

Abstract
We report the optimization of a novel redox-sensitive probe with enhanced dynamic range and an exceptionally well-positioned oxidative midpoint redox potential. The present work characterizes factors that contribute to the improved Förster resonance energy transfer (FRET) performance of this green fluorescent protein (GFP)-based redox sensor. The α-helical linker, which separates the FRET donor and acceptor, has been extended in the new probe and leads to a decreased FRET efficiency in the linker’s reduced, ‘FRET-off’ state. Unexpectedly, the FRET efficiency is increased in the new linker’s oxidized, ‘FRET-on’ state compared with the parent probe, in spite of the longer linker sequence. The combination of a lowered baseline ‘FRET-off’ and an increased ‘FRET-on’ signal significantly improves the dynamic range of the probe for a more robust discrimination of its reduced and oxidized linker states. Mutagenesis of the cysteine residues within the α-helix linker reveals the importance of the fourth, C-terminal cysteine and the relative insignificance of the second cysteine in forming the disulfide bridge to clamp the linker into the high-FRET, oxidized state. To further optimize the performance of the redox probe, various cyan fluorescent protein (CFP)/yellow fluorescent protein (YFP) FRET pairs, placed at opposite ends of the improved redox linker (RL7), were quantitatively compared and exchanged. We found that the CyPet/YPet and ECFP/YPet FRET pairs when attached to RL7 do not function well as sensitive redox probes due to a strong tendency to form heterodimers, which disrupt the α-helix. However, monomeric versions of CyPet and YPet (mCyPet and mYPet) eliminate dimerization and restore redox sensitivity of the probe. The best performing probe, ECFP-RL7-EYFP, exhibits an approximately six-fold increase in FRET efficiency in vitro when passing from the oxidized to the reduced state. We determined the midpoint redox potential of the probe to be −143 ± 6 mV, which is ideal for measuring glutathione (GSH/GSSG) redox potentials in oxidative compartments of mammalian cells (e.g. the endoplasmic reticulum).
Keywords: redox-sensitive linker, green fluorescent protein variants, genetically encoded biosensor, glutathione, redox potential, Förster resonance energy transfer
Introduction
Thiol redox homeostasis is central to the control of cell fate and is associated with various abnormal biochemical processes.1–3 Recent evidence indicates that cells harbor several thiol-disulfide redox couples, which appear to be kinetically controlled and not in equilibrium with each other.4 Glutathione in its reduced form (GSH) is a tripeptide, enzymatically formed from glycine, cysteine and glutamate and is the most abundant non-protein thiol in mammalian cells. Because of its abundance, glutathione is considered to be the major thiol-disulfide redox buffer of the cell and is often used as a proxy of the intracellular redox environment (when measuring multiple redox couples is impractical).4–6 Typically, most (>90%) of the total intracellular glutathione (i.e. GSH + GSSG) is found in the form of GSH in the range of 1–11 mmol/L.5 On average, 85–90% GSH is freely distributed in the cytosol, but can also be compartmentalized in organelles including mitochondria, peroxisomes, the nuclear matrix and the endoplasmic reticulum (ER) after its cytosolic synthesis.4–6 Because of the implications of the thiol redox state, there is a significant need for research tools that permit observation and measurement of intracellular and intraorganellar redox potentials of the GSH/glutathione disulfide couple (GSH/GSSG) in a natural context of living cells or tissues.
Presently, our capabilities to observe clearly and quantify defined redox processes in live cells or tissues are extremely limited. The most advanced and promising tools for non-invasive, specific, quantitative, dynamic and compartment-targeted observations are genetically encoded redox probes that are derived from green fluorescent protein (roGFPs) and yellow fluorescent protein (rxYFP).7–9 Subsequently, the response rate of these probes has been improved via the introduction of positively charged amino acids in the proximity of the two redox-sensitive cysteines.10,11 Recently, the redox-active enzyme glutaredoxin has been coupled directly to redox-sensitive fluorescent proteins to further increase specificity and rate response (thiol–disulfide exchange efficiency).12,13 However, all of the aforementioned redox sensors have a low (relatively reduced) midpoint potential, thereby preventing their use in more oxidizing environments such as the ER. To overcome this problem, a new family of GFP-based redox sensors (roGFP1-iX) with a less negative midpoint potential as low as −229 mV has been developed and successfully tested in the ER of live yeast.14,15 Advances in the development and application of genetically encoded sensors for intracellular redox conditions and many potential pitfalls that should be taken into consideration when venturing into thiol-disulfide redox sensing have been comprehensively covered in recent reviews.12,16,17
The ability to non-invasively observe redox conditions with spatio-temporal resolution would help elucidate the roles of, and the coupling between, the individual components that regulate redox reactions and pathways. To address this need and to complement single protein redox-sensitive sensors, genetically encoded Förster resonance energy transfer (FRET)-based probes have been developed for monitoring intracellular and intraorganellar redox conditions over extended observation periods18,19 The biosensors are fusion proteins constructed from a redox-sensitive peptide – a redox linker (RL) – that is placed between a FRET pair of fluorescent proteins consisting of the enhanced cyan (donor) and yellow (acceptor) variants. A redox event induces a molecular conformational change that alters the distance between the FRET pair, resulting in a measurable change in FRET efficiency. The linker sequence in our bio-sensor is rationally designed to form an α-helix in its reduced state.18 Thiol groups of cysteine residues are strategically placed within the linker to sense the redox potential of the environment and form disulfide bonds upon oxidation. The conformational change of the linker upon oxidation/reduction of the thiol groups (Figure 1) is reversible.
Figure 1.
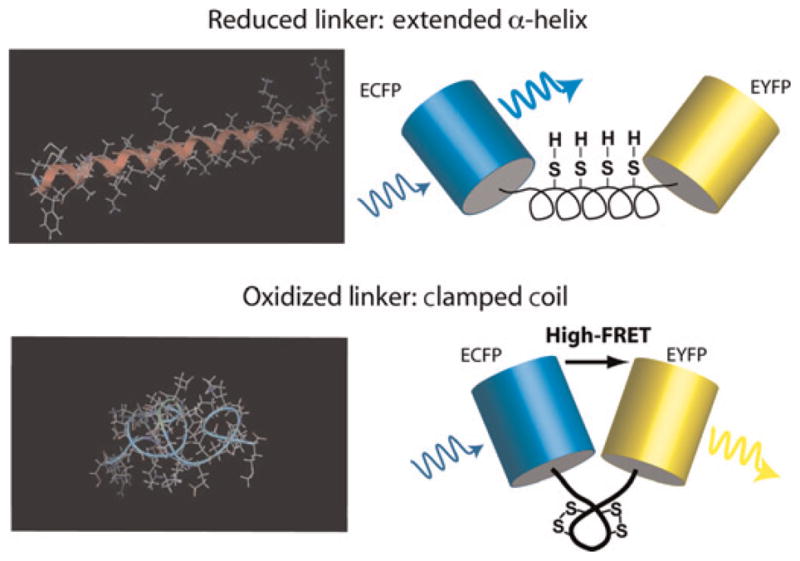
Schematic of the redox biosensor. Minimum energy configurations for dispersed cysteine residues are shown on the left – with (bottom) and without (top) locking the disulfide bonds – and were calculated in MOE (Molecular Operating Environment; Chemical Computing Group, Montreal, Canada). On the right, cartoons of the final redox-sensitive biosensor design (CY-RL7) are shown to illustrate the conformational change that leads to enhanced Förster resonance energy transfer (FRET) in the oxidized ‘clamped coil’ state compared with the reduced α-helical low-FRET state
A distinct advantage of a FRET-based approach is its modularity; i.e. the genetically encoded FRET constructs are cloned, facilitating the rapid inclusion of newly developed fluorescent proteins by exchanging the redox switches and the fluorophores.20–22 Furthermore, recent advances in protein folding indicate that the global protein features contribute little to the formation and prediction of disulfide bonds, while the local sequential and structural information play important roles.23 These findings indicate that the RL is an independent functional polypeptide domain, which provides a stable foundation for tuning the redox sensitivity of the construct. A second distinct advantage of FRET-based approaches is that the FRET measurement is ratiometeric, such that the FRET signal is concentration independent and insensitive to microscope instabilities (such as fluctuations in the excitation light source). This circumvents misinterpretations of fluctuations in local concentration of the probe and the need for monitoring instrument fluctuations.
While we have previously reported the design of several first-generation, FRET-based, redox-sensitive constructs, the present work improves the dynamic range of the FRET-based redox sensor and provides insight into the effectiveness of the imbedded individual cysteine residues to the thiol–disulfide equilibrium. We have engineered a novel redox-sensitive probe with an enhanced dynamic range and an extremely oxidative midpoint redox potential suitable for probing the ER environment. The efficacy of this current FRET sensor has also been recently tested in application to cytosolic redox perturbations of cancer cells.24
Materials and methods
Materials
Reagents of the highest grade available were purchased from Sigma (St Louis, MO, USA), unless otherwise specified. Enzymes for the modification of DNA and pCR2.1 TOPO TA cloning kit were from Invitrogen (Carlsbad, CA, USA) and EarI from NEB (Ipswich, MA, USA). Oligonucleotides were obtained from Integrated DNA Technologies (Coralville, IA, USA). Recombinant plasmids pECFP-C1, pEYFP, pEYFP-N1 were purchased from BD Biosciences (Palo Alto, CA, USA) and pCEP4CyPet-MAMM and pCEP4YPet-MAMM were from Addgene (Cambridge, MA, USA). Ni-NTA agarose, QIAprep spin miniprep and QIAquick polymerase chain reaction (PCR) purification kits were from Qiagen (Valencia, CA, USA), the BSA kit was from Pierce (Rockford, IL, USA) and Taq polymerase was from Eppendorf (Westbury, NY, USA).
Genetic constructs
Redox-sensitive linkers RL7 and a polyproline linker P14 with minor modification were obtained with primer sets RL7 5′-GAAGATCTCCATGTGAAGCAGCTGCGAAGGAGGCAGCAGCTAAGGAGGCTGCAGCTAAGTGCGAGGCAGCTGCAAAATGTGAAGCTG-3′ (forward) and 5′-GGAATTCGCACTTTGCTGCTGCTTCCTTTGCGGCTGCCTCTTTTGCAGCTGCTTCCTTAGCTGCAGCTTCACATTTTGCAGCTGC-3′ (reverse), and P14 5′-GAAGATCTCCACCTCCTCCGCCTCCCCCACCGCCACCA-3′ (forward) and 5′-GGAATTCTGGCGGAGGGGGTGGTGGCGGTGGGGGAGG-3′ (reverse) as described earlier.18 New constructs ECFP-RL7-EYFP (CY-RL7) and ECFP-P14-EYFP (CY-P14) were cloned into a mammalian vector pECFP-CI (Figure 2a) as previously described.18 To build the EYFP-RL7-ECFP construct, the original plasmids pECFP-C1 and pEYFP were used as templates for PCR to obtain EYFP with primers 5′-CCGCTAGCGCTACCGGTCGCCACCATG-3′ and 5′-AAAGATCTCTTGTTACAGCTCGTCCATGCCGAGA-3′ and ECFP with primers 5′-GCGAATTCATGGTGAGCAAGGGCGAGGA-3′ and 5′-TGGATCCTTAATGATGGTGATGGTGGTGCTTGTACAGCTCGTCCATGGCGA-3′. EYFP and ECFP PCR products were ligated into pCR2.1 (TOPO kit, Invitrogen) and confirmed by sequencing. Next, new versions of EYFP and ECFP recovered from recombinant plasmids by digestion with NheI/BglII and EcoRI/BamHI, respectively, replaced the old donor and acceptor in CY-RL7 (Figure 2b). The new FRET pair CyPet/YPet was obtained by PCR from recombinant plasmids pCEP4CyPet-MAMM and pCEP4YPet-MAMM with a set of primers CyPet-For 5′-CCGCTAGCGCTACCGGTCGCCACCATG-3′ with CyPet-Rev 5′-GAAGATCTTTTGTACAGTTCGTCCATGCCGT-3′, and YPet-For 5′-GCGAATTCATGGTGAGCAAAGGCGAAGAG-3′ with YPet-Rev 5′-TGGATCCGTTGGCCGCTTTAATGATGGTGATGGTGGTGCTTATAGAGCTC-3′ respectively. PCR products were cloned into pCR2.1 and confirmed by automated sequencing. FRET constructs CyPet-RL7-YPet, ECFP-RL7-YPet and CyPet-P14-YPet were obtained by replacement of the ECFP/EYFP pair as depicted in Figures 2c and d. To generate monomeric forms mCyPet/mYPet, alanine 206 was substituted with lysine in CyPet/YPet, as reported previously.25,26 This was achieved by PCR-based mutagenesis (see below). Next, the monomeric forms mCyPet and mYPet were cloned into pCR2.1, confirmed by sequencing and used to produce new FRET constructs (Figures 2e and f).
Figure 2.
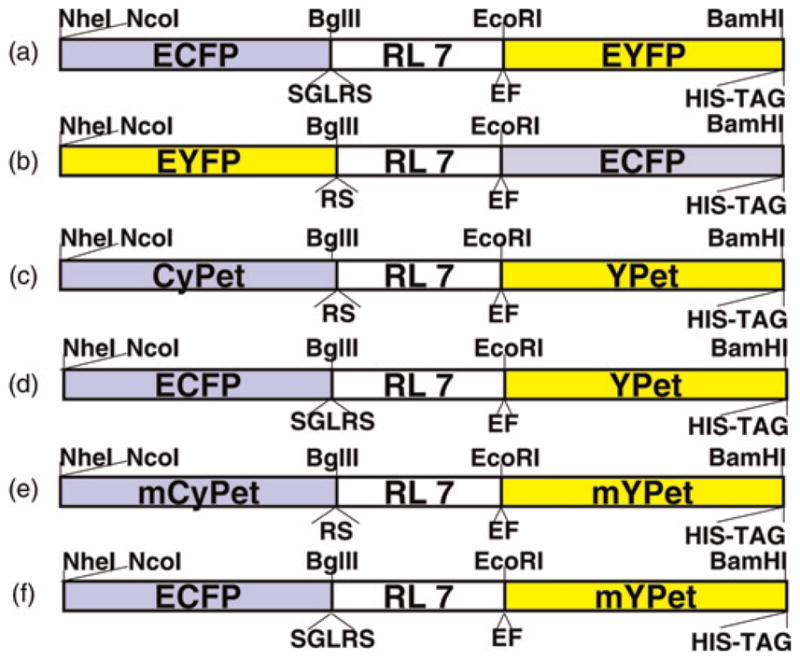
Diagram of the redox-sensitive linker RL7 flanked by various GFP-based FRET pairs. Cloning sites are shown on the top and additional amino acids surrounding the linker are marked on the bottom of each construct. GFP, green fluorescent protein; FRET, Förster resonance energy transfer (A color version of this figure is available in the online journal)
Construction of CY-RL7 mutants
To evaluate the role of cysteine residues on the FRET response, each cysteine residue in RL7 was sequentially substituted with an alanine residue and four mutated FRET constructs, CY-RL7cys1, CY-RL7cys2, CY-RL7cys3 and CY-RL7cys4, were generated. The numerical order of mutated constructs corresponds to the location of cysteine residues in the linker (Figure 6a). Mutagenesis was performed by a PCR-based method described elsewhere with minor modifications.27 Briefly, a complementary DNA (cDNA) of interest was amplified into two separate PCR fragments using four designed primers. The backbone plasmid pECFP-C1 with RL7 cloned in the downstream of ECFP gene between BglII/EcoRI sites served as a template. Each fragment was produced by pairing one anchor primer with one mutagenic primer. As the anchor forward primer was CyPet-For with restriction sites NheI, the anchor reverse primer endowed 5′-ACGCCTTAAGATACATTGATGAGTTTGGAC-3′ and the two mutagenic primers, Cys1for endowed 5′-AACTCTTCAGCCGAAGCAGCTGCGAAGGAGGCAGCAGCT-3′ and Cys1rev endowed 5′-AACTCTTCAGGCTGGAGATCTGAGTCCGGACTTGT-3′, contained a desired mutation near the recognition site of the type II restriction enzyme EarI. The resultant mutated cDNA was obtained after digestion of the two PCR fragments with EarI followed by ligation. The mutated cDNA was cloned into pCR2.1 and confirmed by sequencing. The TA-plasmid, containing the mutated ECFP-RL7 construct, was double digested with NheI and EcoRI followed by agarose gel electrophoresis. The ECFP-RL7 fragment isolated from the gel was ligated to the recombinant plasmid pCY-RL7, which had been previously digested with NheI and EcoRI. The same mutagenic steps were repeated for the next three individual cysteine residues presented in RL7 with the use of primer pair Cys2 5′-AACTCTTCAGCCGAGGCAGCTGCAAAATGTGAAGCT-3′ and 5′-AACTCTTCAGGCCTTAGCTGCAGCCTCCTTAGCTGC-3′, Cys3 pair 5′-AACTCTTCAGCCGAAGCTGCAGCTAAGGAAGCAGCT-3′ and 5′-AACTCTTCAGGCTTTTGCAGCTGCCTCGCACTTAGC-3′, and Cys4 pair 5′-AACTCTTCAGCCGAATTCTGCAGTCGACGGTACCGC-3′ and 5′-AACTCTTCAGGCCTTTGCTGCTGCTTCCTTTGCGGC-3′. The double mutant CY-RL7cys 1.3, containing mutations of cysteine residues in positions 1 and 3, was generated from template CY-RL7cys1 following the steps described above.
Figure 6.
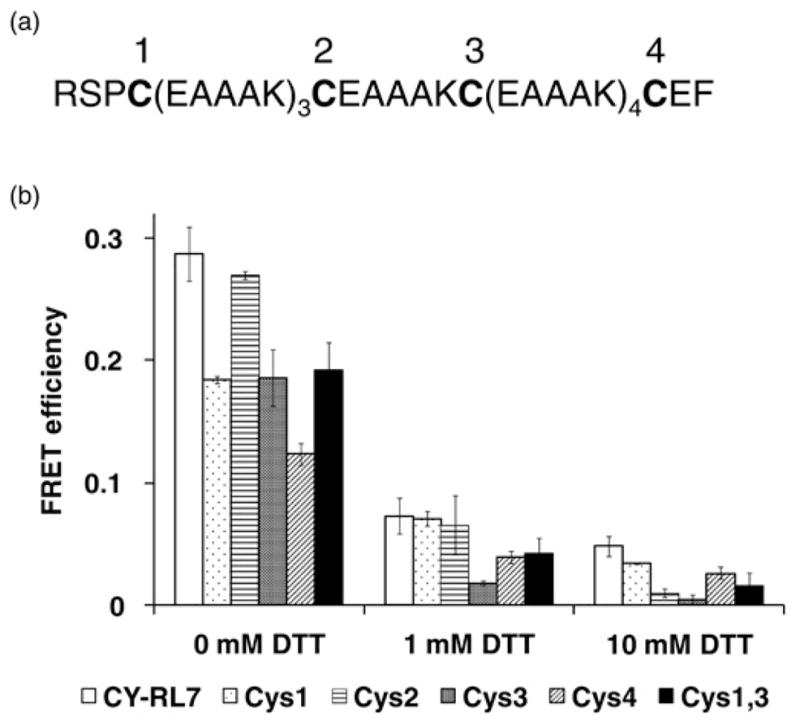
(a) Amino acid sequence for the redox-sensitive linker RL7 where cysteine residues are indicated by numerical order from the N- to the C-terminal end. (b) FRET response to DTT treatment for purified construct CY-RL7, single cysteine mutants CY-RL7cys1 (Cys1) – CY-RL7cys4 (Cys4), and double mutant CY-RL7cys1,3 (Cys1,3) where the cysteine mutant numbers correspond to the numerical order of the mutated cysteines in (a). Differences of calculated FRET efficiencies between untreated (0 mmol/L DTT) and DTT-treated probes were statistically significant (P < 0.05). Values given are the average of six experiments for CY-RL7 and two experiments for others ± SD. DTT, dithiothreitol; FRET, Förster resonance energy transfer
To produce the monomeric form of CyPet, we used the anchor forward primer CyPet-For with the restriction site NheI. The anchor reverse primer CyPet-Rev contained BglII for cloning of a mutated cDNA into a vector, and the two mutagenic primers 5′-AACTCTTCACTTGCTCTGGGTGCTCAGGTAGTGGTTG-3′ and 5′-AACTCTTCAAAGCTGAGCAAGGACCCCAACGAGAAG-3′ contained an EarI site. The mYPet form was generated with the anchor forward primer YPet-For, containing restriction site EcoRI. The anchor reverse primer YPet-Rev contained BamHI, and the two mutagenic primers 5′-AACTCTTCAAAGCTGTTCAAGGACCCCAACGAGAAG-3′ and 5′-AACTCTTCACTTGCTCTGGTAGCTCAGGTAGTGGTTG-3′ were with an EarI site.
Expression and purification
All FRET constructs were subcloned from pECFP-C1 recombinants into NcoI/BamHI sites of pET19b (Novagen, San Diego, CA, USA). Single forms of mCyPet and mYPet with 6xHis tag introduced to the C-terminal end were amplified by PCR with the following set of primers: 5′-GGCGCATATGTCCATGGTGAGCAAGGGAGAGGA-3′ with 5′-GGATCCTTAATGATGGTGATGGTGGTGTTTGTACAGTTCGTCCATG-3′, and 5′-GCGAATCCATGGTGAGCAAAGGCGA-3′ with YPet-Rev, respectively. PCR products were TOPO cloned into pCR2.1, confirmed by sequencing and subsequently cloned into NcoI/BamHI sites of pET19b. Expression and purification of recombinant proteins was performed as described previously.18 Samples were desalted by repeated filtration through centrifugal concentrators Centricon-30 (Millipore, Billerica, MA, USA) and diluted with buffer comprised of 50 mmol/L NaH2PO4, 100 mmol/L NaCl, pH 8.0. Protein content was determined as elsewhere.18
Steady-state fluorescence spectroscopy and the (ratio)A method
All fluorescence and FRET measurements were carried out on 0.5 μmol/L purified proteins as described earlier.18 Collected emission spectra were used for the determination of FRET efficiency via the (ratio)A method. For all FRET constructs (except when CyPet, YPet, mCyPet or mYPet were present), the FRET efficiencies, E, were calculated from the (ratio)A values as described in detail previously:18
| (1) |
The ratio of donor and acceptor extinction coefficients at the relevant excitation wavelengths (1.8 and 0.076) corrects for differences in absorption coefficients of the two molecules.
Measurement of mCyPet and mYPet extinction coefficients
The extinction coefficients for FRET pairs involving CyPet, YPet, mCyPet or mYPet are also required for precise determination of the FRET efficiency. Absorption spectra for mCyPet and mYPet are not commonly available; thus, we measured separately absorption spectra for the mCyPet and mYPet proteins in order to determine the spectral shapes of each and to obtain the extinction coefficient ratios needed for conversion of (ratio)A values to FRET efficiencies. Then, the absorption spectrum for the biosensor with both mCyPet and mYPet was measured. The separate contributions of mCyPet and mYPet can be linearly unmixed by fitting to their individual spectral shapes. This is a convenient method for determining their extinction coefficient ratios empirically. We assumed that extinction coefficients determined for the monomeric, mutated mCyPet and mYPet were the same as for the original CyPet and YPet forms.
The fitting routine was performed with Mathematica (Wolfram Research, Champaign, IL, USA). The absorption spectra of purified mCyPet and mYPet were measured separately and interpolated using the ‘Interpolation’ function in Mathematica. Then the mCyPet-RL-mYPet absorption spectrum was fit using the interpolated mCyPet and mYPet spectra. The fitting function had the following form:
| (2) |
where α and β are the amplitudes of the mCyPet (AmCyPet(λ)) and mYPet (AmYPet(λ)) absorption spectra, respectively. γ is a constant offset to account for any background. The fit was implemented in Mathematica using the ‘FindFit’ function.
In Figure 3, the fit to the absorption spectra of mCyPet-RL7-mYPet is shown. A large absorbance peak found in the ultraviolet (UV) was subtracted from the raw data by fitting the slope of the UV peak and subtracting it from all of the spectra. This subtraction increased the reliability of the final fitting routine. The extinction coefficient ratios were found to be: εmYPet(500 )/εmCYPet(440 nm) = 1.7 and εmYPet(440 nm)/εmYPet(500 nm) = 0.058. Thus, the FRET efficiency is calculated as
Figure 3.
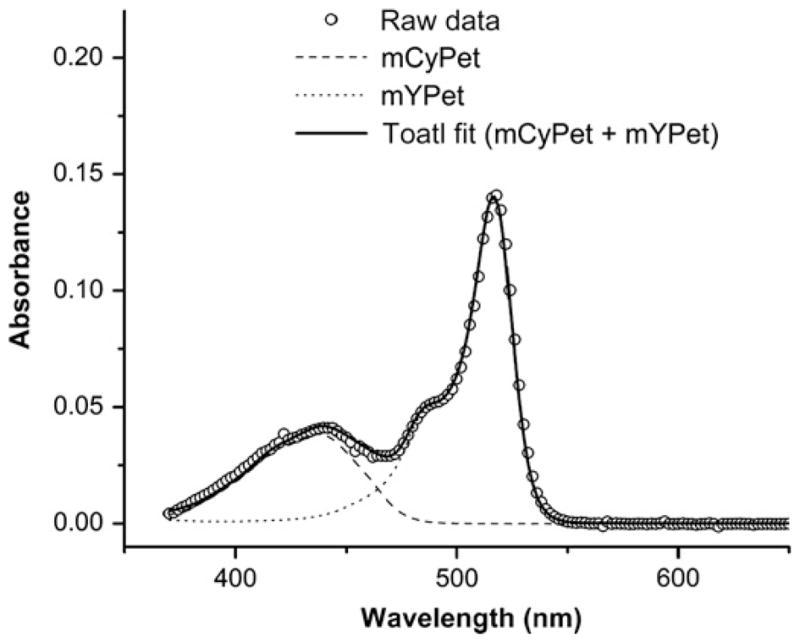
Absorption spectrum of the biosensor with mCyPet and mYPet. The individual spectra of mCyPet and mYPet were measured separately and used to fit the total absorption spectrum of the biosensor. The fit parameters are used to determine the extinction coefficient ratios for mCyPet and mYPet. Since every biosensor is expressed with mCyPet and mYPet, we have assumed that their concentrations are equal and therefore cancel from the calculations of the extinction coefficient ratios
| (3) |
for mCyPet-RL7-mYPet, mCyPet-P14-mYPet, CyPet-RL7-YPet and CyPet-P14-YPet constructs. FRET efficiencies for constructs ECFP-RL7-YPet and ECFP-RL7-mYPet were calculated using Equation (1).
Glutathione measurements and determination of redox potentials
All measurements were performed on the purified CY-RL7 after buffer exchange with Zeba Desalt Spin Columns following the manufacturer’s protocol (Pierce). For determination of the CY-RL7 midpoint potential, oxidized CY-RL7 (0.5 μmol/L) was incubated in degassed buffer composed of 100 mmol/L sodium phosphate, pH 7.0, 100 mmol/L NaCl, 1 mmol/L EDTA and containing different concentrations of GSH (125 μmol/L to 10 mmol/L).28,29 Five millimolar GSSG was added to the reaction mixtures with two additional samples of 10 mmol/L GSH mixed with 1 mmol/L and 0.25 mmol/L GSSG. In addition, CY-RL7 was incubated with 10 mmol/L GSH and 5 mmol/L GSSG only. To reach equilibrium, the redox mixtures were incubated in the dark for 16–20 h at 4°C or alternatively at room temperature. However, incubation for 3–4 h at room temperature was enough to reach equilibrium. Next, fluorometric measurements were carried out at 25°C. The relative amount of oxidized CY-RL7 at equilibrium (R) was calculated according to Equation 4:
| (4) |
where E is the measured FRET efficiency, and Ered and Eox are the FRET efficiencies of completely reduced or oxidized CY-RL7, respectively. Ered was determined by incubating CY-RL7 with 10 mmol/L dithiothreitol (DTT) (FRET efficiency values were identical in both 10 mmol/L DTT and 10 mmol/L GSH incubations). All titrations were performed in five experiments on separate preparations of the CY-RL7 probe.
The equilibrium GSH and GSSG concentrations were determined immediately after fluorescence measurements according to a previously described method with modifications.30 Briefly, samples that were derivatized with iodoacetic acid were analyzed with the QTRAP 5500 liquid chromatography-tandem mass spectrometry system (AB Sciex, Foster City, CA, USA) with a 1200 series high-performance liquid chromatography system (Agilent Technologies, Santa Clara, CA, USA) including a degasser, an autosampler and a binary pump. The liquid chromatography separation was performed on a Platinum EPS C18 100A column (4.6 mm × 150 mm, 3μ) (Grace, Deerfield, IL, USA) with mobile phase A (0.1% formic acid in water) and mobile phase B (0.1% formic acid in acetonitrile). The flow rate was 0.4 mL/min. The linear gradient was as follows: 0–2 min, 95% A; 7–12 min, 5% A; 12.1–20 min, 95% A. The autosampler was set at 5°C and its injection needle was washed with water before each injection to minimize the carryover. The injection volume was 1 μL. Positive mass spectra were acquired with electrospray ionization. The ion spray voltage was 5500 V. The source temperature was 350°C. The curtain gas, ion source gas 1 and ion source gas 2 were 35, 65 and 55, respectively. Multiple reaction monitoring was used to quantify metabolites: GSSG (m/z 613.3→m/z 355.2); GSH (m/z 366.2→m/z 237.2); internal standard N-isobutyryl-D-cysteine (m/z 249.8→m/z 180.0). Reduction potentials in millivolts for varying ratios of GSSG/2GSH were calculated from the Nernst equation at 25°C and pH 7.0.5
Statistical analysis
Results are expressed as mean ± SD. Statistical differences between the treated and control probes were determined by pairwise t-test using the R statistic software package. Differences between means were considered significant if P < 0.05.
Results
The novel redox sensor undergoes a six-fold change in FRET efficiency
Previously engineered RLs did not eliminate FRET completely between the donor and acceptor in the reduced state; that is, in the absence of disulfide bridge(s).18 To achieve a greater separation between the FRET donor/acceptor pair, we introduced three additional α-helical sequences –EAAAK – into the structure of the previously reported RL5 linker (–RSPCAAKEAAAKECAAAKECAAKEAAAKCEF–). Indeed, the RL7 provides a greater separation in the fully reduced state (10 mmol/L DTT) as evidenced by a FRET efficiency decrease from 0.131 (RL5) to 0.048 (RL7). This led to a large change in the sensitized FRET signal, which is apparent from the fluorescence emission spectra of the CY-RL7 acquired from the fully oxidized and reduced states (Figure 4a). FRET efficiency values are shown in Figure 4b. Surprisingly, in addition to the expected decrease in FRET in the reduced state, the CY-RL7 sensor also outperformed CY-RL5 in the oxidized state despite its longer linker (Figure 4b). Furthermore, the CY-RL7 sensor is more sensitive to low concentrations of DTT, with a prominent six-fold change in the FRET signal observed in 1 mmol/L DTT (Figure 4b). Exchanging the positions of ECFP and EYFP at the opposing ends of the linker resulted in almost an eight-fold change in FRET efficiency of the purified sensor between the two redox states (Figure 4b, Table 1). That is, the ECFP/EYFP pair was fused to RL7 by placing the acceptor at the N-terminal side of the sensor (Figure 2b), producing a new variant of the FRET construct EYFP-RL7-ECFP (YC-RL7). Thus, apparently the position of the fluorophores relative to the linker affects the FRET efficiency. In both the fully reduced and oxidized states, the FRET efficiency of YC-RL7 is significantly decreased relative to the CY-RL7 construct (Figure 4b). However, the change in FRET efficiency is slightly improved compared with CY-RL7 (Table 1). Independent of the placement of the fluorophore pair, both sensors are sensitive to low concentrations of DTT (Figure 4b).
Figure 4.
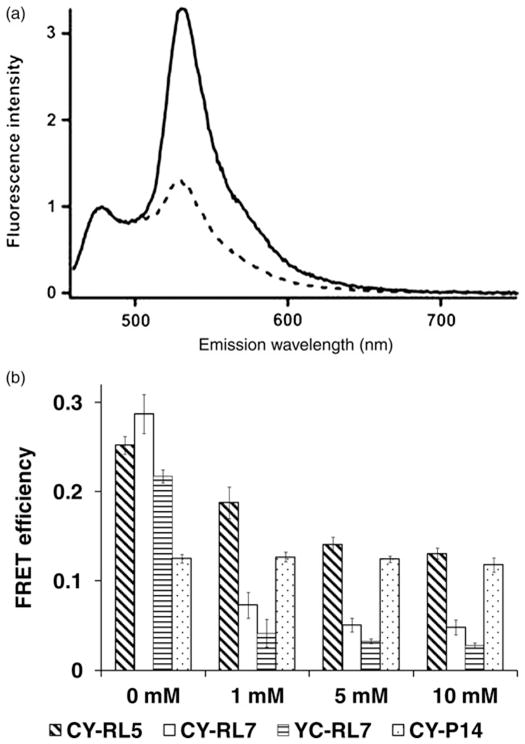
(a) Fluorescence emission spectra of the CY-RL7 sensor at 20°C, where the solid line corresponds to untreated sensor and the dashed line to the protein pretreated with 10 mmol/L DTT. Purified protein was excited at 440 nm and spectra were normalized to the intensity of the ECFP peak (475 nm). (b) FRET response to DTT treatment for purified constructs CY-RL5, CY-RL7 and YC-RL7. Values given are the average of three experiments ± SD. Differences of calculated FRET efficiencies between untreated (0 mmol/L DTT) and DTT-treated sensors CY-RL5, CY-RL7 and YC-RL7 were statistically significant (P < 0.05). Significant differences were not observed for the polyproline CY-P14 construct (snow bars; P > 0.05). DTT, dithiothreitol; FRET, Förster resonance energy transfer
Table 1.
FRET efficiency of various FRET constructs at oxidized and reduced states
| FRET construct | Reduced
|
Oxidized
|
Δr (Å) | ΔE/Ered(%) | ||
|---|---|---|---|---|---|---|
| Ered | rred = R0(1/Ered − 1)1/6 (Å) | Eox | rox = R0(1/Eox − 1)1/6 (Å) | |||
| CY-P14 | 0.118 | 68.5 | 0.125 | 67.8 | 0.7 | 6 |
| CY-RL5 | 0.131 | 67.1 | 0.252 | 58.7 | 8.4 | 92.4 |
| CY-RL7 | 0.048 | 80.6 | 0.287 | 57.0 | 23.6 | 598 |
| EYFP-RL7-ECFP | 0.028 | 88.5 | 0.217 | 60.7 | 27.8 | 775 |
| CyPet-RL7-YPet | 0.265 | 58.1 | 0.270 | 57.8 | 0.2 | 2 |
| CyPet-P14-YPet | 0.375 | 53.4 | 0.371 | 53.5 | 0 | 0 |
| ECFP-RL7-YPet | 0.376 | 53.3 | 0.288 | 57.0 | 3.7 | 23.4 |
| ECFP-RL7-mYPet | 0.062 | 77.1 | 0.289 | 56.9 | 20.1 | 431 |
| mCyPet-RL7-mYPet | 0.051 | 79.8 | 0.211 | 61.0 | 18.7 | 314 |
| mCyPet-P14-mYPet | 0.092 | 71.8 | 0.095 | 71.3 | 0.4 | 3 |
FRET, Förster resonance energy transfer; DTT, dithiothreitol; CFP, cyan fluorescent protein; YFP, yellow fluorescent protein
E, FRET efficiency of various constructs at reduced (10 mmol/L DTT) and oxidized (0 mmol/L DTT) states was determined by the (ratio)A method; r, estimated distance between CFP/YFP pairs assuming their critical Förster distance R0 to be 49 Å (see ref.42); ΔE = ((Eox − Ered)/Ered) × 100%; Δr = rred − rox
To demonstrate that changes in the FRET signal during pretreatment with DTT were caused by the linker only, and not by an environmental sensitivity of the construct, a rigid polyproline linker-based construct, ECFP-P14-EYFP (CY-P14), was used as a control (Figure 4b).
ECFP/EYFP FRET pair outperforms the modified mYPet/mCyPet pair
As recently reported, the CyPet/YPet FRET pair provides seven-fold enhancement of the FRET signal (over the ECFP/EYFP pair) due to its decreased spectral bleed-through in common CFP/YFP filter sets.31 Thus, we reasoned that using the CyPet/YPet pair would increase the dynamic range of our redox probe, and the CyPet/YPet pair was fused to the RL7 and P14 linkers (Figure 2c). In the basal state (0 mmol/L DTT), the purified constructs are found in the fully oxidized state and exhibit a high FRET signal (Figures 5a and b). Surprisingly, redox sensitivity was not observed with the CyPet-RL7-YPet construct (Figure 5a). Moreover, in contrast to the CY-P14 construct, where the linker P14 stably separates the FRET donor and acceptor, the P14 linker is unable to prevent association of CyPet and YPet due to their strong tendency to form heterodimers (under high, local concentrations as part of a single fusion protein).26 The CyPet-P14-YPet construct has a three-fold increased FRET efficiency compared with CY-P14 (Figure 5b). Additionally, in an oxidative environment, the FRET signal for the CyPet-P14-YPet control construct is higher than that of the CyPet-RL7-YPet sensor (0.37 and 0.27, respectively). Taken together, these data show that the P14 linker is ineffective in maintaining FRET donor and acceptor separation for the CyPet/YPet FRET pair. Our data unambiguously demonstrate that CyPet and YPet separated by rigid linkers form heterodimers, which suggests caution for their use in reversible fusion sensors, and corroborates a recent report of similar behavior using unstructured linkers.26 To overcome CyPet/YPet pair dimerization, we performed a monomerizing mutation (A2006K)25,26 to create the monomeric forms mCyPet and mYPet. Unequivocal evidence for elimination of dimerization via the monomerizing mutation is apparent from Figures 5a and b. The response of mCYPet-RL7-mYPet constructs to DTT followed similar patterns observed for RL7-based constructs; however, the FRET dynamic range is moderately less for mCyPet-RL7-mYPet compared with CY-RL7 (Figure 5a).
Figure 5.
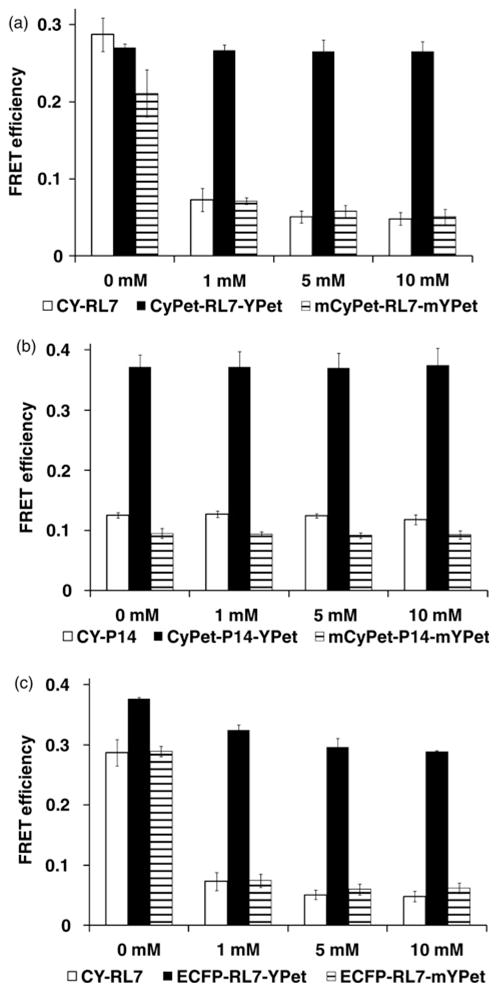
FRET response to DTT treatment for purified constructs. (a) CY-RL7, CyPet-RL7-YPet and mCyPet-RL7-mYPet. Values given are the average of three experiments ± SD. Differences of calculated FRET efficiencies between untreated (0 mmol/L DTT) and DTT-treated probes CY-RL7, and mCyPet-RL7-mYPet were statistically significant (P < 0.05). Significant differences were not observed for the CyPet-RL7-YPet construct (snow bars; P > 0.05). (b) CY-P14, CyPet-P14-YPet and mCyPet-P14-mYPet. Values given are the average of three experiments ± SD. Significant differences were not observed between the treated and control constructs (P > 0.05). (c) CY-RL7, ECFP-RL7-YPet and ECFP-RL7-mYPet. Values given are the average of six experiments for CY-RL7 and two experiments for others ± SD. DTT, dithiothreitol; FRET, Förster resonance energy transfer
In the process of cloning of the FRET constructs, we created and validated mixed donor–acceptor forms (i.e. the ECFP-RL7-YPet and ECFP-RL7-mYPet constructs; Figure 5c). The appearance of moderate redox sensitivity in ECFP-RL7-YPet was still strongly affected by intramolecular (or, possibly intermolecular) dimerization while the ECFP-RL7-mYPet was equivocal to CY-RL7 (Figure 5c).
The individual cysteine residues of the linker make distinct contributions to the FRET response
To gain insight into the roles of the individual cysteine residues of the linker in the performance of the redox sensor, four new FRET constructs, CY-RL7cys1, CY-RL7cys2, CY-RL7cys3 and CY-RL7cys4, were constructed. In each construct, the individual cysteine in RL7 was replaced by an alanine residue. The mutated constructs are named according to the relative placement order of the cysteine residues in RL7 (Figure 6a). The purified mutated proteins were analyzed by steady-state fluorescence spectroscopy. Comparison of FRET efficiency changes in the cysteine mutants in response to DTT treatment are presented in Figure 6b. The data indicate that cysteine in position 2 is the least important cysteine for the formation of disulfide bonds, while cysteines in positions 1 and 3 contribute equally (Figure 6b). The greatest impact was found for the mutation of the fourth cysteine, which decreased the FRET efficiency to half of that of CY-RL7. In an attempt to further explore the role of cysteines 2 and 4 on the sensor performance, a double cysteine mutant CY-RL7cys1.3 was created where only cysteine 2 and cysteine 4 are present. That experiment demonstrated that sensors with only cysteines 2 and 4 are sufficient to induce a measurable FRET response (Figure 6b). However, the greatest FRET response was still achieved with all four cysteines present.
CY-RL7 exhibits the least negative half point redox potential among GFP-based sensors
We chose to investigate the redox sensitivity of CY-RL7 using the physiologically relevant GSH/GSSG redox couple. Early experiments demonstrated that the purified sensor could be fully reduced from its oxidized state with 10 mmol/L GSH. This result suggests that the sensor possesses an extremely oxidative nature, taking into consideration trace amounts of GSSG known to be present in commercially available GSH.13 GSH titrations were then performed in five experiments using purified proteins prepared from separately expressed batches (Figure 7). The midpoint redox potential of the CY-RL7 was found to be −143 ± 6 mV, which indicates that the RL7-based sensor exhibits a lesser negative half point redox potential than the currently available rxYFPs and roGFP1-iX sensors, which possess more negative redox midpoint potentials.7,14,17
Figure 7.
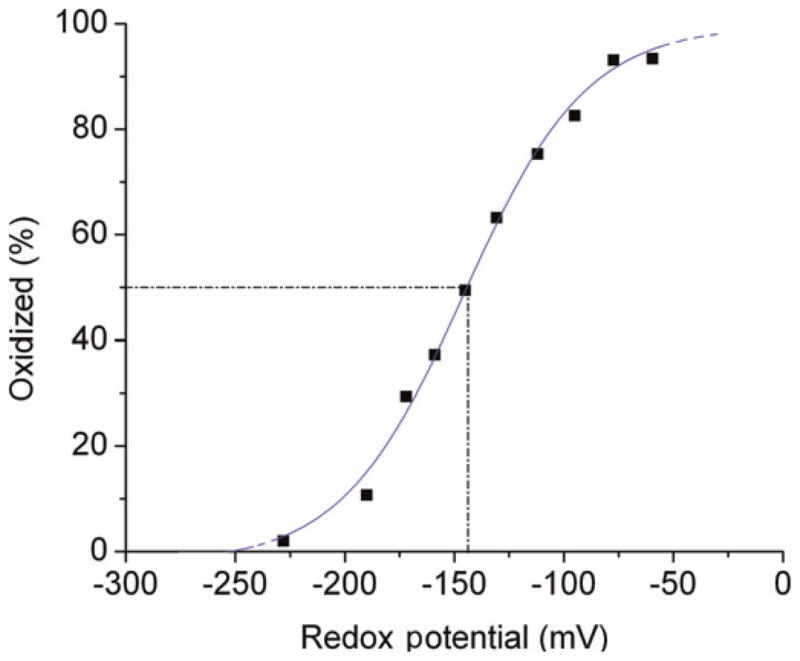
Representative CY-RL7 oxidation curve in response to approximately 10 mmol/L GSSG/2GSH treatment. Redox potentials in mV were calculated with the Nernst equation
Discussion
In this work we have presented significant improvement in the dynamic range of our redox biosensor’s FRET response. Because the FRET efficiency depends very strongly on the distance between the FRET donor and acceptor, we reasoned that varying the distance would lead to a higher dynamic range. In the reduced state, the new redox sensor is characterized by a very low FRET efficiency because the donor and the acceptor are separated by a long distance in the absence of disulfide bonds. We demonstrated that the decrease in the FRET efficiency in the reduced state, compared with the early sensors, can be achieved by simply extending the helix and the oxidized state can still be formed with a significant increase in FRET efficiency. Three more α-helical sequences (EAAAK) were added to the previously described α-helical RL5 to produce RL7, which is comprised of eight α-helical (EAAAK) sequences, and possesses a better separation in its reduced state. Apparently, the rigidity of RL5 comprised of (EAAAK)n (n = 5) was not compromised with extension of the helix. The FRET efficiency of the CY-RL7 sensor decreased 2.5-fold in its fully reduced state (from 0.131 to 0.048, Table 1). Furthermore, we observed a moderate increase (14%) in the FRET signal in the oxidative state, in spite of the greater number of amino acids in the linker sequence (Figure 4b). These data may indicate that the FRET efficiency of the GFP variants is influenced by both the length of the linker and the FRET orientation factor. Overall, an approximately six-fold change in FRET efficiency is observed for the purified sensor between its two extreme redox states (Table 1). Moreover, we observed that the sensitivity of the CY-RL7 sensor is significantly increased at low concentrations of DTT, with the greatest changes in the FRET signal during oxidation/reduction occurring between 0 and 1 mmol/L DTT (Figure 4b). These data demonstrate an improved redox sensitivity of the CY-RL7 construct.
In addition to altering the linker distance, we investigated positional effects of the terminally linked fluorescent proteins on the FRET efficiency. We were motivated by recent observations for fluorescent proteins that suggest a strong contribution of the orientation factor of chromophores to the FRET efficiency, which can occur when the reorientation rate (i.e. the rotational diffusion rate) of the chromophores is slow (relative to the nanosecond timescale of FRET).32 Additionally, it was shown that the contribution of the orientation factor correlates with linker rigidity.32 To probe for the possible positional effects, FRET efficiencies were compared for two redox constructs, which differ by exchanging the locations of the donor and the acceptor chromophores. The YC-RL7 construct, in which the acceptor EYFP was on the N-terminal side of the sensor, revealed decreased FRET efficiency to varying degrees in both the reduced and oxidized states compared with the CY-RL7 construct. Nevertheless, the YC-RL7 demonstrates a larger –7.5-fold – relative change in FRET upon reduction/oxidation mainly due to a minuscule FRET signal in the fully reduced state (Table 1). Thus, the results with the YC-RL7 indicate the importance of the orientation factor on FRET. However, it may be possible to further improve the dynamic range of the FRET redox sensor by systematic, cyclic permutation of the chromophore orientation.33
Furthermore, we used the recently introduced CyPet/YPet FRET pair, which is reported to produce a seven-fold enhancement in dynamic range over the ECFP/EYFP pair for FRET measurements.31 Accordingly, the CyPet/YPet FRET pair was fused with RL7 and P14. However, we determined that CyPet and YPet form heterodimers when incorporated into our constructs, which is in agreement with another report.26 Surprisingly, the rigid P14 linker was less effective in separating donor from acceptor in both reduced and oxidized states than the α-helical RL7, as revealed by higher FRET (Figures 5a and b). Remarkably, the tendency for dimerization was still very strong for the mixed ECFP-RL7-YPet construct. Our data also provide unequivocal evidence of the elimination of dimerization via a monomeric mutation (A2006K).25,26 However, even with the elimination of dimerization, the newly modified mCyPet/mYPet pair under-performed the ECFP/EYFP pair, which has been predicted elsewhere.34 For instance, the FRET efficiency of mCyPet-RL7-mYPet decreased by 25% in its oxidative state compared with CY-RL7 while exhibiting similar redox sensitivity in response to DTT treatments (Figure 5a). In the process of cloning the mCyPet-RL7-mYPet construct we created and validated a mixed form, the ECFP-RL7-mYPet construct (Figure 5b), which had a similar performance to CY-RL7 in our hands but possesses the advantage of the enhanced brightness (i.e. enhanced absorbance and fluorescence quantum yield) of mYPet over EYFP. The (ratio)A method, a ratio-metric measure of sensitized acceptor fluorescence emission due to FRET that employs linear spectral decomposition to isolate the fluorescence emission of the donor and acceptor,35,36 has been used for accurate determination of FRET efficiencies and global comparison of the aforementioned constructs (Figure 8).
Figure 8.
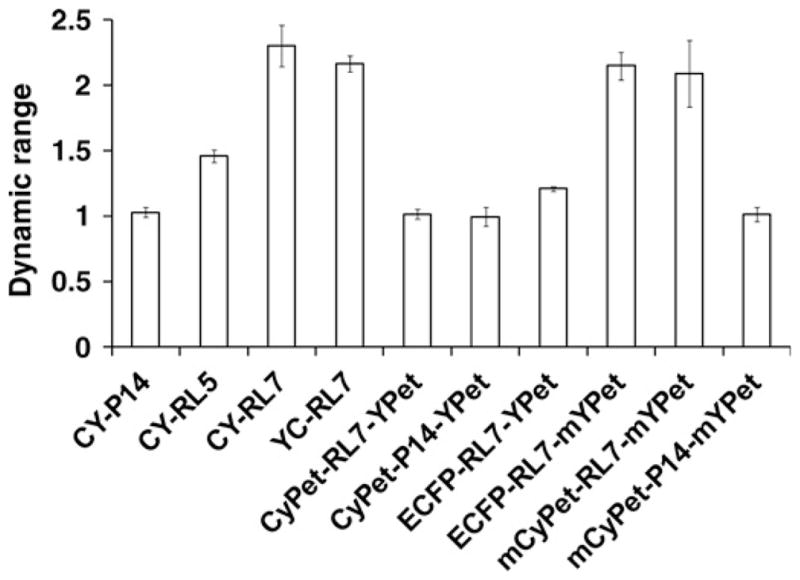
Comparison of various FRET constructs analyzed in a recent study. Dynamic range is [(ratio)A]OX/[(ratio)A]RED. The subscripts OX and RED refer to the oxidized and reduced forms of the constructs. Error bars for the dynamic ranges were calculated by propagating the corresponding standard deviations
In our sensor, reversible formation of disulfide bonds occurs between four cysteine residues embedded within the same α-helix polypeptide chain, RL7.24 The oxidation and reduction of cysteine residues by the reversible formation of disulfide bonds are an essential part of the catalytic functionality of the redox-sensitive sensor. Since the redox biosensor performance is highly sensitive to the ability to form disulfide bonds, optimizing locations of cysteine residues embedded into the linker should improve functionality of our redox probe. The precise placement of the four cysteines within the linker RL7 was based on a disulfide connectivity pattern that was determined with the aid of prediction tools as described previously.18 However, the computational prediction of disulfide connectivity directly from protein primary sequences is challenging.23 Nonetheless, experimental procedures can help identify strategic positioning of the cysteine residues in order to enhance the performance of the sensor.
To determine the strategic sites of the cysteine residues of our sensor, a mutagenesis approach was used whereby the cysteine residues embedded within the α-helical linker were sequentially substituted by alanine. The purified mutated probes were analyzed in vitro by altering the redox buffer conditions with various concentrations of DTT. The oxidative state of the mutants is influenced by the disulfide bonding pattern, while the reductive state evaluates the stiffness of the altered linkers flanked by GFP-based proteins. The FRET efficiency determined for each treatment provided a valuable parameter in understanding these characteristics.
Our results show the critical role of the fourth cysteine and its complex connectivity pattern with other cysteines (Figure 6b). Moreover, the uniformly strong FRET of the CY-RL7cys1 and CY-RL7cys3 mutants in the oxidative state indicates an equivalent role of the first and third cysteines in disulfide bonding. Further, based on a single mutation approach, we speculate that the input of the second cyteine in the bond formation is less significant than the bond formation between the other three cysteines. For instance, the FRET efficiency of the CY-RL7cys2 mutant’s oxidative state is unchanged in comparison with the CY-RL7 (Figure 6b) and, therefore, apparently the second cysteine does not play a significant role in the disulfide connectivity of the CY-RL7 sensor. This result implies either a lessened redox sensitivity for the second cysteine, or that it is in a spatially inactive position. On the other hand, substitution of the second cysteine with alanine amino acid apparently increases the stiffness of the linker. The CY-RL7cys2 mutant, in the fully reduced state, possesses a noticeable decrease in the FRET efficiency compared with the original CY-RL7 probe. A similar effect is also observed for the CY-RL7cys3 mutant. The decline in FRET efficiency may be attributed to an increased rigidity of the mutated linkers, as alanine residues have a greater potential to form helical regions than other amino acids.37,38 This also separates the donor from the acceptor better in mutants CY-RL7cys2 and CY-RL7cys3. Indeed, α-helical propensity of the RL7 is formed by repeats (EAAAK)n (n = 8) and it is not surprising that the second and third cysteines imbedded in the middle of the α-helix may affect α-helix formation. Importantly, the cysteine residues do not perturb α-helical propensity of RL7. In contrast, the first and fourth cysteines do not notably perturb the rigidity of the α-helix. Thus, our data demonstrate the efficacy of longer helical linkers for separating large domains in addition to the rigid peptides (EAAAK)n (n ≤ 6).39,40 This is consistent with recent observations that the linker (EAAAK)n (n = 8) provided better separation of two functional domains over (EAAAK)n (n = 4).41
To test whether the second cysteine is capable of forming disulfide bridges, a double mutant CY-RL7cys1,3 was constructed where only the second and fourth cysteines are present. Surprisingly, the second cysteine participates in the FRET redox response in a double mutant (0 mmol/L DTT, Figure 6b). It should be noted that many considerations based on the FRET efficiency alone are limited by the absence of information about the orientation factor of the GFP-based proteins. In conclusion, the present results demonstrate that the performance of the CY-RL7 sensor could possibly be improved further by repositioning of the second cysteine residue, which plays a negligible role in the current disulfide bonding pattern.
Finally, we determined the midpoint redox potential of the CY-RL7 sensor to be −143 ± 6 mV. Thus, this novel FRET-based sensor has strongly oxidizing potential with a shift of approximately 80 mV toward a less negative midpoint potential than currently developed roGFP-iX thiol probes.14 Therefore, the CY-RL7 sensor extends the measurable range of redox potentials by 80 mV, given that quantitative readout of GFP-based probes may be practically achieved in a range of approximately 40 mV from the midpoint potential.12,14,17 The CY-RL7 sensor should be useful in the range −100 to −180 mV required for monitoring of intraorganellar glutathione potentials in relatively high oxidative environments of ER, Golgi and lysosomes.
Acknowledgments
This work was supported by National Institutes of Health (NIH) grants: R21-EB004513 and R33-CA137719 to PJAK and HRG.
Footnotes
Author contributions: All authors participated in interpretation of the studies, analysis of the data and review of the manuscript. VLK, BQS, PJAK and HRG participated in the experimental design; VLK, BQS, JJH and AS conducted the experiments; and VLK, BQS, RMC and HRG wrote the manuscript.
References
- 1.Estrela JM, Ortega A, Obrador E. Glutathione in cancer biology and therapy. Crit Rev Clin Lab Sci. 2006;43:143–81. doi: 10.1080/10408360500523878. [DOI] [PubMed] [Google Scholar]
- 2.Ballatori N, Krance SM, Notenboom S, Shi S, Tieu K, Hammond CL. Glutathione dysregulation and the etiology and progression of human diseases. Biol Chem. 2009;390:191–214. doi: 10.1515/BC.2009.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Franco R, Cidlowski J. Apoptosis and glutathione: beyond an antioxidant. Cell Death Differ. 2009;16:1303–14. doi: 10.1038/cdd.2009.107. [DOI] [PubMed] [Google Scholar]
- 4.Jones DP. Redefining oxidative stress. Antioxid Redox Signal. 2006;8:1865–79. doi: 10.1089/ars.2006.8.1865. [DOI] [PubMed] [Google Scholar]
- 5.Schafer FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med. 2001;30:1191–212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- 6.Kemp M, Go Y-M, Jones DP. Nonequilibrium thermodynamics of thiol/disulfide redox systems: A perspective on redox systems biology. Free Radic Biol Med. 2008;44:921–37. doi: 10.1016/j.freeradbiomed.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ostergaard H, Henriksen A, Hansen FG, Winther JR. Shedding light on disulfide bond formation: engineering a redox switch in green fluorescent protein. EMBO J. 2001;20:5853–62. doi: 10.1093/emboj/20.21.5853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hanson GT, Aggeler R, Oglesbee D, Cannon M, Capaldi RA, Tsien RY, Remington SJ. Investigating mitochondrial redox potential with redox-sensitive green fluorescent protein indicators. J Biol Chem. 2004;279:13044–53. doi: 10.1074/jbc.M312846200. [DOI] [PubMed] [Google Scholar]
- 9.Dooley CT, Dore TM, Hanson GT, Jackson WC, Remington SJ, Tsien RY. Imaging dynamic redox changes in mammalian cells with green fluorescent protein indicators. J Biol Chem. 2004;279:22284–93. doi: 10.1074/jbc.M312847200. [DOI] [PubMed] [Google Scholar]
- 10.Hansen RE, Ostergaard H, Winther JR. Increasing the reactivity of an artificial dithiol-disulfide pair through modification of the electrostatic milieu. Biochemistry. 2005;44:5899–906. doi: 10.1021/bi0500372. [DOI] [PubMed] [Google Scholar]
- 11.Cannon MB, Remington SJ. Re-engineering redox-sensitive green fluorescent protein for improved response rate. Protein Sci. 2006;15:45–57. doi: 10.1110/ps.051734306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bjornberg O, Ostergaard H, Winther JR. Measuring intracellular redox conditions using GFP-based sensors. Antioxid Redox Signal. 2006;8:354–61. doi: 10.1089/ars.2006.8.354. [DOI] [PubMed] [Google Scholar]
- 13.Gutscher M, Pauleau AL, Marty L, Brach T, Wabnitz GH, Samstag Y, Meyer AJ, Dick TP. Real-time imaging of the intracellular glutathione redox potential. Nature Methods. 2008;5:553–9. doi: 10.1038/nmeth.1212. [DOI] [PubMed] [Google Scholar]
- 14.Lohman JR, Remington SJ. Development of a family of redox-sensitive green fluorescent protein indicators for use in relatively oxidizing subcellular environments. Biochemistry. 2008;47:8678–88. doi: 10.1021/bi800498g. [DOI] [PubMed] [Google Scholar]
- 15.Delic M, Mattanovich D, Gasser B. Monitoring intracellular redox conditions in the endoplasmic reticulum of living yeasts. FEMS Microbiol Lett. 2010;306:61–6. doi: 10.1111/j.1574-6968.2010.01935.x. [DOI] [PubMed] [Google Scholar]
- 16.Jensen KS, Hansen RE, Winther JR. Kinetic and thermodynamic aspects of cellular thiol–disulfide redox regulation. Antioxid Redox Signal. 2009;11:1047–58. doi: 10.1089/ars.2008.2297. [DOI] [PubMed] [Google Scholar]
- 17.Meyer AJ, Dick TP. Fluorescent protein-based redox probes. Antioxid and Redox Signal. 2010;13:621–50. doi: 10.1089/ars.2009.2948. [DOI] [PubMed] [Google Scholar]
- 18.Kolossov VL, Spring BQ, Sokolowski A, Conour JE, Clegg RM, Kenis PJ, Gaskins HR. Engineering redox-sensitive linkers for genetically encoded FRET-based biosensors. Exp Biol Med. 2008;233:238–48. doi: 10.3181/0707-RM-192. [DOI] [PubMed] [Google Scholar]
- 19.Yano T, Oku M, Akeyama N, Itoyama A, Yurimoto H, Kuge S, Fujiki Y, Sakai Y. A novel fluorescent sensor protein for visualization of redox states in the cytoplasm and in peroxisomes. Mol Cellular Biol. 2010;30:3758–66. doi: 10.1128/MCB.00121-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Y, Shyy JY-J, Chien S. Fluorescence proteins, live-cell imaging, and mechanobiology: seeing is believing. Annu Rev Biomed Eng. 2008;10:1–38. doi: 10.1146/annurev.bioeng.010308.161731. [DOI] [PubMed] [Google Scholar]
- 21.Kredel S, Oswald F, Nienhaus K, Deuschle K, Rocker C, Wolff M, Heilker R, Nienhaus GU, Wiedenmann J. Mruby, a bright monomeric red fluorescent protein for labeling of subcellular structures. PloS One. 2009;4:1–7. doi: 10.1371/journal.pone.0004391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ibraheem A, Campbell RE. Designs and applications of fluorescent protein-based biosensors. Curr Opin Chem Biol. 2010;14:30–6. doi: 10.1016/j.cbpa.2009.09.033. [DOI] [PubMed] [Google Scholar]
- 23.Zhu L, Yang J, Song J-N, Chou K-C, Shen H-B. Improving the accuracy of predicting disulfide connectivity by feature selection. J Comput Chem. 2010;31:1478–85. doi: 10.1002/jcc.21433. [DOI] [PubMed] [Google Scholar]
- 24.Lin C, Kolossov VL, Tsvid G, Trump L, Henry JJ, Henderson JL, Rund LA, Kenis PJA, Schook LB, Gaskins HR, Timp G. Imaging in real-time with FRET the redox responses of tumorigenic cells to glutathione petrubations in a microscale flow. Integrative Biology. 2011;3:208–17. doi: 10.1039/c0ib00071j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zacharias DA, Violin JD, Newton AC, Tsien RY. Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science. 2002;296:913–6. doi: 10.1126/science.1068539. [DOI] [PubMed] [Google Scholar]
- 26.Ohashi T, Galiacy SD, Briscoe G, Erickson HP. An experimental study of GFP-based FRET, with application to intrinsically unstructured proteins. Protein Sci. 2007;16:1429–38. doi: 10.1110/ps.072845607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ko JK, Ma J. A rapid and efficient PCR-based mutagenesis method applicable to cell physiology study. Am J Physiol Cell Physiol. 2005;288:C1273–8. doi: 10.1152/ajpcell.00517.2004. [DOI] [PubMed] [Google Scholar]
- 28.Loferer H, Wunderlich M, Hennecke H, Glockshuber R. A bacterial thioredoxin-like protein that is exposed to the periplasm has redox properties comparable with those of cytoplasmic thioredoxins. J Biol Chem. 1995;270:26178–83. doi: 10.1074/jbc.270.44.26178. [DOI] [PubMed] [Google Scholar]
- 29.Watson WH, Pohl J, Montfort WR, Stuchlik O, Reed MS, Powis G, Jones DP. Redox potential of human thioredoxin 1 and identification of a second dithiol/disulfide motif. J Biol Chem. 2003;278:33408–15. doi: 10.1074/jbc.M211107200. [DOI] [PubMed] [Google Scholar]
- 30.Bouligand J, Deroussent A, Paci A, Morizet J, Vassal G. Liquid chromatography-tandem mass spectrometry assay of reduced and oxidized glutathione and main precursors in mice liver. J Chromatogr B. 2006;832:67–74. doi: 10.1016/j.jchromb.2005.12.037. [DOI] [PubMed] [Google Scholar]
- 31.Nguyen AW, Daugherty PS. Evolutionary optimization of fluorescent proteins for intracellular FRET. Nat Biotechnol. 2005;23:355–60. doi: 10.1038/nbt1066. [DOI] [PubMed] [Google Scholar]
- 32.Deuschle K, Okumoto S, Fehr M, Looger LL, Kozhukh L, Frommer WB. Construction and optimization of a family of genetically encoded metabolite sensors by semirational protein engineering. Protein Sci. 2005;14:2304–14. doi: 10.1110/ps.051508105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nagai T, Yamada S, Tominaga T, Ichikawa M, Miyawaki A. Expanded dynamic range of fluorescent indicators for Ca(2+) by circularly permuted yellow fluorescent proteins. Proc Natl Acad Sci USA. 2004;101:10554–9. doi: 10.1073/pnas.0400417101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Piston DW, Kremers GJ. Fluorescent protein FRET: the good, the bad and the ugly. Trends Biochem Sci. 2007;32:407–14. doi: 10.1016/j.tibs.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 35.Clegg RM. Fluorescence resonance energy-transfer and nucleic-acids. Methods Enzymol. 1992;211:353–88. doi: 10.1016/0076-6879(92)11020-j. [DOI] [PubMed] [Google Scholar]
- 36.Clegg RM, Murchie AIH, Zechel A, Lilley DMJ. Observing the helical geometry of double-stranded DNA in solution by fluorescence resonance energy-transfer. Proc Natl Acad Sci USA. 1993;90:2994–8. doi: 10.1073/pnas.90.7.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marqusee S, Baldwin RL. Helix stabilization by Glu−···Lys+ salt bridges in short peptides of de novo design. Proc Natl Acad Sci USA. 1987;84:8898–902. doi: 10.1073/pnas.84.24.8898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Padmanabhan S, Marqusee S, Ridgeway T, Laue TM, Baldwin RL. Relative helix-forming tendencies of nonpolar amino acids. Nature. 1990;344:268–70. doi: 10.1038/344268a0. [DOI] [PubMed] [Google Scholar]
- 39.Arai R, Ueda H, Kitayama A, Kamiya N, Nagamune T. Design of the linkers which effectively separate domains of a bifunctional fusion protein. Protein Eng. 2001;14:529–32. doi: 10.1093/protein/14.8.529. [DOI] [PubMed] [Google Scholar]
- 40.Chang H-C, Kaiser CM, Hartl FU, Barral JM. De novo folding of GFP fusion proteins: high efficiency in eukaryotes but not in bacteria. J Mol Biol. 2005;353:397–409. doi: 10.1016/j.jmb.2005.08.052. [DOI] [PubMed] [Google Scholar]
- 41.Bai Y, Shen W-C. Improving the oral efficacy of recombinant granulocyte colony-stimulating factors and transferrin fusion protein by spacer optimization. Pharmaceutical Res. 2006;23:2116–21. doi: 10.1007/s11095-006-9059-5. [DOI] [PubMed] [Google Scholar]
- 42.Patterson GH, Piston DW, Barisas GB. Förster distances between green fluorescent protein pairs. Anal Biochem. 2000;284:438–40. doi: 10.1006/abio.2000.4708. [DOI] [PubMed] [Google Scholar]