

Abstract
Understanding the control mechanisms of blood flow within the vasculature of skeletal muscle is clearly fascinating from a theoretical point of view due to the extremely tight coupling of tissue oxygen demands and blood flow. It also has practical implications as impairment of muscle blood flow and its prevention/reversal by exercise training has a major impact on widespread diseases such as hypertension and diabetes. Here we analyse the role of mediators generated by skeletal muscle activity on smooth muscle relaxation in resistance vessels in vitro and in vivo. We summarize their cellular mechanisms of action and their relative roles in exercise hyperaemia with regard to early and late responses. We also discuss the consequences of interactions among mediators with regard to identifying their functional significance. We focus on (potential) mechanisms integrating the action of the mediators and their effects among the cells of the intact arteriolar wall. This integration occurs both locally, partly due to myoendothelial communication, and axially along the vascular tree, thus enabling the local responses to be manifest along an entire functional vessel path. Though the concept of signal integration is intriguing, its specific role on the control of exercise hyperaemia and the consequences of its modulation under physiological and pathophysiological conditions still await additional analysis.
Keywords: active hyperaemia, endothelium, metabolic dilation, skeletal muscle, vasodilator
The skeletal muscle is a rather unique organ in as much as blood flow through the muscle can change over an extremely broad range. Compared to resting conditions, blood flow during maximal exercise can increase up to 20-fold on average; in certain, predominantly white, muscles even increases up to 80-fold have been reported (Laughlin et al. 1996, Boushel et al. 2000). This enormous increase in blood flow during maximal exercise is necessary to meet the 20- to 50-fold enhanced oxygen demands of the muscle tissue. Though these extreme demands may be rarely reached in daily life, it remains essential for physical performance that any increase in muscle work is rapidly and reliably matched by adequate increases in muscle blood flow. Under physiological conditions, this matching is achieved perfectly well as shown in exercising humans and in animal experiments (Bockman 1983, Mackie & Terjung 1983, Mohrman & Regal 1988, Saltin et al. 1998), but is impaired for various reasons in vascular diseases such as hypertension (Bertoldi et al. 2006) or diabetes (Frisbee & Delp 2006, Xiang et al. 2006, Lalande et al. 2008), where physical fitness and muscle performance is, therefore, greatly reduced (Ribisl et al. 2007). The failure in disease states emphasizes that the matching of demand and supply requires a vascular network with sufficient capacity and a tight control of the blood vessels regulating capillary perfusion. This control mainly occurs locally but needs to be regionally coordinated to form a functioning network within muscle tissue. These primarily skeletal muscle-related events gain relevance for the global cardiovascular system as soon as a greater set of muscles becomes simultaneously activated. This is not only the case during exercise, involving larger muscle groups, but also when there is a need of complex movements, involving maintenance of balance, and when augmented ventilation becomes necessary. Under such conditions these basically local mechanisms in each single muscle will acutely affect the systemic circulation and its neuronal and hormonal control mechanisms. There is plenty of experimental evidence that the repeated activation of these mechanisms is the basis for the adaptive responses of muscle blood vessels with regard to their growth and their responsiveness to local and central control mechanisms (Bloor 2005, Mueller 2007, Haram et al. 2008, Crimi et al. 2009). This is an important aspect when it comes to the effects of regular training activities on the cardiovascular system and the effects it has on preventing the metabolic syndrome, premature ageing and decrease in muscle tissue mass, having great impact on human health, especially in the elderly population.
Any attempt to understand the mechanisms of matching metabolism by alterations to local blood flow is confronted with a number of questions. What are the cellular mechanisms leading to vasodilation and what are the key mediators of exercise hyperaemia? Which role do muscle activity signals play and on which vascular cells do they exert their stimulatory effects? How and where are the various stimuli and mediators integrated and prioritized? Are there mechanisms to coordinate vascular responses and what is their role as compared with local regulation? How closely are the local increases in blood flow controlled by the local demands?
In this review we aim to briefly summarize the current state of our knowledge on the local and regional regulation of muscle blood flow under conditions of acute dynamic exercise while chronic training effects as well as effects on the systemic circulation are reviewed elsewhere.
Principles of increasing oxygen supply to skeletal muscle
When the oxygen consumption in muscle suddenly increases, an immediate way to increase supply is an enhanced extraction of oxygen from the haemoglobin in the flowing blood. Usually, in resting muscle, oxygen extraction is in the range of only 20–40% (Van Beekvelt et al. 2001), and has been reported to increase to a range of 70–80% with increasing levels of exercise (Proctor et al. 1998). This means that there is a reserve of oxygen to be extracted from the blood that could be used immediately although it can clearly not cover the increased oxygen requirements under heavy exercise. An enhanced extraction can, however, only occur either when the oxygen binding curve is considerably shifted to the right or when the perivascular pO2 decreases. The former requires rapid and significant local increases in pCO2 or H+ concentrations. Increases in the interstitial concentrations of protons can be observed in the tissue even at rather low work loads; however, the changes are comparably small within the first minute (Street et al. 2001). Therefore, most of the increased extraction should result from a decrease in perivascular pO2 due to enhanced oxygen consumption. Measurements of regional tissue oxygenation using near-infrared spectroscopy performed in young volunteers however revealed no decrease in oxygenation in exercising muscle as long as workloads were above the anaerobic threshold. A decrease was seen however at higher workloads (Rao et al. 2009), which implies that at least at moderate exercise, rapid blood flow increases may well match the increased demand without raising a need for enhanced extraction. As any decrease in tissue pO2 puts tissue oxygenation at least locally at risk as tissue distribution of pO2 is inhomogeneous and areas of low pO2 exist already under physiological conditions (cf. Fig. 1), an increase in blood flow is ‘safer’ than an enhanced extraction. Therefore, an enhancement of O2 extraction may be either an instantaneous buffering mechanism or play a role during conditions of maximal perfusion. In either case it can contribute only to a limited extent to matching of enhanced demands under exercise.
Figure 1.
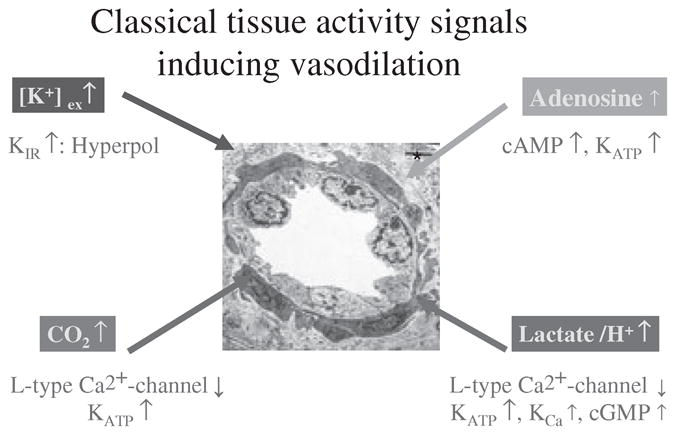
Major ‘classical’ factors mediating vasodilation of skeletal muscle arterioles during exercise. Attributed to each factor are the predominant cellular mechanisms by which the respective factors induce smooth muscle relaxation (upward and downwards arrows indicate increase or decrease in concentration or function). For details, see text.
Thus, the far more important way to meet the increased demands is an appropriate increase in muscle blood flow which also has the effect of increasing the microvascular haematocrit, and hence oxygen flux into the tissue from capillaries (Klitzman & Duling 1979). Significant increases in blood flow can only be obtained with substantial dilation of the small arterioles being associated with enhanced functional capillary density resulting in shorter diffusion distances for oxygen and other substrates. However, theoretical considerations and experimental evidence show that the larger upstream vessels, up to small arteries of about 300 μm in diameter have also to dilate in a coordinated manner to achieve an overall conductivity of the skeletal muscle bed required for the substantial increases in skeletal muscle blood flow (see below). Over the years many potential mediators of this vasodilation have been described and their mechanisms of action elucidated.
Muscle activity-related mediators of vasodilation
Muscle cell derived mediates
Enhanced muscle activity results in the generation of mediators referred to here as ‘muscle activity signals’. Among them are the classical muscle ‘metabolic signals’, including increased pCO2, lactate, K+ and adenosine, as described below but also new mediators whose potential role has become evident only recently.
Augmented muscle activity goes along with enhanced glycolysis and lipolysis (Kiens 2006, Aucouturier et al. 2008), resulting in a higher turnover of the citrate cycle which goes along with enhanced CO2 production. During very intense exercise (15–20 mW g−1) elicited by nerve stimulation in anaesthetized dogs, venous pCO2 levels doubled within 30 s, suggesting the possibility of very rapid changes in pCO2 in tissue at least at high work load (Brechue & Stainsby 1994).
Especially with the onset of heavy exercise, there is also an increase in interstitial and venous concentrations of lactate (Brechue & Stainsby 1994). Particularly in the transition from rest to heavy exercise there is also a net production of lactic acid by muscle. The increase in lactate formation is not necessarily due to a lack of cellular oxygen (Philp et al. 2005). Indeed it has been shown in human muscle under exercise that the lactate efflux is unrelated to cellular pO2 but increases with increasing oxygen consumption (Richardson et al. 1998). Rather, the acceleration of glycolysis is faster than that of the oxidative pathway. Furthermore, the maximal glycolytic capacity of muscle exceeds the maximal oxidative capacity. Under these conditions, glycolysis results in enhanced production of lactic acid which is transported into the interstitial space together with a proton by one of the lactate transporters MCT1 or MCT4 that are expressed in skeletal muscle cells (Juel & Halestrap 1999, Bishop et al. 2008). Again, under conditions of very high workload the arteriovenous difference of lactate concentration has been reported to increase to about 2 mmol L−1 within 30 s (Brechue & Stainsby 1994). With lower levels of exercise, increases in interstitial lactate concentration have also been observed though exhibiting slower kinetics and lower maximal concentrations (Rosendal et al. 2004). Though lactate has been shown to act principally as a dilator of vascular smooth muscle, a further study in humans demonstrated that interstitial lactate concentration as measured with microdialysis did not correlate with the changes in muscle blood flow at 30–60% of maximal work capacity (Lott et al. 2001), thus challenging its role as a major mediator of exercise hyperaemia under submaximal exercise.
Any muscle action potential leads to a release of potassium ions from skeletal muscle cells which under conditions of high action potential frequency cannot be compensated for by the re-uptake by the sodium potassium ATPase or the washout by the blood stream. As a result, interstitial potassium concentrations can quickly increase up to 10 mmol L−1 during early muscle exercise and remain elevated throughout exercise (Juel et al. 2000, Lott et al. 2001, Rosendal et al. 2004). This is reflected by an enhanced K+-efflux from working skeletal muscle which occurs early after initiation of exercise (Hallen 1996). The dilator effect of a moderate elevation of extracellular potassium concentration is, contrary to what one would assume intuitively according to the Nernst equation, due to a hyperpolarization of the smooth muscle cells resulting from an activation of KIR channels and of the Na+/K+ ATPase in smooth muscle cells (Knot et al. 1996, Zaritsky et al. 2000, Burns et al. 2004).
Furthermore, in exercising or hypoxic skeletal muscle an enhanced breakdown of adenosine triphosphate (ATP) occurs during the resynthesis of which, adenosine is formed. Part of it can probably be released from skeletal muscle cells and together with adenosine formed by the degradation of extracellular ATP (see below) may reach the cells of the arteriolar walls (Marshall 2007). Exogenous application of adenosine induces strong dilatation (Radegran et al. 2001, Murrant & Sarelius 2002, Thengchaisri et al. 2009, Heinonen et al. 2010, Roseguini et al. 2010). Furthermore, in humans, a strong correlation between leg blood flow during exercise and interstitial adenosine concentrations as measured by microdialysis has been reported (Hellsten et al. 1998). A mediatory role of adenosine is supported by the observation that the adenosine receptor antagonist theophylline reduced exercise hyperaemia in humans (Radegran & Calbet 2001), whereas in dogs, adenosine receptor blockade did not alter blood flow in exercising muscle (Koch et al. 1990). These discrepancies suggest that adenosine need not necessarily play an essential role in exercise hyperaemia. The role of adenosine as a mediator of reactive hyperaemia has recently also been challenged by the observation that at similar levels of hyperaemia the flow heterogeneity in muscle was significantly higher during adenosine infusion than during voluntary exercise in humans suggesting opening of physiological non-nutritive shunts in muscle by adenosine but not by exercise (Heinonen et al. 2010). Rather it may be an important mediator under experimental conditions that involve skeletal muscle hypoxia (see below).
Clearly, muscle-derived lactate, CO2, adenosine and potassium are formed during exercise and all can reach by diffusion into the vascular smooth muscle of adjacent arterioles as well as the pericytes located in the capillary areas. This makes a contribution to exercise hyperaemia likely and plausible but it is difficult to quantitate their respective contributions. This is at least in part because it seems likely that the local work load and hence oxygen demands are not homogeneously distributed throughout the muscle tissue. In turn, generation of these local signals will be non-homogeneous throughout the muscle, leading to differences in the effectiveness of each of these signalling pathways at different arteriolar or tissue locations. It remains also unclear whether these signalling molecules can reach by diffusion the larger vessels such as small feed arteries in sufficient amounts to allow for a coordinated response in arterioles and their feed arteries, especially as the latter tend to be ensheathed by thick layers of tissue and matrix proteins.
In recent years it has become, however, clear that these classical signals which have been known for many years are not the only ones generated by exercising muscle. Thus it is now established that skeletal muscle cells release increased amounts of cytokines (myokines) in response to long-term strenuous exercise (Pedersen 2009). For example plasma concentrations of IL-6 derived from exercising skeletal muscle cells increase up to a 100-fold during and shortly after exercise (Pedersen & Febbraio 2008). IL-6 has mainly metabolic effects in this context as it increases hepatic glucose production and lipolysis in adipocytes. However, though not yet studied, it may also exert local vasodilation due to its stimulating effects on AMP-activated protein kinase (AMPK) in skeletal muscle and secondary changes of its activity signals (Kelly et al. 2009). Interestingly, endothelial and smooth muscle cells also express AMPK (Goirand et al. 2007, Robertson et al. 2008, Fisslthaler &Fleming 2009, Kreutz&Pohl 2009). This enzyme has effects in both types of vascular cells which may contribute to local vasodilation via endothelial nitric oxide (NO) production (Fisslthaler&Fleming 2009) and decrease in smooth muscle calcium via activation of potassium channels (Kreutz & Pohl 2009), as well as calcium desensitization (Horman et al. 2008). A further muscle-derived myokine, IL-8, may contribute to the long-term effects of exercise on muscle angiogenesis (Li et al. 2003). NO represents a further muscle derived mediator. nNOS-derived NO seems to be involved in enhanced IL-6 production (Vuolteenaho et al. 2009). In addition, studies in mice have shown that the myosin light chain phosphorylation in vessels of isolated fast skeletal muscle was reduced upon electrical stimulation of the muscle. This reduction was significantly weaker in muscles from nNOS knockout mice indicating a direct smooth muscle relaxant effect [due to myosin light chain dephosphorylation (see below) of skeletal musclederived NO on juxtaposed vessels] (Grange et al. 2001).
Mediators from other sources
Nerves and adrenal medulla
Muscle contraction critically depends on the release of acetylcholine (ACh) localized in prejunctional vesicles of the neuromuscular end-plate. As ACh is historically the first compound that has been described being a stimulus of endothelial NO (EDRF) production (Furchgott & Zawadzki 1980) it is tempting to speculate that a spillover of ACh from neuromuscular end-plates during exercise may contribute to exercise hyperaemia in an endothelium-dependent manner. However, this issue is not yet resolved. Welsh & Segal (1998) observed an inhibitory effect of muscarinergic receptor blockade on local and conducted dilation in contracting muscle under certain conditions and the same group presented evidence that, statistically, neuromuscular end-plates in skeletal muscle may be located preferentially nearby arterioles (Pierzga & Segal 1994). In contrast, studies in humans using low concentrations of a competitive neuromuscular blocking agent and a muscarinergic receptor blocker resulted in an augmentation of blood flow rather than the expected reduction. (Hellsten et al. 2009).
Exercise, probably via activation of afferent fibres by mechanical and chemical stimulation, leads to an increase in sympathetic activity (Thomas & Segal 2004, O’Leary 2006). While muscle activity signals such as adenosine and NO inhibit to a large extent sympathetic vasoconstriction in the exercising muscle by prejunctional and postjunctional mechanisms (Cohen & Weisbrod 1988, Thomas & Segal 2004), the simultaneous stimulation of the adrenal medulla and of other vascular beds leads to increases in plasma levels of adrenaline and noradrenaline. Moderately increased concentrations of circulating adrenaline as observed during the initial phases of exercise are known to induce local vasodilation by activation of β2 adrenoceptors. However, as beta adrenergic receptors also play a role in skeletal muscle metabolism and also change cardiovascular adaptation to exercise in general, it is difficult to evaluate the isolated role of vascular β2 receptors in exercise hyperaemia. As non-selective and cardioselective beta-blocking agents exerted similar effects on exercise-induced muscle blood flow, a major role of this pathway is unlikely (Wolfel et al. 1986).
Endothelium and blood cells
Further potential mediators of exercise-induced hyperaemia are endothelium-derived factors like NO, prostacyclin and EDHF, which are known to be released in increased amounts from the endothelium under conditions of increased shear stress and to induce endothelium-dependent vasodilatation (Pohl et al. 1986, 2000, Huang et al. 2001, Watanabe et al. 2005, Loot et al. 2008), and, likely also, enhanced mechanical activation due to compression of vessels during muscle contractions (Sun et al. 2004). It is not clear yet whether the enhanced release upon squeezing of the vessels is a pure effect of endothelial deformation, comparable to that elicited by shear stress or whether this is due to an enhancement of specific integrin-mediated, matrix-dependent signalling. Martinez-Lemus et al. (2003) and Sun et al. (2005) convincingly showed that matrix dependent integrin mediated signalling can induce both vasoconstriction and vasodilation: the mediators, and the signalling mechanisms leading to dilation vs. constriction are still to be characterized. Furthermore, Hocking et al. (2008) have shown that the extracellular matrix protein fibronectin contributes to functional dilation, via a signalling mechanism that is apparently independent of integrin ligation. Thus there is a considerable amount yet to be learned about how mechanical signals originated by some aspect of contraction-related tissue deformation contribute to exercise hyperaemia. Irrespective of the mechanisms of its stimulated production, a role for NO has been clearly identified in local functional (muscle contraction-induced) hyperaemia (Sagach et al. 1992, Hester et al. 1993, Saito et al. 1994, Lau et al. 1998, 2000), but it is also clear from other studies (Barclay & Woodley 1994) that NO-dependent pathways do not account for all of the functional response, or indeed, under some conditions, for any of it. As mentioned before, measurements of cGMP activity and smooth muscle regulatory light chain phosphorylation in contracted skeletal muscle from nNOS and eNOS knockout mice implicate that not only endothelium-derived NO but also NO from the skeletal muscle is involved in functional dilation (Lau et al. 1998, 2000, Grange et al. 2001).
Blood-borne stimuli may also contribute to exercise-induced vasodilation. ATP is released in increased amounts from erythrocytes under conditions of reduced haemoglobin saturation and, possibly, also due to mechanical deformation of erythrocytes in rhythmically contracting muscle (Ellsworth et al. 2009). It is well known that ATP can induce an augmentation of NO production by activation of endothelial purinergic receptors of the P2Y type (Burnstock 2009).
Mediators of early vs. late responses
Blood flow to skeletal muscle increases in less than 5 s after at the onset of exercise (Naik et al. 1999, Clifford & Hellsten 2004, Mihok & Murrant 2004, Tschakovsky & Sheriff 2004). It has been implied that this is mainly due to rapid vasodilatation though it has also been argued that the muscle pump and the resulting changes in venous pressure contribute to the phenomenon as well (Tschakovsky & Sheriff 2004). Microvascular studies however support a role of rapid vasodilation by showing that arterioles dilate within seconds in response to skeletal muscle contraction (Armstrong et al. 2007a,b).
The vasodilator mechanism capable of responding rapidly enough to explain this rapid increase in blood flow with the beginning of exercise is not yet clear. Metabolic processes in general are likely to be too slow to account for the initial blood flow response whereas they may maintain steady-state vasodilation. Though decreases in arterial pO2 can induce rapid dilation (Pohl & Busse 1989), such a mechanism seems possible as a consequence of enhanced oxygen extraction but then the signal should be generated in capillaries which requires signal transfer to upstream vessels (Berg et al. 1997, Mitchell et al. 1997) as further discussed in a later section of this review.
Recently Armstrong et al. (2007b) have published a series of interesting experiments in which short and intermediate-term electrical stimulation of only a few muscle fibres in hamster skeletal muscle were performed. They showed that arterioles dilate even after a single, electrically induced fibre twitch. At low stimulus frequencies (4 Hz) a transient rapid dilatation was observed that peaked within 3–4 s of the contraction whereas higher stimulus frequencies (20–40 Hz) resulted in a biphasic dilatation with a second longer lasting peak occurring at approx. 20 s. Their results suggest that an increase in interstitial potassium could be responsible for the very early responses (Armstrong et al. 2007a). It remains unclear, however, to what extent this is a strictly local response or part of a coordinating mechanism. A more complex pattern emerged after a longer period (2 min) of electrical stimulation where nearly all potential muscle activity signals were shown to be involved in the dilation of nearby arterioles but to different degrees depending on altering stimulus frequency and duration as well as contraction frequency (i.e. metabolism).
Cellular mechanisms of smooth muscle relaxation by mediators
Blood flow in skeletal muscle is mainly regulated via changes in flow resistance in the intramuscular arterioles. The latter is primarily dependent on the arteriolar radius which is controlled by the contractile state (tone) of the vascular smooth muscle cells. Smooth muscle cell contraction occurs, like in other muscles, via interaction of actin and myosin but, unlike in skeletal and heart muscle, this interaction is controlled by the phosphorylation state of the regulatory light chain of the myosin head (Somlyo et al. 2004). There are at least two enzymes controlling the state of myosin light chain phosphorylation: the calcium-calmodulin myosin light chain kinase (MLCK) and the myosin light chain phosphatase (MLCP) which, in contrast to the MLCK, does not require acute increases in intracellular free calcium concentration to be activated. Therefore, vasodilation as a result of the decrease in myosin light chain phosphorylation can be achieved either by inhibiting the MLCK activity, mainly as a result of a decrease in intracellular free calcium concentration (Ca2+i) or by increasing MLCP activity in a calcium-‘independent’ manner (though the enzyme needs some calcium to be active). Consequently, any mechanism that leads to a decrease in Ca2+i will usually be followed by vasodilation. Decreases in Ca2+i are elicited by direct inhibition of L-type calcium channels (Cav1.2 in most arteriolar smooth muscle types) (Jackson 2000, Thorneloe & Nelson 2005), or by their indirect inhibition, e.g. by hyperpolarizing the smooth muscle cells via activation of potassium or chloride channels (Thorneloe & Nelson 2005), or via inhibition of TRPC channels (Dietrich et al. 2010). The latter play an important role in agonist induced (e.g. noradrenaline, UTP) constriction and indirectly activate L-type calcium channels by depolarization elicited by calcium influx (Reading et al. 2005). Furthermore, cyclic nucleotide dependent activation of SERCA (Serca2 isoform in VSM) or plasma membrane Ca2+ pumps as well as sodium calcium exchangers, also localized at the plasma membrane, can reduce Ca2+i (Wray et al. 2005). In particular, microvascular smooth muscle due to the high basal activity of calcium extrusion mechanisms from the cytosolic compartment needs a continuous supply of calcium from extravascular sources, otherwise the Ca2+i decreases.
In microvascular smooth muscle, vasodilation and MLC dephosphorylation can be also achieved when Ca2+i remains unaltered. This is due to an activation of MLCP, e.g. by NO, which, probably in a cGMP-dependent manner (though influences of the calcium level are also likely) elicits a rapid (within seconds) dephosphorylation of the CPI-17 subunit at Thr 38 and with some delay (minutes) phosphorylation of the subunit MYPT1 at Ser 695 thereby deinhibiting MLCP (Ito et al. 2004). It should be noted that cAMP-dependent inhibition ofMLCPhas also been reported (Ito et al. 2004). Conversely, vasoconstriction can be induced without increase in Ca2+i when the MLCP is inhibited, e.g. in a Rho/ROK/MYPT1 or ROK/CPI-17-dependent manner (Bolz et al. 2003, Puetz et al. 2009), as there is always a constitutive activity of MLCK at resting Ca2+i. Interestingly, so far none of the skeletal muscle derived activity signals has been studied with regard to their potential influence on the Rho/Rock pathway.
All these signals synergize in reducing arterial smooth muscle tone by interfering with one of the small number of cellular signalling pathways that produce vasodilation. Figure 1 summarizes the mechanisms of action of the ‘classical’ muscle activity signals at the cellular level. But, in addition, endothelial and blood cell-derived mediators as well as those potentially released from perivascular cells under conditions of exercise will use these cellular signalling pathways in additive, synergistic or competitive manners. It might therefore be more fruitful for our future understanding of the regulatory mechanisms of exercise hyperaemia to study the relative importance of certain smooth muscle-related cellular mechanisms of dilation rather than searching for the ‘principal mediator’.
Interaction of mediators
Considering the convergence of the mediators to so few subcellular mechanisms of vasodilation it is rather difficult to quantify the relative importance of any of these long known activity signals in the process of active or reactive hyperaemia. In fact, studies using inhibitors have consistently revealed that none of the activity signals described above acted as exclusive mediators for the induction or maintenance of exercise hyperaemia (Clifford & Hellsten 2004). Rather, nearly all of them reduced it only to some extent, leaving a very robust ‘remaining dilation’ unaffected; this could not be reduced by further inhibitors (Clifford & Hellsten 2004). A special caveat has to be made when it comes to the interpretation of these inhibitor studies, especially when inhibitors are used in combination. It is possible for example that inhibition of one of the several potent dilator mediators may already limit the capacity to increase blood flow to an extent that oxygen demands cannot be fully covered anymore so that muscle ischaemia occurs, a condition which is normally not seen in exercising muscle. Indeed in skeletal muscle of rats we have found that inhibition of NO production by systemic application of L-NAME induced partial hypoxia already under resting conditions (Pohl et al. 1993, Fig. 2). Therefore, it seems plausible that the robust ‘remaining vasodilation’ that was found to be insensitive to additional inhibitors is due to muscle hypoxia rather than due to exercise-induced release of activity signals. Though some mediators may be the same under both conditions, such as adenosine or lactate, others such as CO2 clearly are not.
Figure 2.

Effect of inhibition of NO synthesis by L-NNA on the pO2 distribution in rat skeletal muscle as assessed by repeated measurements with needle electrodes. (a) pO2 distribution which was obtained in the vastus muscle of anaesthetized rats under control conditions. Following acute inhibition of NO synthesis, the pO2 distribution was shifted to lower values (b) in the same tissue. Mean tissue pO2 decreased from 31.5 to 10.3 mmHg and nearly 25% of the pO2 values were in a very low range between 0 and 5 mm Hg (746 measurements in seven animals) (K.F. Wagner, Ch. Weiss & U. Pohl, unpublished).
Furthermore, any attempt to quantify the role of any single mediator is likely to be hampered by the occurrence of synergistic interactions among them. Surprisingly little is known about additive vs. synergistic effects of vasodilator compounds in the microcirculation, especially for those using the same cellular pathways for dilation or that influence each other’s second messenger levels in smooth muscle cells. An exception to this important lack of specific information is the interaction of cGMP and cAMP in elevating cellular responses to relevant signalling molecules that has been shown to exist in smooth muscle cells as well as in blood platelets (de Wit et al. 1994). For example, it has been shown that prostacyclin and NO, when applied together, induce a dilation of arterioles that exceeds the sum of the effects of each single compound. This is due to the inhibiting effect of cGMP on phosphodiesterase III which normally degrades cAMP, thereby maintaining a higher level of this second messenger. Hence, any inhibition of the prostaglandin effect simultaneously also reduces the effect of NO (de Wit et al. 1994).
From the above it is clear that although the muscle activity signals and endothelial mediators can contribute to active hyperaemia in the steady state of maintained, non-exhausting, exercise, their relative significance and contribution to the overall response remains an open question at present. It seems likely that their relative contributions will indeed vary with variations in physiological state of the tissue, as well as at different locations within the arteriolar tree. This is not to say that understanding these interrelationships among mechanisms will be impossible to elucidate: rather, it emphasizes areas upon which to focus in future studies. An area that we have not addressed in detail, but which contributes to the above considerations, is that of the temporal relationships between the different pathways. For example, it is still not clear what the early signals are immediately after onset of exercise which elicit vasodilation.
Regional inhomogeneities of local control
Even less is known about the local distribution and concentrations of these activity signals in the exercising muscle. Regional differences in oxygen consumption and force generation even in the same myocardial layer (epi- or endocardial) have been described in the heart muscle (Deussen et al. 2001), but not (yet) in the skeletal muscle. A potential source of uneven distribution of work load and oxygen demand is the individual activation of motor units across a muscle with varying recruitment levels. However, it has been suggested by mathematical modelling that an activation of only 20% of all motor units will probably lead to near-homogeneous increase in activity throughout the whole muscle tissue due to the very inhomogeneous distribution of the fibres of one motor unit in the skeletal muscle (Fuglevand& Segal 1997). The latter may be impaired however in older age, a condition of reduced exercise hyperaemia, when the motor units tend to become greater in size (Fuglevand & Segal 1997). However, even when one assumes a more or less homogeneous increase in the interstitial concentration of muscle activity signals, under physiological conditions, this does not necessarily mean homogeneity of vascular responses: some important targets, e.g. L-type calcium channels, seem to be unevenly distributed in skeletal muscle arterioles where they are found to be enriched in vascular branching points (Goligorsky et al. 1995). Furthermore, animal experiments demonstrate considerable differences in the distribution of alpha(1)- and alpha(2)-adrenoreceptors in skeletal muscle vessels, the latter being functionally more important in distal vessels (Anderson & Faber 1991).
Likewise, it is known that the endothelial factors released in response to shear stress and muscle contraction are unevenly distributed along the vascular tree, EDHF being more important in small arterioles than NO (Hoepfl et al. 2002). Finally, moderate concentrations of circulating adrenaline may induce local vasodilation by activation of β2 adrenoceptors. The latter are localized in the smooth muscle, and due to the tight barrier function of the arteriolar endothelial basal membrane to hydrophilic compounds adrenaline may predominantly leave the vascular bed in significant amounts only after having reached the more permeable veins from where it diffuses back into the smooth muscle of resistance arteries at sites of veno-arteriolar juxtaposition (Lew et al. 1989). Therefore, the effect of adrenaline may be inhomogeneous in the vascular network as well.
Thus, it seems reasonable to assume that the local action of activity signals is not sufficient to enable homogeneous increases in muscle blood flow but rather requires additional coordinating processes to achieve this goal.
Local integration of signals at the arteriolar wall
Contributing cell types
While vascular smooth muscle cells (VSMCs) are the primary effectors of vascular responses, it is clear that these cells are intimately influenced by the neighbouring cells of which the arteriolar wall is comprised. The role of endothelial cells (ECs) has been the target of most work (see earlier sections, and below) but it is important to recognize the potential role of other cells, such as pericytes, in contributing to local arteriolar responses. While there is effectively no information available at present whether and to what extent pericytes affect functional dilation in skeletal muscle arterioles, there is considerable evidence delineating the important contribution of this cell type to vascular responses in other tissues, particularly the brain (Fisher 2009). It thus appears likely that a role for these, and possibly other cell types contained within the vessel wall, will be identified once attention is directed towards this gap in our knowledge.
Roles for endothelial cells (ECs) in local arteriolar responses have been defined, and are a target of an ongoing and vigorous research effort, as discussed earlier in this review. As pointed out there, ECs are the source of a variety of signalling molecules that predominantly contribute to vasodilatory mechanisms and thus have the potential to play a significant role in the local arteriolar response to exercise. Though much current work suggests that EDHF(s) contribute more to local responses in small resistance arterioles than does NO, a role for EDHF in exercise hyperaemia still remains undefined. It is of interest that both nNOS and eNOS have been implicated in the local arteriolar response to muscle contraction. Both skeletal and vascular smooth muscle express nNOS (Lau et al. 2000, Han et al. 2007, Zhang et al. 2009), but it is not yet fully clear how this aspect of double NO generation contributes to the integrated response of the arteriolar wall resulting from contraction of skeletal muscle fibres. One ongoing source of uncertainty is that of how eNOS, or for that matter any EC-dependent signalling pathway, can contribute to a response to skeletal muscle contraction via signals arising from the abluminal (tissue) side of the in situ arteriole. For endothelial production of NO, we have mentioned earlier in this review the potential contributions of stimuli such as changed flow-dependent mechanical signals, but a concern that is relevant to this is the relative flow-insensitivity of small arterioles (Pohl & de Wit 1999, Pohl et al. 2000) which nevertheless exhibit an EC (NOS)-dependent component of the integrated response. Are there other signalling mechanisms coupled through ECs but independent of flow changes that could contribute to the exercise-dependent local arteriolar response? In support of a flow independent modulator role of the endothelium, the Sarelius group (Hocking et al. 2008) have recently identified an abluminally generated, tissue (ECM fibronectin)-dependent component of functional hyperaemia that acts via an NO-dependent pathway. In this case it seems somewhat unlikely that signalling molecules released from tissue would diffuse or otherwise be transported through all the structures of the vessel wall, in particular the tight basal lamina, to activate ECs that would then in turn communicate with the adjacent smooth muscle to produce the dilatory response. Nevertheless, there is evidence from direct observations of arterioles in contracting cremaster muscle that indicates that this dilation may be entirely dependent on an intact endothelium (Duza & Sarelius 2003, 2004). Furthermore, the Sarelius group has also shown that purinergic vasodilation, long identified as contributing to functional hyperaemia (Proctor & Duling 1982, Morff & Granger 1983, Murrant & Sarelius 2002, Clifford & Hellsten 2004, Marshall 2007), is mediated via an EC-coupled pathway (Duza & Sarelius 2003). Again, it is an interesting question how this response can be dependent on ECs when the relevant purinergic receptors have also been described on VSMCs, arguing that purines released from contracting skeletal muscle fibres should be available to occupy those receptors on VSMCs. Two, not necessarily mutually exclusive, scenarios might explain the integration of ECs and smooth muscle signals in the final control of vascular tone.
First, local morphological observations, which establish that in small arterioles there are endothelial projections through the basal lamina that enable intimate association with the underlying VSMCs (McSherry et al. 2006, Ledoux et al. 2008, Sandow et al. 2009), point towards a structure in the vessel wall that allows integration of signals coming from either side of the basal membrane. Recent work indicates that these EC processes are the site of localized signalling events that are dependent on the locally high densities of key signalling proteins in these regions. EC projections are enriched in (at least) KCa channels (McSherry et al. 2006, Dora et al. 2008), specific connexins (Isakson & Duling 2005, Isakson 2008, Sandow et al. 2009) and IP3 receptors (Isakson 2008, Ledoux et al. 2008) in a manner that supports their being specialized for myoendothelial communication. This form of local myoendothelial communication has been shown to facilitate transfer of hyperpolarizing signals from ECs to VSMCs and is strongly implicated in myoendothelial communication of ACh-dependent signals from ECs to VSMC. Indeed this constitutes one of the EDHF pathways that is prominent in resistance arterioles (Griffith et al. 2002, Sandow et al. 2002, Dora et al. 2003, 2008, Wolfle et al. 2009). This intimate morphology suggests a way in which signals arriving at the abluminal arteriolar surface might be coupled through an EC-dependent pathway. Thus, in this scenario, material released from contracting skeletal muscle fibres would diffuse into a region of the arteriolar wall occupied by both ECs and VSMCs, facilitating an integrated response from the two cell types. This idea remains to be tested.
Secondly, there is evidence that ECs tonically influence adjacent VSMCs in such a way as to maintain them for example, within an appropriate range of membrane potential that in turn determines their response capability. In fact such a mechanism has been shown to occur in isolated small arteries (Eskinder et al. 1990). While the idea of a tonic modulator or permissive influence of the endothelium is still speculative in part, there is supporting evidence in the literature for individual elements of this concept. It is well established, for example, that blockade of NO or EDHF results in increased vascular tone (de Wit et al. 1993, 1999, Shen et al. 1994), indicating that normally, ECs exert a tonic (in this case paracrine) influence on adjacent smooth muscle cells. This observation thus also serves to remind us of the simple but important concept that under normal physiological conditions, ECs have a role in the maintenance of a basal state of VSMCs. Furthermore, it is also very well established that changes in EC membrane potential can be directly communicated to adjacent VSMCs. This can occur via myoendothelial gap junction communication (Emerson & Segal 2000, Sandow et al. 2002) or, via local, paracrine, release of endothelial K+ into the tightly apposed space between these ECs and the adjacent VSMCs (Edwards et al. 1998). There is also evidence that removal of endothelium alters membrane potential in the underlying VSMCs, and importantly, thus alters their myogenic reactivity (Eskinder et al. 1990, see above). It has also been shown that the dilator effect of adenosine in exercising rat muscle depends on a tonic action but not a mediatory role of NO in smooth muscle (Ray & Marshall 2009). Thus, one can hypothesize that tonic signals from ECs might maintain VSMCs in a state wherein they are able to respond to appropriate occupancy of their surface receptors, e.g. by muscle activity signals. While this still partly speculative concept remains to be tested, it would certainly explain the intriguing observation that vasodilation in response to muscle contraction can be abolished by EC denudation in small arterioles (Duza & Sarelius 2004), when one might expect that muscle products such as adenosine, that are available to occupy receptors on vascular smooth muscle, should still be effective in producing vasodilation despite the absence of the endothelium.
It is established that gap junctional communication between adjacent ECs, between adjacent VSMCs, and between adjacent EC and VSMC pairs occurs readily in small arterioles, although this communication appears to involve different connexins in different sized arterioles and in arterioles from different tissues. This coupling has been shown, for example, from transfer of locally microinjected dyes between adjacent cells (Little et al. 1995), wherein it was also shown that this dye transfer is dependent on both charge and the ionic composition of the dye. This was an early indication that communication between the different cell types can be differentially regulated with respect to which gap junction proteins are activated, and with respect to the directionality of molecular transfer through the gap junction, and thus underlining yet another way in which communication among cells of the arteriolar wall can be regulated. Though it is not clear yet what controls gap junction permeability in vivo and though reportedly myoendothelial gap junctions may exhibit low permeability in vivo under some conditions (Siegl et al. 2005), recent studies in isolated arterioles have intriguingly indicated that communication from VSMCs to ECs can elevate EC calcium. Due to this transfer from contracting VSMC, NO is generated in the endothelium, thus feeding back to modulate VSMC function (Dora et al. 1997, Yashiro & Duling 2000), further illustrating the close coupling between these two cell types when in situ in the arteriolar wall. It is becoming clear that the myoendothelial signalling complex is an important microdomain, with specific expression of KCa channel isoforms, IP3 receptors and connexins as mentioned earlier. Changes in prevalent connexin expression may substantially alter control of the gap junction permeability. In cultured ECs it was shown that NO reduces permeability of Cx37-containing gap junctions whereas it increases that of Cx40-containing gap junctions (Hoffmann et al. 2003, Kameritsch et al. 2003). By far the majority of the in vivo studies define a role of gap junctions in vasodilatory signalling mechanisms that were induced by exposure to ACh, or, occasionally, vasoconstrictions to phenylephrine. Whether this myoendothelial communication occurs as part of the integrated local response to muscle exercise is not known, but it appears likely that some form of myoendothelial signalling will be involved.
Propagation or spread of responses along the arteriolar wall
Particularly during submaximal recruitment of motor units, contraction of the individual fibres of a motor unit, would, if no other integrating pathways were activated, result in local dilations of very restricted length. Such a restricted region of local dilation in turn would not support increased blood flow throughout the length of individual arterioles, let alone through an entire tissue region. However, it is now well established that responses initiated in one restricted arteriolar region can be propagated along the arteriolar wall to remote sites. This axial spread of locally induced dilation enables resistance to be decreased in an entire arteriole and beyond, thus enabling blood flow to be increased in all vessels into which the dilation is propagated. It is established for example, that dilations can be remotely propagated through arteriolar bifurcations into the next vessel upstream or downstream in the network (Berg et al. 1997, Rivers 1997). This conduction allows for a coordinated dilation of arterioles and their feed arteries, which is pivotal for an adequate increase in vascular conductivity, especially under conditions of high flow demands during exercise (de Wit 2004). In this context, conducted dilation can be considered a form of ‘axial integration’.
Most of what is known about mechanisms responsible for this propagated or ‘conducted’ response has been learned from studying responses to locally applied ACh. From this approach it has been shown that EDHF-initiated signals, depending initially on hyperpolarizations via KCa channels, account for a large proportion of the signals conducted axially in small arterioles (Hoepfl et al. 2002). Most often, this conduction appears to be via endothelially dependent, gap junctionally mediated spread of hyperpolarization between cells, but it has also been shown that ACh-dependent vasodilations can be propagated via VSMC-dependent signal transfer pathways (Welsh & Segal 1998, Budel et al. 2003), and it is not clear under what circumstances the endothelial vs. smooth muscle pathway is activated for propagation of a dilation to remote arteriolar sites. Both hyperpolarization and depolarization can be propagated (Xia & Duling 1995, Rodenwaldt et al. 2007), and there is evidence that KCa channels are necessary, at least in some vessels (Dora et al. 2008, Wolfle et al. 2009). There is also evidence for an NO-dependent component and of the NOS substrate, L-arginine in ACh-dependent conducted responses (Frame & Sarelius 1995, Doyle & Duling 1997), and it is also known that the remote dilation initiated by adenosine has features that distinguish it from that produced by ACh (de Wit 2010): Findings such as these indicate that there is more than one mechanism for the dilation to be spread to remote sites. Despite the considerable effort that has been expended in defining this response pathway, and the obvious conceptual utility of the response in enabling integration of local dilations into a wider tissue region, it remains unclear as to how and where these ACh-dependent propagated dilations might fit into the integrated exercise response, especially as ACh spillover from neuromuscular junctions seems not to be a significant source (Hellsten et al. 2009). However, it has been clearly established that propagated responses are indeed produced by muscle contraction (Berg et al. 1997, Cohen et al. 2000, Murrant & Sarelius 2000, Murrant & Sarelius 2002). These have been shown to have NO and KATP channel-dependent components, in contrast to the KCa channel dependence of remote responses to ACh (Hoepfl et al. 2002). Importantly, the remote dilations produced by local contraction of skeletal muscle fibres are not blocked by gap junctional uncouplers such as the glycyrrhetinic acids, whereas ACh-induced remote dilations in the identical vessels are blocked by these compounds (Murrant & Sarelius 2000), providing strong evidence that the mechanisms underlying the contraction-induced remote response are different from those responsible for the remote response to ACh. It is particularly compelling to note that the remote response initiated by contraction of muscle fibres has an apparently completely different K+ channel signature (KATP channel dependence) compared with the widely studied responses to ACh that as described earlier characteristically involve KCa channel activity. Another key feature of remote dilations is that they can be propagated over significant distances (millimetres) and across at least several generations of arterioles (Berg et al. 1997, de Wit et al. 2003), indicating that these responses have the capacity to increase conductance into wider tissue regions. There is also some evidence, in contrast to the ACh-induced response, that the response is unidirectional, being propagated only in an upstream direction (Berg et al. 1997). While this appears sensible with respect to mechanisms that might support the matching of increased flow to active tissue regions, there is as yet no information as to how this kind of directionality would be achieved in an intact network.
Another mechanism for axial integration of functional dilation is found in the flow-dependent responses alluded to earlier in our review. Importantly, these responses are more characteristic of larger arterioles, including the small distributing arteries carrying flow into the muscle. The presence of two complementary mechanisms in series enabling spread of dilator signals from small, to larger, to inflow arterioles clearly enables flow increases to be closely coupled to increased metabolism within the totality of the active tissue. Of note, both potentially coordinating mechanisms have different kinetics and may, therefore, play different roles in the development and maintenance of exercise hyperaemia.
Control of capillary recruitment
It has become widely accepted that the classic ‘capillary sphincter’ that is conceptualized as the controller of capillary perfusion and recruitment resides in the function of small arterioles. In general, the differential sensitivity and receptor distribution of small vs. larger arterioles, as discussed elsewhere in this review, has been cited as a principal mechanism by which the arterioles of the terminal microvascular network can differentially dilate and thus support capillary recruitment and increased capillary perfusion during increased tissue metabolism. Morphological studies of microvascular networks in skeletal muscles show that like other tissues, skeletal muscle capillaries are arranged in groups that are variously called units, networks, modules (Lund et al. 1987, Delashaw & Duling 1988, Berg & Sarelius 1995), and that capillaries are recruited as entire networks (modules) rather than as single capillaries (Berg & Sarelius 1996). This in turn suggests that control of perfusion of these modules likely occurs at the level of the arterioles from which they arise. Indeed, arteriolar controllers of these capillary groups have been identified (Sweeney & Sarelius 1989). Implicit in this scenario is the widely held expectation that changes in metabolism are sensed by these controlling arterioles which then respond with dilation, thus allowing increased perfusion of their downstream capillaries. While this mechanism undoubtedly contributes to capillary recruitment, this scenario necessarily includes the expectation that, particularly during submaximal recruitment of motor units, some of the downstream increase in capillary flow will be perfusing muscle fibre areas from as yet unrecruited motor units, thus effectively resulting in excess perfusion to those regions. And yet classic studies (Stainsby & Otis 1964, Bockman 1983, Saltin et al. 1998) show exquisite matching between increases in muscle work and the resulting blood flow, both in whole contracting muscles, and in regions within a single tissue (Mackie & Terjung 1983). The answer to this apparent contradiction is that an additional mechanism – that of remotely sensed and propagated responses – can also contribute to dilation of the arterioles proximal to areas requiring increased perfusion. It is established that capillaries can themselves sense vasoactive stimuli (Mitchell et al. 1997, McGahren et al. 1998), and that they have the capacity to respond by initiating remote (conducted) dilations in upstream arterioles. Of relevance to this review’s focus on mechanisms underlying functional hyperaemia is that it has also been demonstrated that contraction of a few muscle fibres underlying a single capillary module can initiate dilation in (at least) three generations of arterioles upstream (Berg et al. 1997), thus increasing flow only in capillaries overlying the metabolically active muscle fibres. Importantly, this work also showed that the signal was unidirectional, going upstream but not downstream, and also that no dilator signal was generated if the underlying skeletal muscle fibres were displaced mechanically but not actively contracted, directly implicating some aspect of active muscle contraction (release of metabolites? mechanical transducing signals?) in the signalling pathway for this response. Morphological analysis of microvascular networks indicates that one muscle fibre is likely to be perfused by several capillary networks (Berg & Sarelius 1995, Emerson & Segal 1997), thus this mechanism would serve to maximize flow to each active muscle fibre. Importantly, this mechanism enables capillaries in the vicinity of actively contracting muscle fibres to sense and respond to local ‘metabolic’ signals and thence generate a signal in the microvascular wall that results in upstream dilation, with consequently increased blood flow specifically in the relevant capillary network. In effect, the capillaries become responsible for their own recruitment. This mechanism enables coupling between metabolically generated signals and capillary recruitment to be achieved without the need to postulate a ‘feed forward’ signalling mechanism that would be required if all signal detection and response were initiated at the arteriolar level, making the arteriole both the sensor for, and controller of, the downstream event. In this context it may be of interest that pericytes isolated and cultured from hamster muscle show principally all features enabling them to act as potential ‘metabosensors’ near capillaries and generators of a conducted signal, including expression of hyperpolarizing K-channels which seem to be expressed in response to the muscle activity signal IL-8, and connexin43 potentially enabling them to couple with capillary endothelium (C. Mogensen, B. Bergneo, A. Ritter, S. d’Avis, O. Niuichule, P. Kanneritsche, T. Gloe, W. Nagel, U. Pohl, submiited).
Concluding remarks
This review highlights aspects of exercise-induced vasodilation with special reference to the mediators, their local and functional inhomogeneities and potential integrating mechanisms. We summarize new findings concerning signalling molecules that have been implicated in functional arteriolar dilations, and in particular, make the important point that there is clearly no single signalling molecule that drives the response. Rather it has to be concluded, that different combinations of the several key signalling pathways will dominate the response under different conditions, at different time points, or at different levels of the arteriolar tree or alternatively that their integration may differ. An important goal for future studies will be to understand the determinants that enable certain signalling pathways to differentially dominate the response. We also summarize mechanisms for integration of signals and responses among the cells of the intact arteriolar wall, both within the wall (between ECs and VSMCs) and ‘axially’ along the vascular tree (enabling the response to be manifest along an entire functional vessel path) and thus enabling the arteriole to specifically support increased flow to the relevant tissue region. Much of what is known so far about these integrating mechanisms has been defined in response to local application of ACh which may activate partially different signalling pathways than the signals derived from active muscle fibres. Though the concept of signal integration is intriguing, its specific role on the control of exercise hyperaemia and the consequences of its modulation under physiological and pathophysiological conditions still await additional analysis.
Acknowledgments
This work was supported in part by the 7FP EU integrated Network ‘Health benefits of exercise’ [European Union (FP7 IP Exgenesis LSHM-CT 2004-005272)] (U.P.) and the Deutsche Forschungsgemeinschaft (DFG Po414/2-1) (U.P.) and by National Institutes of Health Grant HL 76414 (I.S.).
Footnotes
Conflict of interest
There is no conflict of interest.
References
- Anderson KM, Faber JE. Differential sensitivity of arteriolar alpha 1- and alpha 2-adrenoceptor constriction to metabolic inhibition during rat skeletal muscle contraction. Circ Res. 1991;69:174–184. doi: 10.1161/01.res.69.1.174. [DOI] [PubMed] [Google Scholar]
- Armstrong ML, Dua AK, Murrant CL. Potassium initiates vasodilatation induced by a single skeletal muscle contraction in hamster cremaster muscle. J Physiol. 2007a;581:841–852. doi: 10.1113/jphysiol.2007.130013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong ML, Dua AK, Murrant CL. Time course of vasodilation at the onset of repetitive skeletal muscle contractions. Am J Physiol Regul Integr Comp Physiol. 2007b;292:R505–R515. doi: 10.1152/ajpregu.00381.2006. [DOI] [PubMed] [Google Scholar]
- Aucouturier J, Baker JS, Duche P. Fat and carbohydrate metabolism during submaximal exercise in children. Sports Med. 2008;38:213–238. doi: 10.2165/00007256-200838030-00003. [DOI] [PubMed] [Google Scholar]
- Barclay JK, Woodley NE. Nitric oxide synthase inhibitors do not alter functional hyperemia in canine skeletal muscle. Can J Physiol Pharmacol. 1994;72:1035–1041. doi: 10.1139/y94-145. [DOI] [PubMed] [Google Scholar]
- Berg BR, Sarelius IH. Functional capillary organization in striated muscle. Am J Physiol. 1995;268:H1215–H1222. doi: 10.1152/ajpheart.1995.268.3.H1215. [DOI] [PubMed] [Google Scholar]
- Berg BR, Sarelius IH. Erythrocyte flux in capillary networks during maturation: implications for oxygen delivery. Am J Physiol. 1996;271:H2263–H2273. doi: 10.1152/ajpheart.1996.271.6.H2263. [DOI] [PubMed] [Google Scholar]
- Berg BR, Cohen KD, Sarelius IH. Direct coupling between blood flow and metabolism at the capillary level in striated muscle. Am J Physiol. 1997;272:H2693–H2700. doi: 10.1152/ajpheart.1997.272.6.H2693. [DOI] [PubMed] [Google Scholar]
- Bertoldi D, Parzy E, Fromes Y, Wary C, Leroy-Willig A, Carlier PG. New insight into abnormal muscle vasodilatory responses in aged hypertensive rats by in vivo nuclear magnetic resonance imaging of perfusion. J Vasc Res. 2006;43:149–156. doi: 10.1159/000090944. [DOI] [PubMed] [Google Scholar]
- Bishop D, Edge J, Thomas C, Mercier J. Effects of high-intensity training on muscle lactate transporters and postexercise recovery of muscle lactate and hydrogen ions in women. Am J Physiol Regul Integr Comp Physiol. 2008;295:R1991–R1998. doi: 10.1152/ajpregu.00863.2007. [DOI] [PubMed] [Google Scholar]
- Bloor CM. Angiogenesis during exercise and training. Angiogenesis. 2005;8:263–271. doi: 10.1007/s10456-005-9013-x. [DOI] [PubMed] [Google Scholar]
- Bockman EL. Blood flow and oxygen consumption in active soleus and gracilis muscles in cats. Am J Physiol. 1983;244:H546–H551. doi: 10.1152/ajpheart.1983.244.4.H546. [DOI] [PubMed] [Google Scholar]
- Bolz SS, Vogel L, Sollinger D, Derwand R, de Wit C, Loirand G, Pohl U. Nitric oxide-induced decrease in calcium sensitivity of resistance arteries is attributable to activation of the myosin light chain phosphatase and antagonized by the RhoA/Rho kinase pathway. Circulation. 2003;107:3081–3087. doi: 10.1161/01.CIR.0000074202.19612.8C. [DOI] [PubMed] [Google Scholar]
- Boushel R, Langberg H, Green S, Skovgaard D, Bulow J, Kjaer M. Blood flow and oxygenation in peritendinous tissue and calf muscle during dynamic exercise in humans. J Physiol. 2000;524 (Pt 1):305–313. doi: 10.1111/j.1469-7793.2000.t01-2-00305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brechue WF, Stainsby WN. Lactate and acid-base exchange during brief intense contractions of skeletal muscle in situ. J Appl Physiol. 1994;77:223–230. doi: 10.1152/jappl.1994.77.1.223. [DOI] [PubMed] [Google Scholar]
- Budel S, Bartlett IS, Segal SS. Homocellular conduction along endothelium and smooth muscle of arterioles in hamster cheek pouch: unmasking an NO wave. Circ Res. 2003;93:61–68. doi: 10.1161/01.RES.0000080318.81205.FD. [DOI] [PubMed] [Google Scholar]
- Burns WR, Cohen KD, Jackson WF. K+-induced dilation of hamster cremasteric arterioles involves both the Na+/K+-ATPase and inward-rectifier K+ channels. Microcirculation. 2004;11:279–293. doi: 10.1080/10739680490425985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnstock G. Purinergic regulation of vascular tone and remodelling. Auton Autacoid Pharmacol. 2009;29:63–72. doi: 10.1111/j.1474-8673.2009.00435.x. [DOI] [PubMed] [Google Scholar]
- Clifford PS, Hellsten Y. Vasodilatory mechanisms in contracting skeletal muscle. J Appl Physiol. 2004;97:393–403. doi: 10.1152/japplphysiol.00179.2004. [DOI] [PubMed] [Google Scholar]
- Cohen RA, Weisbrod RM. Endothelium inhibits norepinephrine release from adrenergic nerves of rabbit carotid artery. Am J Physiol. 1988;254:H871–H878. doi: 10.1152/ajpheart.1988.254.5.H871. [DOI] [PubMed] [Google Scholar]
- Cohen KD, Berg BR, Sarelius IH. Remote arteriolar dilations in response to muscle contraction under capillaries. Am J Physiol Heart Circ Physiol. 2000;278:H1916–H1923. doi: 10.1152/ajpheart.2000.278.6.H1916. [DOI] [PubMed] [Google Scholar]
- Crimi E, Ignarro LJ, Cacciatore F, Napoli C. Mechanisms by which exercise training benefits patients with heart failure. Nat Rev Cardiol. 2009;6:292–300. doi: 10.1038/nrcardio.2009.8. [DOI] [PubMed] [Google Scholar]
- Delashaw JB, Duling BR. A study of the functional elements regulating capillary perfusion in striated muscle. Microvasc Res. 1988;36:162–171. doi: 10.1016/0026-2862(88)90016-7. [DOI] [PubMed] [Google Scholar]
- Deussen A, Lauer T, Loncar R, Kropp J. Heterogeneity of metabolic parameters in the left ventricular myocardium and its relation to local blood flow. Basic Res Cardiol. 2001;96:564–574. doi: 10.1007/s003950170008. [DOI] [PubMed] [Google Scholar]
- Dietrich A, Kalwa H, Gudermann T. TRPC channels in vascular cell function. Thromb Haemost. 2010;103:262–270. doi: 10.1160/TH09-08-0517. [DOI] [PubMed] [Google Scholar]
- Dora KA, Doyle MP, Duling BR. Elevation of intracellular calcium in smooth muscle causes endothelial cell generation of NO in arterioles. Proc Natl Acad Sci USA. 1997;94:6529–6534. doi: 10.1073/pnas.94.12.6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dora KA, Sandow SL, Gallagher NT, Takano H, Rummery NM, Hill CE, Garland CJ. Myoendothelial gap junctions may provide the pathway for EDHF in mouse mesenteric artery. J Vasc Res. 2003;40:480–490. doi: 10.1159/000074549. [DOI] [PubMed] [Google Scholar]
- Dora KA, Gallagher NT, McNeish A, Garland CJ. Modulation of endothelial cell KCa3.1 channels during endothelium-derived hyperpolarizing factor signaling in mesenteric resistance arteries. Circ Res. 2008;102:1247–1255. doi: 10.1161/CIRCRESAHA.108.172379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle MP, Duling BR. Acetylcholine induces conducted vasodilation by nitric oxide-dependent and -independent mechanisms. Am J Physiol. 1997;272:H1364–H1371. doi: 10.1152/ajpheart.1997.272.3.H1364. [DOI] [PubMed] [Google Scholar]
- Duza T, Sarelius IH. Conducted dilations initiated by purines in arterioles are endothelium dependent and require endothelial Ca2+ Am J Physiol Heart Circ Physiol. 2003;285:H26–H37. doi: 10.1152/ajpheart.00788.2002. [DOI] [PubMed] [Google Scholar]
- Duza T, Sarelius IH. Increase in endothelial cell Ca(2+) in response to mouse cremaster muscle contraction. J Physiol. 2004;555:459–469. doi: 10.1113/jphysiol.2003.051029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards G, Dora KA, Gardener MJ, Garland CJ, Weston AH. K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature. 1998;396:269–272. doi: 10.1038/24388. [DOI] [PubMed] [Google Scholar]
- Ellsworth ML, Ellis CG, Goldman D, Stephenson AH, Dietrich HH, Sprague RS. Erythrocytes: oxygen sensors and modulators of vascular tone. Physiology (Bethesda) 2009;24:107–116. doi: 10.1152/physiol.00038.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerson GG, Segal SS. Alignment of microvascular units along skeletal muscle fibers of hamster retractor. J Appl Physiol. 1997;82:42–48. doi: 10.1152/jappl.1997.82.1.42. [DOI] [PubMed] [Google Scholar]
- Emerson GG, Segal SS. Electrical coupling between endothelial cells and smooth muscle cells in hamster feed arteries: role in vasomotor control. Circ Res. 2000;87:474–479. doi: 10.1161/01.res.87.6.474. [DOI] [PubMed] [Google Scholar]
- Eskinder H, Harder DR, Lombard JH. Role of the vascular endothelium in regulating the response of small arteries of the dog kidney to transmural pressure elevation and reduced PO2. Circ Res. 1990;66:1427–1435. doi: 10.1161/01.res.66.5.1427. [DOI] [PubMed] [Google Scholar]
- Fisher M. Pericyte signaling in the neurovascular unit. Stroke. 2009;40:S13–S15. doi: 10.1161/STROKEAHA.108.533117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisslthaler B, Fleming I. Activation and signaling by the AMP-activated protein kinase in endothelial cells. Circ Res. 2009;105:114–127. doi: 10.1161/CIRCRESAHA.109.201590. [DOI] [PubMed] [Google Scholar]
- Frame MD, Sarelius IH. L-arginine-induced conducted signals alter upstream arteriolar responsivity to L-arginine. Circ Res. 1995;77:695–701. doi: 10.1161/01.res.77.4.695. [DOI] [PubMed] [Google Scholar]
- Frisbee JC, Delp MD. Vascular function in the metabolic syndrome and the effects on skeletal muscle perfusion: lessons from the obese Zucker rat. Essays Biochem. 2006;42:145–161. doi: 10.1042/bse0420145. [DOI] [PubMed] [Google Scholar]
- Fuglevand AJ, Segal SS. Simulation of motor unit recruitment and microvascular unit perfusion: spatial considerations. J Appl Physiol. 1997;83:1223–1234. doi: 10.1152/jappl.1997.83.4.1223. [DOI] [PubMed] [Google Scholar]
- Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288:373–376. doi: 10.1038/288373a0. [DOI] [PubMed] [Google Scholar]
- Goirand F, Solar M, Athea Y, Viollet B, Mateo P, Fortin D, Leclerc J, Hoerter J, Ventura-Clapier R, Garnier A. Activation of AMP kinase alpha1 subunit induces aortic vasorelaxation in mice. J Physiol. 2007;581:1163–1171. doi: 10.1113/jphysiol.2007.132589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goligorsky MS, Colflesh D, Gordienko D, Moore LC. Branching points of renal resistance arteries are enriched in L-type calcium channels and initiate vasoconstriction. Am J Physiol. 1995;268:F251–F257. doi: 10.1152/ajprenal.1995.268.2.F251. [DOI] [PubMed] [Google Scholar]
- Grange RW, Isotani E, Lau KS, Kamm KE, Huang PL, Stull JT. Nitric oxide contributes to vascular smooth muscle relaxation in contracting fast-twitch muscles. Physiol Genomics. 2001;5:35–44. doi: 10.1152/physiolgenomics.2001.5.1.35. [DOI] [PubMed] [Google Scholar]
- Griffith TM, Chaytor AT, Taylor HJ, Giddings BD, Edwards DH. cAMP facilitates EDHF-type relaxations in conduit arteries by enhancing electrotonic conduction via gap junctions. Proc Natl Acad Sci USA. 2002;99:6392–6397. doi: 10.1073/pnas.092089799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallen J. K+ balance in humans during exercise. Acta Physiol Scand. 1996;156:279–286. doi: 10.1046/j.1365-201X.1996.187000.x. [DOI] [PubMed] [Google Scholar]
- Han G, Ma H, Chintala R, Miyake K, Fulton DJ, Barman SA, White RE. Nongenomic, endothelium- independent effects of estrogen on human coronary smooth muscle are mediated by type I (neuronal) NOS and PI3-kinase-Akt signaling. Am J Physiol Heart Circ Physiol. 2007;293:H314–H321. doi: 10.1152/ajpheart.01342.2006. [DOI] [PubMed] [Google Scholar]
- Haram PM, Kemi OJ, Wisloff U. Adaptation of endothelium to exercise training: insights from experimental studies. Front Biosci. 2008;13:336–346. doi: 10.2741/2683. [DOI] [PubMed] [Google Scholar]
- Heinonen I, Kemppainen J, Kaskinoro K, Peltonen JE, Borra R, Lindroos MM, Oikonen V, Nuutila P, Knuuti J, Hellsten Y, Boushel R, Kalliokoski KK. Comparison of exogenous adenosine and voluntary exercise on human skeletal muscle perfusion and perfusion heterogeneity. J Appl Physiol. 2010;108:378–386. doi: 10.1152/japplphysiol.00745.2009. [DOI] [PubMed] [Google Scholar]
- Hellsten Y, Maclean D, Radegran G, Saltin B, Bangsbo J. Adenosine concentrations in the interstitium of resting and contracting human skeletal muscle. Circulation. 1998;98:6–8. doi: 10.1161/01.cir.98.1.6. [DOI] [PubMed] [Google Scholar]
- Hellsten Y, Krustrup P, Iaia FM, Secher NH, Bangsbo J. Partial neuromuscular blockade in humans enhances muscle blood flow during exercise independently of muscle oxygen uptake and acetylcholine receptor blockade. Am J Physiol Regul Integr Comp Physiol. 2009;296:R1106–R1112. doi: 10.1152/ajpregu.90477.2008. [DOI] [PubMed] [Google Scholar]
- Hester RL, Eraslan A, Saito Y. Differences in EDNO contribution to arteriolar diameters at rest during functional dilation in striated muscle. Am J Physiol. 1993;265:H146–H151. doi: 10.1152/ajpheart.1993.265.1.H146. [DOI] [PubMed] [Google Scholar]
- Hocking DC, Titus PA, Sumagin R, Sarelius IH. Extracellular matrix fibronectin mechanically couples skeletal muscle contraction with local vasodilation. Circ Res. 2008;102:372–379. doi: 10.1161/CIRCRESAHA.107.158501. [DOI] [PubMed] [Google Scholar]
- Hoepfl B, Rodenwaldt B, Pohl U, de Wit C. EDHF, but not NO or prostaglandins, is critical to evoke a conducted dilation upon ACh in hamster arterioles. Am J Physiol Heart Circ Physiol. 2002;283:H996–H1004. doi: 10.1152/ajpheart.01082.2001. [DOI] [PubMed] [Google Scholar]
- Hoffmann A, Gloe T, Pohl U, Zahler S. Nitric oxide enhances de novo formation of endothelial gap junctions. Cardiovasc Res. 2003;60:421–430. doi: 10.1016/j.cardiores.2003.04.001. [DOI] [PubMed] [Google Scholar]
- Horman S, Morel N, Vertommen D, Hussain N, Neumann D, Beauloye C, El NN, Forcet C, Viollet B, Walsh MP, Hue L, Rider MH. AMP-activated protein kinase phosphorylates and desensitizes smooth muscle myosin light chain kinase. J Biol Chem. 2008;283:18505–18512. doi: 10.1074/jbc.M802053200. [DOI] [PubMed] [Google Scholar]
- Huang A, Sun D, Carroll MA, Jiang H, Smith CJ, Connetta JA, Falck JR, Shesely EG, Koller A, Kaley G. EDHF mediates flow-induced dilation in skeletal muscle arterioles of female eNOS-KO mice. Am J Physiol Heart Circ Physiol. 2001;280:H2462–H2469. doi: 10.1152/ajpheart.2001.280.6.H2462. [DOI] [PubMed] [Google Scholar]
- Isakson BE. Localized expression of an Ins(1,4,5)P3 receptor at the myoendothelial junction selectively regulates heterocellular Ca2+ communication. J Cell Sci. 2008;121:3664–3673. doi: 10.1242/jcs.037481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isakson BE, Duling BR. Heterocellular contact at the myoendothelial junction influences gap junction organization. Circ Res. 2005;97:44–51. doi: 10.1161/01.RES.0000173461.36221.2e. [DOI] [PubMed] [Google Scholar]
- Ito M, Nakano T, Erdodi F, Hartshorne DJ. Myosin phosphatase: structure, regulation and function. Mol Cell Biochem. 2004;259:197–209. doi: 10.1023/b:mcbi.0000021373.14288.00. [DOI] [PubMed] [Google Scholar]
- Jackson WF. Ion channels and vascular tone. Hypertension. 2000;35:173–178. doi: 10.1161/01.hyp.35.1.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juel C, Halestrap AP. Lactate transport in skeletal muscle - role and regulation of the monocarboxylate transporter. J Physiol. 1999;517(Pt 3):633–642. doi: 10.1111/j.1469-7793.1999.0633s.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juel C, Pilegaard H, Nielsen JJ, Bangsbo J. Interstitial K(+) in human skeletal muscle during and after dynamic graded exercise determined by microdialysis. Am J Physiol Regul Integr Comp Physiol. 2000;278:R400–R406. doi: 10.1152/ajpregu.2000.278.2.R400. [DOI] [PubMed] [Google Scholar]
- Kameritsch P, Hoffmann A, Pohl U. Opposing effects of nitric oxide on different connexins expressed in the vascular system. Cell Commun Adhes. 2003;10:305–309. doi: 10.1080/cac.10.4-6.305.309. [DOI] [PubMed] [Google Scholar]
- Kelly M, Gauthier MS, Saha AK, Ruderman NB. Activation of AMP-activated protein kinase by interleukin-6 in rat skeletal muscle: association with changes in cAMP, energy state, and endogenous fuel mobilization. Diabetes. 2009;58:1953–1960. doi: 10.2337/db08-1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiens B. Skeletal muscle lipid metabolism in exercise and insulin resistance. Physiol Rev. 2006;86:205–243. doi: 10.1152/physrev.00023.2004. [DOI] [PubMed] [Google Scholar]
- Klitzman B, Duling BR. Microvascular hematocrit and red cell flow in resting and contracting striated muscle. Am J Physiol. 1979;237:H481–H490. doi: 10.1152/ajpheart.1979.237.4.H481. [DOI] [PubMed] [Google Scholar]
- Knot HJ, Zimmermann PA, Nelson MT. Extracellular K(+)-induced hyperpolarizations and dilatations of rat coronary and cerebral arteries involve inward rectifier K(+) channels. J Physiol. 1996;492(Pt 2):419–430. doi: 10.1113/jphysiol.1996.sp021318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch LG, Britton SL, Metting PJ. Adenosine is not essential for exercise hyperaemia in the hindlimb in conscious dogs. J Physiol. 1990;429:63–75. doi: 10.1113/jphysiol.1990.sp018244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreutz CP, Pohl U. Activation of the AMP-activated protein kinase induces dilation in hamster microvessels. Acta Physiol. 2009;195(Suppl 669):O208. (Abstract) [Google Scholar]
- Lalande S, Gusso S, Hofman PL, Baldi JC. Reduced leg blood flow during submaximal exercise in type 2 diabetes. Med Sci Sports Exerc. 2008;40:612–617. doi: 10.1249/MSS.0b013e318161aa99. [DOI] [PubMed] [Google Scholar]
- Lau KS, Grange RW, Chang WJ, Kamm KE, Sarelius I, Stull JT. Skeletal muscle contractions stimulate cGMP formation and attenuate vascular smooth muscle myosin phosphorylation via nitric oxide. FEBS Lett. 1998;431:71–74. doi: 10.1016/s0014-5793(98)00728-5. [DOI] [PubMed] [Google Scholar]
- Lau KS, Grange RW, Isotani E, Sarelius IH, Kamm KE, Huang PL, Stull JT. nNOS and eNOS modulate cGMP formation and vascular response in contracting fast-twitch skeletal muscle. Physiol Genomics. 2000;2:21–27. doi: 10.1152/physiolgenomics.2000.2.1.21. [DOI] [PubMed] [Google Scholar]
- Laughlin MH, Korthuis RJ, Duncker DJ, Bache RJ. Control of blood flow to cardiac and skeletal muscle during exercise. In: Rowell LB, Shepherd JT, editors. Handbook of Pathophysiology. Regulation and Integration of Multiple Systems. Section 12. Oxford University Press; New York: Oxford: Published for the American Physiological Society; 1996. pp. 705–769. [Google Scholar]
- Ledoux J, Taylor MS, Bonev AD, Hannah RM, Solodushko V, Shui B, Tallini Y, Kotlikoff MI, Nelson MT. Functional architecture of inositol 1,4,5-trisphosphate signaling in restricted spaces of myoendothelial projections. Proc Natl Acad Sci USA. 2008;105:9627–9632. doi: 10.1073/pnas.0801963105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lew MJ, Rivers RJ, Duling BR. Arteriolar smooth muscle responses are modulated by an intramural diffusion barrier. Am J Physiol. 1989;257:H10–H16. doi: 10.1152/ajpheart.1989.257.1.H10. [DOI] [PubMed] [Google Scholar]
- Li A, Dubey S, Varney ML, Dave BJ, Singh RK. IL-8 directly enhanced endothelial cell survival, proliferation, and matrix metalloproteinases production and regulated angiogenesis. J Immunol. 2003;170:3369–3376. doi: 10.4049/jimmunol.170.6.3369. [DOI] [PubMed] [Google Scholar]
- Little TL, Xia J, Duling BR. Dye tracers define differential endothelial and smooth muscle coupling patterns within the arteriolar wall. Circ Res. 1995;76:498–504. doi: 10.1161/01.res.76.3.498. [DOI] [PubMed] [Google Scholar]
- Loot AE, Popp R, Fisslthaler B, Vriens J, Nilius B, Fleming I. Role of cytochrome P450-dependent transient receptor potential V4 activation in flow-induced vasodilatation. Cardiovasc Res. 2008;80:445–452. doi: 10.1093/cvr/cvn207. [DOI] [PubMed] [Google Scholar]
- Lott ME, Hogeman CS, Vickery L, Kunselman AR, Sinoway LI, MacLean DA. Effects of dynamic exercise on mean blood velocity and muscle interstitial metabolite responses in humans. Am J Physiol Heart Circ Physiol. 2001;281:H1734–H1741. doi: 10.1152/ajpheart.2001.281.4.H1734. [DOI] [PubMed] [Google Scholar]
- Lund N, Damon DH, Damon DN, Duling BR. Capillary grouping in hamster tibialis anterior muscles: flow patterns, and physiological significance. Int J Microcirc Clin Exp. 1987;5:359–372. [PubMed] [Google Scholar]
- Mackie BG, Terjung RL. Blood flow to different skeletal muscle fiber types during contraction. Am J Physiol. 1983;245:H265–H275. doi: 10.1152/ajpheart.1983.245.2.H265. [DOI] [PubMed] [Google Scholar]
- Marshall JM. The roles of adenosine and related substances in exercise hyperaemia. J Physiol. 2007;583:835–845. doi: 10.1113/jphysiol.2007.136416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Lemus LA, Wu X, Wilson E, Hill MA, Davis GE, Davis MJ, Meininger GA. Integrins as unique receptors for vascular control. J Vasc Res. 2003;40:211–233. doi: 10.1159/000071886. [DOI] [PubMed] [Google Scholar]
- McGahren ED, Beach JM, Duling BR. Capillaries demonstrate changes in membrane potential in response to pharmacological stimuli. Am J Physiol. 1998;274:H60–H65. doi: 10.1152/ajpheart.1998.274.1.H60. [DOI] [PubMed] [Google Scholar]
- McSherry IN, Sandow SL, Campbell WB, Falck JR, Hill MA, Dora KA. A role for heterocellular coupling and EETs in dilation of rat cremaster arteries. Microcirculation. 2006;13:119–130. doi: 10.1080/10739680500466400. [DOI] [PubMed] [Google Scholar]
- Mihok ML, Murrant CL. Rapid biphasic arteriolar dilations induced by skeletal muscle contraction are dependent on stimulation characteristics. Can J Physiol Pharmacol. 2004;82:282–287. doi: 10.1139/y04-016. [DOI] [PubMed] [Google Scholar]
- Mitchell D, Yu J, Tyml K. Comparable effects of arteriolar and capillary stimuli on blood flow in rat skeletal muscle. Microvasc Res. 1997;53:22–32. doi: 10.1006/mvre.1996.1988. [DOI] [PubMed] [Google Scholar]
- Mohrman DE, Regal RR. Relation of blood flow to VO2, PO2, and PCO2 in dog gastrocnemius muscle. Am J Physiol. 1988;255:H1004–H1010. doi: 10.1152/ajpheart.1988.255.5.H1004. [DOI] [PubMed] [Google Scholar]
- Morff RJ, Granger HJ. Contribution of adenosine to arteriolar autoregulation in striated muscle. Am J Physiol. 1983;244:H567–H576. doi: 10.1152/ajpheart.1983.244.4.H567. [DOI] [PubMed] [Google Scholar]
- Mueller PJ. Exercise training and sympathetic nervous system activity: evidence for physical activity dependent neural plasticity. Clin Exp Pharmacol Physiol. 2007;34:377–384. doi: 10.1111/j.1440-1681.2007.04590.x. [DOI] [PubMed] [Google Scholar]
- Murrant CL, Sarelius IH. Local and remote arteriolar dilations initiated by skeletal muscle contraction. Am J Physiol Heart Circ Physiol. 2000;279:H2285–H2294. doi: 10.1152/ajpheart.2000.279.5.H2285. [DOI] [PubMed] [Google Scholar]
- Murrant CL, Sarelius IH. Multiple dilator pathways in skeletal muscle contraction-induced arteriolar dilations. Am J Physiol Regul Integr Comp Physiol. 2002;282:R969–R978. doi: 10.1152/ajpregu.00405.2001. [DOI] [PubMed] [Google Scholar]
- Naik JS, Valic Z, Buckwalter JB, Clifford PS. Rapid vasodilation in response to a brief tetanic muscle contraction. J Appl Physiol. 1999;87:1741–1746. doi: 10.1152/jappl.1999.87.5.1741. [DOI] [PubMed] [Google Scholar]
- O’Leary DS. Altered reflex cardiovascular control during exercise in heart failure: animal studies. Exp Physiol. 2006;91:73–77. doi: 10.1113/expphysiol.2005.031179. [DOI] [PubMed] [Google Scholar]
- Pedersen BK. Edward F. Adolph distinguished lecture: muscle as an endocrine organ: IL-6 and other myokines. J Appl Physiol. 2009;107:1006–1014. doi: 10.1152/japplphysiol.00734.2009. [DOI] [PubMed] [Google Scholar]
- Pedersen BK, Febbraio MA. Muscle as an endocrine organ: focus on muscle-derived interleukin-6. Physiol Rev. 2008;88:1379–1406. doi: 10.1152/physrev.90100.2007. [DOI] [PubMed] [Google Scholar]
- Philp A, Macdonald AL, Watt PW. Lactate – a signal coordinating cell and systemic function. J Exp Biol. 2005;208:4561–4575. doi: 10.1242/jeb.01961. [DOI] [PubMed] [Google Scholar]
- Pierzga JM, Segal SS. Spatial relationships between neuromuscular junctions and microvessels in hamster cremaster muscle. Microvasc Res. 1994;48:50–67. doi: 10.1006/mvre.1994.1038. [DOI] [PubMed] [Google Scholar]
- Pohl U, Busse R. Hypoxia stimulates release of endothelium-derived relaxant factor. Am J Physiol. 1989;256:H1595–H1600. doi: 10.1152/ajpheart.1989.256.6.H1595. [DOI] [PubMed] [Google Scholar]
- Pohl U, de Wit C. A unique role of NO in the control of blood flow. News Physiol Sci. 1999;14:74–80. doi: 10.1152/physiologyonline.1999.14.2.74. [DOI] [PubMed] [Google Scholar]
- Pohl U, Holtz J, Busse R, Bassenge E. Crucial role of endothelium in the vasodilator response to increased flow in vivo. Hypertension. 1986;8:37–44. doi: 10.1161/01.hyp.8.1.37. [DOI] [PubMed] [Google Scholar]
- Pohl U, Wagner K, de Wit C. Endothelium-derived nitric oxide in the control of tissue perfusion and oxygen supply: physiological and pathophysiological implications. Eur Heart J. 1993;14(Suppl I):93–98. [PubMed] [Google Scholar]
- Pohl U, de Wit C, Gloe T. Large arterioles in the control of blood flow: role of endothelium-dependent dilation. Acta Physiol Scand. 2000;168:505–510. doi: 10.1046/j.1365-201x.2000.00702.x. [DOI] [PubMed] [Google Scholar]
- Proctor KG, Duling BR. Adenosine and free-flow functional hyperemia in striated muscle. Am J Physiol. 1982;242:H688–H697. doi: 10.1152/ajpheart.1982.242.4.H688. [DOI] [PubMed] [Google Scholar]
- Proctor DN, Shen PH, Dietz NM, Eickhoff TJ, Lawler LA, Ebersold EJ, Loeffler DL, Joyner MJ. Reduced leg blood flow during dynamic exercise in older endurance-trained men. J Appl Physiol. 1998;85:68–75. doi: 10.1152/jappl.1998.85.1.68. [DOI] [PubMed] [Google Scholar]
- Puetz S, Lubomirov LT, Pfitzer G. Regulation of smooth muscle contraction by small GTPases. Physiology (Bethesda) 2009;24:342–356. doi: 10.1152/physiol.00023.2009. [DOI] [PubMed] [Google Scholar]
- Radegran G, Calbet JA. Role of adenosine in exercise-induced human skeletal muscle vasodilatation. Acta Physiol Scand. 2001;171:177–185. doi: 10.1046/j.1365-201x.2001.00796.x. [DOI] [PubMed] [Google Scholar]
- Rao RP, Danduran MJ, Frommelt PC, Ghanayem NS, Berger S, Simpson PM, Yan K, offman GM. Measurement of regional tissue bed venous weighted oximetric trends during exercise by near infrared spectroscopy. Pediatr Cardiol. 2009;30:465–471. doi: 10.1007/s00246-009-9393-6. [DOI] [PubMed] [Google Scholar]
- Ray CJ, Marshall JM. Nitric oxide (NO) does not contribute to the generation or action of adenosine during exercise hyperaemia in rat hindlimb. J Physiol. 2009;587:1579–1591. doi: 10.1113/jphysiol.2008.163691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reading SA, Earley S, Waldron BJ, Welsh DG, Brayden JE. TRPC3 mediates pyrimidine receptorinduced depolarization of cerebral arteries. Am J Physiol Heart Circ Physiol. 2005;288:H2055–H2061. doi: 10.1152/ajpheart.00861.2004. [DOI] [PubMed] [Google Scholar]
- Ribisl PM, Lang W, Jaramillo SA, Jakicic JM, Stewart KJ, Bahnson J, Bright R, Curtis JF, Crow RS, Soberman JE. Exercise capacity and cardiovascular/metabolic characteristics of overweight and obese individuals with type 2 diabetes: the Look AHEAD clinical trial. Diabetes Care. 2007;30:2679–2684. doi: 10.2337/dc06-2487. [DOI] [PubMed] [Google Scholar]
- Richardson RS, Noyszewski EA, Leigh JS, Wagner PD. Lactate efflux from exercising human skeletal muscle: role of intracellular PO2. J Appl Physiol. 1998;85:627–634. doi: 10.1152/jappl.1998.85.2.627. [DOI] [PubMed] [Google Scholar]
- Rivers RJ. Cumulative conducted vasodilation within a single arteriole and the maximum conducted response. Am J Physiol. 1997;273:H310–H316. doi: 10.1152/ajpheart.1997.273.1.H310. [DOI] [PubMed] [Google Scholar]
- Robertson TP, Mustard KJ, Lewis TH, Clark JH, Wyatt CN, Blanco EA, Peers C, Hardie DG, Evans AM. AMP-activated protein kinase and hypoxic pulmonary vasoconstriction. Eur J Pharmacol. 2008;595:39–43. doi: 10.1016/j.ejphar.2008.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodenwaldt B, Pohl U, de Wit C. Endogenous and exogenous NO attenuates conduction of vasoconstrictions along arterioles in the microcirculation. Am J Physiol Heart Circ Physiol. 2007;292:H2341–H2348. doi: 10.1152/ajpheart.01061.2006. [DOI] [PubMed] [Google Scholar]
- Roseguini BT, Davis MJ, Harold LM. Rapid vasodilation in isolated skeletal muscle arterioles: impact of branch order. Microcirculation. 2010;17:83–93. doi: 10.1111/j.1549-8719.2009.00005.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosendal L, Blangsted AK, Kristiansen J, Sogaard K, Langberg H, Sjogaard G, Kjaer M. Interstitial muscle lactate, pyruvate and potassium dynamics in the trapezius muscle during repetitive low-force arm movements, measured with microdialysis. Acta Physiol Scand. 2004;182:379–388. doi: 10.1111/j.1365-201X.2004.01356.x. [DOI] [PubMed] [Google Scholar]
- Sagach VF, Kindybalyuk AM, Kovalenko TN. Functional hyperemia of skeletal muscle: role of endothelium. J Cardiovasc Pharmacol. 1992;20(Suppl 12):S170–S175. doi: 10.1097/00005344-199204002-00048. [DOI] [PubMed] [Google Scholar]
- Saito Y, Eraslan A, Hester RL. Role of endothelium-derived relaxing factors in arteriolar dilation during muscle contraction elicited by electrical field stimulation. Microcirculation. 1994;1:195–201. doi: 10.3109/10739689409148274. [DOI] [PubMed] [Google Scholar]
- Saltin B, Radegran G, Koskolou MD, Roach RC. Skeletal muscle blood flow in humans and its regulation during exercise. Acta Physiol Scand. 1998;162:421–436. doi: 10.1046/j.1365-201X.1998.0293e.x. [DOI] [PubMed] [Google Scholar]
- Sandow SL, Tare M, Coleman HA, Hill CE, Parkington HC. Involvement of myoendothelial gap junctions in the actions of endothelium-derived hyperpolarizing factor. Circ Res. 2002;90:1108–1113. doi: 10.1161/01.res.0000019756.88731.83. [DOI] [PubMed] [Google Scholar]
- Sandow SL, Haddock RE, Hill CE, Chadha PS, Kerr PM, Welsh DG, Plane F. What’s where and why at a vascular myoendothelial microdomain signalling complex. Clin Exp Pharmacol Physiol. 2009;36:67–76. doi: 10.1111/j.1440-1681.2008.05076.x. [DOI] [PubMed] [Google Scholar]
- Shen W, Lundborg M, Wang J, Stewart JM, Xu X, Ochoa M, Hintze TH. Role of EDRF in the regulation of regional blood flow and vascular resistance at rest and during exercise in conscious dogs. J Appl Physiol. 1994;77:165–172. doi: 10.1152/jappl.1994.77.1.165. [DOI] [PubMed] [Google Scholar]
- Siegl D, Koeppen M, Wolfle SE, Pohl U, de Wit C. Myoendothelial coupling is not prominent in arterioles within the mouse cremaster microcirculation in vivo. Circ Res. 2005;97:781–788. doi: 10.1161/01.RES.0000186193.22438.6c. [DOI] [PubMed] [Google Scholar]
- Somlyo AV, Khromov AS, Webb MR, Ferenczi MA, Trentham DR, He ZH, Sheng S, Shao Z, Somlyo AP. Smooth muscle myosin: regulation and properties. Philos Trans R Soc Lond B Biol Sci. 2004;359:1921–1930. doi: 10.1098/rstb.2004.1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stainsby WN, Otis AB. Blood flow, blood oxygen tension, oxygen uptake, and oxygen transport in skeletal muscle. Am J Physiol. 1964;206:858–866. doi: 10.1152/ajplegacy.1964.206.4.858. [DOI] [PubMed] [Google Scholar]
- Street D, Bangsbo J, Juel C. Interstitial pH in human skeletal muscle during and after dynamic graded exercise. J Physiol. 2001;537:993–998. doi: 10.1111/j.1469-7793.2001.00993.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun D, Huang A, Kaley G. Mechanical compression elicits NO-dependent increases in coronary flow. Am J Physiol Heart Circ Physiol. 2004;287:H2454–H2460. doi: 10.1152/ajpheart.00364.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Martinez-Lemus LA, Trache A, Trzeciakowski JP, Davis GE, Pohl U, Meininger GA. Mechanical properties of the interaction between fibronectin and alpha5beta1-integrin on vascular smooth muscle cells studied using atomic force microscopy. Am J Physiol Heart Circ Physiol. 2005;289:H2526–H2535. doi: 10.1152/ajpheart.00658.2004. [DOI] [PubMed] [Google Scholar]
- Sweeney TE, Sarelius IH. Arteriolar control of capillary cell flow in striated muscle. Circ Res. 1989;64:112–120. doi: 10.1161/01.res.64.1.112. [DOI] [PubMed] [Google Scholar]
- Thengchaisri N, Miriel VA, Rivers RJ. Multiple receptor subtypes and multiple mechanisms of dilation are involved in vascular network dilation caused by adenosine. Microvasc Res. 2009;77:356–363. doi: 10.1016/j.mvr.2009.01.004. [DOI] [PubMed] [Google Scholar]
- Thomas GD, Segal SS. Neural control of muscle blood flow during exercise. J Appl Physiol. 2004;97:731–738. doi: 10.1152/japplphysiol.00076.2004. [DOI] [PubMed] [Google Scholar]
- Thorneloe KS, Nelson MT. Ion channels in smooth muscle: regulators of intracellular calcium and contractility. Can J Physiol Pharmacol. 2005;83:215–242. doi: 10.1139/y05-016. [DOI] [PubMed] [Google Scholar]
- Tschakovsky ME, Sheriff DD. Immediate exercise hyperemia: contributions of the muscle pump vs. rapid vasodilation. J Appl Physiol. 2004;97:739–747. doi: 10.1152/japplphysiol.00185.2004. [DOI] [PubMed] [Google Scholar]
- Van Beekvelt MC, Shoemaker JK, Tschakovsky ME, Hopman MT, Hughson RL. Blood flow and muscle oxygen uptake at the onset and end of moderate and heavy dynamic forearm exercise. Am J Physiol Regul Integr Comp Physiol. 2001;280:R1741–R1747. doi: 10.1152/ajpregu.2001.280.6.R1741. [DOI] [PubMed] [Google Scholar]
- Vuolteenaho K, Koskinen A, Kukkonen M, Nieminen R, Paivarinta U, Moilanen T, Moilanen E. Leptin enhances synthesis of proinflammatory mediators in human osteoarthritic cartilage – mediator role of NO in leptin-induced PGE2, IL-6, and IL-8 production. Mediators Inflamm. 2009;2009:345838. doi: 10.1155/2009/345838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S, Yashiro Y, Mizuno R, Ohhashi T. Involvement of NO and EDHF in flow-induced vasodilation in isolated hamster cremasteric arterioles. J Vasc Res. 2005;42:137–147. doi: 10.1159/000083652. [DOI] [PubMed] [Google Scholar]
- Welsh DG, Segal SS. Endothelial and smooth muscle cell conduction in arterioles controlling blood flow. Am J Physiol. 1998;274:H178–H186. doi: 10.1152/ajpheart.1998.274.1.H178. [DOI] [PubMed] [Google Scholar]
- de Wit C. Connexins pave the way for vascular communication. News Physiol Sci. 2004;19:148–153. doi: 10.1152/nips.01520.2004. [DOI] [PubMed] [Google Scholar]
- de Wit C. Different pathways with distinct properties conduct dilations in the microcirculation in vivo. Cardiovasc Res. 2010;85:604–613. doi: 10.1093/cvr/cvp340. [DOI] [PubMed] [Google Scholar]
- de Wit C, von Bismarck P, Pohl U. Mediator role of prostaglandins in acetylcholine-induced vasodilation and control of resting vascular diameter in the hamster cremaster microcirculation in vivo. J Vasc Res. 1993;30:272–278. doi: 10.1159/000159006. [DOI] [PubMed] [Google Scholar]
- de Wit C, von Bismarck P, Pohl U. Synergistic action of vasodilators that increase cGMP and cAMP in the hamster cremaster microcirculation. Cardiovasc Res. 1994;28:1513–1518. doi: 10.1093/cvr/28.10.1513. [DOI] [PubMed] [Google Scholar]
- de Wit C, Esser N, Lehr HA, Bolz SS, Pohl U. Pentobarbital-sensitive EDHF comediates ACh-induced arteriolar dilation in the hamster microcirculation. Am J Physiol. 1999;276:H1527–H1534. doi: 10.1152/ajpheart.1999.276.5.H1527. [DOI] [PubMed] [Google Scholar]
- de Wit C, Roos F, Bolz SS, Pohl U. Lack of vascular connexin 40 is associated with hypertension and irregular arteriolar vasomotion. Physiol Genomics. 2003;13:169–177. doi: 10.1152/physiolgenomics.00169.2002. [DOI] [PubMed] [Google Scholar]
- Wolfel EE, Hiatt WR, Brammell HL, Carry MR, Ringel SP, Travis V, Horwitz LD. Effects of selective and nonselective beta-adrenergic blockade on mechanisms of exercise conditioning. Circulation. 1986;74:664–674. doi: 10.1161/01.cir.74.4.664. [DOI] [PubMed] [Google Scholar]
- Wolfle SE, Schmidt VJ, Hoyer J, Kohler R, de Wit C. Prominent role of KCa3.1 in endothelium-derived hyperpolarizing factor-type dilations and conducted responses in the microcirculation in vivo. Cardiovasc Res. 2009;82:476–483. doi: 10.1093/cvr/cvp060. [DOI] [PubMed] [Google Scholar]
- Wray S, Burdyga T, Noble K. Calcium signalling in smooth muscle. Cell Calcium. 2005;38:397–407. doi: 10.1016/j.ceca.2005.06.018. [DOI] [PubMed] [Google Scholar]
- Xia J, Duling BR. Electromechanical coupling and the conducted vasomotor response. Am J Physiol. 1995;269:H2022–H2030. doi: 10.1152/ajpheart.1995.269.6.H2022. [DOI] [PubMed] [Google Scholar]
- Xiang L, Naik JS, Hodnett BL, Hester RL. Altered arachidonic acid metabolism impairs functional vasodilation in metabolic syndrome. Am J Physiol Regul Integr Comp Physiol. 2006;290:R134–R138. doi: 10.1152/ajpregu.00295.2005. [DOI] [PubMed] [Google Scholar]
- Yashiro Y, Duling BR. Integrated Ca(2+) signaling between smooth muscle and endothelium of resistance vessels. Circ Res. 2000;87:1048–1054. doi: 10.1161/01.res.87.11.1048. [DOI] [PubMed] [Google Scholar]
- Zaritsky JJ, Eckman DM, Wellman GC, Nelson MT, Schwarz TL. Targeted disruption of Kir2.1 and Kir2.2 genes reveals the essential role of the inwardly rectifying K(+) current in K(+)-mediated vasodilation. Circ Res. 2000;87:160–166. doi: 10.1161/01.res.87.2.160. [DOI] [PubMed] [Google Scholar]
- Zhang GX, Kimura S, Murao K, Shimizu J, Matsuyoshi H, Takaki M. Role of neuronal NO synthase in regulating vascular superoxide levels and mitogen-activated protein kinase phosphorylation. Cardiovasc Res. 2009;81:389–399. doi: 10.1093/cvr/cvn304. [DOI] [PubMed] [Google Scholar]