 14,15-EET) activate several intracellular protein kinases including tyrosine kinases, the p38 MAP kinase, and extracellular-regulated protein kinases 1 and 2 (Erk1/2) and increase the proliferation of various cell types, including vascular smooth muscle cells and endothelial cells [33, 60, 61].
In isolated human coronary arterioles, CYP450-dependent hyperpolarization plays a significant role in the regulation of smooth muscle tone via activation of KCa + channels [8, 29]. Human coronary arteriolar endothelium-dependent hyperpolarization in response to arachidonic acid is much more dependent on metabolism by CYP450 than by cyclooxygenase or lipoxygenase or activation of NO synthase [8, 62]. The predominant EET synthesized by arteries is 11,12-EET, and its specific inhibition by chemically distinct CYP450 inhibitors impairs EDHF relaxation. 11,12-EET activates large-conductance KCa + channel current and hyperpolarizes arterial smooth muscle. Large-conductance KCa + channels and CYP450-2C mRNA and proteins are less abundant in arteries than saphenous veins explaining the lack of EDHF activity of veins. Large-conductance KCa + channels are primarily present in vascular smooth muscle, whereas the CYP450-2C enzyme is present in both the endothelium and smooth muscle cells. Thus, in human internal mammary arteries, EDHF appears to be 11,12-EET, produced by an EDHF synthase CYP450-2C and accounts for 40% of net endothelial relaxation by stimulating large-conductance KCa + channels [6].
The role of EETs as potential EDHFs can be studied using azoles such as miconazole that selectively inhibit epoxidation (EET generation) of arachidonic acid and have been demonstrated to be partly responsible for endothelium-dependent vasodilation in the human microcirculation [9, 31, 63]. In vivo studies have demonstrated CYP450 inhibition does not alter conductance vessel diameter or resting blood flow [31, 63, 64], but after inhibition of NO and prostacyclin, inhibition of EET synthesis further decreases radial arterial blood flow and diameter [31]. Thus, although it appears that under resting conditions in the healthy human forearm, conductance and resistance vessel tone is not modulated by tonic activity of CYP450-derived epoxides, their role becomes evident after inhibition of NO and prostacyclin synthesis, illustrating the potential compensatory role of EETs on maintenance of basal tone when NO availability is diminished.
In recent studies, we have addressed previous controversies regarding the contribution of EDHF to resting vasodilator tone. In the largest cohort studied to date, an important contribution of EDHF, via activation of KCa + channels, to resting microvascular dilator tone in the human forearm in vivo has been demonstrated [65]. For the first time, we have also demonstrated that cytochrome P450-derived epoxyeicosatrienoic acids also contribute to resting vasodilator tone in the healthy microcirculation with the use of fluconazole to block their action. We found a relatively greater contribution of NO compared to EDHF in the maintenance of resting vasodilator tone in the healthy human forearm microvasculature. The contribution of these two endogenous vasodilators to resting tone differed in subjects exposed to risk factors for atherosclerosis, in whom the vasculature is characterized by decreased NO bioavailability. We found preserved contribution of EDHF that appears to contribute equally as much as NO to resting vasodilator tone in subjects with risk factors. In the presence of NO blockade with L-NMMA, epoxyeicosatrienoic acid-mediated microvascular vasodilation also appeared to be upregulated in healthy subjects [65]. This indicates potential cross-talk between the NO and EET pathways, such that EET activity is upregulated in the setting of NO deficiency which may be demonstrated by blocking NO synthesis in the healthy circulation. Interestingly, in the risk factor group, there was a similar contribution of EETs to resting tone in the presence and absence of NO blockade. Such compensation may be crucial in maintaining normal resting blood flow in nonhypertensive patients with risk factors.
Finally, by conducting experiments using single and combined blockade, we also demonstrated that KCa + channel activation contributes to microvascular dilator tone even after inhibition of epoxyeicosatrienoic acids. This indicates that sources other than epoxyeicosatrienoic acids contribute to hyperpolarization of the resting human forearm microcirculation. Potential candidates which have been investigated in experimental studies, include hydrogen peroxide, potassium ions, and gap junctions. These alternate pathways warrant further investigation in vivo in humans.
2.3. Hydrogen Peroxide
Hydrogen peroxide also activates calcium-dependent potassium channels and remains a contender as an EDHF [66] (Figure 1). Reactive oxygen species can increase K+ channel activity and hyperpolarize smooth muscle [67, 68], and hydrogen peroxide may function as an EDHF [69]. Rubanyi and Vanhoutte reported that superoxide attenuates endothelium-dependent relaxations and that hydrogen peroxide causes endothelium-dependent and -independent relaxations [70]. Matoba et al. utilized catalase, an endogenous peroxidase to show inhibition of EDHF-mediated, endothelium-dependent relaxations and hyperpolarizations, resistant to indomethacin or N(omega)-nitro-l-arginine [71]. These findings have been confirmed in piglet pial arteries, canine subepicardial coronary arteries and arterioles, and during flow-induced vasodilation in human mesenteric arteries and coronary microvessels [66, 72–74].
Vascular endothelial cells have a capacity to produce superoxide and hydrogen peroxide from several intracellular sources, including endothelial NO synthase, cyclooxygenases, lipoxygenases, cytochrome P-450 epoxygenases, NAD(P)H oxidases, and xanthine oxidase [71, 75–78]. Flow-mediated dilation involves generation of superoxide originating from mitochondria and shear stress elicits luminal release of mitochondrial ubisemiquinone, a source for generating superoxide and hydrogen peroxide via metabolic processes occurring between complex I and complex III of the electron transport chain [79]. Although reactive oxygen species appear to fit the profile of EDHF, their physiologic role remains a subject of debate particularly in some human arteries because in human radial and internal mammary arteries, neither catalase nor superoxide dismutase inhibited relaxations to carbachol [80, 81].
Hydrogen peroxide also mediates hyperpolarization via activation of endothelial K+ channels, however, many species variations exist in the type of K+ channels that are activated [67, 68, 72, 73, 82–84]. Importantly, in human coronary microvessels, K+ Ca channels sensitive to charybdotoxin plus apamin appear to mediate hyperpolarization [73, 85]. In mouse mesenteric arteries, the inhibitory effect of catalase was unmasked by the inhibition of NO production, and vice versa, suggesting that NO and EDHF (hydrogen peroxide) compensate for each other [71, 86–89]. In canine subepicardial coronary arteries and arterioles, the response to acetylcholine and hypoxia was inhibited by L-NMMA primarily in subepicardial coronary arteries, whereas combined infusion of L-NMMA plus catalase or tetraethylammonium attenuated the vasodilator responses of coronary arteries of both sizes, demonstrating the predominance of hydrogen peroxide-mediated hyperpolarization in microvessels [74].
2.4. Gap Junctions
The EDHF phenomenon may be further explained by the transmission of endothelial cell hyperpolarization to the vascular smooth muscle via gap junctions [90–92]. These are myoendothelial and heterocellular. They couple endothelial cells to other endothelial cells and to smooth muscle cells, providing a low-resistance electrical pathway between the cell layers. Gap junctions are formed by the docking of two connexons present in adjacent cells that creates an aqueous pore permitting the transfer of ions and electrical continuity that establishes a uniform membrane potential across cells [93, 94]. Their number increases with diminution in the size of the artery [95], paralleling the importance of EDHF to vessel size with a greater influence in the resistance than in the conductance vessels [96].
Investigation of gap junctions as other potential EDHF mechanisms has been limited in man due to the lack of specific pharmacological agents. Rotigaptide, that enhances communication via the connexin 43 gap junction subunit, had no effect on basal vascular tone endothelium-dependent (bradykinin), -independent vasodilation, or t-PA release in the forearm arterial circulation of healthy men [97].
2.5. Potassium (K+)
A moderate increase in the myoendothelial K+ concentration can in some species [98] induce hyperpolarization of vascular smooth muscle cells by activating the inwardly rectifying K+ channels and the Na+/ K+ ATPase [24, 99, 100]. However, it is unlikely that K+ per se is EDHF.
3. Interactions between EDHF, Nitric Oxide, and Prostacyclin
The three main mediators of endothelial vasodilator function, NO, prostacyclin, and EDHF appear not to be mutually exclusive and act synergistically in a complex manner to maintain the health of the vasculature (Figure 1). In conduit arteries, NO is the predominant endothelium-derived vasodilator but has relatively less prominent contribution in the resistance vessels of the microcirculation where EDHF appears to predominate [96]. NO may tonically inhibit EDHF responses as some studies could only demonstrate EDHF responses once NO production had been inhibited [44].
4. EDHF and Disease
Experimental evidence indicates that a shift away from NO-mediated endothelium-dependent relaxation toward EDHF-dependent relaxation occurs in disease states [101, 102]. Alteration of EDHF-mediated responses has been reported with aging, hypertension, atherosclerosis, hypercholesterolemia, heart failure, angioplasty, eclampsia, diabetes, and sepsis. Depending on the vascular bed, this may either contribute to endothelial dysfunction or compensate for the loss of NO bioavailability [103–105]. In the human forearm of hypertensive subjects, Taddei and others demonstrated that endothelium-dependent vasodilation is maintained despite decreased NO bioavailability because of the compensatory increased activity of EDHF [25, 104, 105]. Hypercholesterolemia is generally associated with preserved EDHF responses where its enhanced activity may compensate for the decrease in NO-mediated relaxation [106–108]. Endothelium-dependent hyperpolarization appears to be inhibited in isolated gastroepiploic arteries from atherosclerotic patients, an effect that may be secondary to the duration of hypercholesterolemic injury [4]. In contrast, EDHF-mediated responses are depressed in some models of type I and type II diabetes with the exception of murine models [109].
In the healthy forearm microcirculation, we demonstrated that bradykinin-stimulated vasodilation is in part mediated by activation of KCa + channels and that the magnitude of contribution of NO was less than the contribution of KCa + channel activation [65]. Importantly, we found no contribution of KCa + channel activation to acetylcholine-stimulated vasodilation in healthy subjects, either in the presence or absence of NO-blockade. Thus, acetylcholine predominantly stimulates the release of NO and no EDHF, whereas bradykinin stimulates release of both EDHF and NO. We also demonstrated a similar contribution of NO and EDHF to bradykinin-mediated vasodilation in both groups. In contrast to effects of bradykinin, forearm blood flow response to acetylcholine was diminished in hypercholesterolemic subjects when compared to healthy subjects. Moreover, in hypercholesterolemia, we observed a significant contribution of KCa + channel activation and a lower NO release with acetylcholine that was distinctly different compared to the healthy circulation. Thus, while in health NO is the predominant contributor, in hypercholesterolemia, both NO and KCa + channel activation contribute equally to acetylcholine-mediated microcirculatory vasodilation [65].
Evidence suggests that CYP expression [110–112] and EET generation are increased in hypertension [113, 114], during salt loading [115], and in hypercholesterolemia [116]. Members of the CYP 2C family are inhibited by NO, a phenomenon that may explain why EDHF-mediated responses are barely detectable in the absence of the combined inhibition of NO synthases and cyclooxygenase in normal vessels. Thus the EET/EDHF pathway may be of less importance in healthy vessels and of greater significance in disease states where NO activity is impaired [58]. A similar phenomenon has been described for bradykinin-induced changes in forearm blood flow in essential hypertensive patients [25] and in arterioles removed from patients with coronary artery disease, where vasodilatation is mediated entirely by a mechanism sensitive to both CYP and KCa + channel inhibitors [9]. Such findings indicate that in the absence of NO, vascular tone can be regulated by an EDHF-like mechanism. Thus, whether EDHF plays a causal or compensatory role in the endothelial dysfunction in the human circulation remains to be elucidated.
5. Summary
Absence of consensus regarding the precise identity of EDHFs and a consequent lack of specific inhibitors has long hampered clinical translation of this phenomenon (Table 1). Recently, with improved understanding of the major signaling mechanisms underlying vascular hyperpolarization, the role of EDHF in the human circulation in vivo has begun to be dissected, but experimental pitfalls remain. These include the often nonspecific nature of the antagonists used, the concentrations and duration of action of these blockers are variable, and complete blockade cannot be achieved in vivo, even with high doses, because of the competitive nature of the antagonism. Nevertheless, an impressive body of knowledge has already emerged regarding the role of EDHF in the human circulation (Table 2).
Table 1.
Pharmacological inhibitors of EDHF. Pharmacological agents used as potential inhibitors of EDHF and their targets and limitations (modified from Torondel et al. [117]).
| Pharmacological inhibitors | Targets | Comments |
|---|---|---|
| Apamin | SKCa + | Highly specific |
| Charybdotoxin | IKCa +-BKCa + | Can inhibit some Kv channels |
| Iberiotoxin | BKCa + | Highly specific |
| Tetraethylammonium | SKCa +-IKCa +-BKCa + | Inhibit other K+ channels at >10−2) m |
| Tetraethybutylammonium | SKCa +-IKCa +-BKCa + | Inhibit other K+ channels at >10−2 m |
| BaCl2 | KIR + | — |
| Ouabain | Na+/K+ ATPase | Can affect gap junction activity at >10−4 m |
| KCL | K+ currents | Dilates at >10−2 m through KIR + and Na+/K+ ATPase activation |
| 18 α-glycyrrhetic acid | Gap junctions | Possesses nonjunctional effects on membrane currents |
| Connexin mimetic peptides | Gap junctions | Highly specific |
| Catalase | Hydrogen peroxide | — |
| 17-octadecenoic acid | CYP | Inhibits the synthesis of the vasoconstrictor 20-HETE |
| Clotrimazole | CYP | Can inhibit K+ channels |
| Miconazole | CYP | Can inhibit K+ channels |
| Sulphaphenazole | CYP epoxygenase | Highly specific of CYP 2C9 |
| Fluconazole | CYP epoxygenase | Can inhibit other CYP isoforms at >10−4 m |
| MSPPOH | EETs synthesis inhibitor | Highly specific |
| 14,15-EEZE | EETs antagonist | Inhibits the vasodilator action of all EETs regioisomers |
K+: potassium, SKCa +: small calcium-dependent potassium channels, IKCa +: intermediate calcium-dependent potassium channels, BKCa +: large calcium-dependent potassium channels, Kv: voltage dependent potassium channels, KIR +: inwardly rectifying potassium channels, BaCl2 barium chloride, KCL: potassium chloride, CYP: cytochrome, 20-HETE: 20-hydroxyeicosatetraenoic acids, MSPPOH: N-(methylsulfonyl)-2-(2-propynyloxy)-benzenehexanamide, EETs: epoxyeicosatrienoic acids, and 14,15-EEZE: 14,15-Epoxyeicosa-5(Z)-enoic Acid.
Table 2.
Human vascular territories with characterized EDHF activity.
| Vascular territory | EDHF | Pharmacological agents used |
|---|---|---|
| Preclinical studies | ||
| Coronary arterioles [8, 9, 29, 73, 118] | H2O2, KCa + channels, CYP450 metabolites | Catalase, KCl, charybdotoxin + Apamin, polyethylene glycol catalase, KCl, charybdotoxin, 7-octadecynoic acid |
| Internal Mammary artery [6, 62] | 11,12-EET | 17-octadecynoic acid, N-methylsulfonyl-6-(2-propargyloxyphenyl)hexanamide4,15-epoxyeicosa-5(Z)-enoic acid |
| Gastroepiploic arteries [4] | KCa + channels | KCl |
| Mesenteric artery [66, 119] | H2O2, Gap junctions, superoxide dismutase, H2O2 | Catalase, 18 alpha-glycyrrhetinic acid, Tiron (cell-permeable SOD-mimetic), catalase |
| Renal artery [10] | K+, KCa + channels | KCl, charybdotoxin, and apamin |
| Subcutaneous resistance arteries [7] | CYP450 metabolites, KCa + channels | Ketoconazole |
| Subcutaneous resistance arteries [120] (subcutaneous fat biopsies of healthy pregnant women) | Connexin 43 Gap junctions. | Connexin mimetic peptides |
| Visceral fat arterioles | H2O2 | Polyethylene glycol catalase |
| Umbilical vein endothelial cells [85, 121] | SKCa + channels, IKCa +channels, H2O2 | Apamin and charybdotoxin/triarylmethane-34 |
| Thyroid arteries [122] | KCa + channels, KIR + channels, Na+/K+ ATPase | Iberiotoxin, charybdotoxin, apamin glibenclamide, and barium |
|
| ||
| Clinical studies | ||
| Forearm microvasculature [63] | CYP450 metabolites | KCL, miconazole |
| Forearm microvasculature [30, 32, 123] | KCa + channels | TEA |
| Forearm microvasculature [124] | C-type natriuretic peptide | C-type natriuretic peptide, TEA |
| Forearm microvasculature [104] (hypertensive patients) | CYP450 2C9 | Sulfaphenazole |
| Forearm conductance vessel [31, 125, 126] | CYP 2C9 metabolites, KCa + channels | Sulfaphenazole, TEA, fluconazole |
| Thigh skeletal muscle vessels [127] | CYP450 2C9 | Sulfaphenazole |
H2O2: Hydrogen Peroxide, K+: potassium, SKCa +: small calcium-dependent potassium channels, IKCa +: intermediate calcium-dependent potassium channels, KIR +: inwardly rectifying potassium channels, KCL: potassium chloride, CYP: cytochrome, and EETs: epoxyeicosatrienoic acids.
Apart from its contribution to normal vascular physiology, the accentuated role of EDHF in diseased states is worthy of further investigation because CYP-450 expression and EET generation are increased in hypertension, during salt loading and in hypercholesterolemia. Vasodilation in essential hypertension [25], and in atherosclerotic coronary arterioles, is largely secondary to CYP and KCa + channel stimulation. There are also potential implications regarding disease susceptibility, with some polymorphisms within CYP epoxygenases being associated with an enhanced risk of developing coronary artery disease and hypertension [14]. What may ultimately be of even greater interest is development of specific agents targeting EDHF synthesis, understanding of other biological effects of EETs such as angiogenesis and modulation of cell growth, and their potential role in human disease [14].
6. Implications
Conventionally, endothelial dysfunction is characterized as a deficiency of NO activity, often secondary to exposure to cardiovascular risk factors. This leads to abnormalities in vasodilation and hence blood flow delivery. Because of the known protective role of NO on the vessel wall that impedes thrombosis and atherosclerosis, several strategies have been applied to improve NO availability/activity. Although replacing NO pharmacologically with NO donors is beneficial for symptomatic relief from coronary vasodilation, there appears to be no antiatherosclerotic effect of NO donors. Improving endothelial NO bioavailability with statins and angiotensin antagonists has nevertheless proven to be cardioprotective. What remains unknown is (a) whether enhancing EDHF in conditions with impaired NO activity would also be of therapeutic value, (b) whether agents that improve endothelial dysfunction (acetylcholine responses) such as statins and angiotensin antagonists, also enhance EDHF bioactivity, and (c) whether tissue plasminogen activator release is EDHF-dependent in health and disease. Indeed hypertension is associated with elevated epoxide hydrolase expression [128, 129], angiotensin II increases the expression of the epoxide hydrolase [130], and epoxide hydrolase inhibitors are effective in reversing the hypertensive effects of angiotensin II [128]. Thus, epoxide hydrolase inhibitors that increase epoxide levels and hence aid hyperpolarization need to be further investigated in subjects with endothelial dysfunction.
Experimental studies indicate that cytochrome P450 expression and EET generation are increased in hypertension [104, 113], in hypercholesterolemia [116], and in atherosclerotic coronary arterioles [50, 64, 75]. Moreover, polymorphisms in the cytochrome P450 epoxygenase genes are associated with increased risk of coronary artery disease and hypertension [131, 132]. Thus, understanding the pathophysiology of endothelial dysfunction beyond NO, and in particular with respect to EDHF in these disease states, could be crucial in understanding both the pathophysiology of atherosclerosis and developing novel therapies.
Acknowledgments
Professor A. Quyyumi was supported by National Institutes of Health Research Grant RO1 HL79115, and in part by PHS Grant UL1 RR025008 from the Clinical and Translational Science Award Program, and PHS Grant M01 RR00039 from the General Clinical Research Center program, National Institutes of Health, National Center for Research Resources, and the National Blood Foundation.
References
- 1.Quyyumi AA, Dakak N, Mulcahy D, et al. Nitric oxide activity in the atherosclerotic human coronary circulation. Journal of the American College of Cardiology. 1997;29(2):308–317. doi: 10.1016/s0735-1097(96)00472-x. [DOI] [PubMed] [Google Scholar]
- 2.Ashfaq S, Beinart SC, Abramson JL, et al. Plasma glutathione redox state: a novel marker of oxidative stress, correlates with early atherosclerosis in humans. Journal of the American College of Cardiology. 2003;41:293A–294A. [Google Scholar]
- 3.Quyyumi AA. Endothelial function in health and disease: new insights into the genesis of cardiovascular disease. American Journal of Medicine. 1998;105:32S–39S. doi: 10.1016/s0002-9343(98)00209-5. [DOI] [PubMed] [Google Scholar]
- 4.Urakami-Harasawa L, Shimokawa H, Nakashima M, Egashira K, Takeshita A. Importance of endothelium-derived hyperpolarizing factor in human arteries. Journal of Clinical Investigation. 1997;100(11):2793–2799. doi: 10.1172/JCI119826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Woolfson RG, Poston L. Effect of NG-monomethyl-L-arginine on endothelium-dependent relaxation of human subcutaneous resistance arteries. Clinical Science. 1990;79(3):273–278. doi: 10.1042/cs0790273. [DOI] [PubMed] [Google Scholar]
- 6.Archer SL, Gragasin FS, Wu X, et al. Endothelium-derived hyperpolarizing factor in human internal mammary artery is 11,12-epoxyeicosatrienoic acid and causes relaxation by activating smooth muscle BKCa channels. Circulation. 2003;107(5):769–776. doi: 10.1161/01.cir.0000047278.28407.c2. [DOI] [PubMed] [Google Scholar]
- 7.Coats P, Johnston F, MacDonald J, McMurray JJ, Hillier C. Endothelium-derived hyperpolarizing factor: identification and mechanisms of action in human subcutaneous resistance arteries. Circulation. 2001;103(12):1702–1708. doi: 10.1161/01.cir.103.12.1702. [DOI] [PubMed] [Google Scholar]
- 8.Miura H, Gutterman DD. Human coronary arteriolar dilation to arachidonic acid depends on cytochrome P-450 monooxygenase and Ca2+-activated K+ channels. Circulation Research. 1998;83(5):501–507. doi: 10.1161/01.res.83.5.501. [DOI] [PubMed] [Google Scholar]
- 9.Miura H, Wachtel RE, Liu Y, et al. Flow-induced dilation of human coronary arterioles: important role of Ca2+-activated K+ channels. Circulation. 2001;103(15):1992–1998. doi: 10.1161/01.cir.103.15.1992. [DOI] [PubMed] [Google Scholar]
- 10.Bussemaker E, Popp R, Binder J, Busse R, Fleming I. Characterization of the endothelium-derived hyperpolarizing factor (EDHF) response in the human interlobar artery. Kidney International. 2003;63(5):1749–1755. doi: 10.1046/j.1523-1755.2003.00910.x. [DOI] [PubMed] [Google Scholar]
- 11.Scotland RS, Madhani M, Chauhan S, et al. Investigation of vascular responses in endothelial nitric oxide synthase/cyclooxygenase-1 double-knockout mice: key role for endothelium-derived hyperpolarizing factor in the regulation of blood pressure in vivo. Circulation. 2005;111(6):796–803. doi: 10.1161/01.CIR.0000155238.70797.4E. [DOI] [PubMed] [Google Scholar]
- 12.Wu Y, Huang A, Sun D, Falck JR, Koller A, Kaley G. Gender-specific compensation for the lack of NO in the mediation of flow-induced arteriolar dilation. American Journal of Physiology. 2001;280(6):H2456–H2461. doi: 10.1152/ajpheart.2001.280.6.H2456. [DOI] [PubMed] [Google Scholar]
- 13.Feletou M, Vanhoutte PM. Endothelium-derived hyperpolarizing factor: where are we now? Arteriosclerosis, Thrombosis, and Vascular Biology. 2006;26(6):1215–1225. doi: 10.1161/01.ATV.0000217611.81085.c5. [DOI] [PubMed] [Google Scholar]
- 14.Fleming I, Busse R. Endothelium-derived epoxyeicosatrienoic acids and vascular function. Hypertension. 2006;47(4):629–633. doi: 10.1161/01.HYP.0000208597.87957.89. [DOI] [PubMed] [Google Scholar]
- 15.Busse R, Edwards G, Feletou M, Fleming I, Vanhoutte PM, Weston AH. EDHF: bringing the concepts together. Trends in Pharmacological Sciences. 2002;23(8):374–380. doi: 10.1016/s0165-6147(02)02050-3. [DOI] [PubMed] [Google Scholar]
- 16.Bolton TB, Clapp LH. Endothelial-dependent relaxant actions of carbachol and substance P in arterial smooth muscle. British Journal of Pharmacology. 1986;87(4):713–723. doi: 10.1111/j.1476-5381.1986.tb14589.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen G, Suzuki H, Weston AH. Acetylcholine releases endothelium-derived hyperpolarizing factor and EDRF from rat blood vessels. British Journal of Pharmacology. 1988;95(4):1165–1174. doi: 10.1111/j.1476-5381.1988.tb11752.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cohen RA, Vanhoutte PM. Endothelium-dependent hyperpolarization: beyond nitric oxide and cyclic GMP. Circulation. 1995;92(11):3337–3349. doi: 10.1161/01.cir.92.11.3337. [DOI] [PubMed] [Google Scholar]
- 19.Feletou M, Vanhoutte PM. Endothelium-dependent hyperpolarization of canine coronary smooth muscle. British Journal of Pharmacology. 1988;93(3):515–524. doi: 10.1111/j.1476-5381.1988.tb10306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luckhoff A, Zeh R, Busse R. Desensitization of the bradykinin-induced rise in intracellular free calcium in cultured endothelial cells. Pflugers Archiv. 1988;412(6):654–658. doi: 10.1007/BF00583768. [DOI] [PubMed] [Google Scholar]
- 21.Johns RA, Izzo NJ, Milner PJ, Loeb AL, Peach MJ. Use of cultured cells to study the relationship between arachidonic acid and endothelium-derived relaxing factor. American Journal of the Medical Sciences. 1988;295(4):287–292. doi: 10.1097/00000441-198804000-00012. [DOI] [PubMed] [Google Scholar]
- 22.Chen G, Hashitani H, Suzuki H. Endothelium-dependent relaxation and hyperpolarization of canine coronary artery smooth muscles in relation to the electrogenic Na-K pump. British Journal of Pharmacology. 1989;98(3):950–956. doi: 10.1111/j.1476-5381.1989.tb14625.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Taylor SG, Southerton JS, Weston AH, Baker JR. Endothelium-dependent effects of acetylcholine in rat aorta: a comparison with sodium nitroprusside and cromakalim. British Journal of Pharmacology. 1988;94(3):853–863. doi: 10.1111/j.1476-5381.1988.tb11597.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dawes M, Sieniawska C, Delves T, Dwivedi R, Chowienczyk PJ, Ritter JM. Barium reduces resting blood flow and inhibits potassium-induced vasodilation in the human forearm. Circulation. 2002;105(11):1323–1328. doi: 10.1161/hc1102.105651. [DOI] [PubMed] [Google Scholar]
- 25.Taddei S, Ghiadoni L, Virdis A, Buralli S, Salvetti A. Vasodilation to bradykinin is mediated by an ouabain-sensitive pathway as a compensatory mechanism for impaired nitric oxide availability in essential hypertensive patients. Circulation. 1999;100(13):1400–1405. doi: 10.1161/01.cir.100.13.1400. [DOI] [PubMed] [Google Scholar]
- 26.Corriu C, Feletou M, Canet E, Vanhoutte PM. Endothelium-derived factors and hyperpolarization of the carotid artery of the guinea-pig. British Journal of Pharmacology. 1996;119(5):959–964. doi: 10.1111/j.1476-5381.1996.tb15765.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garland CJ, Plane F. Relative importance of endothelium-derived hyperpolarizing factor for the relaxation of vascular smooth muscle in different arterial beds. In: Vanhoutte PM, editor. Endothelium-Derived Hyperpolarizing Factor. Harwood Academic Publishers; 1996. pp. 173–179. [Google Scholar]
- 28.Zygmunt PM, Hogestatt ED. Role of potassium channels in endothelium-dependent relaxation resistant to nitroarginine in the rat hepatic artery. British Journal of Pharmacology. 1996;117(7):1600–1606. doi: 10.1111/j.1476-5381.1996.tb15327.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miura H, Liu Y, Gutterman DD. Human coronary arteriolar dilation to bradykinin depends on membrane hyperpolarization: contribution of nitric oxide and Ca2+-activated K+ channels. Circulation. 1999;99(24):3132–3138. doi: 10.1161/01.cir.99.24.3132. [DOI] [PubMed] [Google Scholar]
- 30.Honing ML, Smits P, Morrison PJ, Rabelink TJ. Bradykinin-induced vasodilation of human forearm resistance vessels is primarily mediated by endothelium-dependent hyperpolarization. Hypertension. 2000;35(6):1314–1318. doi: 10.1161/01.hyp.35.6.1314. [DOI] [PubMed] [Google Scholar]
- 31.Bellien J, Joannides R, Iacob M, Arnaud P, Thuillez C. Evidence for a basal release of a cytochrome-related endothelium-derived hyperpolarizing factor in the radial artery in humans. American Journal of Physiology. 2006;290(4):H1347–H1352. doi: 10.1152/ajpheart.01079.2005. [DOI] [PubMed] [Google Scholar]
- 32.Inokuchi K, Hirooka Y, Shimokawa H, et al. Role of endothelium-derived hyperpolarizing factor in human forearm circulation. Hypertension. 2003;42(5):919–924. doi: 10.1161/01.HYP.0000097548.92665.16. [DOI] [PubMed] [Google Scholar]
- 33.Fleming I. Cytochrome P450 and vascular homeostasis. Circulation Research. 2001;89(9):753–762. doi: 10.1161/hh2101.099268. [DOI] [PubMed] [Google Scholar]
- 34.Schwartzman M, Ferreri NR, Carroll MA. Renal cytochrome P450-related arachidonate metabolite inhibits (Na+ + K+)ATPase. Nature. 1985;314(6012):620–622. doi: 10.1038/314620a0. [DOI] [PubMed] [Google Scholar]
- 35.Campbell WB, Gebremedhin D, Pratt PF, Harder DR. Identification of epoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factors. Circulation Research. 1996;78(3):415–423. doi: 10.1161/01.res.78.3.415. [DOI] [PubMed] [Google Scholar]
- 36.Hecker M, Bara AT, Bauersachs J, Busse R. Characterization of endothelium-derived hyperpolarizing factor as a cytochrome P450-derived arachidonic acid metabolite in mammals. Journal of Physiology. 1994;481(2):407–414. doi: 10.1113/jphysiol.1994.sp020449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Popp R, Bauersachs J, Hecker M, Fleming I, Busse R. A transferable, beta-naphthoflavone-inducible, hyperpolarizing factor is synthesized by native and cultured porcine coronary endothelial cells. Journal of Physiology. 1996;497(3):699–709. doi: 10.1113/jphysiol.1996.sp021801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nishikawa Y, Stepp DW, Chilian WM. In vivo location and mechanism of EDHF-mediated vasodilation in canine coronary microcirculation. American Journal of Physiology. 1999;277(3):H1252–H1259. doi: 10.1152/ajpheart.1999.277.3.H1252. [DOI] [PubMed] [Google Scholar]
- 39.Widmann MD, Weintraub NL, Fudge JL, Brooks LA, Dellsperger KC. Cytochrome P-450 pathway in acetylcholine-induced canine coronary microvascular vasodilation in vivo. American Journal of Physiology. 1998;274(1):H283–H289. doi: 10.1152/ajpheart.1998.274.1.H283. [DOI] [PubMed] [Google Scholar]
- 40.Fulton D, Mahboubi K, McGiff JC, Quilley J. Cytochrome P450-dependent effects of bradykinin in the rat heart. British Journal of Pharmacology. 1995;114(1):99–102. doi: 10.1111/j.1476-5381.1995.tb14911.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fulton D, Mcgiff JC, Quilley J. Pharmacological evaluation of an epoxide as the putative hyperpolarizing factor mediating the nitric oxide-independent vasodilator effect of bradykinin in the rat heart. Journal of Pharmacology and Experimental Therapeutics. 1998;287(2):497–503. [PubMed] [Google Scholar]
- 42.Quilley J, McGiff JC. Is EDHF an epoxyeicosatrienoic acid? Trends in Pharmacological Sciences. 2000;21(4):121–124. doi: 10.1016/s0165-6147(00)01445-0. [DOI] [PubMed] [Google Scholar]
- 43.Weintraub NL, Fang X, Kaduce TL, Vanrollins M, Chatterjee P, Spector AA. Potentiation of endothelium-dependent relaxation by epoxyeicosatrienoic acids. Circulation Research. 1997;81(2):258–267. doi: 10.1161/01.res.81.2.258. [DOI] [PubMed] [Google Scholar]
- 44.Nishikawa Y, Stepp DW, Chilian WM. Nitric oxide exerts feedback inhibition on EDHF-induced coronary arteriolar dilation in vivo. American Journal of Physiology. 2000;279(2):H459–H465. doi: 10.1152/ajpheart.2000.279.2.H459. [DOI] [PubMed] [Google Scholar]
- 45.Bauersachs J, Hecker M, Busse R. Display of the characteristics of endothelium-derived hyperpolarizing factor by a cytochrome P450-derived arachidonic acid metabolite in the coronary microcirculation. British Journal of Pharmacology. 1994;113(4):1548–1553. doi: 10.1111/j.1476-5381.1994.tb17172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alvarez J, Montero M, Garcia-Sancho J. High affinity inhibition of Ca2+-dependent K+ channels by cytochrome P- 450 inhibitors. Journal of Biological Chemistry. 1992;267(17):11789–11793. [PubMed] [Google Scholar]
- 47.Alvarez DF, Gjerde EA, Townsley MI. Role of EETs in regulation of endothelial permeability in rat lung. American Journal of Physiology. 2004;286(2):L445–L451. doi: 10.1152/ajplung.00150.2003. [DOI] [PubMed] [Google Scholar]
- 48.Imig JD, Falck JR, Wei S, Capdevila JH. Epoxygenase metabolites contribute to nitric oxide-independent afferent arteriolar vasodilation in response to bradykinin. Journal of Vascular Research. 2001;38(3):247–255. doi: 10.1159/000051053. [DOI] [PubMed] [Google Scholar]
- 49.Fisslthaler B, Fleming I, Busse R. EDHF: a cytochrome P450 metabolite in coronary arteries. Seminars in Perinatology. 2000;24(1):15–19. doi: 10.1016/s0146-0005(00)80048-8. [DOI] [PubMed] [Google Scholar]
- 50.Fissithaler B, Popp R, Kiss L, et al. Cytochrome P450 2C is an EDHF synthase in coronary arteries. Nature. 1999;401(6752):493–497. doi: 10.1038/46816. [DOI] [PubMed] [Google Scholar]
- 51.Wu J, Dong H, Cai Z, Yu Y. Stable expression of human cytochrome CYP2B6 and CYP1A1 in Chinese hamster CHL cells: their use in micronucleus assays. Chinese Medical Sciences Journal. 1997;12(3):148–155. [PubMed] [Google Scholar]
- 52.Node K, Huo Y, Ruan X, et al. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science. 1999;285(5431):1276–1279. doi: 10.1126/science.285.5431.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li PL, Campbell WB. Epoxyeicosatrienoic acids activate K+ channels in coronary smooth muscle through a guanine nucleotide binding protein. Circulation Research. 1997;80(6):877–884. doi: 10.1161/01.res.80.6.877. [DOI] [PubMed] [Google Scholar]
- 54.Li PL, Chen CL, Bortell R, Campbell WB. 11,12-epoxyeicosatrienoic acid stimulates endogenous mono-ADP-ribosylation in bovine coronary arterial smooth muscle. Circulation Research. 1999;85(4):349–356. doi: 10.1161/01.res.85.4.349. [DOI] [PubMed] [Google Scholar]
- 55.Gebremedhin D, Ma YH, Falck JR, Roman RJ, VanRollins M, Harder DR. Mechanism of action of cerebral epoxyeicosatrienoic acids on cerebral arterial smooth muscle. American Journal of Physiology. 1992;263(2):H519–H525. doi: 10.1152/ajpheart.1992.263.2.H519. [DOI] [PubMed] [Google Scholar]
- 56.Mancy A, Dijols S, Poli S, Guengerich P, Mansuy D. Interaction of sulfaphenazole derivatives with human liver cytochromes P450 2C: molecular origin of the specific inhibitory effects of sulfaphenazole on CYP 2C9 and consequences for the substrate binding site topology of CYP 2C9. Biochemistry. 1996;35(50):16205–16212. doi: 10.1021/bi961950t. [DOI] [PubMed] [Google Scholar]
- 57.Sai Y, Dai R, Yang TJ, et al. Assessment of specificity of eight chemical inhibitors using cDNA-expressed cytochromes P450. Xenobiotica. 2000;30(4):327–343. doi: 10.1080/004982500237541. [DOI] [PubMed] [Google Scholar]
- 58.Bolz SS, Pieperhoff S, De Wit C, Pohl U. Intact endothelial and smooth muscle function in small resistance arteries after 48 h in vessel culture. American Journal of Physiology. 2000;279(3):H1434–H1439. doi: 10.1152/ajpheart.2000.279.3.H1434. [DOI] [PubMed] [Google Scholar]
- 59.Pratt PF, Li P, Hillard CJ, Kurian J, Campbell WB. Endothelium-independent, ouabain-sensitive relaxation of bovine coronary arteries by EETs. American Journal of Physiology. 2001;280(3):H1113–H1121. doi: 10.1152/ajpheart.2001.280.3.H1113. [DOI] [PubMed] [Google Scholar]
- 60.Fleming I, Fisslthaler B, Michaelis UR, Kiss L, Popp R, Busse R. The coronary endothelium-derived hyperpolarizing factor (EDHF) stimulates multiple signalling pathways and proliferation in vascular cells. Pflugers Archiv. 2001;442(4):511–518. doi: 10.1007/s004240100565. [DOI] [PubMed] [Google Scholar]
- 61.Uddin MR, Muthalif MM, Karzoun NA, Benter IF, Malik KU. Cytochrome P-450 metabolites mediate norepinephrine-induced mitogenic signaling. Hypertension. 1998;31(1):242–247. doi: 10.1161/01.hyp.31.1.242. [DOI] [PubMed] [Google Scholar]
- 62.He GW, Liu ZG. Comparison of nitric oxide release and endothelium-derived hyperpolarizing factor-mediated hyperpolarization between human radial and internal mammary arteries. Circulation. 2001;104(supplement 1):i344–i349. doi: 10.1161/hc37t1.094930. [DOI] [PubMed] [Google Scholar]
- 63.Halcox JPJ, Narayanan S, Cramer-Joyce L, Mincemoyer R, Quyyumi AA. Characterization of endothelium-derived hyperpolarizing factor in the human forearm microcirculation. American Journal of Physiology. 2001;280(6):H2470–H2477. doi: 10.1152/ajpheart.2001.280.6.H2470. [DOI] [PubMed] [Google Scholar]
- 64.Fichtlscherer S, Dimmeler S, Breuer S, Busse R, Zeiher AM, Fleming I. Inhibition of cytochrome P450 2C9 improves endothelium-dependent, nitric oxide-mediated vasodilatation in patients with coronary artery disease. Circulation. 2004;109(2):178–183. doi: 10.1161/01.CIR.0000105763.51286.7F. [DOI] [PubMed] [Google Scholar]
- 65.Ozkor MA, Murrow JR, Rahman AM, et al. Endothelium-derived hyperpolarizing factor determines resting and stimulated forearm vasodilator tone in health and in disease. Circulation. 2011;123(20):2244–2253. doi: 10.1161/CIRCULATIONAHA.110.990317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Matoba T, Shimokawa H, Kubota H, et al. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in human mesenteric arteries. Biochemical and Biophysical Research Communications. 2002;290(3):909–913. doi: 10.1006/bbrc.2001.6278. [DOI] [PubMed] [Google Scholar]
- 67.Barlow RS, White RE. Hydrogen peroxide relaxes porcine coronary arteries by stimulating BKCa channel activity. American Journal of Physiology. 1998;275(4):H1283–H1289. doi: 10.1152/ajpheart.1998.275.4.H1283. [DOI] [PubMed] [Google Scholar]
- 68.Barlow RS, El-Mowafy AM, White RE. H2O2 opens BKCa channels via the PLA2-arachidomic acid signaling cascade in coronary artery smooth muscle. American Journal of Physiology. 2000;279(2):H475–H483. doi: 10.1152/ajpheart.2000.279.2.H475. [DOI] [PubMed] [Google Scholar]
- 69.Beny JL, von der Weid PY. Hydrogen peroxide: an endogenous smooth muscle cell hyperpolarizing factor. Biochemical and Biophysical Research Communications. 1991;176(1):378–384. doi: 10.1016/0006-291x(91)90935-z. [DOI] [PubMed] [Google Scholar]
- 70.Rubanyi GM, Romero JC, Vanhoutte PM. Flow-induced release of endothelium-derived relaxing factor. American Journal of Physiology. 1986;250(6):H1145–H1149. doi: 10.1152/ajpheart.1986.250.6.H1145. [DOI] [PubMed] [Google Scholar]
- 71.Matoba T, Shimokawa H, Nakashima M, et al. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in mice. Journal of Clinical Investigation. 2000;106(12):1521–1530. doi: 10.1172/JCI10506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lacza Z, Puskar M, Kis B, Perciaccante JV, Miller AW, Busija DW. Hydrogen peroxide acts as an EDHF in the piglet pial vasculature in response to bradykinin. American Journal of Physiology. 2002;283(1):H406–H411. doi: 10.1152/ajpheart.00007.2002. [DOI] [PubMed] [Google Scholar]
- 73.Miura H, Bosnjak JJ, Ning G, Saito T, Miura M, Gutterman DD. Role for hydrogen peroxide in flow-induced dilation of human coronary arterioles. Circulation research. 2003;92(2):e31–e40. doi: 10.1161/01.res.0000054200.44505.ab. [DOI] [PubMed] [Google Scholar]
- 74.Yada T, Shimokawa H, Hiramatsu O, et al. Hydrogen peroxide, an endogenous endothelium-derived hyperpolarizing factor, plays an important role in coronary autoregulation in vivo. Circulation. 2003;107(7):1040–1045. doi: 10.1161/01.cir.0000050145.25589.65. [DOI] [PubMed] [Google Scholar]
- 75.Fleming I, Michaelis UR, Bredenkotter D, et al. Endothelium-derived hyperpolarizing factor synthase (cytochrome P450 2C9) is a functionally significant source of reactive oxygen species in coronary arteries. Circulation Research. 2001;88(1):44–51. doi: 10.1161/01.res.88.1.44. [DOI] [PubMed] [Google Scholar]
- 76.Katusic ZS. Superoxide anion and endothelial regulation of arterial tone. Free Radical Biology & Medicine. 1996;20(3):443–448. doi: 10.1016/0891-5849(96)02116-8. [DOI] [PubMed] [Google Scholar]
- 77.Bayraktutan U, Blayney L, Shah AM. Molecular characterization and localization of the NAD(P)H oxidase components gp91-phox and p22-phox in endothelial cells. Arteriosclerosis, Thrombosis, and Vascular Biology. 2000;20(8):1903–1911. doi: 10.1161/01.atv.20.8.1903. [DOI] [PubMed] [Google Scholar]
- 78.Sato A, Sakuma I, Gutterman DD. Mechanism of dilation to reactive oxygen species in human coronary arterioles. American Journal of Physiology. 2003;285(6):H2345–H2354. doi: 10.1152/ajpheart.00458.2003. [DOI] [PubMed] [Google Scholar]
- 79.Liu Y, Zhao H, Li H, Kalyanaraman B, Nicolosi AC, Gutterman DD. Mitochondrial sources of H2O2 generation play a key role in flow-mediated dilation in human coronary resistance arteries. Circulation Research. 2003;93(6):573–580. doi: 10.1161/01.RES.0000091261.19387.AE. [DOI] [PubMed] [Google Scholar]
- 80.Hamilton CA, Mcphaden AR, Berg G, Pathi V, Dominiczak AF. Is hydrogen peroxide an EDHF in human radial arteries? American Journal of Physiology. 2001;280(6):H2451–H2455. doi: 10.1152/ajpheart.2001.280.6.H2451. [DOI] [PubMed] [Google Scholar]
- 81.Hamilton CA, Williams R, Pathi V, et al. Pharmacological characterisation of endothelium-dependent relaxation in human radial artery: comparison with internal thoracic artery. Cardiovascular Research. 1999;42(1):214–223. doi: 10.1016/s0008-6363(98)00316-2. [DOI] [PubMed] [Google Scholar]
- 82.Hayabuchi Y, Nakaya Y, Matsuoka S, Kuroda Y. Hydrogen peroxide-induced vascular relaxation in porcine coronary arteries is mediated by Ca2+-activated K+ channels. Heart and Vessels. 1998;13(1):9–17. doi: 10.1007/BF02750638. [DOI] [PubMed] [Google Scholar]
- 83.Sobey CG, Heistad DD, Faraci FM. Mechanisms of bradykinin-induced cerebral vasodilatation in rats: evidence that reactive oxygen species activate K+ channels. Stroke. 1997;28(11):2290–2294. doi: 10.1161/01.str.28.11.2290. [DOI] [PubMed] [Google Scholar]
- 84.Wei EP, Kontos HA, Beckman JS. Mechanisms of cerebral vasodilation by superoxide, hydrogen peroxide, and peroxynitrite. American Journal of Physiology. 1996;271(3):H1262–H1266. doi: 10.1152/ajpheart.1996.271.3.H1262. [DOI] [PubMed] [Google Scholar]
- 85.Bychkov R, Pieper K, Ried C, et al. Hydrogen peroxide, potassium currents, and membrane potential in human endothelial cells. Circulation. 1999;99(13):1719–1725. doi: 10.1161/01.cir.99.13.1719. [DOI] [PubMed] [Google Scholar]
- 86.Mugge A, Lopez JA, Piegors DJ, Breese KR, Heistad DD. Acetylcholine-induced vasodilatation in rabbit hindlimb in vivo is not inhibited by analogues of L-arginine. American Journal of Physiology. 1991;260(1):H242–H247. doi: 10.1152/ajpheart.1991.260.1.H242. [DOI] [PubMed] [Google Scholar]
- 87.Bauersachs J, Popp R, Hecker M, Sauer E, Fleming I, Busse R. Nitric oxide attenuates the release of endothelium-derived hyperpolarizing factor. Circulation. 1996;94(12):3341–3347. doi: 10.1161/01.cir.94.12.3341. [DOI] [PubMed] [Google Scholar]
- 88.Olmos L, Mombouli JV, Illiano S, Vanhoutte PM. cGMP mediates the desensitization to bradykinin in isolated canine coronary arteries. The American Journal of Physiology. 1995;268(2):H865–H870. doi: 10.1152/ajpheart.1995.268.2.H865. [DOI] [PubMed] [Google Scholar]
- 89.Mombouli JV, Vanhoutte PM. Endothelium-derived hyperpolarizing factor(s): updating the unknown. Trends in Pharmacological Sciences. 1997;18(7):252–256. [PubMed] [Google Scholar]
- 90.Gilligan DM, Panza JA, Kilcoyne CM, Waclawiw MA, Casino PR, Quyyumi AA. Contribution of endothelium-derived nitric oxide to exercise-induced vasodilation. Circulation. 1994;90(6):2853–2858. doi: 10.1161/01.cir.90.6.2853. [DOI] [PubMed] [Google Scholar]
- 91.Joannides R, Haefeli WE, Linder L, et al. Nitric oxide is responsible for flow-dependent dilatation of human peripheral conduit arteries in vivo. Circulation. 1995;91(5):1314–1319. doi: 10.1161/01.cir.91.5.1314. [DOI] [PubMed] [Google Scholar]
- 92.Quyyumi AA, Dakak N, Andrews NP, Gilligan DM, Panza JA, Cannon RO., III Contribution of nitric oxide to metabolic coronary vasodilation in the human heart. Circulation. 1995;92(3):320–326. doi: 10.1161/01.cir.92.3.320. [DOI] [PubMed] [Google Scholar]
- 93.Christ GJ, Spray DC, El-Sabban M, Moore LK, Brink PR. Gap junctions in vascular tissues: evaluating the role of intercellular communication in the modulation of vasomotor tone. Circulation Research. 1996;79(4):631–646. doi: 10.1161/01.res.79.4.631. [DOI] [PubMed] [Google Scholar]
- 94.Griffith TM. Endothelium-dependent smooth muscle hyperpolarization: do gap junctions provide a unifying hypothesis? British Journal of Pharmacology. 2004;141(6):881–903. doi: 10.1038/sj.bjp.0705698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sandow SL, Hill CE. Incidence of myoendothelial gap junctions in the proximal and distal mesenteric arteries of the rat is suggestive of a role in endothelium-derived hyperpolarizing factor-mediated responses. Circulation Research. 2000;86(3):341–346. doi: 10.1161/01.res.86.3.341. [DOI] [PubMed] [Google Scholar]
- 96.Shimokawa H, Yasutake H, Fujii K, et al. The importance of the hyperpolarizing mechanism increases as the vessel size decreases in endothelium-dependent relaxations in rat mesenteric circulation. Journal of Cardiovascular Pharmacology. 1996;28(5):703–711. doi: 10.1097/00005344-199611000-00014. [DOI] [PubMed] [Google Scholar]
- 97.Lang NN, Myles RC, Burton FL, et al. The vascular effects of rotigaptide in vivo in man. Biochemical Pharmacology. 2008;76(10):1194–1200. doi: 10.1016/j.bcp.2008.08.022. [DOI] [PubMed] [Google Scholar]
- 98.Edwards G, Dora KA, Gardener MJ, Garland CJ, Weston AH. K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature. 1998;396(6708):269–272. doi: 10.1038/24388. [DOI] [PubMed] [Google Scholar]
- 99.Nelson MT, Quayle JM. Physiological roles and properties of potassium channels in arterial smooth muscle. American Journal of Physiology. 1995;268(4):C799–C822. doi: 10.1152/ajpcell.1995.268.4.C799. [DOI] [PubMed] [Google Scholar]
- 100.Prior HM, Webster N, Quinn K, Beech DJ, Yates MS. K+-induced dilation of a small renal artery: no role for inward rectifier K+ channels. Cardiovascular Research. 1998;37(3):780–790. doi: 10.1016/s0008-6363(97)00237-x. [DOI] [PubMed] [Google Scholar]
- 101.Clark SG, Fuchs LC. BKCa channels compensate for loss of NOS-dependent coronary artery relaxation in cardiomyopathy. American Journal of Physiology. 2000;279(6):H2598–H2603. doi: 10.1152/ajpheart.2000.279.6.H2598. [DOI] [PubMed] [Google Scholar]
- 102.Malmsjo M, Bergdahl A, Zhao XH, et al. Enhanced acetylcholine and P2Y-receptor stimulated vascular EDHF-dilatation in congestive heart failure. Cardiovascular Research. 1999;43(1):200–209. doi: 10.1016/s0008-6363(99)00062-0. [DOI] [PubMed] [Google Scholar]
- 103.Feletou M, Vanhoutte PM. Endothelium-dependent hyperpolarizations: past beliefs and present facts. Annals of Medicine. 2007;39(7):495–516. doi: 10.1080/07853890701491000. [DOI] [PubMed] [Google Scholar]
- 104.Taddei S, Versari D, Cipriano A, et al. Identification of a cytochrome P450 2C9-derived endothelium-derived hyperpolarizing factor in essential hypertensive patients. Journal of the American College of Cardiology. 2006;48(3):508–515. doi: 10.1016/j.jacc.2006.04.074. [DOI] [PubMed] [Google Scholar]
- 105.Taddei S, Virdis A, Ghiadoni L, Versari D, Salvetti A. Endothelium, aging, and hypertension. Current Hypertension Reports. 2006;8(1):84–89. doi: 10.1007/s11906-006-0045-4. [DOI] [PubMed] [Google Scholar]
- 106.Brandes RP, Behra A, Lebherz C, et al. NG-nitro-L-arginine- and indomethacin-resistant endothelium-dependent relaxation in the rabbit renal artery: effect of hypercholesterolemia. Atherosclerosis. 1997;135(1):49–55. doi: 10.1016/s0021-9150(97)00145-7. [DOI] [PubMed] [Google Scholar]
- 107.Morikawa K, Matoba T, Kubota H, et al. Influence of diabetes mellitus, hypercholesterolemia, and their combination on EDHF-mediated responses in mice. Journal of Cardiovascular Pharmacology. 2005;45(5):485–490. doi: 10.1097/01.fjc.0000159657.93922.cb. [DOI] [PubMed] [Google Scholar]
- 108.Wolfle SE, de Wit C. Intact endothelium-dependent dilation and conducted responses in resistance vessels of hypercholesterolemic mice in vivo. Journal of Vascular Research. 2005;42(6):475–482. doi: 10.1159/000088101. [DOI] [PubMed] [Google Scholar]
- 109.Feletou M, Vanhoutte PM. EDHF: new therapeutic targets? Pharmacological Research. 2004;49(6):565–580. doi: 10.1016/j.phrs.2003.10.017. [DOI] [PubMed] [Google Scholar]
- 110.Yu Z, Huse LM, Adler P, et al. Increased CYP2J expression and epoxyeicosatrienoic acid formation in spontaneously hypertensive rat kidney. Molecular Pharmacology. 2000;57(5):1011–1020. [PubMed] [Google Scholar]
- 111.Frisbee JC, Falck JR, Lombard JH. Contribution of cytochrome P-450 omega-hydroxylase to altered arteriolar reactivity with high-salt diet and hypertension. American Journal of Physiology. 2000;278:H1517–H1526. doi: 10.1152/ajpheart.2000.278.5.H1517. [DOI] [PubMed] [Google Scholar]
- 112.Kroetz DL, Huse LM, Thuresson A, Grillo MP. Developmentally regulated expression of the CYP4A genes in the spontaneously hypertensive rat kidney. Molecular Pharmacology. 1997;52(3):362–372. doi: 10.1124/mol.52.3.362. [DOI] [PubMed] [Google Scholar]
- 113.Omata K, Abraham NG, Escalante B, Schwartzman ML. Age-related changes in renal cytochrome P-450 arachidonic acid metabolism in spontaneously hypertensive rats. American Journal of Physiology. 1992;262(1):F8–F16. doi: 10.1152/ajprenal.1992.262.1.F8. [DOI] [PubMed] [Google Scholar]
- 114.Pomposiello SI, Carroll MA, Falck JR, McGiff JC. Epoxyeicosatrienoic acid-mediated renal vasodilation to arachidonic acid is enhanced in SHR. Hypertension. 2001;37(3):887–893. doi: 10.1161/01.hyp.37.3.887. [DOI] [PubMed] [Google Scholar]
- 115.Holla VR, Makita K, Zaphiropoulos PG, Capdevila JH. The kidney cytochrome P-450 2C23 arachidonic acid epoxygenase is upregulated during dietary salt loading. Journal of Clinical Investigation. 1999;104(6):751–760. doi: 10.1172/JCI7013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Pfister SL, Falck JR, Campbell WB. Enhanced synthesis of epoxyeicosatrienoic acids by cholesterol-fed rabbit aorta. American Journal of Physiology. 1991;261(3):H843–H852. doi: 10.1152/ajpheart.1991.261.3.H843. [DOI] [PubMed] [Google Scholar]
- 117.Bellien J, Thuillez C, Joannides R. Contribution of endothelium-derived hyperpolarizing factors to the regulation of vascular tone in humans. Fundamental and Clinical Pharmacology. 2008;22(4):363–377. doi: 10.1111/j.1472-8206.2008.00610.x. [DOI] [PubMed] [Google Scholar]
- 118.Liu Y, Bubolz AH, Mendoza S, Zhang DX, Gutterman DD. H2O2 is the transferrable factor mediating flow-induced dilation in human coronary arterioles. Circulation Research. 2011;108(5):566–573. doi: 10.1161/CIRCRESAHA.110.237636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Morikawa K, Fujiki T, Matoba T, et al. Important role of superoxide dismutase in EDHF-mediated responses of human mesenteric arteries. Journal of Cardiovascular Pharmacology. 2004;44(5):552–556. doi: 10.1097/00005344-200411000-00006. [DOI] [PubMed] [Google Scholar]
- 120.Lang NN, Luksha L, Newby DE, Kublickiene K. Connexin 43 mediates endothelium-derived hyperpolarizing factor-induced vasodilatation in subcutaneous resistance arteries from healthy pregnant women. American Journal of Physiology. 2007;292(2):H1026–H1032. doi: 10.1152/ajpheart.00797.2006. [DOI] [PubMed] [Google Scholar]
- 121.Sheng JZ, Braun AP. Small- and intermediate-conductance Ca2+-activated K+ channels directly control agonist-evoked nitric oxide synthesis in human vascular endothelial cells. American Journal of Physiology. 2007;293(1):C458–C467. doi: 10.1152/ajpcell.00036.2007. [DOI] [PubMed] [Google Scholar]
- 122.Torondel B, Vila JM, Segarra G, et al. Endothelium-dependent responses in human isolated thyroid arteries from donors. Journal of Endocrinology. 2004;181(3):379–384. doi: 10.1677/joe.0.1810379. [DOI] [PubMed] [Google Scholar]
- 123.Pickkers P, Hughes AD, Russel FG, Thien T, Smits P. Thiazide-induced vasodilation in humans is mediated by potassium channel activation. Hypertension. 1998;32(6):1071–1076. doi: 10.1161/01.hyp.32.6.1071. [DOI] [PubMed] [Google Scholar]
- 124.Honing ML, Smits P, Morrison PJ, Burnett JC, Jr., Rabelink TJ. C-type natriuretic peptide-induced vasodilation is dependent on hyperpolarization in human forearm resistance vessels. Hypertension. 2001;37(4):1179–1183. doi: 10.1161/01.hyp.37.4.1179. [DOI] [PubMed] [Google Scholar]
- 125.Fischer D, Landmesser U, Spiekermann S, et al. Cytochrome P450 2C9 is involved in flow-dependent vasodilation of peripheral conduit arteries in healthy subjects and in patients with chronic heart failure. European Journal of Heart Failure. 2007;9(8):770–775. doi: 10.1016/j.ejheart.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 126.Bellien J, Joannides R, Iacob M, Arnaud P, Thuillez C. Calcium-activated potassium channels and NO regulate human peripheral conduit artery mechanics. Hypertension. 2005;46(1):210–216. doi: 10.1161/01.HYP.0000165685.83620.31. [DOI] [PubMed] [Google Scholar]
- 127.Hillig T, Krustrup P, Fleming I, Osada T, Saltin B, Hellsten Y. Cytochrome P450 2C9 plays an important role in the regulation of exercise-induced skeletal muscle blood flow and oxygen uptake in humans. Journal of Physiology. 2003;546(1):307–314. doi: 10.1113/jphysiol.2002.030833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Imig JD. Epoxide hydrolase and epoxygenase metabolites as therapeutic targets for renal diseases. American Journal of Physiology. 2005;289(3):F496–F503. doi: 10.1152/ajprenal.00350.2004. [DOI] [PubMed] [Google Scholar]
- 129.Yu Z, Xu F, Huse LM, et al. Soluble epoxide hydrolase regulates hydrolysis of vasoactive epoxyeicosatrienoic acids. Circulation Research. 2000;87(11):992–998. doi: 10.1161/01.res.87.11.992. [DOI] [PubMed] [Google Scholar]
- 130.Ai D, Fu Y, Guo D, et al. Angiotensin II up-regulates soluble epoxide hydrolase in vascular endothelium in vitro and in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(21):9018–9023. doi: 10.1073/pnas.0703229104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Spiecker M, Darius H, Hankeln T, et al. Risk of coronary artery disease associated with polymorphism of the cytochrome P450 epoxygenase CYP2J2. Circulation. 2004;110(15):2132–2136. doi: 10.1161/01.CIR.0000143832.91812.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Yu BN, Luo CH, Wang D, et al. CYP2C9 allele variants in Chinese hypertension patients and healthy controls. Clinica Chimica Acta. 2004;348(1-2):57–61. doi: 10.1016/j.cccn.2004.04.028. [DOI] [PubMed] [Google Scholar]