

Abstract
Cultured embryonic and adult skeletal muscle cells have a number of different uses. The micro-dissected explants technique described in this chapter is a robust and reliable method for isolating relatively large numbers of proliferative skeletal muscle cells from juvenile, adult or embryonic muscles as a source of skeletal muscle stem cells. The authors have used micro-dissected explant cultures to analyse the growth characteristics of skeletal muscle cells in wild-type and dystrophic muscles. Each of the components of tissue growth, namely cell survival, proliferation, senescence and differentiation can be analysed separately using the methods described here. The net effect of all components of growth can be established by means of measuring explant outgrowth rates. The micro-explant method can be used to establish primary cultures from a wide range of different muscle types and ages and, as described here, has been adapted by the authors to enable the isolation of embryonic skeletal muscle precursors.
Uniquely, micro-explant cultures have been used to derive clonal (single cell origin) skeletal muscle stem cell (SMSc) lines which can be expanded and used for in vivo transplantation. In vivo transplanted SMSc behave as functional, tissue-specific, satellite cells which contribute to skeletal muscle fibre regeneration but which are also retained (in the satellite cell niche) as a small pool of undifferentiated stem cells which can be re-isolated into culture using the micro-explant method.
Protocol
Two approaches can be employed to isolate proliferative skeletal muscle cells. In the first muscle tissues are enzymatically digested to isolate single cells prior to plating out 1. The second method is to explant pieces of muscle tissue into culture to allow cells to grow out during incubation 2, 3. The second method is described in this protocol. Tissue culture itself has its roots in explant culture. The year 2007 was the 100th anniversary of the classic experiments of Harrison in which he obtained neuron outgrowths by incubating nerve explants in hanging drops of lymph 4. Explant culture techniques have been used and refined in a variety of different contexts in the ensuing 100 years as a means of generating proliferative primary cultures of adult and embryonic cells 4, 5. The principle behind the explant technique, however, remains the same; to minimise the trauma of primary cell isolation by maintaining the three-dimensional structure of the parent tissue during the crucial early stages of cell outgrowth while providing the outgrowing cells with a rich nutritive media in which to proliferate. In skeletal muscle there is an additional advantage to using explant culture because the act of cutting up the muscle tissue mimics muscle fibre trauma, the usual trigger for satellite cell activation, migration and proliferation 3, 6. Adult skeletal muscle satellite cells (also called myoblasts) are the proliferative stem cell population responsible for muscle fibre repair and growth 7.
Skeletal muscle explants thus mimic the in vivo environment of the regenerating muscle and stimulate stem cell migration and division. In the embryo, the majority of vertebrate skeletal muscle (trunk and limb muscles) derives from the somites, although somitomeres and branchial arches give rise to the musculature of the head 8, 9. The myotome can be identified as two distinct groups of Myf-5 expressing stem cells located in the dorsal, medial and lateral edges of the differentiating somite, respectively. Respectively, these cells generate the epaxial muscles of the back, which differentiate in situ, and the ventral and lateral hypaxial musculature (limbs, abdomen and respiratory muscles) which require the migration of muscle stem cells from the somite 10. Embryonic muscle stem cell migration is under the control of Pax 3 11. Myf-5 expression is essential to the establishment of the embryonic musculature and this importance persists to postnatal muscles where over 98% of activated satellite cells express Myf-5 12. Myf-5 is therefore a reliable and specific marker of the proliferating skeletal muscle stem cell population in both adult and embryonic tissues. Embryonic muscle stem cells (also called muscle cell progenitors, skeletal muscle precursors, myoblasts or even embryonic satellite cells) can be isolated from the somites of early-stage mouse, chick and frog embryos 13. In order to isolate myogenic cultures from the embryonic skeletal muscles of older embryos the authors have adapted the microdissected explant technique for embryonic tissues. A similar approach is used by Cossu et al. 14 to generate clonal cell populations from the embryonic somite 14.
1. In vitro Cell Culture of Skeletal Muscle Stem Cells (SMSc)
SMSc are cell lines of single cell origin which have been clonally derived from primary skeletal muscle explant cultures. They can be cultured using standard tissue culture methodology if sufficient care is taken. Note that, unless indicated otherwise, all manipulations described are carried out under aseptic conditions using a laminar flow hood (Class 1 or Class 2 sterile cabinet) and all culture reagents are warmed to 37°C in a water bath before use.
To bring SMSc from liquid nitrogen storage (see Section 1.2 for freezing down method) cryovials should be thawed rapidly and the contents transferred into 5 mL prewarmed (37°C) DF10 culture medium for immediate centrifugation (1,000g for 3 min) to remove DMSO. The best method to thaw cells is by means of repeat pipetting of small quantities of pre-warmed culture medium into the vial before transferring to the centrifuge tube. The process of thawing cells should be carried out very quickly since cryopreserved cells contain 10% DMSO which is toxic to cells at room temperature (LD50 approximately 2 min).
Following centrifugation the supernatant is removed and the cells are washed by re-suspending the cell pellet in a further 5 mL DF10, then centrifuge as before.
The cell pellet is then mixed for the second time with 5 mL of DF10 and the resulting cell suspension is transferred to a small 25 cm2 plastic culture vessel.
Cultures are maintained at 37°C in a humidified incubator containing 5% CO2 in air. Unless vessels are used with a filtered cap, the cap of the flask must be slightly loosened for several hours to allow air in the culture vessel to equilibrate with the incubator and acidify the culture medium. pH of the medium is monitored by means of incorporating a phenol red dye pH indicator into the culture medium.
Thawed cells must always be monitored 24 h after plating and re-fed with fresh DF10 medium to ensure removal of cell debris and residual toxins (see Notes 1 and 2).
1.1. Subculture
For established SMSc lines, when cells reach approximately 95% confluence, they should be removed from their culture vessel, diluted and placed into a fresh vessel to enable further growth. This subculture procedure can be achieved by means of a number of different enzymatic procedures, trypsin/EDTA being the most frequently used (see Note 3). It is usual (and good) practice to grow cells at densities which require them to be subcultured on the third day of growth. For most SMS cell lines this can be achieved by splitting cells 1/10 at each subculture. This allows careful monitoring of cells and enables those performing the tissue culture to immediately identify unusual growth behaviour (for example faster growth) which could indicate phenotypic changes to the cell line such as transformation or reduction in apoptosis caused by adaptation to culture conditions. Additionally, a consistent and careful subculturing routine vastly reduces the incidence of such events.
For subculture using trypsin (trypsinisation) vessels are removed from the incubator and their medium discarded by aspiration.
Cells are then washed twice with sterile calcium- and magnesium-free phosphate-buffered saline (PBS), 10 mL per wash, removed each time by aspiration.
To dissociate the cell monolayer (25 mm2 flask) 1 mL 1 trypsin/EDTA is added and left on the cells at room temperature for 2 3 min until the cells begin to detach (see Note 4). This can be seen by the experienced user as small holes forming in the slightly opaque monolayer when the flask is held to the light (See Note 5). While cells should be trypsinised for sufficient time to ensure a single cell suspension, care should be taken not to overexpose SMSc to trypsinisation since this will result in high levels of cell death and poor attachment when cells are re-plated.
To stop the trypsin reaction, serum-containing medium (DF10) is added at a minimum of 2 volume (i.e. twice the volume of the trypsin solution). When subculturing a 25 mm2 flask it is convenient to add 9 mL DF10 at this stage. A 1/10 split of cells can then be easily made by diluting 1 mL of the resulting cell suspension into a new 25 mm2 culture flask together with a further 9 mL of fresh DF10 medium. The remaining cells can be used for cell expansion (transfer to a larger vessel), cryopreserved (see Section 1.2) or counted and plated in experimental dishes, wells or plates for proliferation and survival assays, differentiation, growth factor treatment or other purposes (see below).
1.2. Cryopreservation of Cell Lines and Primary Cultures
For cryopreservation cells are dissociated from their monolayer for subculture (Section 3.1.1) and pelleted by centrifugation (3 min at 1,000g).
The supernatant is removed by aspiration and cells are carefully and quickly re-suspended in 10 mL of freeze down mix (10% DMSO in DF10) before they are re-pelleted by centrifugation.
This time the pellet is re-suspended in sufficient freeze down mix to allow 0.5 mL of cell suspension per cryovial (see Table 1) and immediately placed into 80°C overnight.
Cryovials are transferred to liquid nitrogen the following day for long-term storage (see Note 6). As with thawing of cells, this process of cryopreservation must be carried out speedily. While DMSO is protective for cell membranes during freezing it is very toxic to cells at non-freezing temperatures.
1.3. Determining Cell Numbers
To determine the cell concentration of a single cell suspension (following subculture) a Neubauer haemocytometer can be used. To ensure accuracy of counting the coverslip must be securely mounted onto the haemocytometer base (see Note 7).
A small drop of cell suspension is then placed close to the edge of the coverslip and will be taken up by capillary action.
Cells are then counted using an inverted microscope with phase-contrast illumination. To increase the accuracy of the final cell concentration, the cells in the counting chamber should not overlap, if they do the original cell suspension should be diluted and the cells re-counted. Clumping should be avoided by thorough dissociation of cells during trypsinisation and 100-200 cells over a known surface area should be counted to obtain an accurate estimate of cell number. A convenient method using the Neubauer haemocytometer is to count cells in 2 or more 16 square sets. Cell density per millilitre is then obtained by dividing the total by the number of sets counted (e.g. 2) and multiplying by 104. For example, 100 cells counted over 2 x 16 square sets = 100/2 = 5 x 105 cells/mL.
2. Establishing Primary Skeletal Muscle Microexplant Cultures
Primary micro-explant culture can be used to isolate SMSc from any accessible skeletal muscle including the individual muscles of the fore and hind limbs, diaphragm, back and abdominal muscles. The method for deriving microexplant cultures from juvenile and adult muscles is described in detail by Smith and Schofield 3 and has subsequently been used extensively to derive SMS cells from juvenile, adult and aged mouse muscles. The method can also be used to derive cultured skeletal muscle cells from fish 24 and human skeletal muscle (Rao and Smith, unpublished). Outgrowth of SMSc from a mouse muscle microexplant is illustrated in Figure 1a, b. The method has been modified for the isolation of embryonic muscle precursor cells (see Section 3). The basic method is as follows:
Aseptic dissection of target muscle(s) from a freshly culled mouse is achieved using sterile instruments, a clean working area and liberal use of 70% ethanol spray.
Isolated muscles are washed through two changes of DF20 medium and are placed into fresh DF20 medium in a 60 mm2 dish. Using a stereo dissection microscope muscles are carefully microdissected under sterile conditions to exclude fat, connective tissue and bone.
Cleaned muscle pieces are then cut into 400 μm3 cubes which, using jeweler s forceps, are placed individually into the centre 60 wells of a 96-well plate containing 50 μL DF20 (see Note 8). Wells are checked under the microscope and placed in the incubator. The outer wells are filled with saline to prevent drying out of wells containing explants (3).
Microexplant attachment and outgrowth is scored after 24-48 h incubation and thereafter at 48-72 h intervals (depending on the rate of growth of the muscles being cultured).
For expansion and isolation of SMS cells, outgrowth cultures should be individually monitored for cells with a predominantly SMSc morphology, i.e. spherical mononucleate cells with high refractivity which grow in aggregating clusters (see Figure 3.1b).
Once explant outgrowth is established individual wells are fed (see Note 9) by the addition of 50 μL increments of medium when the medium acidifies due to increased cell density. When the well is full and cell culture almost confluent, wells are fed by replacement of 50% of the medium each time to ensure the maintenance of "conditioning" factors secreted by the cells (see comments on conditioned medium under cloning; Section 2.1). To suppress differentiation, at 60-70% confluence primary explant outgrowths are switched into calcium-depleted medium by substituting DF20 medium for calcium-depleted DMEM/F12 (all supplements remain the same) for feeding cells 3.
Explant conditioned medium can be prepared from cultures at this stage and stored for use during expansion and cloning of primary SMSc (for cloning method, see Section 2.1, Figure 1c f). Cultures are subcultured using the dispase method (see Section 3.5).
Clonally derived SMSc can be analysed in vivo by intramuscular injection (Figure 3.1 g k) 19.
Karyotyping can be carried out on clonally derived SMSc lines to confirm diploid status (see Section 2.2, Figure 3.1 l) 25.
This method can be adapted for culturing embryonic muscle (Section 3).
2.1. Clonal Derivation
Primary explant myoblast cultures (Figure 1a, b) are a useful and accurate tool for establishing a variety of different growth parameters in wild-type and mutant skeletal muscles. Clonal derivation, the isolation of a cell line from a single cell, is an essential step in the isolation of skeletal muscle stem cells and can also be used to subclone SMSc lines transfected with RNAi constructs or transgenes. Established SMSc and primary explant cultures are highly density dependent and will "crash" (detach from the dish and die) if plated out at too low a cell density. This is because SMSc release soluble factors which are required to maintain growth and cell survival. To simulate a high-density culture and supply these factors during the cloning process, SMSc are cloned in self-conditioned medium. The addition of conditioned medium was found to be essential to allow individual cells to proliferate in an isolated environment.
Conditioned medium is prepared from proliferating SMSc cultured for 48 h between 33 and 75% confluence.
The media in which these cells are grown is removed after 48 h and filtered using a 0.2 μm syringe filter; this ensures sterility of the conditioned medium and removes all residual cells and debris.
This conditioned medium is mixed in a 1:1 ratio with fresh culture medium (cloning medium; see Note 10) and is used as the culture media for single cell cloning.
To achieve single cell dilutions cells are dissociated to a single cell suspension by careful trypsinisation (established SMSc) or dispase treatment (primary explant cultures) and diluted to a concentration of one cell per 100 μL cloning medium.
50 μL of this cell suspension can then be plated into each of the centre 60 wells of a 96-well plate.
Cells are allowed to attach by incubation at 37°C in 5% CO2 for 6 h and each well is then carefully scored for the presence of cells. Wells containing 0 or more than one cell are discounted at this stage.
Wells containing one single attached cell are carefully noted and the colony derived from this single cell is carefully monitored (daily during the first few days) to ensure that only one colony, derived from a single cell, is present. Cell lines were only derived from wells containing 1 cell. Figure 1c-e illustrates the expansion of such a single cell derived colony.
Once the colony reaches confluence in the 96-well plate it can be subcultured into one well of a 48-well plate.
The cloned cell lines can then be carefully expanded into 24- and 6-well plates until sufficient cells are available to plate into a 25 cm2 flask.
The skeletal muscle origin of these cultures can be demonstrated by the expression of Myf-5 (Figure 1f) or other skeletal muscle-specific markers such as MyoD and Pax 7.
At this stage lines are frozen down before further expansion (see Table 1).
2.2. Karyotyping
Karyotyping is an important method of monitoring cell phenotype. Cell lines derived by clonal derivation should be karyotyped to ensure that they have retained a diploid chromosome complement without gross chromosomal rearrangements which could affect their phenotype.
For karyotyping cells are grown to late exponential phase (80% confluent) in 25 cm2 culture vessels (2 days after subculturing) to maximise the proportion of mitotic cells in the culture.
Twenty-four hours before karyotyping the cells are fed with 10 mL fresh culture medium. 0.2 mL of 10 mg/mL colchicine (see Note 11) is then added to the cells which are incubated for a further 1 h at 37°C.
After 1 h, the cells are subject to the standard trypsinisation subculture procedure except that both the culture medium and PBS washes are retained to maximise the number of mitotic cells harvested.
The dissociated cells, retained medium and PBS washes are spun at 1,000g for 3 min to pellet the cells and the supernatant removed and discarded into bleach.
The cell pellet is then re-suspended in 5 mL of 0.0075 M potassium chloride for exactly 4 min, before cells are again pelleted by centrifugation.
Most of the supernatant is aspirated, leaving a small amount (~50-100 μL) in the tube for re-suspension. Resuspend cells by flicking the base of the falcon tube until a cell slurry is achieved. The cells are then placed on ice and fixed in freshly made ice-cold fixative (methanol: glacial acetic acid in a 3:1 ratio) as follows: 10 mL of fixative is slowly added dropwise to the cells using a small glass Pasteur pipette (this prevents cell clumping).
Cells are placed on ice for 30 min and then pelleted by centrifugation after which the cell pellet is re-suspended in 0.5 mL fresh fixative.
Slides are produced by dropping the fixed cell suspension onto prepared slides (see Section 3.2.2.1) held at an angle of 45°. To ensure well-spaced metaphase spreads the pipette should be held at least 30 cm above the slide.
To visualise chromosomes, slides are stained for 2 min in Leishman's Stain, diluted with three volumes of Gurr buffer pH 6.8 just prior to use.
Slides are dried at room temperature and mounted in DePex mounting medium.
2.2.1. Preparation of Slides for Karyotyping
Glass slides (Premium microscope slides, VWR International, UK) are prepared for use in the karyotyping protocol by placing them overnight in a large (glass) container of sulphuric acid.
Slides are then placed under running tap water for 8 h and are then stored in 70% ethanol until required.
Before use, slides should be rinsed under running tap water for a further 30 min and air-dried at room temperature for 1-2 h.
3. Establishing Primary Micro-explant Cultures from Embryos
Three mouse strains were used to validate this method, wild-type (C57BL/10) together with mdx and CAV3KO (both dystrophic mutants). The dystrophin-deficient mdx mouse originated spontaneously in C57BL/10, this line was obtained from the Bullfield laboratory in 1991 and has since been continually maintained in our inbred colony 26. CAV3KO dystrophic mice, which contain a mutation in the caveolin-3 gene, were bred onto the C57BL/10 background for 10 generations before being used in this study 27. Each mouse line generated a robustly reproducible outgrowth, proliferation and survival profile which was embryonic stage specific and different for each strain. The following protocols were adapted for embryos from Smith and Schofield PN (1994) 3, essentially as in Merrick 21.
3.1. Embryo Collection
To obtain staged embryos, pairs are set up as natural (1:1) matings and females checked every morning for vaginal plugs. On the day of plug detection, embryos are counted as E0.5 days (12 h post-fertilization).
Once vaginal plugs have been detected the males are removed from the cage to ensure accuracy of embryonic staging.
When the desired embryonic stage is reached (E11.5 to E17.5) the mothers are killed by cervical dislocation, the abdomen is shaved, the skin and surrounding areas are swabbed with 70% alcohol and the uterus is removed via a horizontal abdominal incision made using sterile dissecting instruments.
The uterus is then washed once in primary explant culture medium (PECM) before being placed into a small dish containing fresh PECM prior to dissection.
E11.5 to E17.5 embryos are dissected from the uterus using a dissecting microscope and placed individually into Petri dishes containing PECM in readiness for detailed microdissection.
3.2. Embryo Microdissection
Individual embryos are further dissected to isolate areas rich in skeletal muscle (see Figure 2a). Hind and forelimbs (hypaxial skeletal muscles) are dissected out, as well as the upper and lower body wall (predominantly epaxial skeletal muscles). To do this an incision along the length of the thorax, abdomen and pelvis is created to allow the internal organs of the embryo to be removed.
To enrich for embryonic skeletal muscle stem cells (eSMSc), the head, spinal cord and all internal organs are then removed.
In older embryos (E15.5-E17.5 embryos) it is also possible to remove skin and cartilage/bone again to increase the proportion of muscle cells in the cultures.
3.3. Setting Up Embryo Microexplant Cultures
Once forelimbs, hindlimbs and upper and lower body walls have been dissected out they are placed into fresh PECM and further microdissected to produce small cubes of tissue of equal size (~0.5 mm3; Figure 2a).
These microexplants are then put into the centre 60 wells of a 96-well plate (one explant per well) containing 50μL PECM per well. A minimum of 60 wells containing 1 explant per well are established, per embryo studied.
For culturing embryos the centre 60 wells can be subdivided into regions denoting where the explant was derived from (Figure 2b). This design allows 15 wells each containing, respectively, forelimb, upper body wall, hindlimb and lower body wall explants 21.
3.4. Monitoring Outgrowth
Outgrowth rate is a reliable measure of the growth rate of embryonic skeletal muscle explants and under the carefully controlled conditions described here is highly reproducible.
Explants are incubated at 37°C and 5% CO2 for 3 weeks and scored on the 3rd, 7th, 14th and 21st day of culturing using an inverted microscope. Explants are scored according to the level of confluence of cells in each individual well (Figure 3a,e).
Photographic images of cultures can be taken, for example, using an SLR camera attached to the microscope and 100 ASA Fuji (colour) or Kodak TMAX (black and white) professional film (Figure 3f).
A skeletal muscle-specific antibody specific for Myf-5 can be used to demonstrate the skeletal muscle origin of eSMSc, depending on strain 80 95% of cells isolated using this method are Myf-5 positive. Other markers such as MyoD and Pax 7 can also be used to demonstrate the skeletal muscle origin of these cell populations. While these cells contain a very high proportion of embryonic skeletal muscle stem cells it cannot be assumed (especially for younger embryos) that they are all of skeletal muscle origin or that they are all stem cells. To isolate pure stem cell populations it is necessary to clonally derive primary explant cultures as described in Section 2.1.
3.5. Subculturing Primary Embryonic Explants
Once confluent, explant cultures displaying the morphological features of SMSc (Figure 3f) can be subcultured as follows 3, 21:
Culture medium is removed from the selected wells, filtered using a 0.2 μm Acrodisc R_ syringe filter and retained for use as conditioned medium. The medium can be stored at 4°C for 1 week.
100 μL of dispase diluted 1:10 in PECM is added to each well and plates then returned to the 37°C incubator for 20 min.
A pipette tip is then used to gently scrape the loosened cells from the surface of the well.
The cell suspension is then centrifuged at 1,000g for 3 min to pellet the cells and the supernatant is removed and discarded.
Cells are re-suspended in 200 μL of 1:1 mix of conditioned medium and PECM.
The cell mix is transferred to 48-well plates for further expansion.
For in vitro analysis cells can be plated at a density of 5 x 103 cells/cm2 either in 48-well plates (each containing a 9 mm2 sterile glass coverslip) or in 8-well-glass chamber slides. For differentiation analysis cells are grown overnight to 50-60% confluence before they are transferred to differentiation permissive medium (see Section 4 for in vitro method details) for 3 days, before fixation.
4. In vitro Analysis of Skeletal Muscle Stem Cells and Primary Cultures
4.1. Preparation of Cells
Dispase subcultured (Section 3.3) primary embryonic explant cultures are plated in PECM/conditioned medium onto coverslips in 48-well plates at a density of 3 x 103 cells/cm2 and allowed to attach.
For assessment of apoptosis and proliferation coverslips are washed twice in PBS, fixed in 4% paraformaldehyde (see Section 4.2) in PBS for 20 min at room temperature, followed by a further 10 min PBS wash.
Coverslips prepared this way can be stored at 4°C for up to 1 week in PBS or PBS/glycine.
4.2. Preparation of Paraformaldehyde Fixative
In a fume hood, weigh 4 g paraformaldehyde (PFA; Sigma-Aldrich, UK) and add to a glass bottle of 100 mL of sterile PBS with a magnetic stirrer. A face mask and gloves should be worn for protection.
In a fume hood, the solution is heated and continually stirred on a magnetic hotplate until the powder dissolves. This takes approximately 5-10 min at 65°C. Care must be taken to prevent the temperature rising above 70°C, as there is a risk of the solution exploding at high temperatures.
4.3. Apoptosis and Proliferation Assay
Fixed coverslips (prepared as in Section 4.1) are stained with 10 μg/mL DAPI for 3 min.
Coverslips are washed once in PBS (5 to10 min) and inverted onto a spot of vectashield mounting medium on a glass slide 17, 18.
The edges of the coverslip are sealed with nail varnish (see Note 12).
For storage, slides are wrapped in foil and placed at 20°C.
For counting, slides are viewed under fluorescence (UV filter) on an upright microscope and scored for apoptotic and mitotic cells using an eyepiece graticule. Twenty randomly distributed grids are counted (representing ~1,000 cells), and cells are morphologically characterised as non-apoptotic, apoptotic or mitotic (Figure 3 g).
Mitotic and apoptotic indices are calculated as a proportion of total cells.
4.4. Immunohistochemistry
Cells fixed onto coverslips can also be used for immunohistochemistry. For antigen retrieval using a pressure cooker coverslips must be firmly attached to glass slides using standard paper clips. Immunostaining can be used to identify proliferating cells, using an antibody to Ki67 (1/1,000 dilution), to establish identity, using an antibody to Myf-5 (1/1,000 dilution), or to investigate gene expression (see Section 4.5). Immunostaining can be achieved using a number of methods, the following (described in (28, 29)) is used routinely by the authors:
Sodium citrate buffer is pre-heated in the pressure cooker. For antigen retrieval, slides containing sectioned tissue are placed into the heated buffer and heated under pressure for 2 min. Pressure is achieved by firmly locking the pressure cooker lid and placing on the weight. Once the 2 min retrieval time has elapsed the pressure cooker is then carefully placed under running cold tap water to reduce pressure. To prevent the buffer boiling up, care should be taken not to remove the lid until pressure has equalized with atmospheric pressure. Pressure is sufficiently reduced when the weight can be removed easily (without force) and the lid removed. Slides are then removed from the buffer and washed in room temperature PBS for 10 min.
Slides are pre-blocked by immersing them in 3% hydrogen peroxide/tap water for 5 min and then washed three times in PBS + 0.05% Tween 20 (10 min per wash).
Blocking is achieved by a 30 min incubation in TNB blocking buffer (supplied in TSA kit) at room temperature.
Primary antibody is diluted in TNB buffer to the appropriate dilution (arrived at by titration, see Note 12) and incubated overnight at 4°C (or alternatively 1 to 2 h at room temperature).
Following three 10 min washes in PBS + 0.05% Tween 20, slides are incubated for 1 h at room temperature in the appropriate biotinylated second antibody diluted in TNB buffer.
Following a further three 10 min washes in PBS + 0.05% Tween 20, slides are incubated for 30 min in Streptavidin-HRP (provided in the TSA kit) diluted 1:100 in TNB buffer and then washed three times (10 min each) in PBS + 0.05% Tween 20.
Biotinyl tyramide (amplification reagent; TSA kit) is then added to each section for between 8 and 15 min (the precise time should be obtained by optimisation experiments).
Following amplification, wash slides three times (10 min each) in PBS + 0.05% Tween 20 and then incubate for 30 min in SA-HRP.
Following three further washes (10 min each) in PBS + 0.05% Tween 20, visualise using 3,3_-diaminobenzidine tetrahydrochloride chromogen (DAB) for 5 to 10 min. Then carry out two final washes in water before counterstaining slides in haematoxylin and coverslipping. DAB is a known carcinogen and should be handled with care (as for Colchicine, Section 3.2.2).
4.5. Differentiation
SMSc plated onto coverslips or chamber slides (see Note 13) can also be differentiated before fixation for myotube analysis.
For these experiments cells are plated at a density of 104/cm2 and allowed to attach for 6 to 8 h.
Cells are then switched in to differentiation permissive conditions for 3 days (see Note 14).
Differentiation medium is composed of DMEM + 0.5% FCS supplemented with 2% horse serum and 1% glutamine. This differentiation permissive culture medium is replaced at 48 h intervals.
Coverslips are then fixed in 4% paraformaldehyde as above (Sections 3.4.1 and 3.4.2).
4.6. Transfection of SMSc: Expression of Transgenes and shRNAi Constructs
Stem cells and primary cultures are refractory to transfection and with a majority of methods the transfection rate in SMSc and primary skeletal muscle cells is very low (<10%), preventing the use of transient transfection methods. To overcome this it has been standard practice in our laboratory to isolate clonal derivates from transgene transfected cultures (see Section 3.2.1) following transfection with calcium phosphate or lipofectamine. Alternatively cells can be efficiently transfected using infection of virally packaged constructs. Figure 1m shows stable expression of β-galactosidase in PD50A, a clonal SMSc derivative isolated under G418 selection following infection with pIRV, a replication defective retrovirus carrying the genes for neo/G418 resistance and β-galactosidase (19). This cell line was used to formally demonstrate that SMSc behave as functional stem cells in vivo (see Figure 3.1). While the generation of a stable clonal cell line expressing a marker gene is desirable for in vivo stem cell transplantation experiments, it is a time-consuming and unsatisfactory method of analysing gene function in vitro. For these reasons the authors have recently developed an optimized modification of the Lipofectamine 2000 transfection reagent which is capable of delivering transfection rates of 60 to 70%. This allows the analysis of gene function using transient transfection of transgenes or RNAi constructs into SMSc or primary explant cultures (Figure 3h, i). The authors use a short hairpin RNAi vector (pSHAG RNAi) (30) to generate shRNAi constructs capable of gene-specific targeting of mRNA expression in SMSc. The success of the shRNAi technique depends on two elements: (a) an efficient transfection method and (b) the design of a short hairpin sequence which specifically recognizes the target gene. A shRNAi construct directed to eGFP can be used to validate the RNAi knockdown method (Figure 3j,m).
4.7. Optimised LipofectamineTM 2000 Transfection Protocol for SMSc
Cells are plated at 5 to 104 cells/cm2 into chamber slides in 250 μL DF10 culture medium and cultured for 18 h to reach 95% confluence (optimal confluence for each cell line was established by assessing transfection rates at different densities).
For each well, 0.5 μg of DNA (shRNAi vectors, transgenes) is added to 33 μL of serum-free DMEM supplemented with 2 mM glutamine and mixed gently in a sterile Eppendorf tube.
For each well, 1.25 μL of Lipofectamine 2000 is separately diluted in a further 33 μL of serum-free medium DMEM + glutamine, gently mixed and retained at room temperature for 5 min
The DNA and lipofectamine mixes are then rapidly added together, mixed gently for 60 s by pipetting and then incubated at room temperature for 19 min to allow DNA Lipofectamine 2000 complexes to form.
For transfection, 66 μL of complex mix is added to each chamber well and slides are gently rocked for 10 s to ensure equal distribution of complexes.
Cells are incubated for 24 to 72 h at 37°C and 5% CO2. Depending on the construct, functional gene expression or shRNAi knockdown is first detected between 8 and 24 h post-transfection.
5. Representative Results
When explants are carefully explanted from adult skeletal muscle or from embryos the explants will begin to generate cells within a few hours to 72 hours of incubation at 37°C (5% CO2/air) (Figure 3A). The time taken for this to occur depends on the source of the explants: embryonic explants will outgrow more quickly than older adult skeletal muscle explants. In our experience the timing of outgrowth is highly reproducible 3, 29. Expansion of the cell population will occur over a period of days (embryo explants) or weeks (older skeletal muscle explants) to generate high density aggregating SMS cell primary cultures (see Figure 3B-F for illustration of these cultures). Figures 1 & 3 show representative results of the successful derivation and culture of skeletal muscle and embryonic explants, clonal derivation and in vivo transplantation of skeletal muscle derived adult stem cells, β-galactosidase labeling, karyotyping and myf-5 immunohistochemistry of embryonic SMSc to illustrate skeletal muscle origin of the cell populations. Figure 3 shows a representative result of outgrowth scoring (using Myf-5 immunohistochemistry to visualize SMSc); the morphology of embryonic muscle primary cells; DAPI staining for apoptosis and the shRNAi transfection protocol. Further details can be found in the figure legends relating to these two figures. Figure 2 illustrates the procedure from embryo micro-dissection to generate enriched populations of embryonic skeletal muscle stem cells.
Table 1: Calculation table to estimate the number of cryovials required for maximum cell viability during cell freeze down protocols
| Plate/Flask/ chamber slide | Volume of Culture medium | PBS washes | Volume of trypsin | Number of freeze down vials (3.1.2) |
| 96-well | 50 μL | 2 X 100 μL | 25 μL | N/A |
| 48-well | 150 μL | 2 X 200 μL | 50 μL | N/A |
| 24-well | 500 μL | 2 X 700 μL | 200 μL | N/A |
| 6-well | 3 mLs | 2 X 3 mLs | 500 μL | 1 |
| Small (25 cm2) | 10 mLs | 2 X 10 mLs | 1 mL | 2 |
| Medium (80 cm2) | 10 mLs | 2 X 10 mLs | 3 mLs | 4 |
| Large (175 cm2) | 10 mLs | 2 X 10 mLs | 5 mLs | 8 |
| Chamber well | 500 μL | 2 X 500 μL | 100 μL | N/A |
N/A = not applicable; cell numbers were too low to freeze down unless multiple wells were frozen down together.
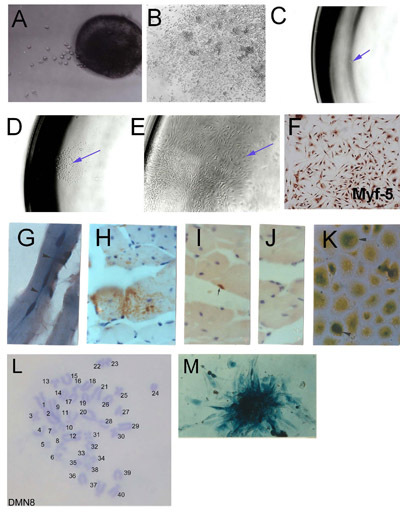 Figure 1. Isolation of skeletal muscle stem cells (SMSc) from microexplants:
(A) Early outgrowth from explanted adult skeletal muscle (day 2). (B) Established explant outgrowth showing aggregated cultures and high cell density. Clonal derivation of SMSc. (C) Single cell isolated into a 96 well plate. (D) Colony of single cell origin. (E) Established clonal population. (F) Verification of SMSc identity using Myf-5 immunohistochemistry. Cells derived from SMSc clone PD50A (expressing β-galactosidase) in host mice at 3 months (G) and (H-J) 14 months post-injection of 2,000 PD50A cells into mouse tibialis anterior muscle. (G) Three recently fused (centrally located nuclei) β-galactosidase-positive cells (blue stain) in muscle fibre (longitudinal section). (H) Extensive contribution of β-galactosidase-positive cells (brown stain, detected by anti-β-galactosidase antibody) in muscle fibres (transverse section). (I) β-Galactosidase-positive satellite cell (brown stain, detected by anti-β-galactosidase antibody). (J) Secondary antibody control (no staining). (K) β-Galactosidase-positive cells (blue stain) proliferate in culture when isolated from injected host muscles 12 months post-injection. (L) Karyotype of a mouse clonal SMSc line (DMN8) showing normal diploid chromosome complement. (M) Histochemistry showing β-galactosidase expression in a colony of PD50A cells (Figure 3.1 g,k, reproduced with permission from AACR press, Smith and Schofield, 1997).
Figure 1. Isolation of skeletal muscle stem cells (SMSc) from microexplants:
(A) Early outgrowth from explanted adult skeletal muscle (day 2). (B) Established explant outgrowth showing aggregated cultures and high cell density. Clonal derivation of SMSc. (C) Single cell isolated into a 96 well plate. (D) Colony of single cell origin. (E) Established clonal population. (F) Verification of SMSc identity using Myf-5 immunohistochemistry. Cells derived from SMSc clone PD50A (expressing β-galactosidase) in host mice at 3 months (G) and (H-J) 14 months post-injection of 2,000 PD50A cells into mouse tibialis anterior muscle. (G) Three recently fused (centrally located nuclei) β-galactosidase-positive cells (blue stain) in muscle fibre (longitudinal section). (H) Extensive contribution of β-galactosidase-positive cells (brown stain, detected by anti-β-galactosidase antibody) in muscle fibres (transverse section). (I) β-Galactosidase-positive satellite cell (brown stain, detected by anti-β-galactosidase antibody). (J) Secondary antibody control (no staining). (K) β-Galactosidase-positive cells (blue stain) proliferate in culture when isolated from injected host muscles 12 months post-injection. (L) Karyotype of a mouse clonal SMSc line (DMN8) showing normal diploid chromosome complement. (M) Histochemistry showing β-galactosidase expression in a colony of PD50A cells (Figure 3.1 g,k, reproduced with permission from AACR press, Smith and Schofield, 1997).
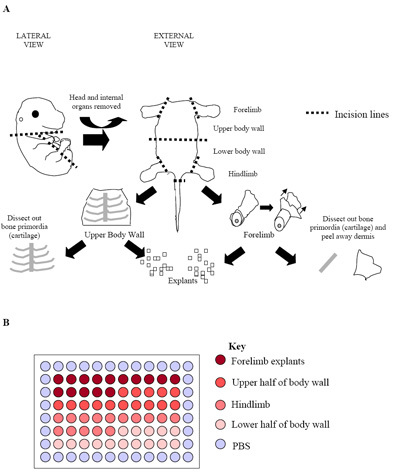 Figure 2. (a) Illustration of the embryo dissection process. The figure represents an E15.5 embryo where bone primordial (cartilage) could easily be identified and dissected free of the surrounding skeletal muscle tissue. At this stage, and in later stage embryos (E15.5 E17.5), dermis was also removed to maximise the proportion of skeletal muscle cells obtained. (B) Setup of primary explant cultures in a 96-well plate. Each embryo was used to produce one plate as seen above. Usual practice is to plate replicates of three separate embryos (three plates = 180 wells) to establish outgrowth rates.
Figure 2. (a) Illustration of the embryo dissection process. The figure represents an E15.5 embryo where bone primordial (cartilage) could easily be identified and dissected free of the surrounding skeletal muscle tissue. At this stage, and in later stage embryos (E15.5 E17.5), dermis was also removed to maximise the proportion of skeletal muscle cells obtained. (B) Setup of primary explant cultures in a 96-well plate. Each embryo was used to produce one plate as seen above. Usual practice is to plate replicates of three separate embryos (three plates = 180 wells) to establish outgrowth rates.
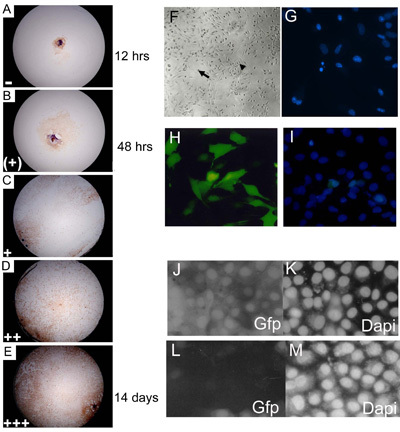 Figure 3. Embryonic primary explant cultures were scored at 3, 7, 14 and 21 days of culture and assigned an outgrowth level representative of confluence level. (A-E) C57BL10 E15.5 primary embryonic explant cultures stained with Myf-5 to illustrate 0 14% ( ); 15 24% (+); 25 49% (+); 50 74% (++); 75 100% (+++) levels of confluence. The proportion of wells showing each level of outgrowth (on each day of scoring) was multiplied by an arbitrary number ( = 1; (+) = 2; + = 3; ++ = 4 and +++ = 5) before collating data to give a final outgrowth value. Approximately 85% of wild-type (C57BL/10) primary eSMSc stain for the skeletal muscle cell marker Myf-5. Magnification is 10. (F) Established embryonic cultures have the morphological characteristics of adult SMSc, bipolar cells (small arrow) and spherical monomorphic cells (large arrow). (G) Identification of fragmenting apoptotic nuclei using DAPI staining. (H-I) High levels (~75%) of transfection of a GFP-expressing construct into SMS cell lines using the optimized Lipofectamine 2000 transfection method. (I) Counting of total cell number is aided by DAPI counterstain. RNAi using pSHAGshRNAigfp (J-M) abolishes GFP expression in SMSc (see (29) for an example of this construct used as a control). (J) Control (mock transfection) showing GFP expression in a GFP SMSc line. (K) DAPI control. (L) shRNAiGfp 24 h after transfection. (M) DAPI control for shRNAiGfp transfected cells in (L).
Figure 3. Embryonic primary explant cultures were scored at 3, 7, 14 and 21 days of culture and assigned an outgrowth level representative of confluence level. (A-E) C57BL10 E15.5 primary embryonic explant cultures stained with Myf-5 to illustrate 0 14% ( ); 15 24% (+); 25 49% (+); 50 74% (++); 75 100% (+++) levels of confluence. The proportion of wells showing each level of outgrowth (on each day of scoring) was multiplied by an arbitrary number ( = 1; (+) = 2; + = 3; ++ = 4 and +++ = 5) before collating data to give a final outgrowth value. Approximately 85% of wild-type (C57BL/10) primary eSMSc stain for the skeletal muscle cell marker Myf-5. Magnification is 10. (F) Established embryonic cultures have the morphological characteristics of adult SMSc, bipolar cells (small arrow) and spherical monomorphic cells (large arrow). (G) Identification of fragmenting apoptotic nuclei using DAPI staining. (H-I) High levels (~75%) of transfection of a GFP-expressing construct into SMS cell lines using the optimized Lipofectamine 2000 transfection method. (I) Counting of total cell number is aided by DAPI counterstain. RNAi using pSHAGshRNAigfp (J-M) abolishes GFP expression in SMSc (see (29) for an example of this construct used as a control). (J) Control (mock transfection) showing GFP expression in a GFP SMSc line. (K) DAPI control. (L) shRNAiGfp 24 h after transfection. (M) DAPI control for shRNAiGfp transfected cells in (L).
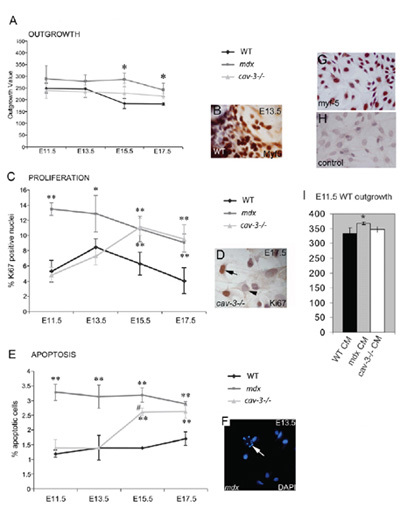 Figure 4. Dystrophic, embryonic Myf5-positive myoblast are hyperproliferative and prone to apoptosis. (A) The outgrowth rate of embryonic myoblasts from muscle explant culture is increased both in mdx mutants from E11.5 and in cav-3(-/-) mutants at E15.5 and E17.5 when compared with WT explants cultured in parallel. (B) A Myf5-immunostained explant. (C) Hyperproliferation of embryonic myoblasts in mdx mutants from E11.5 and in cav-3(-/-) mutants from E15.5, as determined by Ki67-positive immunoreactivity (D). (E) Elevated apoptosis from E11.5 in mdx embryos and from E15.5 in cav-3(-/-) embryos, as shown by DAPI staining (F); the arrow in F points to an apoptotic cell. *p<0.05 compared with WT; **p<0.01 compared with WT; *p<0.05 when comparing mdx with cav-3(-/-). (G,H) E15.5 primary cultured WT embryonic myoblasts with Myf5 staining (G) and a second antibody control (H). (I) The outgrowth rate of E11.5 WT explants increased (*p<0.05) in E11.5 mdx explant-conditioned medium (CM), but not in cav-3(-/-) or WT CM. Error Bars indicate s.d. This figure is reproduced under authors copyright and was first published by the Company of Biologists in Merrick et al., 2009.
Figure 4. Dystrophic, embryonic Myf5-positive myoblast are hyperproliferative and prone to apoptosis. (A) The outgrowth rate of embryonic myoblasts from muscle explant culture is increased both in mdx mutants from E11.5 and in cav-3(-/-) mutants at E15.5 and E17.5 when compared with WT explants cultured in parallel. (B) A Myf5-immunostained explant. (C) Hyperproliferation of embryonic myoblasts in mdx mutants from E11.5 and in cav-3(-/-) mutants from E15.5, as determined by Ki67-positive immunoreactivity (D). (E) Elevated apoptosis from E11.5 in mdx embryos and from E15.5 in cav-3(-/-) embryos, as shown by DAPI staining (F); the arrow in F points to an apoptotic cell. *p<0.05 compared with WT; **p<0.01 compared with WT; *p<0.05 when comparing mdx with cav-3(-/-). (G,H) E15.5 primary cultured WT embryonic myoblasts with Myf5 staining (G) and a second antibody control (H). (I) The outgrowth rate of E11.5 WT explants increased (*p<0.05) in E11.5 mdx explant-conditioned medium (CM), but not in cav-3(-/-) or WT CM. Error Bars indicate s.d. This figure is reproduced under authors copyright and was first published by the Company of Biologists in Merrick et al., 2009.
6. Notes: critical steps and possible modifications
When thawed cells have attached at very low cell density it is prudent to replace only half the medium to prevent culture crash.
SMSc isolated from dystrophic muscle are susceptible to apoptosis and must be treated with particular care. Dystrophic SMSc (such as the dfd-13 cell line, which was established from skeletal muscle obtained from 5-week-old dystrophic (mdx) mice) should be grown at higher cell densities than usual for myoblasts. Such apoptosis-sensitive cell lines are also cryopreserved at higher densities (see Sections 1.1 and 1.2) (19).
An alternative method of removing cells from a monolayer utilises dispase, which provides a gentler method of cell dissociation, with the advantage that it can be carried out in the presence of FCS and calcium (both present in DF10). Dispase can therefore be used to subculture and expand primary skeletal muscle explant cultures and earlystage SMSc clones (see Sections 2 and 3).
For larger flasks the quantity of trypsin/EDTA used should be scaled up as follows: 75 mm2 flask (3 mL trypsin) and 175 mm2 flask (5 mL trypsin). Similarly for smaller surface areas reduce the amount of trypsin used (see Table 1).
Alternatively, dissociation can be monitored using an inverted microscope, this is recommended for beginners.
Cryopreservation of cell lines is usually carried out using a confluent large (175 mm2) plastic vessel. Between 7 and 9 cryovials can be obtained from one such large vessel, depending on the survival profile of the cell line being cryopreserved. Primary cultures and newly established cell lines are often very refractory to freeze down procedures. To improve recovery and success of freezing down such cells two approaches can be used (separately or in combination). (A) The FCS content of the freeze down mixture can be increased from 10% (to a maximum of 50%). (B) The freeze down process can be slowed by placing cryovials in the vapour phase of N2 for 12-24 h before transferring vials to the liquid phase.
A convenient way to check firm attachment of the haemacytometer coverslip is to look for Newton's Rings (rainbow reflections in the glass) on the coverslip or alternatively, to hold the haemacytometer upside down over an open hand.
A variation of the explant method can be employed for short-term cultures for use in immunohistochemistry, proliferation or apoptosis assays. Microdissected explants are placed on glass using 8-well chamber slides. An alternative method is to use 9 mm2 coverslips placed in 24-well plates. In both cases two explants are transferred to each well in 150 μL DF20 medium. Alternatively, primary cultures can be subcultured by the dispase method and plated out onto coverslips placed in 24-well plates or directly into 8-well chamber slides.
For quantitative analysis of outgrowth rates a minimum of 60 wells containing 1 explant per well are established, per muscle/mouse strain. To establish growth parameters for a strain at least three separate animals must be used. Plates and individual wells are not fed while outgrowth is being scored.
For cloning established SMSc lines it is sufficient to culture single cells in a 1:1 mix of conditioned medium and DF10 medium. For primary explants it is necessary to increase the serum content of the culture medium to 20%.
Colchicine is highly toxic and a known carcinogen and should be handled with appropriate care. Double gloving and working in the confines of a designated tray is essential. All disposables (i.e. Gilson tips) are placed in a beaker of bleach (5% sodium hypochlorite) overnight before disposal with copious amounts of water the follo -wing day.
We currently use primary antibodies specific to Ki67 and Myf-5 each at 1/1,000 dilution. The optimal dilution for primary antibodies needs to be established empirically for each antibody to be used and ideally also for different batches of the same antibody, even when obtained from the same source.
Chamber slides can also be used for this assay. Before fixing cells in 4% PFA, the culture medium is removed and cells washed twice with 37◦C sterile PBS. The well chambers, gasket and glue are removed and the glass slides placed in a 50 mL glass Coplin jar containing freshly prepared 4% paraformaldehyde at room temperature, which is then gently rocked on a Gyro-Rocker R_ shaker for 25 min. Slides are then washed twice in PBS (room temperature) and either used immediately or stored in PBS at 4◦C (short term; 1-2 weeks) for immunohistochemical (IHC) analysis.
If more extensive myotube formation is required primary skeletal muscle cells can be differentiated for up to 8 days.
Discussion
Microdissected explant cultures can be used to reliably and reproducibly isolate cell populations containing a very high proportion (~85%) of proliferative Myf-5 positive skeletal muscle stem cells (SMSc). Under the rigorously controlled culture conditions described here primary explant cultures can be used to characterize the growth behaviours of genetically mutant mouse SMSc and can be used as a means of generating myotubes for detailed in vitro analysis of differentiation processes. Careful maintenance and manipulation of these cultures enables long-term culture and expansion. Using the methods described here it is also possible to derive clonal skeletal muscle stem cell lines from explant cultures by means of single cell dilution. To achieve the proliferation of isolated single cells during the cloning procedure, "conditioned medium" is used to mimic the normal requirement of these cells for high-density culture. The method is applicable (with modification) to embryonic, adult and aged-adult tissues and in addition to mouse can be used to isolate cells from the skeletal muscles of other species including human (Rao and Smith, unpublished), chick embryo and fish (salmon) 24. Clonally derived SMSc can be analysed in vivo by intramuscular transplantation and under these conditions injected SMSc will combine with host myotubes to form hybrid muscle fibres. Intramuscularly injected SMSc do not form tumours and have been found in host muscles in the satellite cell position more than a year after injection, suggesting that they are subject to endogenous control by the satellite stem cell niche.These cells can be re-isolated from injected hosts as proliferative SMSc more than 12 months after host injection 19.
Disclosures
No conflicts of interest declared.
Acknowledgments
We thank Patrick Paddison for his gift of the shRNAi shuttle vector. Angela Sloan generated the GFP RNAi image in Figure 3. We also thank the following funding bodies for their support: Muscular Dystrophy Campaign grant number RA2/592/2; SPARKS grant number 02BHM04, The Royal Society grant number 574006.G503/1948./JE and BBSRC grant number 6/SAG10077.
References
- Yaffe D, Saxel O. Serial passaging and differentiation of myogenic cells isolated from dystrophic mouse muscle. Nature. 1997;270:725–727. doi: 10.1038/270725a0. [DOI] [PubMed] [Google Scholar]
- Askanas V, Engel WK. A new program for investigating adult human skeletal muscle grown aneurally in tissue culture. Neurology. 1975;25:58–67. doi: 10.1212/wnl.25.1.58. [DOI] [PubMed] [Google Scholar]
- Smith J, Schofield PN. The effects of fibroblast growth factors in long term primary culture of dystrophic (mdx) mouse muscle myoblasts. Exp Cell Res. 1994;210:86–93. doi: 10.1006/excr.1994.1013. [DOI] [PubMed] [Google Scholar]
- Fell H. The cell in culture. J Clin Pathol. 1958;11:489–495. doi: 10.1136/jcp.11.6.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart DC, Kirk PL. The simultaneous measurement of several parameters of embryo heart explants growth in vitro. J Cell Physiol. 1952;40:183–198. doi: 10.1002/jcp.1030400203. [DOI] [PubMed] [Google Scholar]
- Harvey AL, Robertson JG, Witkowski JA. Maturation of human skeletal muscle fibres in explant tissue culture. J Neurol Sci. 1979;41:115–122. doi: 10.1016/0022-510x(79)90145-x. [DOI] [PubMed] [Google Scholar]
- Zammit PS, Heslop L, Hudon V, Rosenblatt JD, Tajbakhsh S, Buckingham ME, Beauchamp JR, Partridge TA. Kinetics of myoblast proliferation show that resident satellite cells are competent to fully regenerate skeletal muscle fibers. Exp Cell Res. 2002;281:39–49. doi: 10.1006/excr.2002.5653. [DOI] [PubMed] [Google Scholar]
- Konigsberg I. The embryonic origin of muscle. New York: McGraw-Hill; 1986. [Google Scholar]
- Trainor PA, Tan SS, Tam PP. Cranial paraxial mesoderm: regionalization of cell fate and impact on craniofacial development in mouse embryos. Development. 1994;120:2397–2408. doi: 10.1242/dev.120.9.2397. [DOI] [PubMed] [Google Scholar]
- Tajbakhsh S, Rocancourt D, Cossu G, Buckingham M. Redefining the genetic hierarchies controlling skeletal myogenesis Pax-3 and Myf-5 act upstream of MyoD. Cell. 1997;89:127–138. doi: 10.1016/s0092-8674(00)80189-0. [DOI] [PubMed] [Google Scholar]
- Tremblay P, Dietrich S, Mericskay M, Schubert FR, Li Z, Paulin D. A crucial role for Pax3 in the development of the hypaxial musculature and the longrange migration of muscle precursors. Dev Biol. 1998;203:49–61. doi: 10.1006/dbio.1998.9041. [DOI] [PubMed] [Google Scholar]
- Tajbakhsh S, Bober E, Babinet C, Pournin S, Arnold H, Buckingham M. Gene targeting the myf-5 locus with nlacZ reveals expression of this myogenic factor in mature skeletal muscle fibres as well as early embryonic muscle. Dev Dyn. 1996;206:291–300. doi: 10.1002/(SICI)1097-0177(199607)206:3<291::AID-AJA6>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Tam PP. A study of the pattern of prospective somites in the presomitic mesoderm of mouse embryos. J Embryol Exp Morphol. 1986;92:269–285. [PubMed] [Google Scholar]
- Cossu G, Kelly R, Donna SDi, Vivarelli E, Buckingham M. Myoblast differentiation during mammalian somitogenesis is dependent upon a community effect. Proc Natl Acad Sci USA. 1995;92:2254–2258. doi: 10.1073/pnas.92.6.2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shainberg A, Yagil G, Yaffe D. Control of myogenesis in vitro by Ca2+ concentration in nutritional medium. Exp Cell Res. 1969;58:163–167. doi: 10.1016/0014-4827(69)90127-x. [DOI] [PubMed] [Google Scholar]
- Dodson MV, Martin EL, Brannon MA, Mathison BA, McFarland DC. Optimization of bovine satellite cell-derived myotube formation in vitro. Tissue Cell. 1987;19:159–166. doi: 10.1016/0040-8166(87)90001-2. [DOI] [PubMed] [Google Scholar]
- Smith J. Muscle Growth factors, ubiquitin and apoptosis in dystrophic muscle: Apoptosis declines with age in the mdx mouse. B Appl Myol. 1996;6:279–284. [Google Scholar]
- Smith J, Fowke G, Schofield P. Programmed cell death in dystrophic (mdx) muscle is inhibited by IGF-II. Cell Death Differ. 1995;2:243–251. [PubMed] [Google Scholar]
- Smith J, Schofield PN. Stable integration of an mdx skeletal muscle cell line into dystrophic (mdx) skeletal muscle: evidence for stem cell status. Cell Growth Differ. 1997;8:927–934. [PubMed] [Google Scholar]
- O Shea L, Johnson C, Rooney M, Gleeson R, Woods K, Smith J. Adipogenesis and skeletal muscle ageing. Mech Ageing Dev. 2001;122:1354–1355. [Google Scholar]
- Merrick D. A role for Igf-2 in fast skeletal muscle specification during myogenesis in dystrophic and wild type embryos [dissertation] Birmingham: University of Birmingham; 2006. [Google Scholar]
- Stadler LKJ, Merrick D, Smith J. Morphological stem cell and fast myosin abnormalities in the cav-3 / and mdx dystrophic embryo reveal an embryonic basis for muscular dystrophy. Abstr. Genet.Res. 2008;90:281–289. [Google Scholar]
- Merrick D, Stadler LKJ, Larner DP, Smith J. Morphological stem cell and fast myosin abnormalities in the cav-3 / and mdx dystrophic embryo reveal an embryonic basis for muscular dystrophy. Dis Model Mech. 2009;2:374–388. doi: 10.1242/dmm.001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matschak T, Stickland NC, Smith J. Explants of embryonic Atlantic salmon muscle in culture. Proc Soc Exp Biol A. 1997;1:23–23. [Google Scholar]
- Smith J, Hooper ML. Dominance and independent segregation of metabolic cooperation competence and pluripotency in an embryonal carcinoma cell hybrid. Exp Cell Res. 1989;181:40–50. doi: 10.1016/0014-4827(89)90180-8. [DOI] [PubMed] [Google Scholar]
- Bulfield G, Siller WG, Wight PA, Moore KJ. X chromosome linked muscular dystrophy (mdx) in the mouse. Proc Natl Acad Sci USA. 1984;81:1189–1192. doi: 10.1073/pnas.81.4.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagiwara Y, Sasaoka T, Araishi K, Imamura M, Yorifuji H, Nonaka I, Ozawa E, Kikuchi T. Caveolin-3 deficiency causes muscle degeneration in mice. Hum Mol Genet. 2000;9:3047–3054. doi: 10.1093/hmg/9.20.3047. [DOI] [PubMed] [Google Scholar]
- Westbury J, Watkins M, Ferguson-Smith AC, Smith J. Dynamic temporal and spatial regulation of the cdk inhibitor p57 (kip2) during embryo morphogenesis. Mech Dev. 2001;109:83–89. doi: 10.1016/s0925-4773(01)00512-3. [DOI] [PubMed] [Google Scholar]
- Merrick D, Ting T, Stadler LKJ, Smith J. A role for Insulin-like growth factor 2 in specification of the fast skeletal muscle fibre. BMC Dev Biol. 2007;7:65–65. doi: 10.1186/1471-213X-7-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paddison PJ, Caudy AA, Hannon GJ. Stable suppression of gene expression by RNAi in mammalian cells AQ4. Proc Natl Acad Sci USA. 2002;99:1443–1448. doi: 10.1073/pnas.032652399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J, Merrick D. Skeletal Muscle Microexplant Culture and Isolation of Skeletal Muscle Stem Cells. Methods in Molecular Biology. 2010;633 doi: 10.1007/978-1-59745-019-5_3. [DOI] [PubMed] [Google Scholar]