

Abstract
Perinatal ischemia is a common clinical problem with few successful therapies to prevent neuronal damage. Delta-opioid receptor (DOR) activation is a versatile, evolutionarily-conserved, endogenous neuroprotective mechanism that blocks several steps in the deleterious cascade of neurological events during ischemia. DOR activation prior to ischemia or severe hypoxia is neuroprotective in spinal motor networks, as well as cortical, cerebellar, and hippocampal neural networks. In addition to providing acute and long-lasting neuroprotection against ischemia, DOR activation appears to provide neuroprotection when given before, during, or following the onset of ischemia. Finally, DORs can be upregulated by several physiological and experimental perturbations. Potential adverse side effects affecting motor control, such as respiratory depression and seizures, are not well established in young mammals and may be mitigated by altering drug choice and method of drug administration. The unique features of DOR-dependent neuroprotection make it an attractive potential therapy that may be given to at-risk pregnant mothers shortly before delivery to provide long-lasting neuroprotection against unpredictable perinatal ischemic events.
Keywords: perinatal, ischemia, opioid, neuroprotection
Perinatal ischemia is a general term associated with the loss of blood flow to the CNS during the perinatal period; defined as the 20th week of gestation through the 28th postnatal day in humans.1 Other diseases that occur in the perinatal period include hypoxic-ischemic encephalopathy (oxygen deficiency in the whole brain), perinatal asphyxia (lack of oxygen to the fetus during labor and delivery), and perinatal stroke (focal disruption of cerebral blood flow due to arterial or venous thrombosis). The net result of these pathological conditions is lack of oxygen (and glucose in some cases) to the brain and spinal cord, which initiates a cascade of events leading to neuronal damage and cell death. Very few successful treatments exist for perinatal ischemia in humans despite the long list of successful neuroprotective drugs and treatments in animal preparations. This problem has raised the question as to whether neuroprotection in a clinical setting is possible. Thus, there is a compelling need for new strategies and ideas for treating perinatal ischemia (and related diseases). This review suggests that several factors favor the development of successful treatments for perinatal ischemia, and that DOR activation may be a valuable mechanism for providing long-lasting neuroprotection during ischemia or hypoxia in the CNS and especially motor networks.
Perinatal Ischemia in the Clinic
Perinatal ischemia is caused by a wide variety of clinical conditions, such as thrombosis2, perinatal ischemic stroke,2 acute fetal circulatory collapse,3 placental insufficiency,4 forceps application,5 dysfunctional labor,5 inappropriate use of maternal drugs causing pharmacologically-induced fetal depression,5 birth asphyxia,6 and respiratory or cardiac failure.4 Perinatal stroke or birth asphyxia occur at rates of 1 per 2300–5000 and 9.4 per 1,000 live births, respectively7,8,9 while hypoxic-ischemic encephalopathy occurs at a rate of 1.4 per 1,000 live births.9 Perinatal ischemia can lead to life-long conditions such as serious motor disabilities, seizure disorders, cerebral palsy, and respiratory difficulties.10,4,11,2 In one study of 46 neonates with hypoxic-ischemic encephalopathy, 44% of surviving children had significantly delayed motor abilities.12 A retrospective study of children with perinatal spinal cord injury suggests that ischemia accounts for ~23% of spinal cord injuries, but that spinal cord injury in the perinatal period is likely an under-diagnosed clinical condition.13 Thus, perinatal ischemia can produce damage to central motor networks as well as cortical networks involved in higher cognitive functions. Hypothermia is the only treatment routinely used in term infants with moderate to severe encephalopathy to decrease excitatory neurotransmitter release, free radical production, edema, neutrophil infiltration and cytokine release.14 Hypothermic protection, however, depends on body size (smaller patients are more susceptible to over-cooling) and cooling is tissue-dependent (cortex needs more cooling than deep gray matter).15 Tailoring cooling treatment to each patient is a challenge and can only be used postnatally. Thus, no treatment for perinatal ischemia exists that is effective, standard, and readily administered throughout the entire perinatal period.
Ischemia-Induced Cascade
Reduced blood flow to the brain impairs delivery of oxygen and glucose, which then reduces ATP availability, and initiates a cascade of events (Fig. 1). Energy depletion results in dysfunctional ATP-dependent ion channels and ion exchangers causing cellular depolarization and excessive excitatory neurotransmitter release--extracellular glutamate concentration can increase 3–10 fold during ischemia.16 Excitotoxic injury is further compounded since energy-dependent glutamate re-uptake is compromised. Postsynaptic glutamate receptors differentially mediate ischemic injury in the perinatal brain.16 Cortical injury is mediated mostly by NMDA receptors whereas brainstem injury is mainly AMPA receptor-dependent.17 Activation of postsynaptic glutamate receptors produces a transmembrane flux of sodium and calcium cations, which contributes to depolarization and neuronal excitation.16 Water passively follows sodium and calcium ion influx and contributes to brain swelling. Along with glutamatergic excitotoxicity and calcium influx, there is free radical attack and prolonged seizure activity, which causes further neuronal damage.18 High intracellular calcium levels activate numerous signaling cascades that cause further tissue damage. Following excitotoxic injury and loss of synaptic connectivity, apoptosis or programmed cell death is initiated.16 Inflammation and apoptosis increase over hours to days after the initial ischemic event, and neurotrophic factors are down-regulated.18, 19
Figure 1.
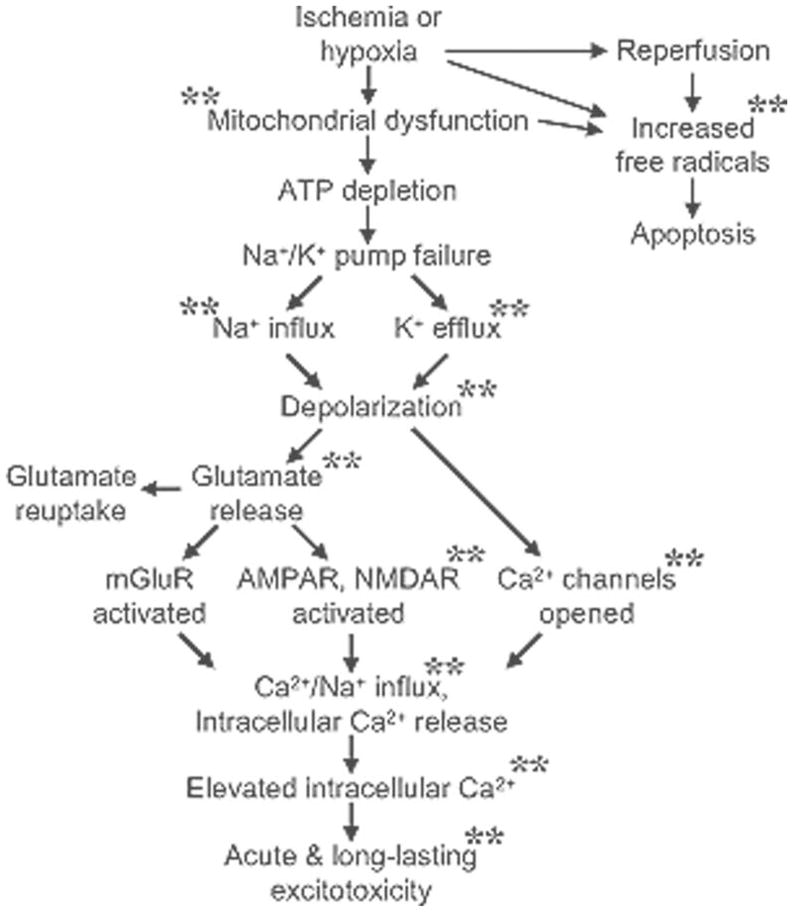
Ischemia or severe hypoxia reduces ATP production and initiates a series of deleterious cascade of events that lead to neuronal damage or death. Experimental evidence shows that DOR activation has to the capacity to attenuate or block several steps in the cascade.** Indicates a step that disrupted by DOR activation.
Neuroprotection Against Perinatal Ischemia
The ischemic cascade in the brain is well understood based on data from several mammalian models.20 However, translating animal model results to clinical practice has proven to be highly problematic.21 In animal studies, neuroprotective drugs are typically given in healthy rats shortly after (or prior to) administering the ischemic insult (usually blood vessel occlusion) that results in a reproducible ischemic lesion. In contrast for human clinical trials, neuroprotective drugs are given at various times following strokes that produced highly variable brain lesions in aged humans who often have significant co-morbidity. Also, brain reperfusion is usually well controlled in animal studies, whereas reperfusion in humans is left to chance (except for studies testing thrombolytic drugs). Likewise, it’s difficult to attain therapeutic levels of neuroprotective agents within the poorly perfused brain tissue. Animal studies often use infarct volume as an outcome measure whereas human clinical trials focus on functional outcomes.22 Finally, in studies of non-adult mammals there are many difficulties translating the developmental period of the animal to human development. A detailed analysis of neurodevelopmental events suggests that a P14 rat corresponds most closely to a G211 human fetus, which represents the early third trimester.23
Accordingly, expectations and goals for neuroprotection research need to be adjusted to reflect these realities. Although ischemic events during the perinatal period are unpredictable, a woman in labor represents a clearly defined time when the mother and fetus are at risk for ischemic events that tend to occur during delivery and early postnatal life. Thus, it may be possible to prophylactically administer a drug combination to women to provide neuroprotection for the fetus before, during, and after parturition. Alternatively, a neuroprotective drug combination could be developed that would be available during an otherwise healthy delivery for use at the first sign of an ischemic event. The ideal neuroprotective agent against perinatal ischemia should be easy to administer, rapidly absorbed, and able to cross the placental and fetal blood-brain barriers with minimal or no adverse side effects. The ideal agent should also activate endogenous neuroprotective mechanisms, disrupt the ischemic cascade at multiple points, and provide long-lasting (>24 h) protection no matter if given before, during, or after an unexpected perinatal ischemic event.
DOR-Dependent Neuroprotection
DOR activation is a unique form of neuroprotection because it appears to be a highly conserved, inducible mechanism, which is used by vertebrate extremophiles (e.g., mammalian hibernators and hypoxia resistant vertebrates). Also, DOR-dependent protection is observed in various tissues other than brain, which makes it an attractive candidate for providing systemic protection during whole-body ischemia or hypoxia. The key features of DOR-dependent neuroprotection are discussed below:
DOR-dependent neuroprotection in extremophile vertebrates
Hibernating animals exemplify natural tolerance to oxygen-, blood-, or energy- deprivation.24,25 During hibernation, blood flow to the brain is severely reduced but central neurons remain viable24 and cardiorespiratory function is still regulated during torpor.26 Hibernation-induced neuroprotection is not simply due to colder brain temperatures, but appears to be due to increased resistance to ischemic conditions.26,27 In summer active ground squirrels, intravenous infusion of DADLE (DOR agonist) induces hibernation.28 Similarly, injections of Deltorphin-Dvariant (DOR agonist) as well as hibernating woodchuck plasma into mice induced neuroprotection prior to undergoing focal ischemia.29 Likewise, hypoxia-resistant red-eared slider turtles can hold their breath for up to 48 h.30 This ability is hypothesized, in part, to be due to endogenous DOR activation because hypoxia-resistant red eared slider turtles have greater DOR expression in the CNS compared to rats31 and endogenous DOR activation protects against NMDA-dependent excitotoxicity in anoxic turtle cortical slices.32
DOR drugs disrupt several steps in the acute ischemic cascade
The goal of influencing multiple complex signaling pathways simultaneously over different time frames is unlikely to be achieved by a single drug. Thus, some argue for the introduction of pleiotropic drugs (i.e., single drugs that produce multiple effects) for the treatment of ischemia-reperfusion damage.33 DOR agonist drugs are pleiotropic because they disrupt several steps in the acute phases of the ischemic cascade (see asterisks in Fig. 1) via different mechanisms (Table 1). Although some mechanistic features may be tissue-specific and species-specific, Table 1 illustrates DOR agonists’ capacity to attenuate multiple deleterious ischemic events. During clinical perinatal ischemic events, DOR agonists will likely need to be used in combination with other drugs that complement DOR-activation’s beneficial effects.
Table 1.
Potential mechanisms underlying DOR-dependent neuroprotection.
| Ischemic Cascade Event | DOR-Dependent Action | Key Features | References |
|---|---|---|---|
| Mitochondrial Depolarization | KATP channel activation | Protects cultured cortical neurons against NaN3;reduces NaN3-induced downregulation of DOR. | 64 |
| Na+ influx | Decreases Na+ influx | Blocks voltage-gated Na+ channels and reduces Na+ influx via NMDA channels in cortical slices exposed to anoxia. | 65,66 |
| K+ Efflux | Decreases K+ efflux | Attenuates K+ efflux from cortical slices exposed to anoxia or OGD via PKC-dependent, PKA-independent pathway. | 67,68 |
| Inhibition of Ca2+ influx reduces activation of Ca2+-activated K+ (BK) channels in cortical slices exposed to anoxia. | 68 | ||
| Ca2+ influx | Decreases Ca2+ influx | Indirect evidence of reduced Ca2+ influx cortical slices exposed to anoxia. | 68 |
| Hypoxia-induced Ca2+ influx reduced in adrenal medulla cells by decreasing voltage-gated Ca2+ currents. | 69 | ||
| Increased Glutamate Release | Inhibits glutamate release presynaptically | Decreased amplitude of AMPA EPSCs/EPSPs in lamina II of lumbar spinal cord slices without altering responses to pressure-ejected AMPA. | 70 |
| Decreased frequency, but not amplitude, of mEPSCS in amygdala slices. | 71 | ||
| Increased AMPA and NMDA Receptor Activation | Decreases NMDA-dependent currents | Reduces Na+ influx via NMDA channels in cortical slices exposed to anoxia. | 66 |
| Reduces NMDA currents during anoxia in turtle cortical slices. | 32 | ||
| Increased Free Radical Production | Decreases free radical release or impact | Hypoxic preconditioning attenuates decrease in antioxidant scavengers and increase in oxidant proteins via DOR-dependent mechanism in retinal cells. | 39 |
| DOR agonist drug acts as free radical scavenger. | 72 | ||
| DOR agonist and plasma from hibernating woodchuck reduces NO release in microglia cell culture via DOR mechanism | 29 |
Role of DOR in neuroprotective preconditioning treatments
Animals or tissues that are exposed to sublethal ischemic challenges become resistant to subsequent severe ischemic exposures due to a process referred to as preconditioning. Ischemic preconditioning in the brain represents a form of endogenous neuroprotection that can be triggered by numerous factors and involves several signaling pathways.34,35 Although DOR activation is required for the expression of ischemic preconditioning in the heart,36 there are currently no examples of DOR activation involvement in neuronal ischemic preconditioning. On the other hand, DORs are involved in hypoxic preconditioning where animal or tissue hypoxia is used to induce neuroprotection against lethal challenges. For example, hypoxia (1% O2 for 30 min) applied 30 min before glutamate-induced excitotoxicity attenuates neuronal damage in cultured cortical neurons, an effect that is blocked by prior application of a DOR antagonist.37 Also, hypoxic preconditioning (5% O2 for 6 h) in cultured cortical neurons increased DOR expression and reduced damage to sustained hypoxic exposures.38 Finally, hypoxia-induced preconditioning in rat retinal cells requires the increased expression and activation of DORs for neuroprotection against later ischemia.39 Thus, hypoxic preconditioning in neurons induces a DOR-dependent neuroprotection.
DOR activation provides long-lasting neuroprotection
In addition to protecting against acute excitotoxicity during ischemia, DOR activation attenuates signaling pathways that continue for hours to days after the initial ischemic event. For example, Tan-67 (DOR agonist) administration 24 h prior to OGD solution application reduces cell death in organotypic hippocampal cell cultures.40 Similarly, Tan67 administration 24 h prior to right middle cerebral artery occlusion reduces infarct size and improves functional outcome.40 Thus, DOR activation can induce neuroprotection lasting for at least one day.
DORs are expressed in developing mammals
DOR receptors are expressed in neonatal rat spinal cord41 and DOR-dependent neuroprotection is demonstrated in neonatal rat spinal cord (see below). Binding affinities for DOR in the rat brain or spinal cord are constant or increase from the first postnatal day42,41,43 and DOR expression is postulated to increase 40 fold between neonates and adults.44 The location of DORs in the neonatal rat spinal cord is not known, but DOR immunoreactivity is located in the ventral horn of adult rat spinal cords.45 Thus, the substrate for DOR-dependent neuroprotection is present in the neonatal spinal cord, and may be located both pre- and post-synaptically in the ventral horn throughout development. Future studies need to determine whether DORs are expressed in the perinatal human CNS before testing whether DOR activation induces neuroprotection.
DOR agonist drugs cross the blood-brain and placental barriers
Any DOR agonist that proves to be neuroprotective has to reach its target tissues in the brain and spinal cord. For peptidergic DOR agonists, the placental and fetal blood-brain barriers represent significant obstacles to cross before activating central DORs. However, the peptidergic DOR agonist DPDPE, crosses the blood-brain barrier in rats via a carrier-mediated mechanism,46 and DPDPE crosses the placental barrier via a paracellular route based on studies in cultured cells.47 Alternatively, non-peptidergic synthetic DOR agonists, such as SNC80, Tan67 and BW373U86, easily cross these barriers and have increased systemic distribution. Thus, lipophilic DOR agonists administered to pregnant mothers or newborn infants will likely reach their target tissues in the CNS.
DOR-Dependent Neuroprotection in Motor Networks
Since cortical and hippocampal tissues are highly sensitive to ischemia, most information on DOR-dependent neuroprotection is derived from studies on these tissues. With respect to motor networks, a robust literature on spinal cord ischemia in adults exists due to the problem of spinal cord ischemia occurring during surgical aortic aneurysm repair. In addition to several studies on ischemic preconditioning in spinal cord, DOR activation is neuroprotective in adult spinal cord. Intrathecal SNC80 (DOR agonist; 40 mM) protects against spinal cord ischemia administered 9 min later in adult rat lumbar spinal cord.48 Forty-eight hours afterwards, hind limb motor function is improved and significantly more neurons are uninjured compared to sham rats.48 Although DOR activation is neuroprotective in mature spinal cords, it is important to understand how ischemia alters motor network function in younger mammals since perinatal ischemia causes significant morbidity with respect to motor function.
In isolated in vitro neonatal rat (P4–P6) preparations, electrically-evoked responses in spinal motoneurons are rapidly depressed and abolished within 30 min when exposed to ischemia-like solutions that lack oxygen and glucose (OGD solutions).49 The potential role of DOR activation in providing neuroprotection in the neonatal spinal cord during exposure to OGD solutions is not known. To address this question in our laboratory, the neuroprotective effects of DOR activation on spinal respiratory motor circuits were studied in neonatal rat brainstem-spinal cord preparations (S.M. Freiberg and S.M. Johnson, unpublished observations). Instead of electrically evoking spinal motoneurons responses, we examined the effects of spinal OGD solutions on spontaneously-produced, quantifiable respiratory motor output on cervical and thoracic spinal ventral roots. We tested whether cervical and thoracic respiratory motor output are equally sensitive to OGD, and whether neuroprotection is provided in the following conditions: 1) sustained spinal DOR activation prior to and during spinal OGD, 2) brief spinal DOR activation several minutes prior to spinal OGD (i.e., a form of neuroplasticity), and 3) spinal DOR activation following the onset of OGD exposure. Preliminary data show that cervical and thoracic motoneurons pools are equally sensitive to OGD exposure, and that DOR activation before, during, and after OGD onset prolongs spontaneous respiratory motor output. Thus, DOR-dependent neuroprotection induced in neonatal rat spinal cord protects motor networks controlling breathing.
Potential Side Effects of DOR Activation
There are reports of DOR activation being associated with respiratory depression and seizures, but there is a wide range of conflicting data in the literature that is likely due to differences in species, drug specificity, dosage, method (bolus vs. infusion) and route (intravenous vs. intracerebroventricular) of drug administration, and animal state (awake vs. anesthetized). There is no clear consensus that most DOR agonist drugs cause respiratory depression and seizures in either the mother or offspring during the perinatal period. Even if mild side effects are present following DOR activation, the potential benefits of DOR-dependent neuroprotection during perinatal ischemia are likely to outweigh the consequences. Also, there may be several ways to overcome negative side effects, such as administering the safest and most effective DOR agonists over time in low dosages, or by combining the DOR agonist with other drugs that attenuate or block the side effects.
For respiratory depression in adult mammals, many drugs used in earlier experiments lacked specificity for DOR and crossed over to activate mu-opioid receptors. Despite the development of DOR selective drugs, there is still a large amount of variation in respiratory responses to DOR activation. For example, intracerebroventricular injection of DPDPE does not alter respiration rate or blood gases in adult rabbits50 and decreases arterial pH by only 0.05 pH units in adult rats.51 Also, BW373U86 (DOR agonist) has no effect on pCO2 levels in awake rats.52 In pregnant ewes, an intravenous bolus (0.3 mg/kg) of DPDPE (0.3 mg/kg), deltorphin I (DOR agonist), or SNC80 causes respiratory depression in the ewes that lasts for only 15 min and resolves within 30–60 min.53,54 Thus, the bulk of the evidence suggests that DOR activation causes only transient mild respiratory depression in adult mammals.
With respect to younger mammals, studies performed on neonatal rat and fetal sheep have strikingly different results. DPDPE injections (0.1 mg/kg, intraperitoneally) do not cause respiratory depression in P1 neonatal rats, but older pups (P17) have decreased breath amplitude and frequency.55 Likewise, bath-applied DPDPE does not alter spinal respiratory motor output or bulbospinal respiratory neuronal discharge in neonatal rat (P0–P4) brainstem-spinal cord preparations.55,56,57 In contrast, intracerebroventricular infusion of DPDPE or [D-Ala2]deltorphin I (DOR agonist) in third-trimester fetal lambs increases breath frequency in a dose-dependent manner.58,59,60 Thus, DOR activation appears to stimulate breathing movements in utero, have little or no effect on breathing in newborns, and cause mild respiratory depression during maturation in juveniles. Testing this hypothesis in several mammalian species, including primates, will be an important step towards understanding how DOR activation alters breathing during early development.
A second adverse side effect attributed to DOR activation is increased neuronal activity, which can lead to seizures or spasticity. In fetal lambs, intracerebroventricular administration of DPDPE stimulates EEG activity, but there were reports of seizures.58,59,61 In adult rhesus monkeys, SNC80 administration (10 mg/kg, intramuscular) at doses 3–10 fold greater than that required to produce behavioral effects produced convulsions and EEG seizures in only one of four monkeys.62 A repeat challenge at the same dosage in the same monkey one year later had no effect. The authors concluded that the 10 mg/kg dosage of SNC80 was, at worst, a threshold dosage for inducing seizures in rhesus monkeys. The only other negative study showed that intrathecal administration of DPDPE in adult rats 30 min after spinal ischemia caused increased spasticity during the 30–60 min period following spinal ischemia.63 Although DOR activation increases cortical and spinal activity, the drug dosage that causes significant seizure activity is likely to depend on species, drug, dosage, and rate/route of drug administration.
Future Directions
DOR-dependent neuroprotection holds promise for attenuating ischemic injury to the CNS throughout the perinatal period. Considerable work, however, is required to adequately test this hypothesis and produce meaningful results. One of the most important problems to consider is the choice of animal model and how the ischemic event is experimentally performed. Rodent models are valuable because they are inexpensive and readily available for high throughput studies. Progression to other species, such as sheep and non-human primates, should proceed only after careful assessment of the rodent data. For example, it is too laborious and time-consuming to run dose-response curves for several DOR agonist drugs in fetal lambs. The age of the rodent used in high throughput studies, however, is highly critical since newborn rats are not equivalent to newborn humans. The age of the rodents should be determined after careful consideration of the experimental question and the region of interest to be studied (e.g., cortex vs. spinal cord) since different parts of the CNS mature at different rates. The model of ischemia is also important depending on the experimental question: artery clamping or occlusion mimics stroke, clamping the umbilical artery mimics placental and umbilical cord pathologies, systemic hypoxia can mimic cardiac shunt and asphyxia.
As discussed above, drawing solid conclusions from studies that use a wide range of drugs and drug administration protocols is difficult. The choice of drug (peptide vs. non-peptidergic synthetic), route of administration (intravenous vs. central), dosage, and timing are all critical to producing the most neuroprotection with the least side effects. Timing is particularly important since clinical conditions that cause perinatal ischemia are highly unpredictable. DOR agonists appear to provide substantial protection whether they are administered before or after the ischemic event, and likely have the ability to induce long-lasting neuroprotection (i.e., neuroplasticity). Therefore, DOR agonists might be administered in low dosages (to minimize side effects) intermittently to elicit long-lasting neuroprotection.
Assessment of neuroprotection needs to shift from morphological to functional experimental approaches. A large portion of the DOR-dependent neuroprotection literature to date has measured the number of live vs. dead cells, or the amount of lactate dehydrogenase released by damaged cells. More recent studies are quantifying behavior, motor function, ion shifts, electrophysiological responses, breathing, and EEG to better understand DOR-dependent effects on intact network function and system physiology.
Finally, a unique promise of the DOR system is the capacity to physiologically up-regulate DOR expression and provide greater neuroprotection. Various physiological mechanisms to induce greater DOR expression should be explored and tested. For example, a single bout or several short bouts of mild hypoxia may greatly up-regulate DOR expression within hours and last for several days, and thereby allow lower dosages of DOR agonist drugs to be effective. Ideally, such maneuvers may also enhance endogenous DOR-dependent neuroprotection, an area well explored for cardiac muscle, but only recently tested in central neural networks.
Acknowledgments
This work was supported by the National Institute of Neurological Disorders and Stroke (NS-051580)
References
- 1.Raju TN, Nelson KB, Ferriero D, et al. NICHD-NINDS Perinatal stroke workshop participants: Ischemic perinatal stroke: summary of a workshop sponsored by the National Institute of Neurological Disorders and Stroke. Pediatrics. 2007;120:609–616. doi: 10.1542/peds.2007-0336. [DOI] [PubMed] [Google Scholar]
- 2.Nelson KB. Perinatal Ischemic Stroke. Stroke. 2007;38:742–745. doi: 10.1161/01.STR.0000247921.97794.5e. [DOI] [PubMed] [Google Scholar]
- 3.Rennie JM, Hagmann CF, Robertson NJ. Outcome after intrapartum hypoxic-ischemia at term. Semin in Fetal & Neonatal Med. 2007;12:398–407. doi: 10.1016/j.siny.2007.07.006. [DOI] [PubMed] [Google Scholar]
- 4.Badr LK, Purdy I. Brain injury in the infant: The old, the new and the uncertain. J Perinat Neonat Nurs. 2006;20:163–175. doi: 10.1097/00005237-200604000-00011. [DOI] [PubMed] [Google Scholar]
- 5.Volpe JJ. Brain injury in the premature infant--current concepts. Prev Med. 1994;23:638–645. doi: 10.1006/pmed.1994.1106. [DOI] [PubMed] [Google Scholar]
- 6.Whitelaw A, Thoresen M. Clinical trials of treatments after perinatal asphyxia. Curr Opin Pediatr. 2002;14:664–668. doi: 10.1097/00008480-200212000-00002. [DOI] [PubMed] [Google Scholar]
- 7.Legido A, Katsetos CD, Mishra OP, et al. Perinatal hypoxic ischemic encephalopathy: Current and future treatments. Intl Ped. 2000;15:143–151. [Google Scholar]
- 8.Laugesaar R, Kolk A, Tomberg T, et al. Acutely and retrospectively diagnosed perinatal stroke: a population-based study. Stroke. 2007;38:2234–2240. doi: 10.1161/STROKEAHA.107.483743. [DOI] [PubMed] [Google Scholar]
- 9.Palsdottir K, Dagbjartsson A, Thorkelsson T, et al. Birth Asphyxia and hypoxic ischemic encephalopathy, incidence and obstertric risk. Laeknabladid. 2007;93:595–601. [PubMed] [Google Scholar]
- 10.Volpe JJ. Perinatal brain injury: from pathogenesis to neuroprotection. Ment Retard Dev Disabil Res Rev. 2001;7:56–64. doi: 10.1002/1098-2779(200102)7:1<56::AID-MRDD1008>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 11.Sotero de Menezes M, Shaw DWW. Hypoxic-ischemic brain injury in the newborn. eMedicine. 2006:139. [Google Scholar]
- 12.van Schie PEM, Becher JG, Dallmeijer AJ, et al. Motor outcome at the age of one after perinatal hypoxic-ischemic encephalopathy. Neuropediatrics. 2007;38:71–77. doi: 10.1055/s-2007-984449. [DOI] [PubMed] [Google Scholar]
- 13.Ruggieri M, Smarason A, Pike M. Spinal cord insults in the prenatal, perinatal, and neonatal periods. Dev Med & Child Neurol. 1999;41:311–317. doi: 10.1017/s0012162299000699. [DOI] [PubMed] [Google Scholar]
- 14.Barnette AR, Inder TE. Evaluation and management of stroke in the neonate. Clin Perinatol. 2009;36:125–136. doi: 10.1016/j.clp.2008.09.008. [DOI] [PubMed] [Google Scholar]
- 15.Robertson NJ, Iwata O. Bench to bedside strategies for optimizing neuroprotection following perinatal hypoxia-ischemia in high and low resource settings. Early Hum Dev. 2007;83:801–811. doi: 10.1016/j.earlhumdev.2007.09.015. [DOI] [PubMed] [Google Scholar]
- 16.Sanders RD, Manning HJ, Ma D, et al. Perinatal neuroprotection. Current Anaesthesia & Critical Care. 2007;18:215–224. [Google Scholar]
- 17.Johnston MW. Excitotoxicity in perinatal brain injury. Brain Pathol. 2005;15:234–240. doi: 10.1111/j.1750-3639.2005.tb00526.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hagberg H, Blomgren K, Mallard C. Neuroprotection of the fetal and neonatal brain. In: Levene MI, Cherenak FA, Whittle M, editors. Fetal and neonatal neurology and neurosurgery. 3. London: Churchill Livingstone; 2001. pp. 505–520. [Google Scholar]
- 19.van Bel F, Groenendaal F. Long-term pharmacologic neuroprotection after birth asphyxia: Where do we stand? Neonatology. 2008;94:203–210. doi: 10.1159/000143723. [DOI] [PubMed] [Google Scholar]
- 20.Hoyte L, Kaur J, Buchan AM. Lost in translation: taking neuroprotection from animal models to clinical trials. Exp Neurol. 2004;188:200–204. doi: 10.1016/j.expneurol.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 21.Röther J. Neuroprotection does not work! Stroke. 2008;39:523–524. doi: 10.1161/STROKEAHA.107.494799. [DOI] [PubMed] [Google Scholar]
- 22.Hussain MS, Shuaib A. Research into neuroprotection must continue … But with a different approach. Stroke. 2008;39:521–522. doi: 10.1161/STROKEAHA.107.494781. [DOI] [PubMed] [Google Scholar]
- 23.Clancy B, Kersh B, Hyde J, et al. Web-based method for translating neurodevelopment from laboratory species to humans. Neuroinformatics. 2007;5:79–94. doi: 10.1385/ni:5:1:79. [DOI] [PubMed] [Google Scholar]
- 24.Drew KL, Rice ME, Kuhn TB, et al. Neuroprotective adaptations in hibernation: therapeutic implications for ischemia-reperfusion, traumatic brain injury and neurodegenerative diseases. Free Radic Biol Med. 2001;31:563–573. doi: 10.1016/s0891-5849(01)00628-1. [DOI] [PubMed] [Google Scholar]
- 25.Borlongan CV, Wang Y, Su TP. Delta opioid peptide [D-Ala2 D-Leu5] enkephalin: linking hibernation and neuroprotection. Frontiers in Biosci. 2004;9:3392–3398. doi: 10.2741/1490. [DOI] [PubMed] [Google Scholar]
- 26.Drew KL, Buck CL, Barnes BM, et al. Central nervous system regulation of mammalian hibernation: implications for metabolic suppression and ischemia tolerance. J Neurochem. 2007;102:1713–1726. doi: 10.1111/j.1471-4159.2007.04675.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bullard RW, David G, Nichols CT. The mechanisms of hypoxic tolerance in hibernating and non-hibernating mammals. Mammilian Hibernation. Bulletin of the Museum of Comparative Zoology at Havard College. 1960;24:321–335. [Google Scholar]
- 28.Oeltgen PR, Nilekani SP, Nuchols PA, et al. Further studies on opioids and hibernation: delta opioid receptor ligand selectively induced hibernation in summer-active ground squirrels. Life Sci. 1988;43:1565–1574. doi: 10.1016/0024-3205(88)90406-7. [DOI] [PubMed] [Google Scholar]
- 29.Govindaswami M, Brown SA, Yu J, et al. Delta 2-specific opioid receptor agonist and hibernating woodchuck plasma fraction provide ischemic neuroprotection. Acad Emerg Med. 2008;15:265–266. doi: 10.1111/j.1553-2712.2008.00048.x. [DOI] [PubMed] [Google Scholar]
- 30.Musacchia X. The viability of Chrysemys picta submerged at various temperatures. Physiol Zool. 1959;32:47–50. [Google Scholar]
- 31.Xia Y, Haddad GG. Major difference in the expression of delta- and mu-opioid receptors between turtle and rat brain. J Comp Neurol. 2001;436:202–210. [PubMed] [Google Scholar]
- 32.Pamenter ME, Buck LT. delta-Opioid receptor antagonism induces NMDA receptor-dependent excitotoxicity in anoxic turtle cortex. J Exp Biol. 2008;211:3512–3517. doi: 10.1242/jeb.021949. [DOI] [PubMed] [Google Scholar]
- 33.Menger MD, Vollmar B. Pathomechanisms of ischemia-reperfusion injury as the basis for novel preventive strategies: is it time for the introduction of pleiotropic compounds? Transplant Proc. 2007;39:485–488. doi: 10.1016/j.transproceed.2007.01.022. [DOI] [PubMed] [Google Scholar]
- 34.Davis DP, Patel PM. Ischemic preconditioning in the brain. Curr Opin Anaesthesiol. 2003;16:447–452. doi: 10.1097/00001503-200310000-00002. [DOI] [PubMed] [Google Scholar]
- 35.Dirnagl U, Simon RP, Hallenbeck JM. Ischemic tolerance and endogenous neuroprotection. Trends Neurosci. 2003;26:248–254. doi: 10.1016/S0166-2236(03)00071-7. [DOI] [PubMed] [Google Scholar]
- 36.Gross GJ. Role of opioids in acute and delayed preconditioning. J Mol Cell Cardiol. 2003;35:709–718. doi: 10.1016/s0022-2828(03)00135-4. [DOI] [PubMed] [Google Scholar]
- 37.Zhang J, Qian H, Zhao P, et al. Rapid hypoxia preconditioning protects cortical neurons from glutamate toxicity through delta-opioid receptor. Stroke. 2006;37:1094–1099. doi: 10.1161/01.STR.0000206444.29930.18. [DOI] [PubMed] [Google Scholar]
- 38.Ma MC, Qian H, Ghassemi F, et al. Oxygen-sensitive {delta}-opioid receptor-regulated survival and death signals: novel insights into neuronal preconditioning and protection. J Biol Chem. 2005;280:16208–16218. doi: 10.1074/jbc.M408055200. [DOI] [PubMed] [Google Scholar]
- 39.Peng PH, Huang HS, Lee YJ, et al. Novel role for the delta-opioid receptor in hypoxic preconditioning in rat retinas. J Neurochem. 2009;108:741–754. doi: 10.1111/j.1471-4159.2008.05807.x. [DOI] [PubMed] [Google Scholar]
- 40.Zhao P, Huang Y, Zuo Z. Opioid preconditioning induces opioid receptor-dependent delayed neuroprotection against ischemia in rats. J Neuropathol Exp Neurol. 2006;65:945–952. doi: 10.1097/01.jnen.0000235123.05677.4b. [DOI] [PubMed] [Google Scholar]
- 41.Attali B, Saya D, Vogel Z. Pre- and postnatal development of opiate receptor subtypes in rat spinal cord. Brain Res Dev Brain Res. 1990;53:97–102. doi: 10.1016/0165-3806(90)90128-l. [DOI] [PubMed] [Google Scholar]
- 42.McDowell J, Kitchen I. Ontogenesis of delta-opioid receptors in rat brain using [3H][D-Pen2, D-Pen5]enkephalin as a binding ligand. Eur J Pharmacol. 1986;128:287–289. doi: 10.1016/0014-2999(86)90780-6. [DOI] [PubMed] [Google Scholar]
- 43.Szucs M, Coscia CJ. Evidence for delta-opioid binding and GTP-regulatory proteins in 5-day-old rat brain membranes. J Neurochem. 1990;54:1419–1425. doi: 10.1111/j.1471-4159.1990.tb01978.x. [DOI] [PubMed] [Google Scholar]
- 44.Milligan G, Streaty RA, Gierschik P, et al. Development of opiate receptors and GTP-binding regulatory proteins in neonatal rat brain. J Bio Chem. 1987;262:8626–8630. [PubMed] [Google Scholar]
- 45.Mailly P, Gastard M, Cupo A. Subcellular distribution of delta-opioid receptors in the rat spinal cord: an approach using a three-dimensional reconstruction of confocal series of immunolabelled neurons. J Neurosci Methods. 1999;87:17–24. doi: 10.1016/s0165-0270(98)00149-6. [DOI] [PubMed] [Google Scholar]
- 46.Williams SA, Abbruscato TJ, Hruby VJ, et al. Passage of a delta-opioid receptor selective enkephalin, [D-penicillamine2,5] enkephalin, across the blood-brain and the blood-cerebrospinal fluid barriers. J Neurochem. 1996;66:1289–99. doi: 10.1046/j.1471-4159.1996.66031289.x. [DOI] [PubMed] [Google Scholar]
- 47.Ampasavate C, Chandorkar GA, Vande Velde DG, et al. Transport and metabolism of opioid peptides across BeWo cells, an in vitro model of the placental barrier. Int J Pharm. 2002;233:85–98. doi: 10.1016/s0378-5173(01)00929-2. [DOI] [PubMed] [Google Scholar]
- 48.Horiuchi T, Kawaguchi M, Sakamoto T, et al. The effects of the delta-opioid agonist SNC80 on hind-limb motor function and neuronal injury after spinal cord ischemia in rats. Anesth Analg. 2004;99:235–240. doi: 10.1213/01.ANE.0000130389.77859.1C. [DOI] [PubMed] [Google Scholar]
- 49.Jha A, Das Gupta S, Deshpande SB. Deprenyl blocks the aglycemia-induced depression of the synaptic transmission but not the ischemia-induced depression in neonatal rat spinal cord in vitro. Neurosci Res. 2003;47:23–29. doi: 10.1016/s0168-0102(03)00159-7. [DOI] [PubMed] [Google Scholar]
- 50.May CN, Dashwood MR, Whitehead CJ, et al. Differential cardiovascular and respiratory responses to central administration of selective opioid agonists in conscious rabbits: correlation with receptor distribution. Br J Pharmacol. 1989;98:903–913. doi: 10.1111/j.1476-5381.1989.tb14620.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kiritsy-Roy JA, Marson L, van Loon GR. Sympathoadrenal, cardiovascular and blood gas responses to highly selective Mu and Delta opioid peptides. J Pharmacol Exp Ther. 1989;251:1096–1103. [PubMed] [Google Scholar]
- 52.Su YF, McNutt R, Chang KJ. Delta-opioid ligand reverse alfentanil-induced respiratory depression not not antinociception. J Pharmacol Exp Ther. 1998;287:815–823. [PubMed] [Google Scholar]
- 53.Clapp JF, Kett A, Olariu N, et al. Cardiovascular and metabolic responses to two receptor-selective opioid agonists in pregnant sheep. Am J Obstet Gynecol. 1997;178:397–401. doi: 10.1016/s0002-9378(98)80032-x. [DOI] [PubMed] [Google Scholar]
- 54.Szeto HH, Soong Y, Wu D, et al. Respiratory depression after intravenous administration of δ-selective opioid peptide analogs. Peptides. 1999;20:101–105. doi: 10.1016/s0196-9781(98)00141-7. [DOI] [PubMed] [Google Scholar]
- 55.Greer JJ, Carter JE, al-Zubaidy Z. Opioid depression of respiration in neonatal rats. J Physiol. 1995;485:845–855. doi: 10.1113/jphysiol.1995.sp020774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Takita K, Herlenius EA, Lindahl SG, et al. Actions of opioids on respiratory activity via activation of brainstem mu-, delta-, and kappa-receptors; an in-vitro study. Brain Res. 1997;778:233–241. doi: 10.1016/s0006-8993(97)01105-0. [DOI] [PubMed] [Google Scholar]
- 57.Takeda S, Eriksson LI, Yamamoto Y, et al. Opioid action on respiratory neuron activity of the isolated respiratory network in newborn rats. Anesthesiology. 2001;95:740–749. doi: 10.1097/00000542-200109000-00029. [DOI] [PubMed] [Google Scholar]
- 58.Cheng PY, Wu DL, Decena J, et al. Opioid-induced stimulation of fetal respiratory activity by [D-Ala2]deltorphin I. Eur J Pharmacol. 1993;230:85–88. doi: 10.1016/0014-2999(93)90413-c. [DOI] [PubMed] [Google Scholar]
- 59.Cheng PY, Wu DL, Soong Y, et al. Role of μ1 and δ-opioid receptors in modulation of fetal EEG and respiratory activity. Am J Physiol. 1993;265:R433–438. doi: 10.1152/ajpregu.1993.265.2.R433. [DOI] [PubMed] [Google Scholar]
- 60.Szeto HH, Cheng PY, Soong Y, et al. Lack of relationship between opioid-induced changes in fetal breathing and plasma glucose levels. Am J Physiol. 1995;269:R702–707. doi: 10.1152/ajpregu.1995.269.3.R702. [DOI] [PubMed] [Google Scholar]
- 61.Szeto HH, Cheng PY, Wu DL, et al. Effects of the delta-opioid agonist, [D-Pen2, D-Pen5]-enkephalin, on fetal lamb EEG. Pharmacol Biochem Behav. 1994;49:795–800. doi: 10.1016/0091-3057(94)90225-9. [DOI] [PubMed] [Google Scholar]
- 62.Danielsson I, Gasior M, Stevenson GW, et al. Electroencephalographic and convulsant effects of the delta opioid agonist SNC80 in rhesus monkeys. Pharmacol Biochem Behav. 2006;85:428–434. doi: 10.1016/j.pbb.2006.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kakinohana M, Nakamura S, Fuchigami T, et al. Mu and delta, but not kappa, opioid agonists induce spastic paraparesis after a short period of spinal cord ischaemia in rats. Br J Anaesth. 2006;96:88–94. doi: 10.1093/bja/aei285. [DOI] [PubMed] [Google Scholar]
- 64.Zhu M, Li MW, Tian XS, et al. Neuroprotective role of delta-opioid receptors against mitochondrial respiratory chain injury. Brain Res. 2009;1252:183–191. doi: 10.1016/j.brainres.2008.11.030. [DOI] [PubMed] [Google Scholar]
- 65.Chao D, Bazzy-Asaad A, Balboni G, et al. Activation of DOR attenuates anoxic K+ derangement via inhibition of Na+ entry in mouse cortex. Cereb Cortex. 2008;18:2217–2227. doi: 10.1093/cercor/bhm247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chao D, Balboni G, Lazarus LH, et al. Na+ mechanism of delta-opioid receptor induced protection from anoxic K+ leakage in the cortex. Cell Mol Life Sci. 2009;66:1105–1115. doi: 10.1007/s00018-009-8759-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chao D, Donnelly DF, Feng Y, et al. Cortical delta-opioid receptors potentiate K+ homeostasis during anoxia and oxygen-glucose deprivation. J Cereb Blood Flow Metab. 2007;27:356–368. doi: 10.1038/sj.jcbfm.9600352. [DOI] [PubMed] [Google Scholar]
- 68.Chao D, Bazzy-Asaad A, Balboni G, et al. delta-, but not mu-, opioid receptor stabilizes K(+) homeostasis by reducing Ca(2+) influx in the cortex during acute hypoxia. J Cell Physiol. 2007;212:60–67. doi: 10.1002/jcp.21000. [DOI] [PubMed] [Google Scholar]
- 69.Keating DJ, Rychkov GY, Adams MB, et al. Opioid receptor stimulation suppresses the adrenal medulla hypoxic response in sheep by actions on Ca(2+) and K(+) channels. J Physiol. 2004;555:489–502. doi: 10.1113/jphysiol.2003.056176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Glaum SR, Miller RJ, Hammond DL. Inhibitory actions of delta 1-, delta 2-, and mu-opioid receptor agonists on excitatory transmission in lamina II neurons of adult rat spinal cord. J Neurosci. 1994;14:4965–4971. doi: 10.1523/JNEUROSCI.14-08-04965.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bie B, Zhu W, Pan ZZ. Rewarding morphine-induced synaptic function of delta-opioid receptors on central glutamate synapses. J Pharmacol Exp Ther. 2009;329:290–296. doi: 10.1124/jpet.108.148908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tsao LI, Ladenheim B, Andrews AM, et al. Delta opioid peptide [D-Ala2,D-leu5]enkephalin blocks the long-term loss of dopamine transporters induced by multiple administrations of methamphetamine: involvement of opioid receptors and reactive oxygen species. J Pharmacol Exp Ther. 1998;287:322–331. [PubMed] [Google Scholar]