

Abstract
The ability to genetically remove specific components of various cell signalling cascades has been an integral tool in modern signal transduction analysis. One particular method to achieve this conditional deletion is via the use of the Cre-loxP system. This method involves flanking the gene of interest with loxP sites, which are specific recognition sequences for the Cre recombinase protein. Exposure of the so-called floxed (flanked by loxP site) DNA to this enzyme results in a Cre-mediated recombination event at the loxP sites, and subsequent excision of the intervening gene3. Several different methods exist to administer Cre recombinase to the site of interest. In this video, we demonstrate the use of an adenovirus containing the Cre recombinase gene to infect primary mouse embryonic fibroblasts (MEFs) obtained from embryos containing a floxed Rac1 allele1. Our rationale for selecting Rac1 MEFs for our experiments is that clear morphological changes can be seen upon deletion of Rac1, due to alterations in the actin cytoskeleton2,5. 72 hours following viral transduction and Cre expression, cells were stained using the actin dye phalloidin and imaged using confocal laser scanning microscopy. It was observed that MEFs which had been exposed to the adeno-Cre virus appeared contracted and elongated in morphology compared to uninfected cells, consistent with previous reports2,5. The adenovirus method of Cre recombinase delivery is advantageous as the adeno-Cre virus is easily available, and gene deletion via Cre in nearly 100% of the cells can be achieved with optimized adenoviral infection.
Keywords: Cellular Biology, Issue 43, Cre-loxP, andenovirus, MEF, actin cytoskeleton, cell culture
Protocol
Thawing cells
Obtain frozen mouse embryonic fibroblasts (MEFs) (generated previously from individual embryo primary cultures) from liquid nitrogen.
Thaw cells by immersing vial in 37°C water with swirling until contents are completely thawed.
Add 9mL of DMEM that has been supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin to a 10cm cell culture plate.
Add the thawed cell suspension drop-wise to the plate, rock the plate gently to disperse cells and place plate in a 37°C, 5% CO2 incubator overnight. Note that if cells are sensitive to DMSO (from the freezing process), initial media can be replaced with fresh media after cells have adhered to the plate (approximately four hours post plating). Also note that confluence of the cells after thawing depends on the number of cells frozen, the recovery rate of the cells after freezing, and the growth rate of the cells.
Passaging cells
When cells have grown to essentially 100% confluency (ie: the next day), aspirate off the media and wash cells with 5mL of sterile PBS.
Remove PBS and add 1mL of trypsin containing 10mM EDTA and place plate in incubator for approximately 5 minutes to allow cells to detach from plate.
When cells have detached, add 9mL of media to the trypsinized cells, pipette 2-3 times to disperse clumps, add 1mL of this suspension drop-wise onto a new plate containing 9mL of media, and place this into the 37°C incubator overnight. The presence of FBS in the media will inactivate the trypsin – alternatively, cells can be washed first to dilute out the trypsin.
Counting and plating cells onto cover slips
Remove cells from incubator, wash and trypsinize as before, except this time add 5-6mL media back to the trypsinized cells instead of 9mL.
Pipette cells thoroughly to ensure complete dissociation into a single cell suspension, and remove 15μL to combine with 15μL of trypan blue in a separate microfuge tube.
Mix this cell/ trypan blue suspension by pipetting up and down several times, and add 15μL to the hemocytometer.
Under the microscope, dead cells will appear blue. Count the number of clear/colourless (living) cells in one of the 4X4 grids on the hemocytometer. Repeat this count on the three other 4X4 grids (denoted A, B, C and D) and use the following equation to calculate the number of cells per μL of suspension:
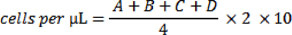
Through protocol optimization, we have determined that plating 30,000 cells results in appropriate cell density for visualization at the conclusion of the experiment.
To calculate the volume of cell suspension needed for 30,000 cells, use the following equation:
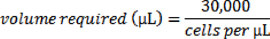
Now place a sterile glass cover slip into each well of a six well dish and add 2mL of media to each well, and then add drop-wise from your cell suspension the volume required for 30,000 cells into each well. Note that glass cover slips are used for this experiment as we will be visualizing the cells using fluorescence microscopy at a later time point; not all applications will necessarily require microscopy, and thus cover slips may not be required.
Virus Transduction
After several hours or overnight (sufficient time to ensure that the cells adhere to the cover slips), remove media and wash cells with 1mL of serum-free DMEM.
Add 1mL of serum-free media to each well for the addition of the virus. For this experiment, adeno-Cre virus was obtained commercially from Vector Biolabs.
Previous protocol optimization has shown that a multiplicity of infection (MOI) of 500 provides highly efficient gene transduction into primary MEFs with little or no effect on cell viability. MOI can be calculated as follows:

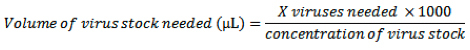
Add the calculated volume of virus directly to each well that is to be infected, gently rock the plate to disperse the virus and place cells into the 37°C incubator overnight.
The next morning, remove the virus-containing media from the cells and wash cells in 1mL sterile PBS and then add 2mL of regular media to each well.
In the case of the Rac1 cells exemplified in this experiment, morphological changes occurred in the cells at approximately 72 hours post transduction.
Representative Results
Cells were visualised by staining with the actin dye phalloidin (for imaging purposes only) and imaged using the Leica TCS-SP5 multiphoton confocal laser scanning microscope at the Advanced Analysis Centre at the University of Guelph. Figure 1 shows the contracted and elongated morphology of the Rac1flox/flox cells that were exposed to adeno-Cre virus compared to the same cells not exposed to virus.
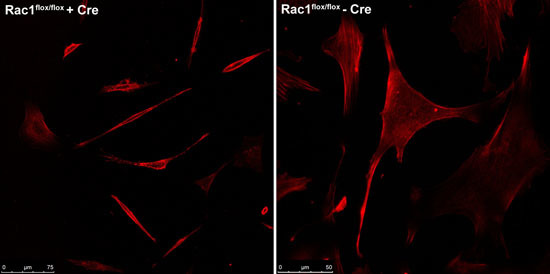 Figure 1. Comparison between Rac1flox/flox cells infected with adeno-Cre virus (left) compared to uninfected cells from the same source (right).
Figure 1. Comparison between Rac1flox/flox cells infected with adeno-Cre virus (left) compared to uninfected cells from the same source (right).
Discussion
The accompanying video exemplifies how a recombinant virus can be used to excise a specific gene in vitro using Cre recombinase. The development of this protocol however did reveal some particular points worth addressing.
Proper safety procedures should always be employed when working with adenoviruses. A lab coat and gloves should be worn at all times while working with the virus. Media and plastic ware should be treated with 10% bleach for at least 30 minutes, and plastic ware should be autoclaved thereafter.
Avoid repeated freeze-thawing of the virus stock as this can affect virus titer and thus reduce transduction efficiency. Small volumes of virus stock should initially be aliquoted into separate tubes.
MOIs ranging from 50-500 were tested during method development and efficient transduction rates were observed in most cases, without negative effects on cell viability.
Adeno-GFP was also used during method development to quickly screen transduction efficiencies. This control (or an empty vector) should be conducted during the experiment to ensure that viral infection alone does not result in cellular changes. Similarly, if excising the floxed gene of interest in your cells does not result in any cellular changes (i.e. changes in morphology or viability), then infecting the same cell type with adeno-GFP is an important control to use to demonstrate that virus transduction is occurring.
Beyond the examination of signalling cascades in culture, the adeno-Cre approach has also been employed in vivo to conditionally remove genes from experimental animals3,4, and to label specific populations of cells within an organism6. The adenovirus system can also be adapted to introduce genes other than Cre recombinase into host cells. Two principal advantages of using this delivery vector are its high transduction and gene delivery efficiency, and lack of integration with host DNA. However, the generation of novel recombinant adenoviruses can be labour intensive. The protocol described here therefore harnesses the power of the adenoviral system by exploiting the previously developed adeno-Cre virus, and coupling that with our floxed allele of interest, Rac1. Through this conditional knock out experiment, we have confirmed that transduction using the adeno-Cre virus of MEFs carrying a floxed Rac1 allele does indeed cause excision of Rac1 leading to the changes in cellular morphology characteristic of Rac1-deficient cells2,5.
Disclosures
Experiments on animals were performed in accordance with the guidelines and regulations set forth by the Canadian Council on Animal Care.
Acknowledgments
The authors would like to thank Dr. Rizaldy Scott at Mount Sinai Hospital in Toronto, Ontario for his gift of the primary Rac1flox/flox mouse embryonic fibroblasts. This work was supported by operating grants to NJ from the Canadian Institutes of Health Research (CIHR, MOP 2009-93526 ) and the Natural Science and Engineering Research Council of Canada (NSERC, RG 327372-06). SH and MW are supported by CGSMA and CGS-M awards from CIHR and NSERC, respectively.
Steve Hawley and Melanie Wills contributed equally to this paper.
References
- Guo F, Debidda M, Yang L, Williams DA, Zheng Y. Genetic Deletion of Rac1 GTPase Reveals Its Critical Role in Actin Stress Fiber Formation and Focal Adhesion Complex Assembly. J. Biol. Chem. 2006;281:18652–18659. doi: 10.1074/jbc.M603508200. [DOI] [PubMed] [Google Scholar]
- Glogauer M, Marchal CC, Zhu F, Worku A, Clausen BE, Foerster I, Marks P, GP Downey, Dinauer M, Kwiatkowski DJ. Rac1 deletion in mouse neutrophils has selective effects on neutrophil functions. J. Immunol. 2003;170:5652–5657. doi: 10.4049/jimmunol.170.11.5652. [DOI] [PubMed] [Google Scholar]
- Prost S, Sheahan S, Rannie D, Harrison DJ. Adenovirus-mediated Cre deletion of floxed sequences in primary mouse cells is an efficient alternative for studies of gene deletion. Nucleic Acids Res. 2001;29:E80–E80. doi: 10.1093/nar/29.16.e80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rijnkels M, Rosen JM. Adenovirus-Cre-mediated recombination in mammary epithelial early progenitor cells. J. Cell. Sci. 2001;114:3147–3153. doi: 10.1242/jcs.114.17.3147. [DOI] [PubMed] [Google Scholar]
- Vidali L, Chen F, Cicchetti G, Ohta Y, Kwiatkowski DJ. Rac1-null mouse embryonic fibroblasts are motile and respond to platelet-derived growth factor. Mol. Biol. Cell. 2006;17:2377–2390. doi: 10.1091/mbc.E05-10-0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Xie H, Zhang H, Das SK, Dey SK. Conditional gene recombination by adenovirus-driven Cre in the mouse uterus. Genesis. 2006;44:51–56. doi: 10.1002/gene.20185. [DOI] [PMC free article] [PubMed] [Google Scholar]