

Abstract
Several well-known morphogenetic gradients and cellular movements occur along the dorsal/ventral axis of the Drosophila embryo. However, the current techniques used to view such processes are somewhat limited. The following protocol describes a new technique for mounting fixed and labeled Drosophila embryos for coronal viewing with confocal imaging. This method consists of embedding embryos between two layers of glycerin jelly mounting media, and imaging jelly strips positioned upright. The first step for sandwiching the embryos is to make a thin bedding of glycerin jelly on a slide. Next, embryos are carefully aligned on this surface and covered with a second layer of jelly. After the second layer is solidified, strips of jelly are cut and flipped upright for imaging. Alternatives are described for visualizing the embryos depending upon the type of microscope stand to be used. Since all cells along the dorsal-ventral axis are imaged within a single confocal Z-plane, our method allows precise measurement and comparison of fluorescent signals without photobleaching or light scattering common to 3D reconstructions of longitudinally mounted embryos.
Keywords: Developmental Biology, Issue 43, Drosophila, Confocal imaging, Multiplex in situ hybridization, Embryo, Insect Development
Protocol
1. Imaging Methodology
Melt glycerin jelly in a 55° C degree water bath for 20-30 minutes until it becomes a homogenous liquid. Swirl, but do not shake the bottle to avoid the formation of bubbles.
While the jelly melts, apply a thin layer of glycerol using a Kimwipe on a clean glass slide (optional).
Pipette 1 to 1.5 mL of melted glycerin jelly using a cut P1000 pipette tip on the surface of the slide to create a thin first layer. Pipette carefully to avoid bubbles.
Let the jelly solidify for 5 min on a horizontal surface, then place the slide at -20°C for 5 min and proceed to embryo alignment. Alternatively, cover the slide with Saran Wrap and place it at 4°C for up to 5 days.
Pipette a small drop of stained embryos in 70% glycerol/PBS or other antifade glycerol-based mounting media (e.g. Slowfade or Vectashield) on the side of the jelly bedding.
Begin pre-aligning the embryos by individually transferring them from the drop to the jelly surface. Transfer them using a slanted hypodermic needle, using the slanted side as a spoon.
Roughly place the embryos along 2 to 3 columns. If an embryo gets stuck in the needle, stir it in the drop containing the other embryos to release it. Do not waste time carefully aligning them at this point. Leave some space between columns.
Align the embryos by sliding them horizontally on the jelly medium with the aid of the hypodermic needle. While sliding, orient the heads and tails in the same direction. Make them parallel to each other along the column. Remove excess of PBS glycerol left as a trail with a piece of twisted Kimiwipe.Note: Excess glycerol will cause embryos to be loosely encased within gel, resulting in separation of the two jelly layers, making it difficult to cut and mount. Conversely, removing too much glycerol will dry and flatten embryos.
Use a cut yellow tip to spread a thin layer of jelly over the embryos (50-150 μL, depending on how many were aligned).
Place slide in -20°C freezer for 10-20 min or store it at 4°C overnight. Make sure the jelly is completely rigid before cutting. Otherwise, the block of embryos will be very difficult to excise. Best results for cutting are obtained on the following day.
Under a dissecting scope, use a razor blade to cut completely through the jelly on both sides of the aligned embryos. Hold the razor blade at a 90° angle to make straight cuts. The cuts should be made very close to the anterior and posterior poles of the embryos to avoid excess jelly that may obstruct imaging.
Add 100-200 μL of cold PBT (4°C, PBS+0.1% Tween 20) next to the cuts and carefully scrape off the jelly between embryos using a spatula.Note: The detergent in PBT helps the removal of the jelly from the slide without damaging the embryos.
Using a needle, transfer the block of jelly with embedded embryos to a clean slide for further cutting. Cut smaller blocks with 5-7 embryos. Use PBT to easily move around the jelly block. Alternatively, place the block on a dry glass surface to let it attach to the glass, which creates more stability for precisely trimming the jelly, if necessary.
Flip upright the embryos embedded in the jelly with the aid of a razor blade and a needle. For visualization under microscope, proceed with either one of the two steps below, depending on the type of microscope stand being used.
Inverted microscope: place finished embryo blocks onto a long coverslip, with embryos in an upright position. Ensure that the side of the embryo with the least amount of jelly will be in closer contact with objective.
Upright microscope: make two stacks with four #1 coverslips glued with superglue to be used as "supports" and place the embryo block in glycerol in between the "supports". Bridge the stacks using a long coverslip.
For confocal analysis, check the working distance of the objectives to be used to find out whether you can image all the way through the embryo or only partially. For example, D. melanogaster embryos have ~ 0.55 mm in length and can be entirely imaged using a Zeiss 20x Plan-Apo or a 40x LD Neo-Fluor objective. The following website has information on available Zeiss objectives and gives you the option to search objectives according to working distances: https://www.micro-shop.zeiss.com/us/us_en/objektive.php
2. Representative Results
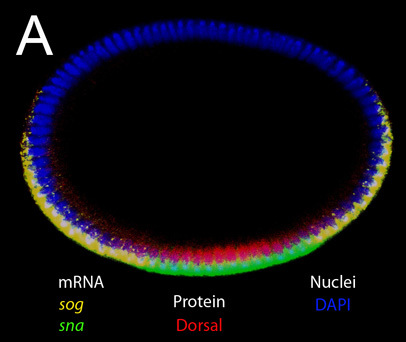 Figure 1. Upright optical slice of an intact embryo in blastoderm stage. Double staining for mRNA and protein. Target mRNAs for in situ probes (sog and sna) and protein stained by primary antibody (anti-Dorsal) are indicated in the figure. DAPI was used to label the nuclei. Note that the expression of sog and sna are located apically in the cytoplasm, while the expression of Dorsal is nuclear. Strong signals from sog nascent transcripts that are located in nuclei can also be seen at this magnification.
Figure 1. Upright optical slice of an intact embryo in blastoderm stage. Double staining for mRNA and protein. Target mRNAs for in situ probes (sog and sna) and protein stained by primary antibody (anti-Dorsal) are indicated in the figure. DAPI was used to label the nuclei. Note that the expression of sog and sna are located apically in the cytoplasm, while the expression of Dorsal is nuclear. Strong signals from sog nascent transcripts that are located in nuclei can also be seen at this magnification.
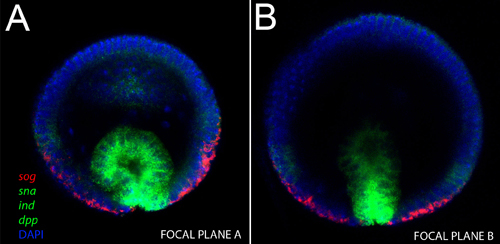 Figure 2. Upright imaging of an intact gastrulating embryo. mRNA probes used and their respective colors are indicated in the figure. A) Note that in this stage, three genes stained with same fluor (sna, sog and dpp, in green), are expressed in spatially separated domains. The invaginating mesoderm expresses sna, the neuroblast layer expresses ind and the ectoderm expresses dpp, while sog expression is still present in ventral regions of the nervous system. B) In a deeper confocal plane of the same embryo, we note the base of the invaginating mesoderm strongly expresses sna, but its apical region expresses lower levels.
Figure 2. Upright imaging of an intact gastrulating embryo. mRNA probes used and their respective colors are indicated in the figure. A) Note that in this stage, three genes stained with same fluor (sna, sog and dpp, in green), are expressed in spatially separated domains. The invaginating mesoderm expresses sna, the neuroblast layer expresses ind and the ectoderm expresses dpp, while sog expression is still present in ventral regions of the nervous system. B) In a deeper confocal plane of the same embryo, we note the base of the invaginating mesoderm strongly expresses sna, but its apical region expresses lower levels.
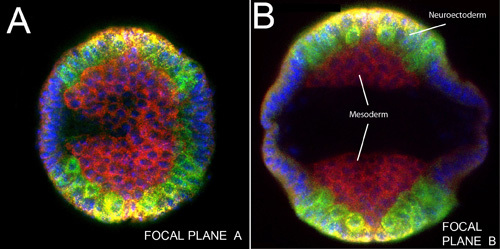 Figure 3. Upright imaging of an intact embryo with germ band extended. sog RNA(yellow); snail, ind and dpp mRNAs (green), Dorsal protein (red). Nuclei were stained with DAPI (blue). A) Focal plane near embryo head. B) Another focal plane in the trunk region of same embryo. Note mass of mesodermal cells before lateral migration (compare to Figure 4) lying close to the ventral midline on both sides of a germ-band extended embryo. Delaminating neuroblasts are labeled in green (ind and sna).
Figure 3. Upright imaging of an intact embryo with germ band extended. sog RNA(yellow); snail, ind and dpp mRNAs (green), Dorsal protein (red). Nuclei were stained with DAPI (blue). A) Focal plane near embryo head. B) Another focal plane in the trunk region of same embryo. Note mass of mesodermal cells before lateral migration (compare to Figure 4) lying close to the ventral midline on both sides of a germ-band extended embryo. Delaminating neuroblasts are labeled in green (ind and sna).
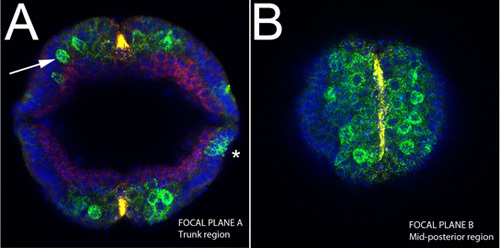 Figure 4. Upright imaging of an intact embryo during germ band extension.sog RNA(yellow); snail, ind and dpp mRNAs (green), Dorsal protein (red). A) Note the delaminating neuroblasts from the epithelium (arrow), stained both for ind and sna at this stage (green). Note also the restricted expression of sog to the ventral midline (yellow). Expression of dpp at this stage can be seen at lateral sides of the embryo (asterisk). B) Optical section in the same embryo shown in A near the end of the embryo length, which corresponds to mid-posterior region. The ventral midline cells are stained for sog.
Figure 4. Upright imaging of an intact embryo during germ band extension.sog RNA(yellow); snail, ind and dpp mRNAs (green), Dorsal protein (red). A) Note the delaminating neuroblasts from the epithelium (arrow), stained both for ind and sna at this stage (green). Note also the restricted expression of sog to the ventral midline (yellow). Expression of dpp at this stage can be seen at lateral sides of the embryo (asterisk). B) Optical section in the same embryo shown in A near the end of the embryo length, which corresponds to mid-posterior region. The ventral midline cells are stained for sog.
Discussion
Taking cross section images of Drosophila embryos can be challenging, since it requires either a careful hand dissection of slices 2 or the use of embedded media for microtome processing, which is usually detrimental for fluorescent stainings. Alternatively, Z-stacks made in a Confocal microscope can be used to make 3D reconstruction of cross-section images. However, it is time consuming to make several thin Confocal slices for an accurate 3D reconstruction and photobleaching becomes problematical. Another concern when imaging embryos that are mounted longitudinally is the light scattering that occurs when reaching deeper sections of the embryo, which also complicates taking precise measurements of fluorescent signals for the purpose of quantification of protein or mRNA expression levels1. Even with a two-photon confocal microscope, light scattering is still a concern due to the opacity of yolk material in the middle of the embryo, which makes selection of mounting media for optimal transparency of sample to be limited.
To circumvent these problems, a new technique called "End-on" imaging of positioning embryos vertically was recently developed to image both live and fixed samples 4. In this paper, we developed a new protocol for vertically mounted embryos that allows a simple preparation and visualization of fixed embryos or tissues of any size and shape that are stained with multiple in situ probes 3. Our method consists of using a mounting media with gelatinous consistency that is used to encase aligned embryos within it. Cut blocks of this mounting media containing embedded embryos can be positioned upright before imaging in any type of Confocal microscope.
By embedding the embryos into the jelly and positioning them vertically, we are able to take horizontal cross-sections using Confocal microscopy, in which cells along the dorsal-ventral axis of the embryo are presented simultaneously. This is a significant improvement for quantification purposes since problems of light scattering and photobleaching of fluorescent dyes that occurs in conventional Z-stack reconstruction of longitudinally mounted embryos are now eliminated. Thus, quantification of gene expression or protein levels along the dorsal-ventral axis is accurate, since cells located at opposite dorsal-ventral positions are imaged at the same time and at the same confocal Z-position. In addition, the described method allows us to obtain a complete 2-D image across the dorsal-ventral axis from a 3-D embryo without using complex computational unrolling methods to visualize cells at different positions along the DV axis 6.
Some of the critical steps include removing excess glycerol after aligning embryos, to avoid split between the two layers of jelly. However, if the sample is too dehydrated, the resulting image will be misshapen and have incorrect morphology. In addition, if the jelly medium around the embryo is cut at an angle rather than straight, the resulting image may appear less round and more ovular in shape, due to an incorrect positioning of the embryos at an angle other than 90 degrees. Finally, if the jelly is not cut closely enough to the embryo on the anterior/posterior ends, it is not possible to obtain a proper image because the objective's working distance would be mainly used to focus into the jelly and not the embryo.
Another important step that may be necessary when imaging late gastrulating embryo is to carefully sort the stages of interest under the scope before aligning them. Usually, embryos are staged according to morphological characteristics that can be most easily recognized when in a longitudinal position.
Among alternative modifications that can be made with this method is to include an anti-fade reagent (e.g. p-Phenylenediamine or PPD) into the second layer of jelly to help protect against photobleaching of fluorescent dyes.
Disclosures
No conflicts of interest declared.
Acknowledgments
The authors are especially indebted to Deborah Harris and Danielle MacKay. Support for this work was provided by the Department of Biology and the College of Arts and Sciences of CWRU, and by HHMI grant number 52005866 for support of undergraduate education in the biological sciences.
References
- Ay A, Fakhouri WD, Chiu C, Arnosti DN. Image processing and analysis for quantifying gene expression from early Drosophila embryos. Tissue Engineering. 2008;14:1517–1526. doi: 10.1089/ten.tea.2008.0202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grβhans J, Wieschaus E. A genetic link between morphogenesis and cell division during formation of the ventral furrow in Drosophila. Cell. 2000;101:523–531. doi: 10.1016/s0092-8674(00)80862-4. [DOI] [PubMed] [Google Scholar]
- Kosman D, Mizutani CM, Lemons D, Cox WG, McGinnis W, Bier E. Multiplex detection of RNA expression in Drosophila embryos. Science. 2004;305:846–846. doi: 10.1126/science.1099247. [DOI] [PubMed] [Google Scholar]
- Witzberger MM, Fitzpatrick JAJ, Crowley JC, Minden JS. End-on imaging: a new perspective on dorsoventral development in Drosophila embryos. Developmental Dynamics. 2008;237:3252–3259. doi: 10.1002/dvdy.21752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zander RH. On mounting delicate bryophytes in glycerol. The Bryologist. 1997;100:380–382. [Google Scholar]
- Liberman LM, Reeves GT, Stathopoulos A. Quantitative imaging of the Dorsal nuclear gradient reveals limitations to threshold-dependent patterning in Drosophila. Proc Natl Acad Sci U S A. 2009;106(52):22317–2222. doi: 10.1073/pnas.0906227106. [DOI] [PMC free article] [PubMed] [Google Scholar]