

Abstract
The adenosine agonist 2-(4-(2-carboxyethyl)phenylethylamino)-5′-N-ethylcarboxamidoadenosine (CGS21680) was recently reported to be selective for the A2A adenosine receptor subtype, which mediates its hypotensive action. To investigate structurelactivity relationships at a distal site, CGS21680 was derivatized using a functionalized congener approach. The carboxylic group of CGS21680 has been esterified to form a methyl ester, which was then treated with ethylenediamine to produce an amine congener. The amine congener was an intermediate for acylation reactions, in which the reactive acyl species contained a reported group, or the precursor for such. For radioiodination, derivatives of p-hydroxyphenylpropionic, 2-thiophenylacetic, and p-aminophenylacetic acids were prepared. The latter derivative (PAPA-APEC) was iodinated electrophilically using [125I]iodide resulting in a radioligand which was used for studies of competition of binding to striatal A, adenosine receptors in bovine brain. A biotin conjugate and an aryl sulfonate were at least 350-fold selective for A, receptors. For spectroscopic detection, a derivative of the stable free radical tetramethyl-1-piperidinyloxy (TEMPO) was prepared. For irreversible inhibition of receptors, meta- and para-phenylenediisothiocyanate groups were incorporated in the analogs. We have demonstrated that binding at A2A receptors is relatively insensitive to distal structural changes at the 2-position, and we report high affinity molecular probes for receptor characterization by radioactive, spectroscopic and affinity labelling methodology.
INTRODUCTION
Adenosine acts as a modulator of activity in the cardiovascular system, nervous system, immune system, and other physiological systems (Williams, 1987). Adenosine receptors are subdivided into two subclasses, the A1 and A2 receptors, which are in general inhibitory and stimulatory, respectively, towards adenylate cyclase (Londos et al., 1980). A, receptors in brain and adipocytes have been characterized through photoaffinity (Stiles et al., 1985; Stiles et al., 1986; Linden et al., 1988; Stiles et al., 1987), and chemical affinity labelling (Stiles et al., 1988; Jacobson et al., 1989a) and through enzymatic digests (Stiles, 1986), as a glycoprotein of molecular weight 38 000–40 000.
A2A Adenosine receptors mediate the following physiological effects of adenosine: inhibition of platelet aggregation (Lohse et al., 1988), immunosuppression (Polmar et al., 1988), vasodilation (Olsson et al., 1986) and antipsychotic-like action (Bridges et al., 1987). Few selective agonists (Bridges et al., 1988; Bruns et al., 1986) or antagonists (Shamim et al., 1989) for A2A adenosine receptors are known. Recently an adenosine agonist, CGS21680, 2-(4-(2-carboxyethyl)phenylethylamino)-5′-N-ethylcarboxamidoadenosine, 1, was reported to be 170-fold A2A selective in binding assays (Jarvis et al., 1989), and the only adenosine agonist found to be A2A selective in vivo in cardiovascular models (Hutchison et al., 1989). [3H]CGS21680 has been synthesized and utilized as a radioligand displaying 80–90% specific binding at A2A adenosine receptors in rat striatum (Jarvis et al., 1989).
In this study, structural modifications of CGS21680 are made at a distal site, using a functionalized congener approach. By this approach, analogs containing chains terminating structurally in chemical function groups are synthesized. An alkyl diamino spacer chain serves as a general site for derivatization, with retention of high affinity for A2A adenosine receptors.
MATERIALS AND METHODS
CGS21680, 1, (Hutchison et al., 1989), and the methyl ester derivative, 2 (prepared by the action of diazomethane on CGS21680), were synthesized by one of the authors (A.J.H.). 1,3-phenylenediisothiocyanate (Stiles et al., 1988) was prepared as described. New compounds were characterized (and resonances assigned) by 300 MHz proton nuclear magnetic resonance spectroscopy using a Varian XL-300 FT-NMR spectrometer (Varian Associates Inc., Sunnyvale, CA, USA). Unless noted, chemical shifts are expressed as ppm downfield from tetramethylsilane. Synthetic intermediates were characterized by NMR and by chemical ionization mass spectroscopy (CIMS, NH,) using a Finnigan 1015 mass spectrometer modified with Extrel electronics or on a Finnigan 4500 MS (Finnigan Corp., San Jose, CA, USA). Adenosine analogs were characterized additionally by plama desorption mass spectroscopy (Jacobson et al., 1986), and were identified by the presence of positive ion peaks observed at m + 23. C, H, and N analysis was carried out by Atlantic Microlabs (Norcross, GA, USA) (±0.4% acceptable). N6-cyclopentyladenosine, XAC and ADAC were obtained from Research Biochemicals Inc., Natick, MA, USA. The 2-alkylamino-5′-carboxamidoadenosine derivatives were generally soluble in methanol, or mixtures of alcohol and acetonitrile, and were insoluble in ether or pure acetonitrile.
Synthesis of derivatives
Synthesis of 2-(4-(2-(2-aminoethylaminocarbonyl)ethyl)phenylethylamino)-5′-N-ethylcarboxamidoadenosine, 3, APEC
Compound 2 (100 mg, 0.195 mmol) was dissolved in ethylenediamine and heated at 50° C for 12 h. The solution was evaporated under a stream of nitrogen. Methanol and ether were added to give an oily precipitate, which solidified in vacuo. The solid was dissolved in methanol and reprecipitated with ether. The amorphous product (yield 96 mg, 91%) melted at 113–117° C. Characteristic ‘H NMR resonances in d6-DMSO occurred at δ 8.02 (s, IH, aromatic, C-8); 7.75 (m, lH, NHγ to 1° amine); 7.1 1 (m, 4H, phenyl ring); 5.83 (d, lH, J = 6.3 Hz, ribose C,); 4.71, 4.24, and 4.17, (each 1H, ribose); 3.00 (m, 2H, CH, p to I° amine); 2.7 (m, 4H, α to phenyl ring); 2.33 (t, 2H, J = 7Hz, CH, 3 to CO); and 0.96 (t, J = 7 Hz, CH,).
Synthesis of 2-(4-(2-(2-(4-aminophenyl)methylcarbonyl)amino)-ethylaminocarbonyl)ethyl)phenylethylamino)-5′-N~thylcarboxamidoadenosine, 6
Compound 7 (4.0 mg, 4.9 μmol) and 5% palladium on 3 mg charcoal (Engelhard, Edison, NJ, USA) were added to 0.3 mL of an equivolume mixture of methanol, dimethylformamide, and acetic acid. The mixture was hydrogenated at 40 lb/in2 for 6 h. The catalyst was removed by centrifugation. The title compound was isolated in 68% yield as a white solid (2.3 mg). An additional purification, by preparative TLC (silica, chloroform + methanol + acetic acid, 70: 25: 5 v/v) was necessary.
Radioiodination of 2-(4-(2-(2-(4-aminophenyl)methylcarbonylamino) ethylaminocarbnyl)ethyl)phenylethylamino)-5′-~-ethylcarboxamidoadenosine
Ten μL of a solution of compound 6 (0.1 mg/mL) in methanol was placed in a microcentrifuge tube and dried completely under a stream of nitrogen. The residue was dissolved in 30pL of 0.5 M sodium phosphate (pH 7.39, and mixed well with 1.5 mCi of [125I]Na. The reaction was initiated by the addition of 10 μL of aqueous chloramine T (1 mg/mL), and the entire reaction mixture was mixed by pipette aspiration for 4 min. The reaction was terminated by the addition of 10 μL of aqueous sodium metabisulfite (2 mg/ mL).
The product, [125I]PAPA-APEC, was purified by (Milford, MA, USA) HPLC apparatus utilizing a C,, μBondapak column, and a mobile phase consisting of methanol and 20 mM ammonium formate at pH 8.1. A shallow concave gradient pattern (curve No. 8 on Waters model 680 Automated Gradient Controller) was used. The percent methanol was varied from 60% at zero time to 50% after IOmin, with the remainder being ammonium formate solution. The flow rate was 1 .O mL/min, and the UV detector (254 nm) was set on the 0.1 absorbance unit scale. Four major absorbance peaks (Fig. 1) were detected at retention times of approximately 4 (two peaks), 5 (peak A), and 7 (peak B) minutes after injection. Unreacted compound 6 (peak A) and [125I]PAPAAPEC (peak B), were identified by TLC on silica plates, in which the mobile phase consisted of a chloroform + methanol + acetic acid mixture in the ratio 85: 105 v/v/v. The R, values of compound 6 and the product were 0.11 and 0.23, respectively. Peak B was the only radioactive fraction to display specific binding and the appropriate A2A receptor pharmacology. A small radioactive contaminant was seen at approximately 6.5 min, but was distinguished from [125I]PAPA-APEC by a Rf value of 0.36.
Figure 1.

HPLC elution profile for the synthesis of [125I]PAPA-APEC. Peaks A and B correspond to compound 6, and [125I]PAPA-APEC, respectively.
Synthesis of 2-(4-(2-(2-((4-(benzyloxycarbonylamino)phenyl)-methylcarbonylamino)ethylaminocarbonyl)ethyl)-. phenylethyIamino)-5′-N-ethylcarboxamidoadenosine, 7
2-(4-(2-Carboxyethyl)phenyl)ethylamino-5′-N-ethylcarboxamidoadenosine sodium salt (CGS2168OC), 7.2 mg, 13 μmol) was suspended in 0.5 mL of dimethylformamide and treated with 1-hydroxybenzotriazole (20 mg) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (13 mg, 68 μmol). The mixture was stirred for several min and then treated with 2-((4-(benzyloxycarbonylamino) phenylacetylamino)ethylamine (compound 18, 5.5mg, 17 μmol). After 24 h, 4 mL of water was added and a precipitate was separated by centrifugation, washed with water, and dried at 50° C in vacuo. The title compound was obtained in 65% yield (7.0mg), and was shown to be homogeneous by TLC (R, = 0.18 on silica, chloroform +methanol + acetic acid, 85:10:5 v/v). The 300MHz ‘H NMR spectrum was consistent with the assigned structure.
Representative acylation of APEC (used to synthesize compounds 4, 5, 9, 10–13, and 15)
APEC (3, 10 μmol) was dissolved in 0.5 mL of dimethylformamide or in a 1: 1 v/v mixture of isopropanol+ acetonitrile. To form amides, an active ester (20 μmol) was added. To form thioureas (compounds 11–13, and 15), the appropriate isothiocyanate derivative (20 μmol, or 50 μmol if bifunctional), was added. The reaction was followed by TLC, and generally was complete within several min. The solvent was evaporated under a stream of nitrogen. Acetonitrile and ether were added causing the product to precipitate. The product was recrystallized sequentially from DMF + acetonitrile+ ether and from methanol + ether.
Synthesis of N-benzyloxycarbonyl derivatives
Methyl 4-aminophenylacetate hydrochloride (1.01 g, 5.0 mmol) was acylated using carbobenzoxy chloride (0.72 mL, 5.0 mmol) in a methanol + aqueous sodium bicarbonate mixture. The product, methyl 4-(benzyloxycarbonylamino) phenylacetate, 17, was extracted into methylene chloride and isolated as an oil in quantitative yield. m-Aminobenzoic acid (2.94 g, 21 mmol) was suspended in 50 mL of methanol and treated with carbobenzoxy chloride (3.0 mL, 21 mmol). The mixture was sonicated for 10 min and water was added, resulting in the crystallization of the product, 3-(benzyloxycarbony1amino)benzoic acid, 19 (m.p. 218–219° C), obtained in 67% yield. C, H, and N analysis.
Synthesis of 2-(4(benzyloxycarbonylamino)phenylacetylamino)-ethylamine, 18
Methyl 4-(benzyloxycarbonylaminopheny1) acetate (compound 17, 1.3 g, 4.3 mmol) was treated with ethylenediamine (4 mL) and heated at 50° C for 10 min. Several cycles of evaporation under vacuum and addition of methanol, left an oil which could be crystallized from methanol/ether. Yield 0.80 g (56% yield), m.p. 186–190° C. C, H, and N analysis was correct for the 3/2 hydrate.
Synthesis of N-succinimidyl 3-(benzyloxycarbonylamino)benzoate, 20
Compound 19 (3-(benzyloxycarbonylamino)-benzoic acid, 0.30 g, 1.1 mmol), N-hydroxysuccinimide (0.30 g, 2.6 mmol), and EDAC (0.36 g, 1.9 mmol) were combined in 10 mL DMF. After being stirred for 12h, the reaction mixture was treated with water and ethyl acetate. The organic layer was separated, washed (0.1 M HCl / 1 M bicarbonate), and evaporated leaving the product (m.p. 110–115°C) in 89% yield. C, H, and N analysis.
Biochemical assays
Radioligands ([3H]PIA, ([3H]NECA, and ([3H]CGS21680) were obtained from DuPont NEN Products Boston, MA, USA. Stock solutions of adenosine derivatives in the millimolar concentration range in dimethylsulfoxide were prepared for binding assays. The solutions were diluted as necessary and were stable to storage in the frozen state. For each assay at A1 or at A2A receptors, inhibition of binding by a range of concentrations of xanthines was assessed in triplicate in at least three separate experiments. Protein was determined using the BCA (based on the complex with cuprous ions and bicinchoninic acid) protein assay reagents purchased from Pierce Chemical Co., Rockford, IL, USA. Increase in cyclic AMP in human platelets was measured using the method described in Newman et al. (1978).
Competitive binding assay in rat brain using [3H]PIA, [3H]CGS21680, and [3H]NECA
Inhibition of binding of 1 nM [3H]N6-phenylisopropyladenosine (specific activity 42.5 Ci/mmol), to A1 adenosine receptors in rat cerebral cortex membranes, was assayed as described in Jacobson et al. (1987). Inhibition of binding by a range of concentrations of an adenosine derivative was assessed in triplicate in at least three separate experiments. At least seven different concentrations spanning three orders of magnitude (adjusted appropriately for the IC,, of each compound), were used. IC,, values, which were computer-generated using a nonlinear regression formula on the GraphPAD program (Institute for Scientific Information), were converted to Ki values using a KD value for [3H]PIA of 1.0 nM (Jacobson et al. 1987) and the Cheng-Prusoff equation (Cheng et al. 1973).
Affinity at rat striatal A2A receptors was measured by two methods, using either [3H]NECA or [3H]CGS21680. Inhibition of binding of 5′-N-[3H]ethylcarboxamidoadenosine (specific activity 18 Ci/mmol) to A2A adenosine receptors in rat striatal membranes was measured as described (Bruns et al., 1986), except that 5 mM theophylline was used to define non-specific binding. N6-cyclopentyladenosine was present at 50 nM to inhibit binding of the ligand at A1 adenosine receptors. IC50 values were converted to Ki values as described (Bruns et al., 1986). Use of [3H]CGS21680 (specific activity 48.1 Ci/mmol) as an A2A radioligand, precluded the need for adding N6-cyclopentyladenosine (Hutchison et al, 1989). Rat striatum was homogenized in 25 volumes of ice cold 50 mM Tris adjusted to pH 7.4 with hydrochloric acid containing 10 mM magnesium chloride, using a polytron (Kinematica, GmbH., Luzerne, Switzerland) at a setting of 2–3 for 10s. The membrane suspension was then centrifuged at 37 000 × g for 20 min at 4° C. The pellet was resuspended in buffer containing 2 IU/mL adenosine deaminase Type VI from calf intestinal mucosa (Sigma, St. Louis, MO, USA) to 20 mg/mL original tissue weight, and incubated at 37°C for 30min. The membrane homogenate was recentrifuged as before, and the pellet was stored frozen at −70°C until use.
For competitive binding assays using [3H]CGS21680, a volume of I mL was used in each 13 × 100 mm glass tube. The unlabelled competing ligand of 2-chloroadenosine, for determination of nonspecific binding, was dissolved in 25 pL of DMSO. Then 50pL of 200 m~MgCl,, 725pL of 50mM Tris (pH7.4 at room temperature) and 1OOpL of radioligand were added to produce a final concentration of 5 nM. Finally 100 pL of a striatal tissue suspension (final concentration of 150–200 μg protein/mL) was added. The mixture was incubated with shaking for 90 min at 24° C. The samples were filtered on a Brandel Cell Harvester (Brandel, Gaithersburg, MD, USA) with Whatman GF/B filters, and washed rapidly twice with 5 mL of ice cold 50 mM Tris, pH 7.4. Each filter disc was added to 4mL of scintillation fluid, vortexed, and counted after 6 h.
Competitive binding assay in bovine brain using [‘*’I]PAPAAPEC
Bovine striatal membranes were prepared as described in Barrington et al. (1989). 150 μL of striatal membranes (approximately 0.5 mg protein/mL, suspended in 50 m ~ HEPES buffer at pH 7.2, containing 10 mM MgCl2) was combined with 50 μL of the indicated competitor and 50 μL of [125I]PAPA-APEC to yield a final concentration of radioligand of 1 nM. After a 1 h incubation at 37° C, the mixture was filtered rapidly over No. 32 Schleicher and Scheull glass fiber filters, which had been pretreated for 1 h with 0.3% polyethyleneimine. The filters were washed with three × 3 mL aliquots of buffer (pH 7.2), containing 50 mM HEPES, 10 mM MgCl,, and 0.05% CHAPS. The filters were placed in polypropylene tubes and counted in a Packard gamma counter.
aminoethylaminocarbonylethylphenylethylamino-5′-Nethylcarboxamidoadenosine), was prepared as a common synthetic intermediate for molecular probes of A2a adenosine receptors. The amine was synthesized (Scheme 1 and Table 1) through aminolysis by ethylene diamine of the methyl ester derivative of CGS21680, compound 2. The amine congener, 3, was readily acylated, with various activated carboxylic active esters containing prosthetic groups, or with aryl isothiocyanates. The new derivatives were then assayed for activity in binding studies (Table 2) and in a functional assay (Fig. 2).
Scheme 1.

Synthetic routes to derivatives of CGS21680 as adenosine receptor probes.
Table 1.
Structures of 2,5′-disubstituted adenosine derivatives synthesized, and their characterization by californium plasma desorption mass spectroscopy.
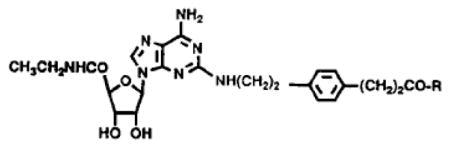 | |||
|---|---|---|---|
| R | %Yield) | MS Peaks | |
| 1 | OH (CGS21680) | - | 544[M + Na2–H]+,522,#393,c371c |
| 2 | -OCH3 | - | 536,a514,b341,c336,163 |
| 3 | -NH(CH2)2NH2(APEC) | 91 | 564,a542,b369,c336,163 |
| 4 |
|
82 | 712,a690,b539,c517,c336, 163 |
| 5 |
|
96 | 689,a667,b163 |
| 6 |
|
68 | 697,a546,c524,c163 |
| 7 |
|
65 | 831,a809,b658,c336 |
| 8 |
|
87 | 697b |
| 9 |
|
70 | 817,a795,b336,163 |
| 10 |
|
82 | 791,a769b |
| 11 |
|
47 | 756,a734,b724 [M + Na − S],702 [M + H+ − S] |
| 12 |
|
44 | 734,b702 |
| 13 |
|
85 | 802,a764c |
| 14 | -NH(CH2)2NHCOCH2Br | 57 | 685a |
| 15 |
|
69 | 724 [M + H − S], 564,550,c336,163 |
| 16 |
|
81 | 728,a 706, b555, c534, c336, 163,130 |
[M + Na]+
[M + H]+
Loss of 173(5′-N-ethylcarboxamidoribose) from [M + Na]+, [M + H+−S], or from [M + Na2−H]+
Table 2.
Potencies of adenosine derivatives and xanthines at adenosine A1 and at A2 adenosine receptors in binding assaysa
| Compound | A1 receptors | A2 receptors |
|
|||
|---|---|---|---|---|---|---|
| (3H)PIA rat | [125I] PAPA APEC bovine | [3H)NECA rat | [3H]CGS21680 rat | |||
| NECA | 6.26 ± 0.52b | 55 ±26 | 10.3 ± 0.5b | 12 ± 1 | 0.11 | |
| R-PIA | 1.17 ± 0.16b | 870 ± 270 | 124 ± 9b | 410 ± 32 | 0.0013 | |
| S-PIA | 49.3 ± 2.4b | 10 300 ± 2800 | 1820 ± 380b | 3 020 ±210 | 0.0048 | |
| ADAC | 0.85 | (Nd)d | 210 | 218 ± 28 | - | |
| Theophylline | 8470 ± 1490b | 20 300 ± 2700 | 25 300 ± 200b | 20 800 ± 1540 | 0.417 | |
| XAC | 1.2 ± 0.5 | (Nd)d | 44 | 30.7 ± 1.7 | - | |
| 1 | 2600 ± 300c | 14.1 ± 2.1 | 15 | 14 ± 1 | 184 | |
| 2 | 2260 ± 440c | 6.1 ± 1.5 | 17.5 ± 1.6 | 5 ± 2 | 290 | |
| 3 | 99.4 ±11.6 | 6.1 ± 1.3 | 5.73 ± 0.52 | 12± 3 | 16 | |
| 4 | 950 ± 100c | 13.2 ± 1.5 | (Nd)d | 13 ± 4 | 72 | |
| 5 | 1400 ± 100c | 21.3 ± 7.9 | (Nd)d | 15± 1 | 66 | |
| 6 | 1340 | 43 ± 14 | (Nd)d | 28 ± 7 | 31 | |
| 7 | 278 ± 18.7 | 6.2 ± 0.9 | 5.59 • 0.99 | 37 ± 5 | 45 | |
| 8 | 1340 ± 28 | 43.6 ± 5.2 | (Nd)d | 16.5 ±2.8 | 31 | |
| 9 | 680 ± 110c | 8.7 ± 2.0 | (Nd)d | 12 ± 2 | 78 | |
| 10 | > 5000c | 14.3 ± 3.3 | (Nd)d | 55 • 30 | >350 | |
| 11 | 69.7 ± 7.9 | 6.2 ± 2.1 | 2.82 ± 0.38 | 22 ± 7 | 11 | |
| 12 | 276 ± 75c | 7.1 ± 2.3 | (Nd)d | 35 ± 10 | 39 | |
| 13 | 3780 ± 800 | 10.9 ± 2.3 | (Nd)d | 26 ± 10 | 347 | |
| 14 | 1980 ± 102 | 15.8 ± 4.8 | (Nd)d | 69.7 | 125 | |
| 15 | 177 ± 38c | 14.8 ± 3.6 | (Nd)d | 27 ± 3 | 13 | |
| 16 | 1450 ± 67 | 25.4 ± 7.6 | (Nd)d | 18.9 ± 4.9 | 57 | |
Unless noted, expressed as the Ki value in nM, for inhibition of binding of [3 H] PIA at A1 receptors, inhibition of binding of [125I] PAPA-APEC, [3H]NECA, or [3H]CGS21680 at A2 receptors. Compounds 11, 12, and 14 are potential irreversible inhibitors of A2 receptors, thus the values given represent apparent Ki’s. Values are the means ± SEM for three or more determinations in triplicate. The A2 selectivity ratio derived from Ki values for [125I] PAPA-APEC binding at A2 receptors.
Data from Bruns et al. (1986); inhibition of binding of N6 -[3H] cyclohexyladenosine at A1, receptors, inhibition of binding of [3H]NECA at A2 receptors.
Using N6 -[3H] cyclohexyladenosine.
Not determined
The ratio of Ki values at A1, receptors vs the Ki values in the [125I] PAPA-APEC assay.
Figure 2.

Dose-dependent increase in cyclic AMP in human platelets produced by a potent A2 adensoine agonist, compound 6. This curve is typical of three experiments, each being a triplicate assay. Basal cyclic AMP production was 28.3 pmol mg−1 min−1 At 1 μM, NECA gave a response of 38.1 pmol mg−1 min−1 representing a 35% increase in cyclic AMP.
The small quantities synthesized and the high molecular weights, necessitated the use of high field NMR and californium plasma desorption mass spectroscopy (Jacobson et al., 1986) for characterization of the homogeneous products. The mass spectra (Table 1) for positive ions show either the [M + H]+ or [M + Na]+ ions, or peaks resulting from the loss of the 5′-N-ethylcarboxamidoribose moiety. Thiourea derivatives, obtained from reaction of APEC with isothiocyanates and model compounds for comparison, tended to lose sulfur during measurement of mass spectra. As was observed previously, some of the mass spectra showed the sodium replacement ion of the sodium salt, [M + 2Na −H]+.
Radioactive probes
Towards the goal of radioiodination of functionalized congeners, both phenolic prosthetic groups (Jacobson et al., 1985), such as p-hydroxyphenylpropionic acid (Bolton-Hunter group), and aryl amine-containing prosthetic groups (Stiles et al., 1987), such as p-aminophenylacetic acid (PAPA) have been utilized. An additional prosthetic group for iodination, a 2-substituted thienyl group, which does not contain a hydroxyl or amino group, has been shown to iodinate readily (and selectively in the presence of phenols) via its easily formed mercury adduct (Jacobson et al., 1989b). Thus, the p-hydroxyphenylpropionyl-, 4, 2-thienylacetyl-, 5, and p-aminophenylacetyl-, 6, derivatives of APEC were synthesized as radioiodination substrates (Scheme 1). Compounds 4 and 5 were synthesized via the direct acylation of APEC. Compound 6 was synthesized via the condensation of the
RESULTS
In the development of ligands for molecular characterization of the A2A receptor protein, we adopted a functionalized congener approach, utilizing the carboxyl group of CGS2I680 (Compound 1, Table 1) for attachment of a chemically reactive chain designed not to preclude receptor binding of the derivative. A conceptually similar series of functionalized congeners has been developed for the characterization of A1 adenosine receptors by radioactive and spectroscopic methods (Jacobson et al., 1987). A carboxyl or amino group at a newly created distal site on an adenosine (agonist) or xanthine (antagonist) derivative, was covalently linked to a reporter group, for probing the receptor binding interaction. Similarly, in the present series of adenosine derivatives, a pivotal amine analog, compound 3 (APEC, carboxylic acid CGS21680 with the appropriate amine, i.e., compound 18. The resulting benzyloxycarbonylprotected intermediate, 7, was deprotected through hydrogenolysis. An additional aryl amine, 8, was prepared via the corresponding N-carbobenzyloxy protected intermediate 9.
Radioiodinated aryl amine derivatives have an advantage over radioidinated phenols as molecular probes, in that they may be crosslinked to the receptor protein. Aryl amines, following iodination with I, have been photoaffinity crosslinked to A1 adenosine receptors (Stiles et al., 1985) or converted to azido derivatives which were photolyzed in the receptor bound state (Stiles et al., 1986; Linden et al., 1988). Compounds 6 and 8 are designed for both radioiodination and photoaffinity crosslinking. Compound 8 was designed in an attempt to overcome low yield in the iodination step (see below), due to possible oxidation at a benzylic methylene group of 6. Compound 8 is a benzoic acid derivative in which one potentially susceptible a-methylene group is absent. The meta aniline derivative was selected over the para, because of its greater nucleophilicity (required for the crosslinking reaction) based on electronic effects and pK, data in 3-aminobenzoyl derivatives vs the corresponding 4-aminobenzoyl derivatives.
PAPA-APEC, 6, was radioiodinated by the chloramine T method in 70% radiochemical yield. The product, [1251]PAPA-APEC, having a specific activity of approximately 2200 Ci/mmol, was purified by reverse phase high pressure liquid chromatography (Fig. 1). The two main radioactive peaks were identified using TLC as [125I]PAPA-APEC (peak B) and recovered [1251]iodide. The major ultraviolet absorbing peak (A) corresponded to unreacted compound 6.
[125I]PAPA-APEC displayed a 60–80% degree of specific binding to striatal A2A adenosine receptors in the bovine brain with a Kd value of 1.5 nM (Barrington et al., 1989. [125I]PAPA-APEC was used in photoaffinity crosslinking followed by SDS gel electrophoresis, determining the molecular weight of this receptor to be 45 000 (Barrington et al., 1989). Here we further demonstrated the utility of this A2 selective iodinated radioligand in competitive binding assays. Typical binding curves for displacement of [125I]PAPA-APEC for bovine brain membranes are shown in Fig. 3.
Figure 3.
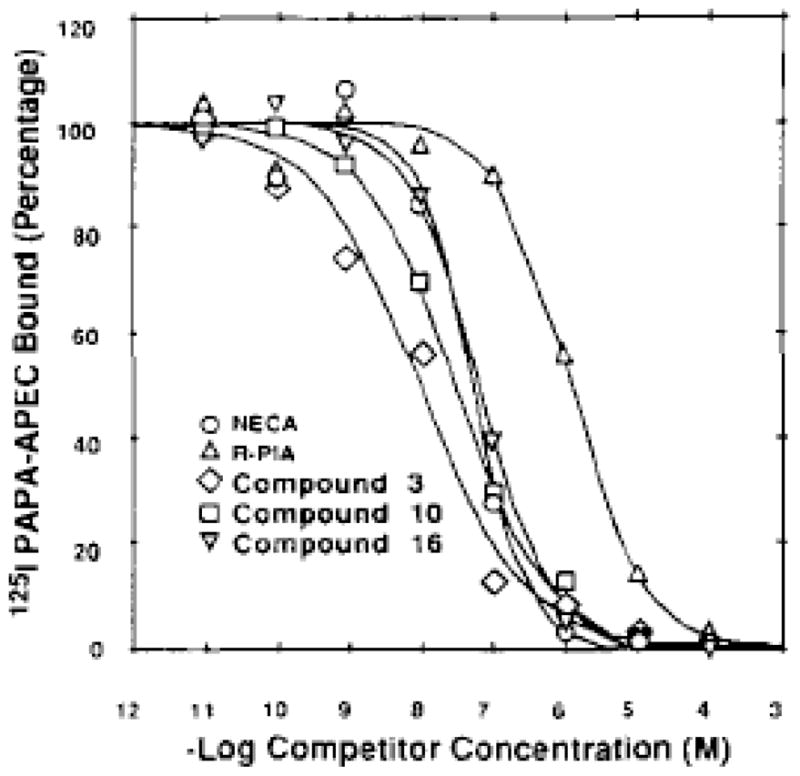
Competition curves for the displacement of [125I]PAPA-APEC from A2 adenosine receptors in bovine brain membranes The concentration of [125I]PAPA-APEC was 1 nM. IC50 values were as follows: NECA, 51 nM; R-PIA, 1290nM; compound 6, 9.1 nM; compound 10, 28.9 nM; compound 16, 62.2 nM.
A prosthetic group for radiofluorination of functionalized drugs and peptides was recently reported (Shai et al., 1989). This group consisted of a 4-fluoromethylbenzoyl moiety (FMB) obtained through nucleophilic fluorination (with 18F) of the corresponding 4-bromomethylbenzoyl derivatives. As a potential “F probe for in vivo positron emission tomography (PET) of A2A adenosine receptors, an FMB derivative of CGS21680, compound 16, in which the spacer chain consists of 1,4-diaminobutane, was prepared. It was synthesized by condensing the 4-(4-fluoromethyl)benzoylamino)-butaneamino (Shai et al., 1989) with CGS21680.
Non-radioactive probes
APEC, 3, was acylated with other reported groups suitable for non-radioactive methods of receptor characterization. Biotin/avidin technology has been used to isolate receptors by affinity chromatography on immobilized avidin columns (Finn et al., 1988) and for histochemistry. A biotin conjugate of APEC, 10, was synthesized for this purpose.
The amine-functionalized congeners XAC and ADAC, which are antagonist and agonist probes, respectively, for A1 receptors, have been converted to irreversibly binding ligands for the receptor (Stiles et al., 1988; Jacobson et al., 1989a) through chemical activation using hetero- and homo-bifunctional crosslinking reagents. Examples of bifunctional reagents used successfully in that capacity are the p- and m- isomers of phenylenediisothiocyanate (DITC). Here, we synthesized m- and p-DITC-APEC, compounds 11 and 12, respectively, as potentially chemically reactive affinity labels. A bromoacetyl N6-substituted derivative of adenosine was found to inhibit A1 adenosine receptors irreversibly (Jacobson et al., 1989a). Here we have prepared a bromoacetyl derivative, 14, designed to inhibit A2A adenosine receptors.
A charged thiourea derivative, 13, containing a sulfonate salt, was prepared. This conjugate, which is approximately as potent and selective as CGS21680, is expected to be prevented from crossing the blood/brain barrier, due to its completely charged state at physiological pH.
Spin label and fluorescent analogs of purine functionalized congeners, having Ki values at A1 adenosine receptors in rat brain ranging from 4 to 40 nM, have been synthesized as A1 receptor probes (Jacobson et al., 1987). A spin label probe, compound 15, containing the stable free radical TEMPO (2,2,6,6-tetramethyl- l-piperidinyloxy), for detection using electron spin resonance spectroscopy (Blanton et al., 1988), was prepared in an analogous fashion from APEC, and the isothiocyanate derivative of TEMPO.
Assays of receptor affinity and adenylate cyclase
The adenosine analogs were assayed for affinity at A1 adenosine receptors using the radioligands N6[3H]-phenylisopropyladenosine and N6-[3H]cyclohexyladenosine. At A2A receptors, three methods were used for comparison: 1, inhibition of binding of 5′-N-[3H]ethylcarboxamidoadenosine to rat striatal membranes, according to the method of Bruns et al. (1986), in which the A1-selective ligand N6-cyclopentyladenosine is added to eliminate the A1 receptor component of specific binding; 2, inhibition of binding of [3H]CGS21680 to rat striatal membranes, and 3, inhibition of binding of [125I]-PAPA-APEC to bovine brain striatal membranes. Striatal membranes from the calf were used for the following reasons: 1, bovine striatum has a higher density of A, adenosine receptors than does the rat (1.1 vs 0.5 pmol receptors per mg of protein); 2, larger quantities of striatum are more readily dissected from calf brains than from rat brains; 3, bovine A2A receptors are more stable to storage and have more favorable levels of nonspecific binding of radioligands.
At A2A adenosine receptors, the derivatives retained high affinity, comparable to the agonist CGS21680, from which they were derived. CGS21680 had a Ki value of 14 nM in inhibition of binding of [125I]PAPA-APEC. Compounds 2,3,7,9, 11, and 12 displayed Ki values less than l0 nM in displacement of binding of [1251]PAPAAPEC from bovine brain A2A adenosine receptors. Compounds 6, 8, and 16 were less potent in displacing [125I]PAPA-APEC binding. The affinity of the aminefunctionalized congener, compound 3 (APEC), was unaffected at A2A receptors but was enhanced substantially at A1 receptors. At A2A adenosine receptors, the amine congener APEC, 3, is 26-fold more potent than CGS21680, 1, a carboxylic acid derivative. At A2A adenosine receptors, APEC is similar in potency to CGS21680. Thus, in binding assays the amine congenerAPEC is not as A2A-selective as CGS21680 or the immediate precursor, the methyl ester of CGS21680, 2. A distal benzyloxycarbonyl group was well tolerated at A2A receptors, as evidenced from the affinity of compounds 7 and 9, in comparison to the corresponding free aryl amines, compounds 6 and 8. The aryl amine derivatives both displayed moderate affinity (Ki values approximately 40 nM) at A, receptors, which would likely be enhanced upon iodination. An iodo substituent on the aniline ring probably enhances potency, in view of the Kd value of 1.4 nM observed for [1251]PAPA-APEC (derived from compound 6). Among the iodinatable analogs synthesized, the p-hydroxyphenylpropionyl (Bolton-Hunter) derivative, 4, had the highest affinity at A, receptors (equal to the affinity of CGS21680). Thus, the reaction of APEC with the Bolton-Hunter reagent (commercially available in the radioiodinated form) is a viable route to another iodinated radioligand.
The aryl isothiocyanates (DITC conjugates), 11 and 12, and the bromoacetyl derivative, 14, are potential affinity labels for A2A receptors. Both isothiocyanate isomers displayed relatively high affinity at A2A receptors, with apparent Ki values nearly equal to the Ki value for the synthetic precursor, APEC. The meta isomer, 11, was particularly potent in [3H]NECA binding.
The biotin conjugate, 10, and a p-sulfonate derivative, 13, displayed the highest A2A selectivity (at least 350-fold) of the compounds prepared. The precise selectivity value of 10 was not determined, since at A, receptors, the highest concentration used in the binding assay (M) caused less than 50% inhibition of specific binding of the radioligand.
A comparison of Ki values derived from displacement of binding of [125I] PAPA-APEC and [3H]NECA, show some difference (eg., for NECA and compounds 2 and ll), but no clear trend is apparent. In spite of the species difference (Stone et al., 1988), there is a fair correspondence in Ki values using the two methods. The differences in Ki values are generally less than threefold, except for NECA and R-PIA, which appear to be fivefold and sevenfold less potent, respectively using I-PAPA-APEC in the bovine brain. At A1 receptors (Ukena et al., 1986) a large species dependence has been observed for certain N6 -substituted adenosine analogs as well, and also for many C-8 aryl xanthine derivatives.
Although the high affinity at A2A receptors is maintained in general as the functionalized chain is lengthened, the high A2A selectivity observed for CGS21680 and the methyl ester, 2, is often diminished. This occurs mainly as a result of increased affinity at A1 receptors rather than loss of affinity at A2A receptors. In general, the A2A affinity of the conjugates synthesized was relatively constant, with Ki values for [1251]PAPA-APEC binding in the range of 6 to 43 nM. Conversely, the affinity at A1 receptors was subject to greater variation, with K, values ranging from 70 to > 5000 nM. Thus, the A2A adenosine receptor is less sensitive to structural changes at this distal site than is the A1 adenosine receptor. Biological activity of the adenosine agonists in a functional assay was also examined. The ability of compound 6 to increase cyclic AMP in human platelets (Newman et al., 1978) is shown in Fig. 2. A dose-dependent increase in cyclic AMP was observed (Fig. 2) over the range of concentration of compound 6 of 1 nM to 10 μM. At 10−6 M compound 6 produced a 40% increase in the level of cyclic AMP. At the same concentration, NECA produced a comparable 35% rise in cyclic AMP.
DISCUSSION
We have shown that CGS21680 may be derivatized as an amine derivative (APEC) through the inclusion of an aminoethylamide group. This amine derivative retains high affinity for A, receptors, albeit with diminished selectivity. APEC has been subsequently coupled to a variety of reporter groups, resulting in high affinity probes for detection and characterization of A, adenosine receptors. The new conjugates of high molecular weight were active in receptor binding assays and in a functional assay as A, adenosine agonists (stimulation of cyclic AMP in platelets). These probes include analogs for radiolabelling, a biotin conjugate, ligands for chemical affinity labelling (bearing electrophilic groups), photoaffinity crosslinking (aryl amines), and a free radical derivative for electron spin resonance spectroscopy. This series includes chemically reactive chains potentially of use in anchoring the high affinity ligands to a solid matrix, for isolation of A, receptors by affinity chromatography. One aryl amine derivative was iodinated using 125I, to afford an A2A-selective agonist radioligand of high affinity and high specific activity.
As with previous findings with ligands selective for A1 receptors (Jacobson et al., 1985), the presence of a bulky functionalized chain on adenosine analogs need not interfere with a high affinity interaction at A2A receptors. This chain, here located at the C-2 position of adenosine 5′-carboxamides (based on CGS21680) may be extended, in effect creating new distal sites for derivatization. The desirable pharmacological potency in this series of derivatives is not eliminated upon structural extension. Moreover, the conjugates reported here are potentially useful as high affinity molecular probes for receptor characterization by radioactive, spectroscopic, or affinity labelling methodology, and for receptor isolation by affinity chromatography.
Acknowledgments
G.L.S. is an established investigator of the American Heart Association and is supported in part by the NIH grant No. ROlH35134 and supplement from NHLBI and a grand-in-aid from the American Heart Association and 3M Riker.
Abbreviations used
- CGS21680
2-(4-(2-carboxyethyl)phenylethylamino)-5′-N-ethylcarboxamidoadenosine
- NECA
5′-N-ethylcarboxamidoadenosine
- PIA
N6-phenylisopropyladenosine
- E DAC
1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride
- CHAPS
3-((3-cholamidopropyI)dimethylammonio)- I -propane sulfonate
Contributor Information
Kenneth A. Jacobson, National Institute of Diabetes, and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, MD 20892, USA.
Lewis K. Pannell, National Institute of Diabetes, and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, MD 20892, USA.
Xiao-duo Ji, National Institute of Diabetes, and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, MD 20892, USA.
Michael F. Jarvis, Research Department, Pharmaceuticals Division, CIBA-Geigy Corporation, Summit, NJ 07901, USA
Michael Williams, Research Department, Pharmaceuticals Division, CIBA-Geigy Corporation, Summit, NJ 07901, USA.
Alan J. Hutchison, Research Department, Pharmaceuticals Division, CIBA-Geigy Corporation, Summit, NJ 07901, USA.
William W. Barrington, Departments of Medicine and Biochemistry, Duke University Medical Center, Durham, NC 27710, USA
Gary L. Stiles, Departments of Medicine and Biochemistry, Duke University Medical Center, Durham, NC 27710, USA
References
- Barrington WW, Jacobson KA, Hutchison AJ, Williams M, Stiles GL. Identification of the A, adenosine receptor binding subunit by photoaffinity crosslinking. Proc Natl Acad Sci, USA. 1989;86:6572–6576. doi: 10.1073/pnas.86.17.6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanton M, McCardy E, Gallaher T, Wang HH. Noncompetitive inhibitors reach their binding site in the acetylcholine receptor by two different paths. Mol Pharmacol. 1988;33:634–642. [PubMed] [Google Scholar]
- Bridges AJ, Moos WH, Szotek DL, Trivedi BK, Bristol JA, Heffner TG, Bruns RF, Downs DA. N6 -(2,2-diphenylethyl)adenosine, a novel adenosine receptor agonist with antipsychotic-like activity. J Med Chem. 1987;30:1709–1771. doi: 10.1021/jm00393a003. [DOI] [PubMed] [Google Scholar]
- Bridges AJ, Bruns RF, Ortwine DF, Priebe SR, Szotek DL, Trivedi BK. N6 -[2-(3,5-dimethoxyphenyl)-2-(2-methylphenyl)ethyl]adenosine and its uronamide derivatives. Novel adenosine agonists with both high affinity and high selectivity for the adenosine A, receptor. J Med Chem. 1988;31:1282–1285. doi: 10.1021/jm00402a004. [DOI] [PubMed] [Google Scholar]
- Bruns RF, Lu GH, Pugsley TA. Characterization of the A, adenosine receptor labelled by [3H]NECA in rat striatal membranes. MoI Pharmacol. 1986;29:331–346. [PubMed] [Google Scholar]
- Cheng Y-C, Prusoff WH. Relationship between the inhibition constant (K,) and the concentration of inhibitor which causes 50 percent inhibition (I&,) of an enzyme reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Finn FM, Hoffmann K. Biotinylated insulins for the purification of insulin receptors. In: Kahn CR, Harrison LC, editors. Insulin Receptors Part A. Alan R. Liss; New York: 1988. pp. 3–14. [Google Scholar]
- Hutchison AJ, Williams M, deJesus R, Yokoyama R, Oei HH, Ghai GR, Webb RL, Zoganas HC, Stone GA, Jarvis MF. 2-Arylalkylamino-adenosine-5′-uronamides: a new class of highly selective adenosine A, receptor ligands. J Med Chem. 1990 doi: 10.1021/jm00169a015. (in press) [DOI] [PubMed] [Google Scholar]
- Jacobson KA, Kirk KL, Padgett WL, Daly JW. Functionalized congeners of adenosine: preparation of analogues with high affinity for A, adenosine receptors. J Med Chem. 1985;28:1341–1346. doi: 10.1021/jm00147a039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson KA, Pannell LK, Fales HM, Sokoloski EA, Kirk KL. Californium plasma desorption mass spectrometry in following the synthesis of polar, high molecular weight compounds. J Chem SOC Perkin I. 1986:2143–2149. [Google Scholar]
- Jacobson KA, Ukena D, Padgett W, Kirk KL, Daly JW. Molecular probes for extracellular adenosine receptors. Biochem Pharmacol. 1987;10:1697–1707. doi: 10.1016/0006-2952(87)90056-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson KA, Barone S, Kammula U, Stiles GL. Electrophilic derivatives of purines as irreversible inhibitors of A, adenosine receptors. J Med Chem. 1989a;32:1043–1051. doi: 10.1021/jm00125a019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson KA, Kiriasis L, Barone S, Bradbury B, Kammula U, Campagne JM, Daly JW, Neumeyer JL, Pfleiderer W. Sulfur-containing xanthine derivatives as selective antagonists at A, adenosine receptors. J Med Chem. 1989b;32:1873–1879. doi: 10.1021/jm00128a031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis MF, Schutz R, Hutchison AJ, Do E, Sills MA, Williams M. [3H]CGS21680, a selective A2 adenosine receptor agonist directly labels A2 receptors in rat brain. J Pharmacol Exp Ther. 1989;251:888–893. [PubMed] [Google Scholar]
- Linden J, Patel A, Earl CQ, Craig RH, Daluge SM. 1251-Labelled 8-phenylxanthine derivatives: antagonist radioligands for adenosine A, receptors. J Med Chem. 1988;31:745–751. doi: 10.1021/jm00399a010. [DOI] [PubMed] [Google Scholar]
- Lohse MJ, Elger B, Lindenborn-Fotinos J, Klotz KN, Schwabe U. Separation of solubilized A2A adenosine receptors of human platelets from non-receptor [3H]NECA binding sites by gel filtration. Naunym Schmiedeberg’s Arch Pharmacol. 1988;337:64–68. doi: 10.1007/BF00169478. [DOI] [PubMed] [Google Scholar]
- Londos C, Cooper DMF, Wolff J. Subclasses of external adenosine receptors. Proc Natl Acad Sci USA. 1980;77:2551–2254. doi: 10.1073/pnas.77.5.2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman KD, Williams KT, Bishopric NH, Lefkowitz RJ. Identification of a-adrenergic receptors in human platelets by [3H]dihydroergocryptine binding. J Clin Invest. 1978;61:395–402. doi: 10.1172/JCI108950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson RA, Kusachi S, Thompson RD, Ukena D, Padgett W, Daly JW. N′-Substituted N-alkyladenosine-5′ wonamides: bifunctional ligands having recognition groups for A, and A, adenosine receptors. J Med Chem. 1986;29:1683–1689. doi: 10.1021/jm00159a020. [DOI] [PubMed] [Google Scholar]
- Polmar SH, Fernandez-Mejia C, Birch RE. Adenosine receptors: immunological aspects. In: Cooper DMF, Londos C, editors. Adenosine Receptors. Alan R. Liss; New York: 1988. pp. 97–112. [Google Scholar]
- Shai Y, Kirk KL, Channing MA, Dunn BB, Lesniak MA, Eastman RC, Finn RD, Roth J, Jacobson KA. ‘*F labelled insulin: a prosthetic group methodology for incorporation of a positron emitter into peptides and proteins. Biochemistry. 1989;28:4801–4806. doi: 10.1021/bi00437a042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamim MT, Ukena D, Padgett WL, Daly J. Effects of 8-phenyl and 8-cycloalkyl substituents on the activity of mono-, di-, and trisubstituted alkylaxanthines with substitution at the 1-, 3-, and 7-positions. J Med Chem. 1989:32. doi: 10.1021/jm00126a014. [DOI] [PubMed] [Google Scholar]
- Stiles GL. Photoaffinity crosslinked A, adenosine receptor-binding subunits. Homologous glycoprotein expression by different tissues. J Biol Chem. 1986;261:10839–10843. [PubMed] [Google Scholar]
- Stiles GL, Daly DT, Olsson RA. The A1 adenosine receptor. Identification of the binding subunit by photoaffinity crosslinking. J Biol Chem. 1985;260:10806–10811. [PubMed] [Google Scholar]
- Stiles GL, Daly DT, Olsson RA. Characterization of the A, adenosine receptor-adenylate cyclase system of cerebral cortex using an agonist photoaffinity ligand. J Neurochem. 1986:47. doi: 10.1111/j.1471-4159.1986.tb00715.x. [DOI] [PubMed] [Google Scholar]
- Stiles GL, Jacobson KA. A new high-affinity, iodinated adenosine receptor antagonist as a radioligand/ photoaffinity crosslinking probe. Mol Pharmacol. 1987;32:184–188. [PMC free article] [PubMed] [Google Scholar]
- Stiles GL, Jacobson KA. High affinity acylating antagonists for the A, adenosine receptor: identification of binding subunit. Mol Pharmacol. 1988;34:724–728. [PMC free article] [PubMed] [Google Scholar]
- Stone GA, Jarvis MF, Sills MA, Weeks B, Snowhill EW, Williams M. Species differences in high-affinity adenosine A, binding sites in striatal membranes from mammalian brain. Drug Devel Res. 1988;15:31–46. [Google Scholar]
- Ukena D, Jacobson KA, Padgett W, Ayala C, Shamim MT, Kirk KL, Olsson RA, Daly JW. Species differences in structure/activity relationships of adenosine agonists and xanthine antagonists at brain A, adenosine receptors. FEBS Letts. 1986;209:122–128. doi: 10.1016/0014-5793(86)81096-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams M. Purine receptors in mammalian tissues: pharmacology and physiological significance. Ann Rev Pharmacol Toxicol. 1987;27(315):1020–1025. doi: 10.1146/annurev.pa.27.040187.001531. [DOI] [PubMed] [Google Scholar]