

Abstract
Creation of transgenic animals is a standard approach in studying functions of a gene of interest in vivo. However, many knockout or transgenic animals are not viable in those cases where the modified gene is expressed or deleted in the whole organism. Moreover, a variety of compensatory mechanisms often make it difficult to interpret the results. The compensatory effects can be alleviated by either timing the gene expression or limiting the amount of transfected cells.
The method of postnatal non-ventricular microinjection and in vivo electroporation allows targeted delivery of genes, siRNA or dye molecules directly to a small region of interest in the newborn rodent brain. In contrast to conventional ventricular injection technique, this method allows transfection of non-migratory cell types. Animals transfected by means of the method described here can be used, for example, for two-photon in vivo imaging or in electrophysiological experiments on acute brain slices.
Protocol
1. Introduction
Creation of transgenic animals is a powerful method of investigation of gene functions in living animals1 and for unraveling disease mechanism2,3 as well as for manipulating properties of cells4. However, the procedure is rather laborious, extremely time consuming and expensive, thus warranting the use of alternative gene delivery methods such as viral injection5, neonatal ventricular injection with electroporation6 and in utero electroporation7,8. The method of postnatal non-ventricular injection and electroporation has a unique set of advantages: use of non-viral genetic constructs; possibility to create a local expression pattern in the area of interest; in situ transfection of non-migratory cell types, e.g. cortical astrocytes.
In this video, we demonstrate the detailed procedure of postnatal gene delivery to the neonatal rat brain using a plasmid encoding for the enhanced green fluorescent protein under chicken beta actin promoter with CMV enhancer (pCAG-EGFP)9. We illustrate the preparation of the plasmid-containing injection solution, manufacturing of thin glass pipettes and assembling of the microinjector on a stereotactic instrument. Then we talk about anaesthetizing rat pups with isoflurane, about performing the surgery, about the injection procedure and about the electroporation using forceps electrodes and the in vivo porator. Finally, we briefly discuss the anticipated results, perspectives and difficulties that may arise during these experiments.
2. Plasmid Preparation for Electroporation
We usually prepare 10 μl of the plasmid injection solution in a 200 μl thin wall tube. This solution is sufficient for several experiments.
We mix 8 μl of the plasmid solution (see Materials and equipment) with 1 μl of the 10X PBS (see Materials and equipment) and 1 μl of 0,1% Fast Green water solution (see Materials and equipment).
The final concentration of the plasmid in the injection solution should be between 1 and 3 μg/μl. Lower concentrations of plasmid DNA will reduce the transfection efficiency, while the higher DNA concentration will be too viscous for injection through the thin tip of the glass capillary.
3. Glass Needle Preparation
For the glass pipette preparation, we use borosilicate glass capillary with a filament (see Materials and equipment) and a vertical electrode glass puller (see Materials and equipment) on maximum pulling and heating parameters.
We then break the tip of the pipette using a piece of paper to achieve a tip 10-20 μm in diameter.
We check the tip diameter and shape under the microscope objective.
4. Microinjector Assembling
Using a thin polymer capillary (see Materials and equipment) we fill approximately 1/3 of the volume of a 10 μl Hamilton syringe (see Materials and equipment) with mineral oil (see Materials and equipment). Avoid air bubbles!
Then we fill the glass pipette with mineral oil and insert the pipette into the Hamilton syringe. Avoid air bubbles!
The syringe with glass pipette is then fixed on the microinjector connected to a controller (see Materials and equipment).
The microinjection setup fixed on a stereotactic instrument (see Materials and equipment) allows us to safely immerse the tip of the glass pipette into the tube with the plasmid injection solution.
When the tip touches the surface of the solution, dip it further by 1 or 2 mm and fill the pipette with approximately 1 μl of the injection mix using the micro injector controller.
5. Anaesthetizing animal
All the procedures presented here were performed according to the University of Helsinki regulations for animal experiments.
For each experiment, we fill a gas-tight syringe (see Materials and equipment) with approximately 2 ml of isoflurane (see Materials and equipment).
Than the syringe is fixed on the anesthesia unit (see Materials and equipment) connected to the air flow source, the animal box and the mask fixed on stereotactic setup.
On the anesthesia unit, we adjust the airflow to approximately 250 ml/min and the isoflurane level to 4 %.
We place the rat pup into the animal box for 2-5 minutes.
When the pup stops moving, place it on the heating pad (see Materials and equipment) attached to the stereotactic setup.
Place a rostral part of pup's head into the anaesthetizing mask.
It takes 5 to 10 minutes for the pup to enter the deep anesthesia.
Check anesthesia deepness with a tail pinch and decrease the isoflurane inflow to about 1.5 to 2.0 %.
6. Surgery, Microinjection and Electroporation
Treat the skin on the pup's head with the 70% ethanol.
Using small scissors (see Materials and equipment) and thin forceps (see Materials and equipment), cut the skin from a scruff of the pup to its forehead.
Bend skin pieces sideways and pull ear bars slightly into aural orifices of the skull for head and skin fixation.
Using a binocular microscope, we locate the bregma point on the skull.
Identify the region of interest using the stereotactic coordinates.
Position the injection pipette above this region and mark it by dropping 25-50 nl of the injection solution onto the skull surface.
Drill the skull gently and carefully at the marked point using a high speed surgical drill (see Materials and equipment) under the microscope until liquid appears in the drilled area.
Fix an electropotation forceps electrodes (see Materials and equipment) on the sides of the skull with current conductive gel (see Materials and equipment) to achieve better conductivity.
Remove the liquid drop from the drilled hole using small tampon (see Materials and equipment) and dip the glass needle to the hole according to the coordinates of the injection site.
Infuse 25 to 100 nl of the injection solution at the rate of 5 to 20 nl/sec.
Quickly remove the pipette and electroporate immediately. Electroporation is done by applying five rectangle pulses with the duration of 50 ms and amplitude of 99 V at the frequency of 1 Hz.
Remove electrodes and ear bars.
Sew the cut skin using a combination of a dumb (see Materials and equipment) and sharp forceps and surgical threads (see Materials and equipment).
Place the pup into the warm chamber for 15-30 minutes to aid its recovery after anaesthesia.
Then put the pup back to its mother's cage.
7. Representative Results
The expression of the transgene in the successfully transfected cells will appear approximately 10 hours after the electroporation, and it will remain stable for several weeks.
We microinjected the plasmid either into deep cortical layers or striatum region of postnatal day 2 (P2) Wistar rat pups and performed in vivo electroporation as described above. Brain slices (400 μm in thickness) were cut after 2 to 6 days (P4-P8) using a vibrotome in cold slicing medium (Hepes buffered Earle's Balanced Salt Solution)10. Image acquisition was performed using Zeiss Axioplan 2 LSM5 Pascal confocal microscope (Zeiss, Germany) in the slicing medium at room temperature.
The injection sites in deep cortical layers and striatum contained many transfected cells expressing EGFP that were located in a compact region of approximately 200-300 μm in diameter (Figure 1A, B). Within transfected cells, the EGFP expression level is sufficiently high to allow imaging of thin cellular processes (Figure 1C) including dendrites with spines of different shapes (Figure 1D) and axon bundles (Figure 1E). The robust stability of EGFP as well as cell viability after electroporation allowed tracing distal parts of axons (up to 1.5-2 mm from cell bodies) such as cortical terminals (Figure 1F).
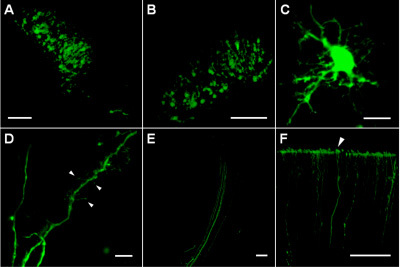 Figure 1. (A) Successfully transfected cells in the striatum region of P4 rat brain delineating the plasmid injection site. (B) Transfected cells in deep cortical layers (approximately 1000 μm below cortical surface) of P5 rat brain. (C) A transfected cell in the striatum region of P8 rat brain. (D) Dendritic processes of a transfected cell in the striatum region of P8 rat brain; arrows indicate different types of dendritic spines and filopodia. (E) Axons of transfected cells within collateral pathway. (F) Axons terminating at the pial surface of P8 rat cortex (arrowhead). Scale bars are 100 μm on A, B, E and F; 10 μm on C and D.
Figure 1. (A) Successfully transfected cells in the striatum region of P4 rat brain delineating the plasmid injection site. (B) Transfected cells in deep cortical layers (approximately 1000 μm below cortical surface) of P5 rat brain. (C) A transfected cell in the striatum region of P8 rat brain. (D) Dendritic processes of a transfected cell in the striatum region of P8 rat brain; arrows indicate different types of dendritic spines and filopodia. (E) Axons of transfected cells within collateral pathway. (F) Axons terminating at the pial surface of P8 rat cortex (arrowhead). Scale bars are 100 μm on A, B, E and F; 10 μm on C and D.
Discussion
Methods of gene delivery into living rodent brain are well established for in utero electroporation7,8,11,12 and, more recently, for postnatal electroporation6. However, these methods are based on intraventricular injection of the plasmid DNA, which may be limiting for several applications. For example, these methods do not allow targeting cells in certain brain areas such as hippocampus, nor transfection of such non-migratory cell types as cortical astrocytes. Non-ventricular injection coupled with electroporation was first used for gene delivery into cerebellar neurons13. Our protocol illustrates the extension of non-ventricular method for other regions of rat brain. We propose that our methodological approach is a useful alternative to viral methods of gene delivery to restricted areas of interest within postnatal rat brain5.
Despite the advantages of the present method, some difficulties may be expected as discussed below.
1.Optimal age of animal
When possible, younger neonatal rat pups should be use, as a means to increase both transfection efficacy and pup survival after surgery. In our hands, the P0 pups are too small as it is difficult to target reproducibly a certain area in the brain using stereotactic coordinates obtained from the brain atlas available for this age14. In addition, the P0 pups have thin and tender skin that may impede sewing. The P3 and P4 are most convenient in terms of surgical and stereotactic manipulations as compared to P0-P2 animals, but they have hypersensitivity to isoflurane anesthesia that may cause seizures and breathing interruptions during surgery. Thus, the best option is P1-P2 rat pups which are predictable during surgery and large enough for reproducible microinjections.
2.Possible problems with microinjection
When small volumes of solution (25-100 nl) are injected through thin glass tip to the brain tissue exerts an opposing pressure, it is critical that air bubbles or leakages are carefully avoided. Failure to avoid these may cause a dramatic decrease in transfection efficiency.
3.Precision limits for stereotactic coordinates
Despite the precision of stereotactic coordinates, they have a limit at 0.3 mm reproducibility15. In our experience using animals of the same age and weight it is possible to decrease this limit to 0.2−0.1 mm which is sufficient for reproducible targeting of the CA1 hippocampal area.
Acknowledgments
We thank Ekaterina Karelina for help with soundtrack recording for the video, Ivan Molotkov for 3D animation and Dr. Peter Blaesse for CAG-EGFP plasmid preparation.
The work was supported by grants from the Centre of International Mobility of Finland, Finnish Cultural Foundation and the Academy of Finland.
References
- Gerlai R, Clayton NS. Analyzing hippocampal function in transgenic mice: an ethological perspective. Trends Neurosci. 1999;22:47–51. doi: 10.1016/s0166-2236(98)01346-0. [DOI] [PubMed] [Google Scholar]
- McGowan E, Eriksen J, Hutton M. A decade of modeling Alzheimer's disease in transgenic mice. Trends Genet. 2006;2:281–289. doi: 10.1016/j.tig.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Cryan JF, Holmes A. The ascent of mouse: advances in modeling human depression and anxiety. Nat. Rev. Drug Discov. 2005;4:775–790. doi: 10.1038/nrd1825. [DOI] [PubMed] [Google Scholar]
- Wells T, Carter DA. Genetic engineering of neural function in transgenic rodents: towards a comprehensive strategy. J. Neurosci. Methods. 2001;108:111–130. doi: 10.1016/s0165-0270(01)00391-0. [DOI] [PubMed] [Google Scholar]
- Pilpel N. reproducible transduction of select forebrain regions by targeted recombinant virus injection into the neonatal mouse brain. J. Neurosci. Methods. 2009;182:55–63. doi: 10.1016/j.jneumeth.2009.05.020. [DOI] [PubMed] [Google Scholar]
- Boutin C. Efficient in vivo electroporation of the postnatal rodent forebrain. PLoS One. 2008;3:e1883–e1883. doi: 10.1371/journal.pone.0001883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T, Nakatsuji N. Efficient gene transfer into the embryonic mouse brain using in vivo electroporation. Dev. Biol. 2001;240:237–246. doi: 10.1006/dbio.2001.0439. [DOI] [PubMed] [Google Scholar]
- Saito T. In vivo electroporation in the embryonic mouse central nervous system. Nat. Protoc. 2006;1:1552–1558. doi: 10.1038/nprot.2006.276. [DOI] [PubMed] [Google Scholar]
- Matsuda T, Cepko CL. Electroporation and RNA interference in the rodent retina in vivo and in vitro. Proc. Natl. Acad. Sci. U S A. 2004;101:16–22. doi: 10.1073/pnas.2235688100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Simoni A, Yu LM. Preparation of organotypic hippocampal slice cultures: interface method. Nat. Protoc. 2006;1:1439–1445. doi: 10.1038/nprot.2006.228. [DOI] [PubMed] [Google Scholar]
- Walantus W. In utero intraventricular injection and electroporation of E15 mouse embryos. J. Vis. Exp. 2007 doi: 10.3791/239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walantus W, Elias L, Kriegstein A. In utero intraventricular injection and electroporation of E16 rat embryos. J. Vis. Exp. 2007 doi: 10.3791/236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umeshima H, Hirano T, Kengaku M. Microtubule-based nuclear movement occurs independently of centrosome positioning in migrating neurons. Proc. Natl. Acad. Sci. U S A. 2007;104:16182–16187. doi: 10.1073/pnas.0708047104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashwell K, Paxinos G. Atlas of the Developing Rat Nervous System. 3rd edition. Academic Press; 2008. [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. 6th edition. Academic Press; 2007. [Google Scholar]