

Abstract
Vestibular schwannomas (VSs) result from inactivating mutations in the merlin tumor suppressor gene. The merlin protein suppresses a variety of progrowth kinase–signaling cascades, including extracellular regulated kinase/mitogen-activated protein kinase (ERK/MAPK), c-Jun N-terminal kinase (JNK), and phosphatidyl-inositol 3-kinase (PI3-K)/Akt. Recent studies indicate that ERKs and Akt are active in human VSs, and here we show that JNKs are also persistently active in human VS cells. With use of cultures of human VSs, we investigated the contribution of each of these signals to the proliferative and survival response of VS cells. Inhibition of ERK or Akt signaling reduced VS cell proliferation but did not increase apoptosis, whereas inhibition of JNK with SP600125, I-JIP, or siRNA knock-down reduced VS cell proliferation and survival by inducing apoptosis. By contrast, JNK activity promotes apoptosis in normal Schwann cells. Inhibition of JNK increased the fluorescence intensity of VS cells loaded with 5-(and-6)-chloromethyl-2',7'-dichlorodihydrofluorescein diacetate (H2DCFDA), a fluorescent probe for reactive oxygen species (ROS). Furthermore, ebselen, a ROS scavenger, rescued VS cells with suppressed JNK from apoptosis, suggesting that JNK activity protects VS cells from apoptosis by limiting accumulation of ROS. VS cultures treated with JNK inhibitors demonstrated significantly higher levels of MitoSOX Red fluorescence, implying that persistent JNK activity specifically suppresses superoxide production in the mitochondria. Overexpression of superoxide dismutase 2 (MnSOD; mitochondrial SOD) prevented apoptosis in VS cells with suppressed JNK signaling. Taken together, these results indicate that persistent JNK activity enhances VS cell survival, at least in part, by suppressing accumulation of mitochondrial superoxides.
Keywords: acoustic neuroma, apoptosis, cell proliferation, cell signaling, merlin, reactive oxygen species
Vestibular schwannomas (VSs) represent benign tumors that arise from the Schwann cells (SCs) lining the vestibular nerve and comprise 10% of all intracranial neoplasms. VSs occur in either sporadic or familial (neurofibromatosis type 2 [NF2]) forms. Both are associated with defects in the tumor suppressor gene, schwannomin (Sch)/merlin.1–3 Germline defects in merlin, as occur in NF2, result in bilateral VSs and multiple other intracranial and spinal tumors, including schwannomas.2,4
Recent investigations have identified potential mechanisms by which lack of functional merlin protein promotes tumor growth. Merlin inhibits several intracellular signals implicated in cell proliferation and tumor formation, including Ras, Rac1/Cdc42, RhoA, Src, Raf, p21-activated kinases 1 and 2 (PAK1/2), mTORC1, extracellular regulated kinase/mitogen-activated protein kinase (ERK/MAPK), c-Jun N-terminal kinase (JNK), and phosphatidyl-inositol 3-kinase (PI3-K)/Akt.5–15 In addition to its effects on intracellular signals, merlin also regulates receptor tyrosine kinase trafficking and activity, including platelet-derived growth factor (PDGF) receptor,8 epidermal growth factor receptor (EGFR/ErbB1),16 and ErbB2.13,17 Because of limited access and ability to culture primary human tumors, most of these studies used transformed cell lines or cultures derived from merlin-deficient mice to examine the consequences of merlin deficiency on cell growth. Thus, the extent to which these mechanisms contribute to the development and growth of human VSs, the primary tumors resulting from merlin deficiency, is unknown.
We have begun to investigate the merlin-sensitive signaling cascades that contribute to the growth potential of human VS cells. With use of tumor lysates and primary cultures derived from acutely resected human VSs, we found that JNK is persistently phosphorylated in VS cells and confirmed that this phosphorylation is regulated by merlin status. Along with mitogen activated protein kinase kinase (MAPKK, MEK) MEK/ERK and PI3-K/Akt signaling, JNK activity promotes VS cell proliferation. In addition, JNK activity appears to protect VS cells from apoptosis by suppressing superoxide production in the mitochondria. Thus, in contrast to normal SCs, in which activation of JNK leads to apoptosis after denervation,18–20 persistent JNK activity in VS cells appears to contribute to their ability to grow and survive in the absence of axons.
Experimental procedures
VS Collection
The institutional review board of the University of Iowa (Iowa City) approved the study protocol. VS and greater auricular nerve specimens from separate patients undergoing neck dissection were collected after surgical removal and immediately placed in ice-cold Hank's balanced salt solution until used for cultures or snap frozen in liquid nitrogen until used for protein extracts.
Primary VS Cell Cultures
Primary VS cultures were prepared as described elsewhere.21,22 Cultures were maintained in serum-free conditions for at least 24 h before experimental use. More than 90% of the cells in the cultures were S100 positive. Ebselen (EMD Bioscience) and/or kinase inhibitors (including PD98059, U0126, LY294002, and SP600125 [JNK Inhibitor II]; cell permeable I-JIP [JNK Inhibitor VII] and PD158780 [all from EMD Bioscience]) were added to the indicated cultures 4 h before experimental manipulation and were maintained throughout the duration of the experiment.
For adenoviral-mediated gene transfer, VS cultures were incubated with AdCMVEmpty (AdCon), AdSOD2 (superoxide dismutase 2, MnSOD), AdGFP, or Ad.Merlin.GFP (each at 2 × 108 pfu/mL) manufactured by Viraquest. Treatment of parallel cultures with AdGFP confirmed that >85% of VS cells are transduced by adenoviral vectors at this titer (data not shown). Experimental manipulation was started 48 h after viral transduction to allow time for transgene expression.
siRNA knock-down of JNK1/2 was accomplished by transfecting cultures with an oligonucleotide that targets both JNK1 and 2 (#6232; Cell Signaling) using Lipofecta mine RNAiMax Transfection Reagent (#13778-075; Invitrogen), as described elsewhere.23 The siRNA sequence is GGAGCUCAAGGAAUAGUAU. Control cultures were transfected with a scrambled oligonucleotide (#6568; Cell Signaling). The scrambled RNA sequence is CGUACGCGGAAUACUUCGA. The final RNA concentration was 10 nM. Protein lysates were prepared 48 h after transfection, and the cultures were fixed 48 h after transfection prior to immunolabeling. The experiment was repeated on cultures derived from 4 separate patients.
Western Blotting
Western blots of protein extracts prepared from VS tissue or culture lysates were preformed as described elsewhere.22,24 The primary antibodies used were anti-phosphorylated JNK (pJNK; Cell Signaling #9251), pERK (Cell Signaling #9106), pAKT (Cell Signaling #9721), JNK1 (Santa Cruz sc-474), JNK2 (Cell Signaling #4672), ERK(Cell Signaling #9102), Akt (Cell Signaling #9272), pJUN (Cell Signaling #9261), and Rho-GDI (dilution, 1:1000). Secondary antibodies (dilution,1:5000–1:50,000; Santa Cruz) were conjugated with horseradish-peroxidase. Blots were developed using Super Signal West Femto kit (Thermo Scientific) and exposed to film (Amersham Hyperfilm TM ECL; GE Healthcare). As needed, membranes were stripped and re-probed with other antibody combinations. Digital images of gels were captured on an Alpha Innotech gel imaging system, and band pixel intensity was quantified using Image J software (National Institutes of Health).
Immunostaining
Small fragments of VS or normal vestibular nerve specimens (removed for treatment of intractable Meniere's disease or temporal bone malignancies not affecting the inner ear) were fixed in 4% paraformaldehyde, cryoprotected in serial sucrose gradients, mounted in OCT, and frozen sections (6–10 µm) were made on a cryostat. Immunostaining was performed as described elsewhere21,22 (Hansen 2006, Hansen 2008) with the following primary antibodies: anti-pJNK (1:500), anti-pJUN (1:500), anti-BrdU (1:800; clone G3G4, Hybridoma core, University of Iowa, Iowa City, IA), anti-cleaved caspase 3 (1:400; Cell Signaling #9664S), and anti-S100 (dilution, 1:400; Sigma # S-2644). Alexa 488–, Alexa 546–, Alexa 568–, and Alexa 647–labeled secondary antibodies (dilution, 1:800; Invitrogen) were used as indicated. Nuclei were labeled with Hoechst 33342 (10 µg/mL; Sigma) for 15 min.
Fluorescence images were captured using an inverted Leica DMRIII microscope (Leica) equipped with epifluorescence filters and a charge-coupled device camera using Leica FW4000 software or with a Leica SP5 confocal microscope and prepared for publication using Adobe Photoshop.
VS Cell Proliferation, Apoptosis, and Survival
Because of the limited number of cells that can be cultured from each tumor, we were not able to use flow cytometry or to perform clonogenic or 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assays to assay cell proliferation or apoptosis. Rather we scored the percent of BrdU-positive and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)–positive VS cells to determine the extent of proliferation and apoptosis, respectively, as described elsewhere.21,22 Only S100-positive cells were scored. Given the variability in basal proliferation and apoptotic rates for each primary tumor, the percent of BrdU-positive and TUNEL-positive VS cells was expressed as a percentage of the control condition, defined as 100%. Apoptosis was confirmed in a subset of cultures by immunostaining for cleaved caspase 3. Each condition was repeated on ≥3 VS cultures derived from separate tumors.
For cell survival, cultures were maintained in the presence or absence of the inhibitors for 5 days total, with culture medium exchanged every 2 days. After fixation and immunostaining, the number of S100-positive cells with intact nuclei was scored for 10 randomly selected 20× fields for each well. The percentage of surviving VS cells was expressed as a percentage of the control condition, defined as 100%. The conditions were performed in duplicate and repeated on ≥3 VS cultures derived from separate tumors.
Detection of Reactive Oxygen Species
VS cultures in 4-well slides were loaded with 25 µM of CM-H2DCFDA (Invitrogen) for 30–45 min at 37°C, in accordance with the manufacturer's instructions. During the final 5 min, Hoechst 3342 (1.0 µM) was added to label nuclei. JNK inhibitors (SP600125 or I-JIP), with or without ebselen, were added to the indicated cultures 4–5 h before imaging. Digital epifluorescent images were captured from live cells using a Leica DMRIII inverted scope, with exposure times set to a linear range on the basis of cultures in control conditions and cells from cultures treated with 0.03% H2O2 or antimycin A (10 µM; Sigma). Fluorescence intensity was quantified from a circular selection in the cytoplasm of at least 30–50 cells using Image J software for each condition, and the experiment was repeated on cultures from ≥4 separate tumors. Background fluorescence, determined from a similarly sized circle placed outside cell boundaries, was subtracted from each image.
To detect mitochondrial superoxide accumulation, VS cultures on 4 well glass coverslips were loaded with MitoSOX Red mitochondrial superoxide indicator (1 µM; Invitrogen) for 10 min at 37°C in accordance with the manufacturer's instructions. JNK inhibitors were added to the indicated cultures 4 h before MitoSOX loading and were maintained throughout the experiment. Fluorescent digital images were captured using a Leica SP5 confocal microscope, and fluorescent intensity was quantified as above using Image J software.
Data and Statistical Analysis
Numerical data were managed and graphed in Excel (Microsoft), and SigmaStat (Systat Software) was used for statistical analyses.
Results
JNK is Persistently Phosphorylated in VS Cells
Merlin suppresses the activity of several kinase-signaling cascades implicated in tumorigenesis, including MEK/ERK, PI3-K/Akt, and JNK.5–11 Recent studies have demonstrated that PI3-K/Akt, MEK/ERK, and JNKs are persistently active in human VS cells that lack functional merlin.3,25–27 To confirm that JNK is active in VS cells, we immunostained frozen sections from acutely resected VSs and normal vestibular nerve with antibodies that recognize phosphorylated JNK (pJNK) and with anti-S100 antibodies to specifically label VS cells. S100-positive cells displayed pJNK labeling, indicating that JNK is phosphorylated in VS cells in vivo (Fig. 1). Conversely, pJNK immunoreactivity was not detectable in the SCs of the normal vestibular nerve. Scarpa's ganglion (vestibular) neurons demonstrated punctate pJNK immunoreactivity (Fig. 1). Immunostaining with anti-neurofilament 200 (NF200) antibodies verified that the cells with punctate pJNK immunoreactivity were neuronal. Thus, JNK is constitutively phosphorylated in vestibular schwannoma cells and in vestibular neurons, but not normal vestibular SCs, in vivo.
Fig. 1.
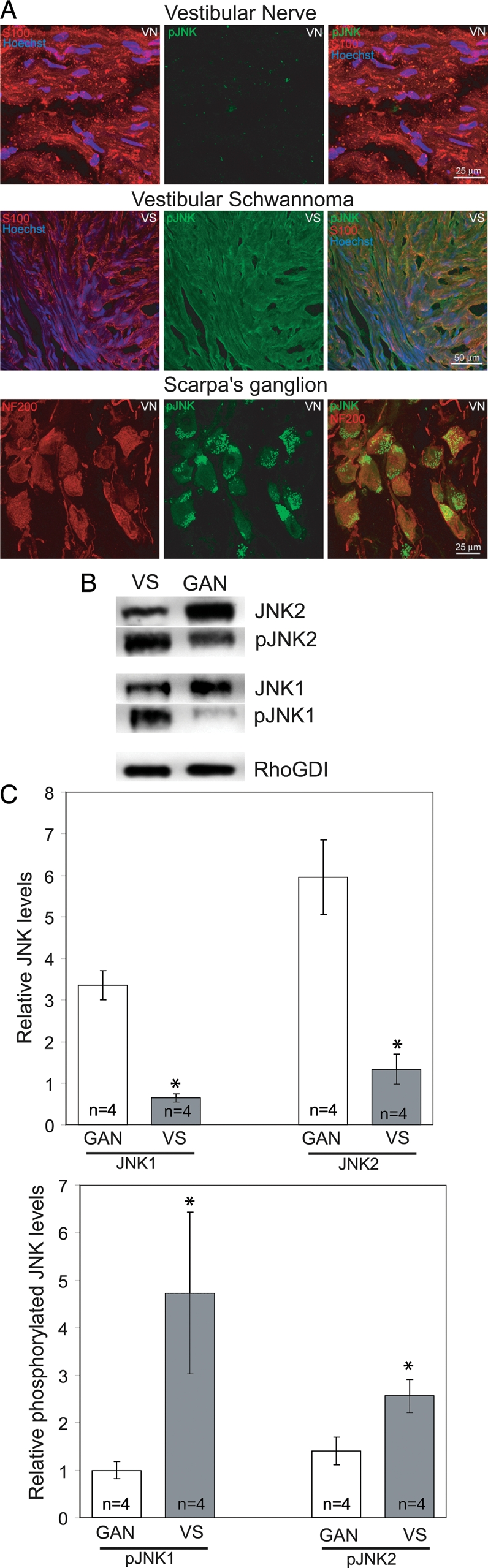
c-Jun N-terminal kinase (JNK) is phosphorylated in vestibular schwannoma (VS) tissue but not Schwann cells (SCs) from mature nerve. (A) Confocal images of VS or vestibular nerve (VN) frozen sections immunostained with anti-phosphorylated JNK (anti-pJNK; green) and anti-S100 (red) or anti-NF200 (red) antibodies. pJNK immunoreactivity is detected in VS cells but not VN SCs. Punctate pJNK immunoreactivity appears in the neurons but not the SCs of the VN in the region of Scarpa's ganglion. Scale bars = 25 µm. (B) Representative western blots of VS or great auricular nerve (GAN) lysates probed with antibodies against pJNK, JNK1/2, or Rho-GDI. (C) Mean relative expression levels of JNK1/2 and pJNK from 4 VS and 4 GAN specimens. Error bars present standard error of the mean. *P < .05, by the Student's 2-tailed t test.
To compare the extent of JNK phosphorylation in VSs, we probed immunoblots of protein lysates from acutely resected VSs with anti-pJNK antibodies. Protein lysates from acutely resected great auricular nerve (GAN) specimens served as a control to determine the extent of JNK phosphorylation in mature SCs. The blots were stripped and reprobed with non–phospho-specific antibodies and subsequently with an antibody against Rho-GDI, a protein with comparable expression in SCs and schwannoma cells.25 Band intensity was quantified by densitometry, and the levels of pJNK1 and pJNK2 were compared with those of nonphosphorylated JNK 1 and 2. The overall levels of JNK 1 and 2 were compared to Rho-GDI to determine whether there are differences in kinase expression in VS tissue versus in normal nerves. Data were quantified for 4 VS and 4 GAN specimens and are representative of results from 4 additional VS and 3 GAN specimens. As shown in Fig. 1, both JNK1 and JNK2 are expressed at significantly higher levels in the GAN versus VS tissue. The extent of JNK1—and, to a lesser extent, JNK2 phosphorylation—was significantly greater in VS than GAN tissue. Thus, JNK is persistently active in VS cells, compared with normal SCs.
Increased JNK phosphorylation in VS compared with normal nerve tissue suggests that merlin suppresses JNK activity. To confirm that merlin regulates JNK activity, we treated primary VS cultures with an adenoviral vector expressing wild-type merlin (Ad.Merlin.GFP). Control cultures were treated with AdGFP. Immunoblots of protein lysates from the cultures were probed with anti-pJNK to evaluate the extent of JNK activity. Blots were stripped and reprobed with non–phospho-specific anti-JNK1/2, anti-merlin, and anti-RhoGDI antibodies to confirm equal protein loading and successful gene transfer. As shown in Fig. 2, replacement of wild-type merlin in primary VS cells suppresses JNK phosphorylation.
Fig. 2.
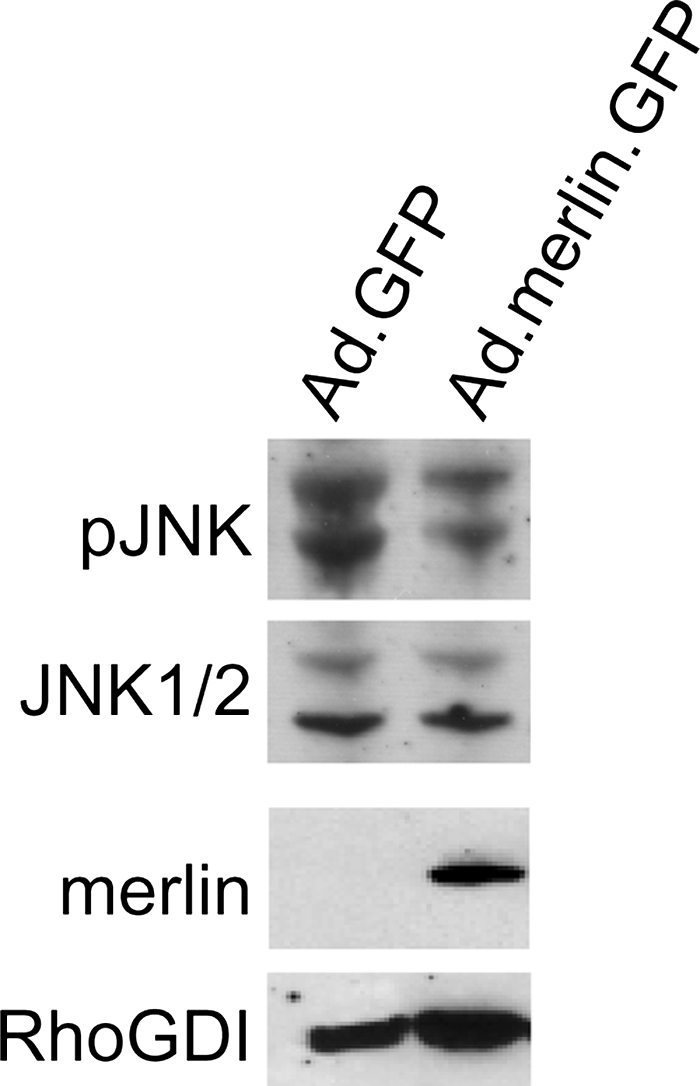
Replacement of merlin in vestibular schwannoma (VS) cells reduces c-Jun N-terminal kinase (JNK) phosphorylation and activity. Western blot of VS cell lysates from cultures treated with Ad.GFP or Ad.Merlin.GFP vectors and probed with the indicated antibodies.
To further explore ERK, Akt, and JNK signaling in VS cells, we also probed immunoblots prepared from primary human VS cell cultures maintained in basal medium without serum with anti-pERK, anti-pAkt, and anti-pJNK antibodies. Consistent with observations in tumor lysates (Fig. 1),26 cultured VS cells demonstrate persistent basal phosphorylation of these kinases (Fig. 3). ERK phosphorylation was specifically suppressed by the MEK inhibitor U0126 (10 µM), and Akt phosphorylation was suppressed by the PI3-K inhibitor LY294002 (20 µM). Because we could not obtain sufficient amount of protein lysate from primary VS cultures to probe multiple panels of antibodies, we stripped and reprobed the blots with either ERK (Fig. 3) or AKT (Fig. 3) non–phospho-specific antibodies to verify equal expression of the most relevant kinases. We also probed lysates with anti-β-actin to verify equal protein loading (Fig. 3). These blots are representative of similar results obtained from cultures derived from ≥3 separate VSs for each condition. On the basis of immunostaining findings, pJNK distributes diffusely in the cytoplasm and nucleus in cultured VS cells (Fig. 3). Thus, cultured primary VS cells retain active ERK, Akt, and JNK similar to their counterparts in vivo, suggesting that they represent a realistic model to study the contribution of these signals to VS tumorigenesis.
Fig. 3.
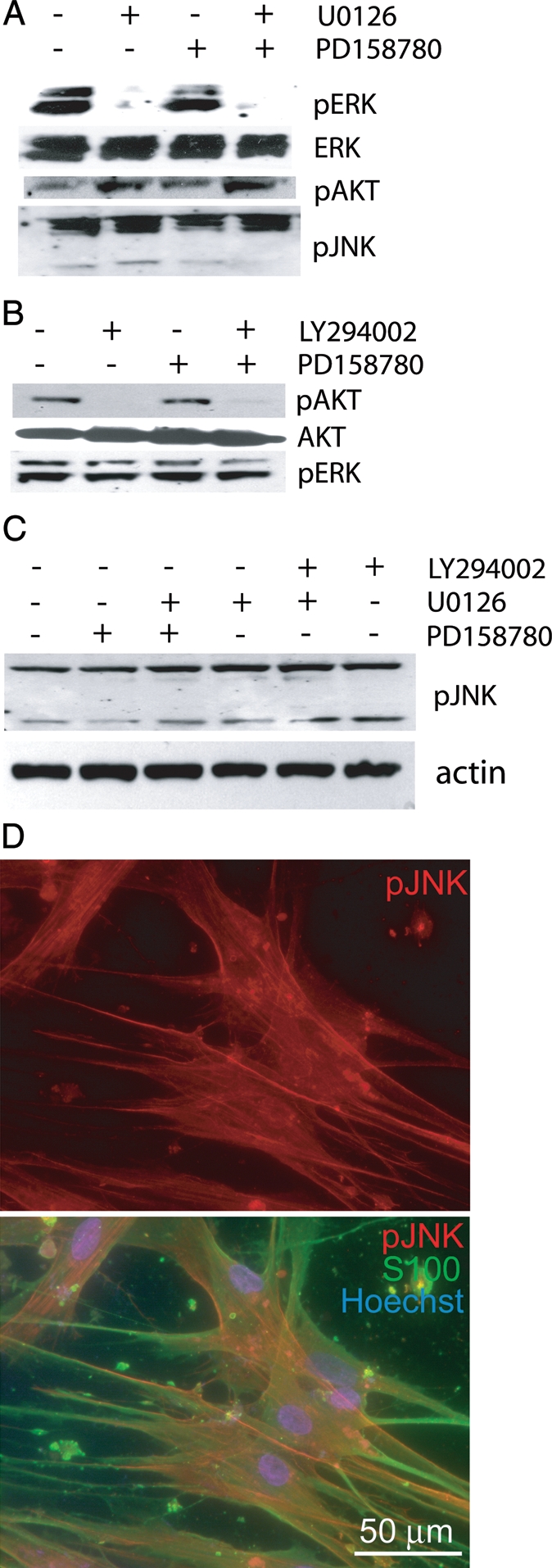
Extracellular regulated kinases (ERKs), Akt, and c-Jun N-terminal kinase (JNK) are persistently phosphorylated in human vestibular schwannoma (VS) cells. (A–C) Western blots of VS cell lysates from cultures maintained in serum-free, basal medium treated with the mitogen activated protein kinase kinase (MAPKK, MEK) MEK inhibitor U0126 (10 µM); the PI3-K inhibitor, LY294002 (20 µM); and/or the ErbB2 inhibitor PD158780 (100 nM) as indicated and probed with the indicated antibodies. (D) Immunostaining of cultured VS cells with anti-pJNK (red; upper and lower panel) and anti-S100 (green; lower panel). Scale bar = 50 µm.
ErbB2, a receptor tyrosine kinase required for normal SC development and survival, constitutively resides in lipid rafts in VS cells and is active.22,24 This constitutive ErbB2 activity contributes to the proliferative potential of VS cells and is required for Src activation.13,22 Furthermore, merlin regulates ErbB2 trafficking and signaling.17 Because MEK/ERK, PI3-K/Akt, and JNKs are activated by ErbB2,28 it is possible that the activation of these kinases in VS cells results from constitutive ErbB2 signaling. We compared ERK, Akt, and JNK phosphorylation status in protein lysates prepared from primary VS cultures maintained in the presence or absence of the ErbB2 inhibitor PD158780 (100 nM).21 Lysates from primary rat sciatic nerve SC cultures confirmed that PD158780 inhibits the ability of NRG-1, an ErbB2 ligand, to induce ErbB2 phosphorylation (Supplemental Fig. 1). PD158780 failed to reduce pERK, pAkt, and pJNK levels in primary VS cells (Fig. 2). Similar results were obtained from ≥3 VS cultures derived from separate tumors. Similar results were obtained with AG825 (1 µM), a different ErbB2 inhibitor (not shown). These data suggest that, in contrast to Src phosphorylation in merlin-deficient glial cells,13 persistent phosphorylation of ERKs, Akt, and JNK in VS cells does not require ErbB2 activity.
MEK/ERK and PI3-K/Akt Promote VS Proliferation
Given that MEK/ERK, PI3-K/Akt, and JNKs are active in VS cells, we sought to determine the extent to which these pro-growth signals contributed to the proliferative and survival response of primary VS cells. We scored BrdU-positive VS cells in cultures treated with either LY294002 (2, 20 µM), a PI3-K inhibitor, or with U0126 (10 µM) or PD98059 (2, 20 µM), MEK inhibitors. VS cell apoptosis was determined by scoring TUNEL–positive VS cells in parallel cultures. Inhibition of MEK with U0126 or PD98059 significantly reduced VS cell proliferation but did not result in a significant difference in the percent of TUNEL-positive cells (Fig. 4). Similarly, the PI3-K inhibitor, LY294002, significantly reduced VS cell proliferation but did not increase the percent of TUNEL-positive cells (Fig. 4). These data suggest that both MEK/ERK and PI3-K/Akt activity contribute to VS cell proliferation in vitro but are not independently required for cell survival.
Fig. 4.

MEK/ extracellular regulated kinase (ERK) and PI3-K/Akt activity contributes to vestibular schwannoma (VS) cell proliferation. Primary VS cultures were maintained in the presence of the MEK inhibitors, U1026 or PD98059, or the PI3-K inhibitor, LY294002, for 24 h. (A and B) Cell proliferation was determined by scoring the percent of BrdU-positive, S100-positive VS cells from 10 randomly selected fields and is expressed as a percentage relative to control cultures, defined as 100%. (C and D) Apoptosis was scored as the percent of terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)–positive, S100-positive VS cells with condensed nuclei. Each condition was performed on at least 4 cultures derived from separate tumors. Error bars represent standard errors of the mean. *P < .05, by 1-way analysis of variance with post-hoc Tukey analysis.
JNK Promotes VS Proliferation and Survival
SCs critically depend on axons for long-term survival, and activation of JNK contributes to the apoptosis of denervated SCs following axotomy.18–20 Conversely, JNK activity promotes survival of transformed rat RN22 schwannoma cells.29 Given that JNK remains active in primary human VS cells, we sought to determine whether persistent JNK activity contributes to the ability of VS cells to proliferate and survive in the absence of axons. We quantified the percent of BrdU-positive and TUNEL-positive VS cells in cultures treated with cell-permeant peptide I-JIP (10, 30, or 100 µM) or SP 600125 (20 µM). I-JIP is a peptide that competitively inhibits the binding of JNKs to the JIP scaffolding protein, thereby inhibiting JNK activation.30–32 SP600125 is a small-molecule inhibitor of JNK.31 We confirmed the ability of these compounds to inhibit JNK signaling by probing protein lysates of cultures treated with SP600125 (2 or 20 µM) or I-JIP (30 or 100 µM) with anti-pJNK and pJun antibodies (Fig. 5). For each culture derived from separate tumors, the percentage of BrdU-positive VS cells was expressed as a percentage of the control condition, defined as 100%. In control conditions, 5.5% ± 2.5% (mean ± standard deviation) of VS cells were BrdU positive. VS cell apoptosis was determined by scoring TUNEL-positive VS cells in parallel cultures. The percentage of TUNEL-positive VS cells was likewise expressed as a percentage of the control condition. Overall, 3.0% ± 1.6% (mean ± standard deviation) of VS cells were TUNEL positive in control conditions. Treatment with either I-JIP or SP600125 significantly decreased the percentage of BrdU-positive and increased the percentage of TUNEL-positive VS cells (Fig. 6). Thus, inhibition of persistent JNK activity reduces VS cell proliferation and survival.
Fig. 5.

SP600125 and I-JIP each suppress c-Jun N-terminal kinase (JNK) signaling in vestibular schwannoma (VS) cells. Western blots of VS cell lysates from cultures maintained in serum free, basal medium treated with the JNK inhibitors, SP600125 (2, 20 µM) or I-JIP (30, 100 µM), as indicated and probed with anti-pJNK (A) or pJun (B) antibodies. The blots were stripped and reprobed with non–phospho-specific antibodies. The blots are from separate cultures, and the order of the lanes is different for each blot. The blots are representative of cultures derived from 3 separate tumors.
Fig. 6.

c-Jun N-terminal kinase (JNK) activity promotes vestibular schwannoma (VS) cell proliferation and survival. Primary VS cultures were cultured in the presence or absence of JNK inhibitors (I-JIP or SP600125) for 24 h (A and B) or 5 days (C). (A) Representative cultures immunolabeled with anti-BrdU (red) and anti-S100 (green) antibodies. Nuclei were identified with Hoescht 3342 (blue). Arrows indicate BrdU-positive nuclei determined by overlap of red and blue channels. Scale bar = 100 µm. Cell proliferation was determined by scoring the percentage of BrdU-positive, S100-positive VS cells and is expressed as a percentage relative to control cultures, defined as 100%. (B) Apoptosis was scored as the percentage of terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)–positive, S100-positive VS cells with condensed nuclei and is expressed as a percentage relative to control cultures, defined as 100%. VS cultures treated with SP600125 or I-JIP and labeled with TUNEL (red), anti-cleaved caspase 3 antibody (green), and Hoechst (blue) demonstrating activation of caspase 3 in TUNEL-positive cells. Scale bar = 20 µm. (C) VS cell survival, as determined by counting the number of S100-positive cells remaining after 5 days, is expressed as a percentage relative to control cultures, defined as 100%. Error bars represent standard errors of the mean. *P < .05 by 1-way analysis of variance with post-hoc Tukey analysis.
Inhibition of JNK results in RN22 schwannoma cell necrosis but apparently not apoptosis.29 However, we observed an increase in TUNEL-positive human primary VS cells in the presence of JNK inhibitors, implying that JNK inhibition leads to apoptosis in these cells. To verify that JNK inhibition results in VS cell apoptosis, we stained primary VS cell cultures maintained in I-JIP (30 µM) or SP600125 (20 µM) with anti-active caspase-3 antibody, which detects cleaved caspase-3, a hallmark of apoptosis,33 and TUNEL. As shown in Fig. 6, TUNEL-positive VS cells demonstrated cleaved caspase-3 (active caspase) immunoreactivity in their cytoplasm, confirming that the TUNEL-positive cells are apoptotic. Finally, to verify that the increase in apoptosis leads to an overall decrease in cell survival, we counted the number of surviving cells 5 days after treatment with I-JIP and SP600125. Although TUNEL and active caspase-3 immunolabeling only detect the fraction of cells undergoing apoptosis at the time that the cultures are fixed, counting the number of surviving cells accounts for the cumulative cellular death over the culture interval. Both JNK inhibitors significantly reduced the number of surviving VS cells (Fig. 6). Thus, inhibition of JNK decreases VS cell survival at least in part by inducing apoptosis.
The results above imply that persistent JNK activity promotes VS cell survival. To verify that VS cells with persistent JNK activity are those that survive, we labeled primary VS cultures maintained in the presence or absence of I-JIP or SP600125 with anti-phosphorylated Jun (pJUN) antibodies and TUNEL. pJun immunofluorescence was determined from digital, epifluorescent images for each VS cell by measuring the mean pixel density in each nucleus.34 Background fluorescence/nonspecific labeling was determined by measuring the mean pixel intensity in the cytoplasm, taking advantage of the fact that pJun is an obligatorily nuclear protein, such that any cytoplasmic fluorescence represents background labeling.34 Cells with a ratio of nuclear to cytoplasmic pJun immunofluorescence intensity ratio ≥5 were considered pJun positive. We then compared the pJun and TUNEL status for each of >300 cells per condition, and the results were verified in 3 VS cultures from separate tumors. As shown in Fig. 7, I-JIP and SP600125 both decreased the percentage of pJun-positive cells and increased the percentage of TUNEL-positive cells. None of the TUNEL-positive cells in any condition were also pJun positive. These observations indicate that susceptibility to apoptosis highly correlates with loss of pJun immunoreactivity and support the hypothesis that persistent JNK activity protects VS cells from apoptosis.
Fig. 7.

Vestibular schwannoma (VS) cell apoptosis due to inhibition of c-Jun N-terminal kinase (JNK) correlates with loss of c-JUN phosphorylation (pJUN). VS cultures were treated with JNK inhibitors (I-JIP, 100 µM or SP600125, 20 µM) for 24 h. (A) Representative images of cultures treated with I-JIP and immunostained with anti-pJUN (Alexa 488 secondary antibody, psuedocolored green, right panels) and S100 antibodies (Alexa 647 secondary antibody, psuedocolored green, left panels). Apoptotic cells were detected by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) (red) and nuclei were labeled with Hoechst (blue). Scale bar = 100 µm. (B) The percent of pJUN-negative and TUNEL-positive, pJUN-negative and TUNEL-negative, and pJUN-positive and TUNEL-negative cells were scored. Cells were considered pJUN positive if the ratio of nuclear to cytoplasmic ratio of pJUN immunofluorescence intensity exceeded 5. No cells were simultaneously pJUN positive and TUNEL positive in any condition. At least 300 cells were scored per condition, and the results were repeated in cultures from 3 separate tumors. JNK inhibitors decreased the percentage of pJUN-positive cells and increased the percentage of TUNEL-positive cells.
To further confirm that JNK activity promotes VS cell survival, we transfected VS cultures with a siRNA oligonucleotide targeting both JNK1 and JNK2 or with a scrambled oligonucleotide. Protein lysates were probed with anti-JNK and pJun antibodies to confirm JNK1/2 knock-down and suppression of JNK signaling (Fig. 8). Transfection of the JNK1/2-targeted oligonucleotide significantly increased the percentage of TUNEL-positive VS cell nuclei, compared with cultures transfected with a scrambled oligonucleotide (P = .035, by the Student's 2-tailed t test) (Fig. 8).
Fig. 8.

siRNA-mediated c-Jun N-terminal kinase (JNK) 1/2 knock-down increases vestibular schwannoma (VS) cell apoptosis. VS cultures were transfected with a siRNA oligonucleotide targeting JNK1 and JNK2 (JNK1/2) or with a scrambled (Scr) oligonucleotide. (A) Western blot probed with the indicated antibodies demonstrating decreased JNK expression and Jun phosphorylation in cultures expressing JNK1/2-targeted siRNA oligonucleotide. (B) Apoptosis was scored as the percentage of terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)–positive, S100-positive VS cells with condensed nuclei. The data are from cultures derived from 4 separate tumors. Error bars represent standard errors of the mean. P = .035, by the Student's 2-tailed t test.
JNK Activity Protects VS Cells from Apoptosis by Limiting Accumulation of Reactive Oxygen Species (ROS)
We next sought to identify mechanisms by which JNK activity contributes to VS cell survival. Accumulation of ROS regulates a variety of cellular functions relevant to SC neoplasia, including proliferation, apoptosis, and senescence,35 and inhibition of JNK with SP600125 in RN22 schwannoma cells cultured in the absence of serum results in increased ROS and necrotic cell death.29 To determine the effect of JNK activity on the ROS status of primary human VS cells, we loaded primary cultures with CM-H2DCFDA, a fluorescent probe for ROS. Oxidation of the nonfluorescent CM-H2DCFDA yields DCF, a highly fluorescent product that detects reactive oxygen intermediates in intact cells.29 After loading with CM-H2DCFDA, the cells were maintained in the presence or absence of I-IJP (30 or 100 µM) or SP600125 (20 µM) for 4 h. DCF fluorescence intensity was quantified from digital images of live cultures by measuring the mean pixel density in the perinuclear cytoplasm of each cell. In cultures derived from 4 separate tumors, inhibition of JNK significantly increased DCF fluorescence (Fig. 9). Thus, JNK inhibitors increase ROS in VS cultures.
Fig. 9.

Inhibition of c-Jun N-terminal kinase (JNK) increases vestibular schwannoma (VS) cell oxidative stress leading to cell death. (A) Oxidative status of VS cells in the presence or absence of SP600125, I-JIP, or ebselen (Eb) was determined by measuring the DCF fluorescence intensity in cultures loaded with CM-H2DCFDA. Thirty to 50 cells were imaged per condition. Results are representative of 4 cultures derived from separate tumors. Error bars represent standard errors of the mean. *P < .05, by 1-way analysis of variance with post-hoc Tukey analysis. (B) Representative images of DCF fluorescence in the indicated conditions. Scale bar = 100 µm. (C) VS cultures were treated with SP600125 or I-JIP in the presence or absence of Eb (20 µM) and the percent of TUNEL-positive cells was determined as before. *P < .05, by 1-way analysis of variance with post-hoc Tukey analysis.
To determine whether the increase in ROS accounts for the pro-apoptotic effects of JNK inhibitors, we treated 5 VS cultures derived from different tumors with ebselen, a seleno-organic compound that acts as a glutathione peroxidase mimic and is an effective ROS scavenger (Fig. 9),36 in combination with JNK inhibitors and determined the percentage of apoptotic VS cells as above. Ebselen significantly protected VS cells from apoptosis due to JNK inhibition (Fig. 9). Taken together, these observations suggest that persistent JNK activity protects VS cells from apoptosis, at least in part by limiting the accumulation of ROS.
To further explore the mechanisms of ROS suppression by JNK activity, we sought to identify the subcellular source and type of ROS in VS cells treated with JNK inhibitors. Mitochondria are a major source of ROS, and JNK regulates several mitochondrial activities, including ROS production.29 To determine whether JNK regulates mitochondrial ROS accumulation in VS cells, primary VS cultures were loaded with MitoSOX Red, a fluorogenic dye that specifically detects superoxide in the mitochondria of live cells.37 It fails to detect other ROS- or reactive nitrogen species–generating systems. Cells were maintained in the presence or absence of SP600125 or I-IJIP for 4 h, and MitoSOX Red fluorescence was quantified. VS cultures treated with JNK inhibitors demonstrated significantly higher levels of MitoSOX Red fluorescence implying that persistent JNK activity suppresses superoxide accumulation in VS cell mitochondria (Fig. 10).
Fig. 10.

Inhibition of c-Jun N-terminal kinase (JNK) increases vestibular schwannoma (VS) cell mitochondrial superoxide accumulation leading to cell death. (A and B) Mitochondrial superoxide accumulation was determined by measuring MitoSOX Red fluorescent intensity in VS cultures treated with SP600125 (20 µM) or I-JIP (100 µM). (A) MitoSOX Red fluorescence is presented as a scaled image, with intensity levels as indicated. Scale bar = 25 µm. (B) Mean MitoSOX Red fluorescence in indicated conditions. Thirty to 50 cells were imaged per condition. The results are representative of 3 cultures derived from separate tumors. Error bars represent standard errors of the mean. *P < .05 by 1-way analysis of variance with post-hoc Tukey analysis. (C) VS cultures maintained in the presence or absence of JNK inhibitors were transduced with adenoviral vector expressing superoxide dismutase 2 (AdSOD2) or control vector (AdCon), and the percentage of terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)–positive cells were scored as above. The results are from 4 cultures derived from separate tumors. Error bars represent SEM. *P < .05 by 1-way analysis of variance with post-hoc Tukey analysis. (D) Representative images of VS cultures treated with I-JIP and either AdCon (left panel) or AdSOD2 (right panel). Cultures were immunostained with anti-S100 (green) antibody and labeled with TUNEL (red) and Hoechst (blue). Scale bar = 100 µm.
To determine whether this increase in mitochondrial superoxide accumulation contributes to VS cell apoptosis, VS cultures were treated with an adenoviral vector–encoding superoxide dismutase 2 (SOD2; MnSOD), a mitochondrially localized superoxide scavenger, or a control vector. Cultures were subsequently treated with I-JIP or SP600125, and the percentage of TUNEL-positive VS cells was determined. Expression of SOD2 significantly suppressed VS cell apoptosis in the presence of JNK inhibitors (Fig. 10). Taken together, these results suggest that JNK activity protects primary human VS cells from apoptosis specifically by suppressing mitochondrial superoxide accumulation.
Discussion
Progrowth Signals in VSs
As a tumor suppressor, the merlin protein regulates a wide variety of signaling events implicated in tumorigenesis, including Ras, Rac1/Cdc42, RhoA, Src, Raf, PAK1/2, ERK/MAPK, JNK ); and PI3-K/Akt, ErbB2, and PDGF receptor signaling; mTORC1; and the E3 ubiquitin ligase CRL4(DCAF1).5–15,25,38 Limited access to human specimens has hampered identification of which of these merlin-sensitive signals contribute to VS formation and growth. With use of primary cultures derived from human VSs, we found that MEK/ERK, PI3-K/Akt, and JNK activity each promote VS cell proliferation. Consistent with these observations, recent studies found that proliferation of human VS cells depends on MEK/ERK and Akt signaling.25,39
Merlin also regulates ErbB2 localization, and activity and inhibition of ErbB2 reduces VS cell proliferation and growth of human VS xenografts in nude mice.13,17,22,24 Nevertheless, basal levels of ERK, Akt, and JNK activity appear to be independent of ErbB2 signaling in human VS cells (Fig. 2). By contrast, Src activity in merlin-deficient central nervous system glial cells depends on ErbB2.13 Recent evidence indicates that the persistent basal ERK phosphorylation in human VS cells likely results from focal adhesion kinase (FAK)/Src/Ras, but not Rac/PAK signaling.25
JNK Expression and Activity in VSs
JNK isoforms 1–3 present diverse and often opposing effects on cell survival, apoptosis, and tumorigenesis.40 JNK1 and 2 are expressed ubiquitously, whereas JNK 3 is primarily expressed in neurons. We found that JNK2 and, to a lesser extent, JNK1 are expressed at higher levels in normal nerve tissue than in VSs. The significance of the lower levels of JNK1 and JNK2 expression in VS tissue versus normal nerve remains unknown. In some cellular responses, JNK1 and JNK2 function redundantly, whereas in other cases, they differentially regulate cellular responses. For example, fibroblasts lacking JNK1 exhibit proliferation defects, whereas a lack of JNK2 confers a proliferative advantage.41 Complete absence of both JNKs results in a dramatic proliferation defect, similar to cells lacking c-Jun.42 Despite increased levels of JNK expression, JNK is not phosphorylated in mature SCs, whereas it is phosphorylated in VS cells. This persistent JNK phosphorylation in VS cells is related to merlin status, because overexpression of merlin suppressed JNK activity in our primary VS cultures, and in HEI 193 cells, a human schwannoma cell line transformed with viral oncogenes.7
Depending on the cell type and context, JNK activity can either promote or suppress cell survival, proliferation, and tumorigenesis.40,43,44 For example, JNKs suppress ras-induced tumorigenesis in fibroblasts but promote Bcr-Abl induced B-cell lymphoma.44 Both the physiological context and time course of kinase activation appear to determine whether JNK signaling promotes survival or apoptosis.40,45 In SCs, JNKs are activated in response to nerve injury and promote SC apoptosis.18–20 Conversely, JNK activity promotes survival of transformed schwannoma cells and other glial neoplasms.29,46 Here, we show that JNK promotes primary cultured human VS cell proliferation and survival. Thus, JNK represents a potential specific therapeutic target for treatment of VSs that would likely reduce VS cell growth yet not harm normal SCs. Such treatment strategies may be especially suitable for patients with NF2 and multiple schwannomas that are difficult to manage with current treatment options.
ROS in VS Cells
In many cells, JNK is activated by elevated ROS, contributing to cell death.47 However, JNK appears to suppress ROS accumulation in rat RN22 schwannoma cells,29 and here we show that JNK specifically suppresses mitochondrial superoxide accumulation in human VS cells. Three enzyme activities function to scavenge cellular ROS. SOD enzymes convert superoxides to hydrogen peroxide (H2O2), whereas catalase and glutathione peroxidase convert H2O2 to O2 and H2O.48 SOD2 (MnSOD), a nuclear-encoded and mitochondria-localized homotetrameric enzyme, is the primary defense against mitochondrially generated ROS, whereas SOD1 (Cu/ZnSOD) is distributed in the cytosol and accounts for 70%–80% of cellular SOD activity.49 Overexpression of SOD2 protects human VS cells from apoptosis due to JNK inhibition, confirming that the increase in mitochondrial superoxide contributes to the death of VS cells with suppressed JNK activity. Taken together, these observations suggest that the reduction in ROS stress due to persistent JNK activity may account, at least in part, for the ability of VS cells to grow and survive in the absence of axons, in contrast to their normal SC counterparts, which critically depend on axonal contact for long-term survival.
Supplementary Material
Funding
National Institutes of Health/National Institutes on Deafness and Other Communication Disorders-(R01DC009801) and the Department of Defense (NF050193)
Supplementary Material
References
- 1.Rouleau GA, Merel P, Lutchman M, et al. Alteration in a new gene encoding a putative membrane-organizing protein causes neuro-fibromatosis type 2. Nature. 1993;363(6429):515–521. doi: 10.1038/363515a0. doi:10.1038/363515a0. [DOI] [PubMed] [Google Scholar]
- 2.Trofatter JA, MacCollin MM, Rutter JL, et al. A novel moesin-, ezrin-, radixin-like gene is a candidate for the neurofibromatosis 2 tumor suppressor. Cell. 1993;72(5):791–800. doi: 10.1016/0092-8674(93)90406-g. doi:10.1016/0092-8674(93)90406-G. [DOI] [PubMed] [Google Scholar]
- 3.Stemmer-Rachamimov AO, Xu L, Gonzalez-Agosti C, et al. Universal absence of merlin, but not other ERM family members, in schwannomas. Am J Pathol. 1997;151(6):1649–1654. [PMC free article] [PubMed] [Google Scholar]
- 4.Baser ME, DG RE, Gutmann DH. Neurofibromatosis 2. Curr Opin Neurol. 2003;16(1):27–33. doi: 10.1097/01.wco.0000053583.70044.ab. doi:10.1097/00019052-200302000-00004. [DOI] [PubMed] [Google Scholar]
- 5.Kissil JL, Wilker EW, Johnson KC, Eckman MS, Yaffe MB, Jacks T. Merlin, the product of the Nf2 tumor suppressor gene, is an inhibitor of the p21-activated kinase, Pak1. Mol Cell. 2003;12(4):841–849. doi: 10.1016/s1097-2765(03)00382-4. doi:10.1016/S1097-2765(03)00382-4. [DOI] [PubMed] [Google Scholar]
- 6.Lim JY, Kim H, Kim YH, et al. Merlin suppresses the SRE-dependent transcription by inhibiting the activation of Ras-ERK pathway. Biochem Biophys Res Commun. 2003;302(2):238–245. doi: 10.1016/s0006-291x(03)00124-4. doi:10.1016/S0006-291X(03)00124-4. [DOI] [PubMed] [Google Scholar]
- 7.Chadee DN, Kyriakis JM. MLK3 is required for mitogen activation of B-Raf, ERK and cell proliferation. Nat Cell Biol. 2004;6(8):770–776. doi: 10.1038/ncb1152. doi:10.1038/ncb1152. [DOI] [PubMed] [Google Scholar]
- 8.Fraenzer JT, Pan H, Minimo L, Jr., Smith GM, Knauer D, Hung G. Overexpression of the NF2 gene inhibits schwannoma cell proliferation through promoting PDGFR degradation. Int J Oncol. 2003;23(6):1493–1500. [PubMed] [Google Scholar]
- 9.Kaempchen K, Mielke K, Utermark T, Langmesser S, Hanemann CO. Upregulation of the Rac1/JNK signaling pathway in primary human schwannoma cells. Hum Mol Genet. 2003;12(11):1211–1221. doi: 10.1093/hmg/ddg146. doi:10.1093/hmg/ddg146. [DOI] [PubMed] [Google Scholar]
- 10.Rong R, Tang X, Gutmann DH, Ye K. Neurofibromatosis 2 (NF2) tumor suppressor merlin inhibits phosphatidylinositol 3-kinase through binding to PIKE-L. Proc Natl Acad Sci USA. 2004;101(52):18200–18205. doi: 10.1073/pnas.0405971102. doi:10.1073/pnas.0405971102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chadee DN, Xu D, Hung G, et al. Mixed-lineage kinase 3 regulates B-Raf through maintenance of the B-Raf/Raf-1 complex and inhibition by the NF2 tumor suppressor protein. Proc Natl Acad Sci USA. 2006;103(12):4463–4468. doi: 10.1073/pnas.0510651103. doi:10.1073/pnas.0510651103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yi C, Wilker EW, Yaffe MB, Stemmer-Rachamimov A, Kissil JL. Validation of the p21-activated kinases as targets for inhibition in neurofibromatosis type 2. Cancer Res. 2008;68(19):7932–7937. doi: 10.1158/0008-5472.CAN-08-0866. doi:10.1158/0008-5472.CAN-08-0866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Houshmandi SS, Emnett RJ, Giovannini M, Gutmann DH. The neurofibromatosis 2 protein, merlin, regulates glial cell growth in an ErbB2- and Src-dependent manner. Mol Cell Biol. 2009;29(6):1472–1486. doi: 10.1128/MCB.01392-08. doi:10.1128/MCB.01392-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Flaiz C, Chernoff J, Ammoun S, Peterson JR, Hanemann CO. PAK kinase regulates Rac GTPase and is a potential target in human schwannomas. Exp Neurol. 2009;218(1):137–144. doi: 10.1016/j.expneurol.2009.04.019. doi:10.1016/j.expneurol.2009.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.James MF, Han S, Polizzano C, et al. NF2/merlin is a novel negative regulator of mTOR complex 1, and activation of mTORC1 is associated with meningioma and schwannoma growth. Mol Cell Biol. 2009;29(15):4250–4261. doi: 10.1128/MCB.01581-08. doi:10.1128/MCB.01581-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Curto M, Cole BK, Lallemand D, Liu CH, McClatchey AI. Contact-dependent inhibition of EGFR signaling by Nf2/Merlin. J Cell Biol. 2007;177(5):893–903. doi: 10.1083/jcb.200703010. doi:10.1083/jcb.200703010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fernandez-Valle C, Tang Y, Ricard J, et al. Paxillin binds schwannomin and regulates its density-dependent localization and effect on cell morphology. Nat Genet. 2002;31(4):354–362. doi: 10.1038/ng930. [DOI] [PubMed] [Google Scholar]
- 18.Parkinson DB, Bhaskaran A, Droggiti A, et al. Krox-20 inhibits Jun-NH2-terminal kinase/c-Jun to control Schwann cell proliferation and death. J Cell Biol. 2004;164(3):385–394. doi: 10.1083/jcb.200307132. doi:10.1083/jcb.200307132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parkinson DB, Dong Z, Bunting H, et al. Transforming growth factor beta (TGFbeta) mediates Schwann cell death in vitro and in vivo: examination of c-Jun activation, interactions with survival signals, and the relationship of TGFbeta-mediated death to Schwann cell differentiation. J Neurosci. 2001;21(21):8572–8585. doi: 10.1523/JNEUROSCI.21-21-08572.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng HL, Steinway ML, Xin X, Feldman EL. Insulin-like growth factor-I and Bcl-X(L) inhibit c-jun N-terminal kinase activation and rescue Schwann cells from apoptosis. J Neurochem. 2001;76(3):935–943. doi: 10.1046/j.1471-4159.2001.00110.x. [DOI] [PubMed] [Google Scholar]
- 21.Hansen MR, Clark JJ, Gantz BJ, Goswami PC. Effects of ErbB2 signaling on the response of vestibular schwannoma cells to gamma-irradiation. Laryngoscope. 2008;118(6):1023–1030. doi: 10.1097/MLG.0b013e318163f920. doi:10.1097/MLG.0b013e318163f920. [DOI] [PubMed] [Google Scholar]
- 22.Hansen MR, Roehm PC, Chatterjee P, Green SH. Constitutive neuregulin-1/ErbB signaling contributes to human vestibular schwannoma proliferation. Glia. 2006;53(6):593–600. doi: 10.1002/glia.20316. doi:10.1002/glia.20316. [DOI] [PubMed] [Google Scholar]
- 23.Cioffi JA, Yue WY, Mendolia-Loffredo S, Hansen KR, Wackym PA, Hansen MR. MicroRNA-21 overexpression contributes to vestibular schwannoma cell proliferation and survival. Otol Neurotol. 2010;31(9):1455–1462. doi: 10.1097/MAO.0b013e3181f20655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brown KD, Hansen MR. Lipid raft localization of erbB2 in vestibular schwannoma and Schwann cells. Otol Neurotol. 2008;29(1):79–85. doi: 10.1097/mao.0b013e31815dbb11. doi:10.1097/mao.0b013e31815dbb11. [DOI] [PubMed] [Google Scholar]
- 25.Ammoun S, Flaiz C, Ristic N, Schuldt J, Hanemann CO. Dissecting and targeting the growth factor-dependent and growth factor-independent extracellular signal-regulated kinase pathway in human schwannoma. Cancer Res. 2008;68(13):5236–5245. doi: 10.1158/0008-5472.CAN-07-5849. doi:10.1158/0008-5472.CAN-07-5849. [DOI] [PubMed] [Google Scholar]
- 26.Jacob A, Lee TX, Neff BA, Miller S, Welling B, Chang LS. Phosphatidylinositol 3-kinase/AKT pathway activation in human vestibular schwannoma. Otol Neurotol. 2008;29(1):58–68. doi: 10.1097/mao.0b013e31816021f7. doi:10.1097/mao.0b013e31816021f7. [DOI] [PubMed] [Google Scholar]
- 27.Hilton DA, Ristic N, Hanemann CO. Activation of ERK, AKT and JNK signalling pathways in human schwannomas in situ. Histopathology. 2009;55(6):744–749. doi: 10.1111/j.1365-2559.2009.03440.x. doi:10.1111/j.1365-2559.2009.03440.x. [DOI] [PubMed] [Google Scholar]
- 28.Nuntharatanapong N, Chen K, Sinhaseni P, Keaney JF., Jr. EGF receptor-dependent JNK activation is involved in arsenite-induced p21Cip1/Waf1 upregulation and endothelial apoptosis. Am J Physiol Heart Circ Physiol. 2005;289(1):H99–H107. doi: 10.1152/ajpheart.00901.2004. doi:10.1152/ajpheart.00901.2004. [DOI] [PubMed] [Google Scholar]
- 29.Lopez-Sanchez N, Rodriguez JR, Frade JM. Mitochondrial c-Jun NH2-terminal kinase prevents the accumulation of reactive oxygen species and reduces necrotic damage in neural tumor cells that lack trophic support. Mol Cancer Res. 2007;5(1):47–60. doi: 10.1158/1541-7786.MCR-06-0233. doi:10.1158/1541-7786.MCR-06-0233. [DOI] [PubMed] [Google Scholar]
- 30.Barr RK, Kendrick TS, Bogoyevitch MA. Identification of the critical features of a small peptide inhibitor of JNK activity. J Biol Chem. 2002;277(13):10987–10997. doi: 10.1074/jbc.M107565200. doi:10.1074/jbc.M107565200. [DOI] [PubMed] [Google Scholar]
- 31.Kuan CY, Burke RE. Targeting the JNK signaling pathway for stroke and Parkinson's diseases therapy. Curr Drug Targets CNS Neurol Disord. 2005;4(1):63–67. doi: 10.2174/1568007053005145. doi:10.2174/1568007053005145. [DOI] [PubMed] [Google Scholar]
- 32.Dickens M, Rogers JS, Cavanagh J, et al. A cytoplasmic inhibitor of the JNK signal transduction pathway. Science. 1997;277(5326):693–696. doi: 10.1126/science.277.5326.693. doi:10.1126/science.277.5326.693. [DOI] [PubMed] [Google Scholar]
- 33.Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407(6805):770–776. doi: 10.1038/35037710. doi:10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- 34.Alam SA, Robinson BK, Huang J, Green SH. Prosurvival and proapoptotic intracellular signaling in rat spiral ganglion neurons in vivo after the loss of hair cells. J Comp Neurol. 2007;503(6):832–852. doi: 10.1002/cne.21430. doi:10.1002/cne.21430. [DOI] [PubMed] [Google Scholar]
- 35.Genestra M. Oxyl radicals, redox-sensitive signalling cascades and antioxidants. Cell Signal. 2007;19(9):1807–1819. doi: 10.1016/j.cellsig.2007.04.009. doi:10.1016/j.cellsig.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 36.Nakamura Y, Feng Q, Kumagai T, et al. Ebselen, a glutathione peroxidase mimetic seleno-organic compound, as a multifunctional antioxidant. Implication for inflammation-associated carcinogenesis. J Biol Chem. 2002;277(4):2687–2694. doi: 10.1074/jbc.M109641200. doi:10.1074/jbc.M109641200. [DOI] [PubMed] [Google Scholar]
- 37.Robinson KM, Janes MS, Beckman JS. The selective detection of mitochondrial superoxide by live cell imaging. Nat Protoc. 2008;3(6):941–947. doi: 10.1038/nprot.2008.56. doi:10.1038/nprot.2008.56. [DOI] [PubMed] [Google Scholar]
- 38.Li W, You L, Cooper J, et al. Merlin/NF2 suppresses tumorigenesis by inhibiting the E3 ubiquitin ligase CRL4(DCAF1) in the nucleus. Cell. 2010;140(4):477–490. doi: 10.1016/j.cell.2010.01.029. doi:10.1016/j.cell.2010.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee TX, Packer MD, Huang J, et al. Growth inhibitory and anti-tumour activities of OSU-03012, a novel PDK-1 inhibitor, on vestibular schwannoma and malignant schwannoma cells. Eur J Cancer. 2009;45(9):1709–1720. doi: 10.1016/j.ejca.2009.03.013. doi:10.1016/j.ejca.2009.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bode AM, Dong Z. The functional contrariety of JNK. Mol Carcinog. 2007;46(8):591–598. doi: 10.1002/mc.20348. doi:10.1002/mc.20348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tournier C, Hess P, Yang DD, et al. Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science. 2000;288(5467):870–874. doi: 10.1126/science.288.5467.870. doi:10.1126/science.288.5467.870. [DOI] [PubMed] [Google Scholar]
- 42.Schreiber M, Kolbus A, Piu F, et al. Control of cell cycle progression by c-Jun is p53 dependent. Genes Dev. 1999;13(5):607–619. doi: 10.1101/gad.13.5.607. doi:10.1101/gad.13.5.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu J, Lin A. Role of JNK activation in apoptosis: a double-edged sword. Cell Res. 2005;15(1):36–42. doi: 10.1038/sj.cr.7290262. doi:10.1038/sj.cr.7290262. [DOI] [PubMed] [Google Scholar]
- 44.Kennedy NJ, Davis RJ. Role of JNK in tumor development. Cell Cycle. 2003;2(3):199–201. [PubMed] [Google Scholar]
- 45.Ventura JJ, Hubner A, Zhang C, Flavell RA, Shokat KM, Davis RJ. Chemical genetic analysis of the time course of signal transduction by JNK. Mol Cell. 2006;21(5):701–710. doi: 10.1016/j.molcel.2006.01.018. doi:10.1016/j.molcel.2006.01.018. [DOI] [PubMed] [Google Scholar]
- 46.Tsuiki H, Tnani M, Okamoto I, et al. Constitutively active forms of c-Jun NH2-terminal kinase are expressed in primary glial tumors. Cancer Res. 2003;63(1):250–255. [PubMed] [Google Scholar]
- 47.Shen HM, Liu ZG. JNK signaling pathway is a key modulator in cell death mediated by reactive oxygen and nitrogen species. Free Radic Biol Med. 2006;40(6):928–939. doi: 10.1016/j.freeradbiomed.2005.10.056. doi:10.1016/j.freeradbiomed.2005.10.056. [DOI] [PubMed] [Google Scholar]
- 48.Halliwell B, Gutteridge JMC. Free Radicals in Biology and Medicine. 3rd ed. New York: Oxford University Press, Inc.; 1999. [Google Scholar]
- 49.Zelko IN, Mariani TJ, Folz RJ. Superoxide dismutase multigene family: a comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radic Biol Med. 2002;33(3):337–349. doi: 10.1016/s0891-5849(02)00905-x. doi:10.1016/S0891-5849(02)00905-X. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.