

Abstract
The magnetosome, a biomineralizing organelle within magnetotactic bacteria, allows their navigation along geomagnetic fields. Magnetosomes are membrane-bound compartments containing magnetic nanoparticles and organized into a chain within the cell, the assembly and biomineralization of magnetosomes are controlled by magnetosome-associated proteins. Here, we describe the crystal structures of the magnetosome-associated protein, MamA, from Magnetospirillum magneticum AMB-1 and Magnetospirillum gryphiswaldense MSR-1. MamA folds as a sequential tetra-trico-peptide repeat (TPR) protein with a unique hook-like shape. Analysis of the MamA structures indicates two distinct domains that can undergo conformational changes. Furthermore, structural analysis of seven crystal forms verified that the core of MamA is not affected by crystallization conditions and identified three protein–protein interaction sites, namely a concave site, a convex site, and a putative TPR repeat. Additionally, relying on transmission electron microscopy and size exclusion chromatography, we show that highly stable complexes form upon MamA homooligomerization. Disruption of the MamA putative TPR motif or N-terminal domain led to protein mislocalization in vivo and prevented MamA oligomerization in vitro. We, therefore, propose that MamA self-assembles through its putative TPR motif and its concave site to create a large homooligomeric scaffold which can interact with other magnetosome-associated proteins via the MamA convex site. We discuss the structural basis for TPR homooligomerization that allows the proper function of a prokaryotic organelle.
The biomineralization of iron oxides or iron sulfides takes place in unique prokaryotic organelles assembled by magnetotactic bacteria, termed magnetosomes. Magnetosomes originate from invaginations of the inner membrane and are organized into a chain. Each magnetosome contains a magnetite (iron oxide) or greigite (iron sulfide) nano-crystal (Fig. S1). The linear arrangement of these nano-crystals creates a single and stable magnetic dipole, allowing magnetotactic bacteria to navigate along geomagnetic fields. This trait assists the bacteria in their search for transition environments, such as the oxic-anoxic transition zone (1) more effectively. Magnetosome assembly and function are governed by a unique set of soluble and integral membrane proteins encoded by a distinct genomic region referred to as the magnetosome island (2, 3). Deletion of the magnetosome island results in the lack of magnetosomes and a loss of the ability of the bacteria to orient themselves along a magnetic field (4, 5). The magnetosome island contains four main operons that control the biomineralization process, namely mamAB, mamCD, mms6, and mamXY (6). The mamAB operon contains several highly conserved genes which are assumed to be essential for magnetosomal membrane biogenesis, magnetosome alignment, and iron transport (2, 4).
One of the most abundant and conserved magnetosome-associated proteins is MamA [also known as Mms24 (7) and Mam22 (8)]. While deletion of mamA has no effect on membrane invagination, most of the invaginations formed in strains lacking the gene do not biomineralize iron oxide crystals, such that iron accumulation throughout the magnetosome chain is altered (9). MamA - green fluorescent protein (GFP) complementation of a ΔmamA mutant shows that MamA changes its subcellular localization during growth phase (9). Other studies showed MamA to exist in homooligomeric complexes in vivo, to localize to the magnetosomal matrix, and to dissociate from this matrix upon treatment with alkaline solution (pH 11.0) (8, 10) (Fig. S1).
MamA was predicted to fold into a structure containing five sequential tetra-trico-peptide repeat (TPR) motifs with an additional putative TPR motif at the N terminal (11). TPR represents a structural motif consisting of 34 amino acids, sharing a degenerate consensus sequence defined by a pattern of small and large amino acids. The canonical unit of the TPR motif adopts a helix-turn-helix fold. Adjacent TPR units packed in parallel create a series of repeating antiparallel α-helices that give rise to an overall superhelix structure. This superhelix forms a pair of concave and convex curved surfaces that display some extent of flexibility, as well as amino acid variety. This conformation permits binding of diverse ligands, usually via the concave TPR surface. Repetitive TPR motifs found in this format or as part of a bigger fold, exist in a wide range of proteins and organisms, where they promote complex formation through TPR-target protein interactions. Proteins containing TPR motifs thus participate in a variety of biological processes, such as synaptic vesicle fusion (12), peroxisomal targeting and import (13, 14), mitochondria and chloroplast import (15, 16), and others (17). In addition, such motifs were found to be involved in outer membrane assembly (18) and bacterial pathogenesis (19, 20). While TPR motifs have been shown to form small homooligomers in vitro, no evidence exists for the formation of large homooligomers either in vivo or in vitro.
Magnetosome formation is presumed to involve protein sorting and the assembly of multiprotein complexes. Therefore, we initiated a biochemical and structural study of MamA, a predicted TPR-containing protein expected to mediate magnetosomal protein–protein interactions and to form multiprotein complexes (3). In this study, we present structures of MamA from Magnetospirillum magneticum AMB-1 and Magnetospirillum gryphiswaldense MSR-1. We confirm that MamA folds as a sequential TPR-containing protein. Surprisingly, MamA displays a unique separation between two distinct TPR-containing domains involved in ligand binding and recognition. In addition, we identify three protein–protein interaction sites involved in MamA homooligomerization and complex formation. Based on our structural and biochemical studies, we propose that the putative TPR motif is responsible for homooligomerization via its interaction with the TPR-based MamA concave site that undergoes conformational change upon ligand binding.
Results
MamA Purification and Complex Formation.
To understand MamA structure–function relationship, we expressed and purified recombinant versions of MamA from M. magneticum AMB-1 and M. gryphiswaldense MSR-1 in Escherichia coli. MamA, a 24 kDa protein, was successfully expressed in the soluble fraction. Earlier studies had demonstrated that M. magneticum AMB-1 MamA displays partial solubility-dependence on NaCl concentration and is highly soluble at ∼1 mM NaCl (11). In contrast, our solubility tests for M. gryphiswaldense MSR-1 MamA indicated solubility not to be NaCl-dependent, as we could detect the protein in the soluble fraction of samples containing between 1 mM and 1 M NaCl. To fully purify MamA, we employed a construct in which MamA was fused to a cleavable C-terminal histidine10 tag. Purification initially involved immobilized metal ion affinity chromatography, followed by ion exchange chromatography on MonoQ resin. An additional size exclusion chromatography purification step yielded a single population of ∼500 kDa MamA protein (Fig. 1). Extended purification performed using various conditions further showed the tendency of MamA to homooligomerize into highly stable complexes. The MamA complexes formed in vitro were partially disassembled using previously reported reagents which dissociated MamA from magnetosome membranes (i.e., Tween-20, pH 11) (8, 21) (Fig. 1A) but not by other common aggregate-dissociating conditions, such as EDTA, 1 mM–2 M NaCl, DNase I, glycerol, arginine, Triton X-100, buffers with a pH of 4–9, or ATP. Interestingly, MamA incubated at pH 11 displayed multiple populations on the anion exchange column, which we assume to be the result of partial protein unfolding. Different types of culture media, induction methods (e.g., IPTG or auto induction), cultivation temperature, and duration of incubation all resulted in the appearance of the same stable oligomers. In addition, the MamA oligomerization state was not altered by the position of the poly histidine tag (i.e., N or C-terminal), its length (i.e., 6 vs. 10 histidine residues), or its removal via thrombin cleavage.
Fig. 1.

Analysis and visualization of MamA oligomerization. (A) Size exclusion (Superdex 200) chromatograms of wild type MamA and the MamAΔ41 truncation mutant under various conditions, revealing alterations in the MamA oligomeric state. Wild type M. magneticum MamA (AMB-1) and M. gryphiswaldense MamA (MSR-1), colored in blue and pink, respectively, both eluted at a volume corresponding to ∼500 kDa. MamA, after an incubation of 2 h with 2% Tween-20 (colored in green), shows a dissociation profile. MamA, after incubation of 2 h with 0.1 M N-cyclohexyl-3-aminopropanesulfonic acid (CAPS), pH 11.5 (in orange), appears as three peaks, respectively corresponding to aggregates (elution at 7 mL), a complex (elution at 8 mL) and a pentamer (elution at 13 mL). The MamAΔ41 mutant (colored in red) eluted at a volume appropriate for the monomer. (B) TEM images of negatively stained MamA complexes. (B1) M. gryphiswaldense MamA and (B2) M. magneticum MamA both appear as 14–20 nm globular complexes with a central pore cavity. (B3 and B4) The M. gryphiswaldense MSR-1 MamAΔ26 truncation mutant appears as broken and distorted complexes with a wider pore cavity.
To further investigate the overall spatial shape of MamA homooligomers, we employed electron microscopy imaging. Samples containing purified MamA homooligomers were placed on carbon-coated grids and negatively stained for transmission electron microscopy (TEM) analysis. MamA from both species formed ∼20 nm (diameter) round-shaped complexes with a central pore cavity (Fig. 1B). Because MamA oligomerizes into highly stable complexes, we chose to destabilize these protein–protein interactions using various truncation mutants. MamA contains a putative N-terminal TPR motif assumed to bear an amphiphilic character (11) and which might contribute to complex stabilization. To test this hypothesis, we designed MamA N-terminal truncation mutants based on multiple sequence alignment and secondary structure prediction (Fig. S2). We tested two mutants, i.e., MamAΔ26, in which the first predicted helix of the MamA N-terminal region was truncated, and MamAΔ41, in which the full putative TPR motif was absent. Partial disassembly, as well as broken and asymmetric MamA complexes, were observed for M. gryphiswaldense MSR-1 MamAΔ26 upon TEM analysis (Fig. 1B), while complex formation was completely abolished for the M. gryphiswaldense MSR-1 and M. magneticum AMB-1 MamAΔ41 truncation mutants. These results suggest that the putative TPR motif is involved in complex formation, with the first 26 amino acids being responsible for the protein–protein interaction normally needed for the formatjon of stable complexes.
To assess the role of the first putative TPR motif in MamA oligomerization, we preformed Isothermal Titration Calorimetry (ITC), in which we examined the binding affinity of two synthetic peptides to MamAΔ41. These two peptides were chemically synthesized according to the amino acid sequence of the two predicted helices comprising the putative MamA TPR motif (first putative helix,  ; second putative helix,
; second putative helix,  ). The ITC results revealed a single binding site with a dissociation constant (KD) of 1.58 ± 0.46 μM between the first putative helix and MamAΔ41 (Fig. S3). In contrast, the ITC results of MamAΔ41 and the second putative helix peptide did not reveal any significant binding. According to these results, we conclude that MamA complex formation involves binding of the first putative helix from one monomer to a binding site located on another MamA molecule.
). The ITC results revealed a single binding site with a dissociation constant (KD) of 1.58 ± 0.46 μM between the first putative helix and MamAΔ41 (Fig. S3). In contrast, the ITC results of MamAΔ41 and the second putative helix peptide did not reveal any significant binding. According to these results, we conclude that MamA complex formation involves binding of the first putative helix from one monomer to a binding site located on another MamA molecule.
In Vivo Subcellular Localization of MamA from M. magneticum AMB-1.
Due to the large distortion undergone by MamA complexes upon deletion of the putative TPR helices, we wanted to assess the behavior of the MamA truncation mutants in terms of subcellular localization in vivo. Accordingly, we replaced the wild type mamA gene in the mamA-gfp-bearing plasmid, pAK20 (9), with the mamAΔ26 and mamAΔ41 sequences. These plasmids were then transformed into a mamA null mutant of M. magneticum AMB-1 and subcellular localization was assessed by fluorescence microscopy, as previously described. We found that MamA-GFP localized to a thin spotted line extending from pole-to-pole, as observed earlier (9), indicating that MamA localized to the magnetosome chain (Fig. 2). In contrast, MamAΔ26-GFP and MamAΔ41-GFP displayed a diffuse pattern throughout the cell with no apparent localization reminiscent of the magnetosome chain. Surprisingly, higher fluorescence intensity was observed for MamAΔ41-GFP, as opposed to MamAΔ26-GFP (Fig. 2, Fig. S4). Western blotting with GFP-recognizing antibodies revealed that MamAΔ26-GFP undergoes degradation and that its cellular expression level is significantly lower than that of MamA-GFP or MamAΔ41-GFP (Fig. S5). As we deleted a single helix from a full putative TPR fold in MamAΔ26, leaving an unstable N-terminal tail, expression alteration or protein degradation may result in the cell.
Fig. 2.

Localization of MamA, its N-terminally-truncated versions and mutants fused to GFP in a M. magneticum AMB-1 ΔmamA strain. Shown is a representative cell for each strain. For each strain, 300 cells were analyzed in independent experiments (see Fig. S4 for more cell images). MamA displays a linear localization pattern reminiscent of the magnetosome chain reported before (9). MamAΔ26 and MamAΔ41 share a diffuse cellular localization pattern in all analyzed cells, while the fluorescence intensity of MamAΔ26 is significantly lower. Note that the image of the MamAΔ26 cell was electronically enhanced (green frame) to show fluorescence. MamA R50E displays a largely diffuse localization pattern (∼80% of all cells) but occasionally also a linear pattern, whereas MamA D159K displays a linear localization pattern alone.
In addition, we conducted a functional complementation experiment in which we replaced the AMB-1 mamA gene in plasmid pAK20 with its MSR-1 homolog. Although MamA proteins from these two species share a common ancestor and display 92% amino acid identity, a similar structure and similar biochemical properties, the one cannot restore complete, proper, subcellular localization in place of the other, as both linear and diffuse patterns were observed in complemented cells (Fig. S4).
MamAΔ41 Folds as a TPR.
To obtain structural information on MamA, we determined the structure of MamAΔ41. To this end, we initiated crystallization trials using a sitting drop vapor diffusion method (22). These trials resulted in the appearance of several crystal forms which diffracted to a resolution of 2.0 Å (see Table S1 for data collection and refinement statistics and Table S2 for crystallization conditions). In an attempt to resolve the phase problem, we unsuccessfully attempted molecular replacement experiments using previously determined structure templates (22). As such, our initial structure, 3AS5 (M. magneticum AMB-1 MamAΔ41), was constructed using experimental phases that were obtained by single-wavelength anomalous dispersion (SAD) from sodium iodide-soaked crystals (quick soak). Structures of all other MamAΔ41 crystal forms [PDB codes: 3AS4, 3AS8, 3ASF, 3ASD, 3ASH, and 3ASG] were determined by molecular replacement techniques, with 3AS5 serving as template. The N- and C-terminal extremities were disordered in some of the structures (Table S2), depending on the different packing properties of the crystal forms (Fig. S6).
The overall structure of the MamAΔ41 monomer is composed of 10 antiparallel α-helices and turn motifs that form a hook-like structure containing concave and convex surfaces. These helices are folded as five TPR motifs, namely TPR1 (H1 and H2), TPR2 (H3 and H4), TPR3 (H5 and H6), TPR4 (H7 and H8), and TPR5 (H9 and H10). All MamAΔ41 structures adopted the same fold, despite the amino acid sequence difference between the M. magneticum AMB-1- and M. gryphiswaldense MSR-1-derived proteins (Fig. S2). The average root mean square deviation (rmsd) between all of the monomeric structures is ∼1.2 Å. Because TPR domains share a common structural motif, a DALI search (http://www.ebi.ac.uk/dali/) yielded 665 PDB entries significantly similar to MamAΔ41, with Z scores larger than 2.0 (23). We decided to narrow these results by comparing the MamAΔ41 structures to other TPR-containing proteins with similar curvature angles and a number of motif repeats larger than four. A total of eight PDB entries representing six different proteins fit these criteria, including: YrrB (PDB entry 2Q7F, Z score = 18.7) (24), synthetic consensus TPR protein (PDB entries 2FO7 and 2hyz, Z score = 17.4) (25), O-linked GlcNAc transferase (OGT, PDB entry 1W3B, Z score = 16.5) (26), the Type 4 fimbrial biogenesis protein, PilF (PDB entry 2FI7-A and 2HO1-A, Z score = 16.4) (27), the thermophilic TPR protein, TTC0263 (PDB entry 2PL2-B, Z score = 16.4) (28), and putative fimbrial biogenesis and twitching protein (PDB entry 2VQ2, Z score = 16) (29). The rmsd between the Cα positions was calculated using MSDfold (http://www.ebi.ac.uk/msd-srv/ssm/). The rmsd values for DALI-obtained structures against 3AS5 chain A ranged from 1.537 to 3.211 Å, indicating that these TPR-containing proteins are structurally similar to one another. While most sequential TPR-containing proteins share the similar overall fold of a superhelix, such as YrrB and synthetic TPR proteins, MamA structures adopt a unique overall hook-shaped structure.
In addition to the ten TPR helices, a C-terminal helix was observed in the 3AS5 chain A and 3AS8 structures (Fig. 3). This helix (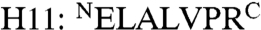 ) consists of a His-tag linker sequence that remained after thrombin proteolysis of MamAΔ41 and which adopted a helical conformation only in 3AS5 and 3AS8. Helix stabilization in these crystal forms arises from crystal packing, as well as due to crystal contacts with other MamA monomers. Such interactions are not favored in the other crystal forms as the angles and distances between H11 and its interaction sites are different. H11 does not seem to affect MamA oligomerization or its solution properties in vivo and in vitro, as we observed MamAΔ41 to remain a monomer in solution (Fig. 1A). In addition, large MamA complexes were detected when using purification tags that do not contain the H11 sequence, indicating that oligomerization is not H11-dependent.
) consists of a His-tag linker sequence that remained after thrombin proteolysis of MamAΔ41 and which adopted a helical conformation only in 3AS5 and 3AS8. Helix stabilization in these crystal forms arises from crystal packing, as well as due to crystal contacts with other MamA monomers. Such interactions are not favored in the other crystal forms as the angles and distances between H11 and its interaction sites are different. H11 does not seem to affect MamA oligomerization or its solution properties in vivo and in vitro, as we observed MamAΔ41 to remain a monomer in solution (Fig. 1A). In addition, large MamA complexes were detected when using purification tags that do not contain the H11 sequence, indicating that oligomerization is not H11-dependent.
Fig. 3.

Overall MamAΔ41 structures and TPR representations. (A) The representative MamAΔ41 structure (3AS4 in cartoon drawing) contains five sequential TPR motifs. The molecule is shown in two views, related by a 90° rotation. (B) Overlay of all MamAΔ41 monomers (in cartoon drawing) reveals the high degree of structural similarity, apart from the His-tag linker sequence remaining after thrombin proteolysis (H11: ELALVPR), which appears in the 3AS5 chain A and 3AS8 structures. (C) Overlay of all five TPR units within the 3AS5 chain A structure (in ribbon drawing) shows a conserved, typical TPR fold. All structural images were prepared using PyMOL.
MamA Comprises Two Distinct Domains.
In depth analysis of all six MamAΔ41 monomeric structures and their rmsd values (Table S3) revealed that each structure comprises two distinct domains, i.e., a N-terminal domain (NTD), comprising TPR motifs 1–2 (amino acids 41–112), and a C-terminal domain (CTD), comprising TPR motifs 3–5 (amino acids 113–217), with an average carbon α rmsd of 0.56 Å and 0.77 Å, respectively, between all MamA structures (Table S3). Comparing 3AS5 chain A to chain B suggested that the NTD undergoes an induced-fit while wrapping around H11 (Fig. 4B). A radial movement of ∼3° was observed between the NTDs of 3AS5 chain A and chain B when overlapping their CTDs as reference. The hinge region is at Asp112, located in the loop connecting TPR2 to TPR3. A maximum shift of ∼3.5 Å was observed as a result of hook movement (Fig. 4B). A similar movement of the NTD, although smaller, is observed upon overlaying the 3AS4 and 3AS8 CTDs (M. magneticum AMB-1 and M. gryphiswaldense MSR-1, respectively), both without H11 at the concave surface. Additional support for the existence of these two domains in sequential TPR-containing proteins can be seen upon surface rendering, which indicates a narrow gap between the two folded domains (Fig. 4D).
Fig. 4.

MamAΔ41 displays two separate domains which adopt a hook-like overall shape. (A) Crystal packing in the 3AS5 asymmetric unit in which chain B (orange) binds chain A H11 (marine blue). (B) Structural superposition of the two polypeptide chains present in the 3AS5 asymmetric unit (rmsd of 1.3 Å), demonstrating the protein hook-like shape and movement upon ligand binding (The ligand, H11, is colored in red). Upon binding, chain B residues 41–112 wrap around H11, resulting in a 3° turn and 3.5 Å movement, relative to the nonbinding chain A. (C) Improved chain superposition is achieved by dividing the protein into two distinct domains, the N-terminal domain (NTD—residues 41–112) and the C-terminal domain (CTD—residues 113–217). Superposition of these separate domains reveals a higher degree of structural similarity between chain A and B, with a rmsd value of 0.62 Å for the NTD and 0.79 Å for the CTD. (D) Surface charge representations of 3AS5 chain B, with blue and red colors representing regions of positive and negative electrostatic potential, respectively. The molecule is shown in three views, related by 90° rotations. (Top) The narrow area between the two domains is marked by a green arrow. (Center) The concave surface displays an extreme positive charge distribution. (Bottom) The convex surface displays mainly negative charge distribution. All electrostatic surfaces representations were produced with the APBS plug-in of PyMOL.
MamAΔ41 Binding Sites.
MamA self-assembly and its binding of other magnetosome-associated proteins (10) points to the existence of at least three MamA-associated protein–protein interaction sites. A minimum of two sites are needed to create the observed round complexes (Fig. 1B) and at least one site is needed to bind these complexes to the magnetosome chain. In crystal forms 3AS5 and 3AS8, we identified two possible binding sites at the concave and convex TPR surfaces. These sites are involved in the binding of a H11 entity that may imitate the natural ligand. In 3AS5, the binding of H11 in the concave binding pocket induces the helical conformation of the peptide (Fig. 4A), which is unstructured in the other crystal forms. 3AS5-H11 binding is achieved by a salt bridge (Glu218-Asn147B), hydrogen bond formation (through the backbone of Ala220 to Lys92B), hydrophobic interactions (Leu221 to Ile88B, Val119B and Val91B, and Val217 to Ile88B), and highly ordered water molecules (Wat1-8) that all compensate for imperfect binding (Fig. 5A, Fig. S7). The overall interactions create a binding surface of 327 Å2, which accounts for ∼40% of the total 3AS5-H11 surface. Peptide binding also involves electrostatic potential charge interaction, given that the concave binding pocket is highly positive and that H11 is mainly negative.
Fig. 5.

Helix binding at the concave and convex surfaces, mimicking natural MamA ligands. (A) The 3AS5 chain B concave binding site and 3AS5 chain BH11. (Top) 3AS5 chain B, in electrostatic surface display, binds H11 (green) at the central concave pocket. (Center) Detailed representation of interactions between 3AS5 chain B (light pink) and H11 (blue) residues. Water molecules involved in ligand binding are represented as red spheres. A 2.0 Å 2Fo - Fc electron density omit map was calculated and is presented around H11. The map is countered at 1.0σ (light blue). (Bottom) Interaction scheme between H11 (backbone in black) and 3AS5 chain B (backbone in brown). (B) 3AS8 convex binding site and the symmetry-related H11. (Top) 3AS8, in electrostatic surface display, binds H11 (yellow) at the TPR convex surface. (Center) Detailed representation of interactions between 3AS8 monomer (pink) and H11 (deep blue) residues. A 2.0 Å 2Fo - Fc electron density omit map was calculated and presented around H11. The map is countered at 1.0σ (light brown). (Bottom) Interaction scheme between H11 (backbone in black) and 3AS8 (backbone in brown). Interaction schemes were produced using LigPlot (35). An enlarged and detailed binding site image can be found in Fig. S7.
The convex binding site of 3AS8 binds the crystallographic symmetry-related (S) H11 that appears as a helical extension of TPR5 H10. This extended helix binds TPR3 H6 and the TPR4 loop region. H11 interactions include a single salt bridge (Glu218-Arg163S), as well as hydrophobic interactions (Leu221 to Glu166S and Val158S, Val217 to Leu161S and Arg163S, Ala220 to Val138S and Pro135S, and Glu213 to Leu161S) (Fig. 5B, Fig. S7). The overall interactions of 3AS8-H11 create a binding surface of 163 Å2, representing ∼20% of the total peptide surface, consistent with the transient binding of the peptide.
To validate the observed concave and convex binding sites, we overlaid the 3AS5 and 3AS8 structures onto those of previously determined ligand-binding TPR-containing proteins. These MamAΔ41-homologous structures demonstrate the ability of TPR proteins to bind an unstructured peptide, a helix, or an entire TPR motif (Fig. S8) at similar sites (30–33). Such ligand binding versatility might explain the self-oligomerization and complex formation of full-length MamA.
To date, TPR-containing proteins have been reported to present a moderately positive or highly negative binding pocket at their concave surface. MamA displays a unique charge distribution, as it is highly positive over the entire concave surface and mainly negative over the convex surface (Fig. 4D). Given the dimensions of the positively charged concave surface and the fact that it is able to accommodate a helix (3AS5-H11), we speculate that this surface might bind the putative TPR motif, containing a mixture of hydrophobic and negatively charged residues (Fig. S2).
Conserved Residues Contribute to Complex Formation.
Multiple sequence alignment reveals high similarity of MamA (Fig. S2). Of all the conserved MamA amino acids, only three were not related to the TPR consensus sequence, i.e., Arg50, Asp79, and Asp159. To assess the importance of the conserved residues in structure complex and stabilization, opposite charge mutations were introduced into both MamA and the MamAΔ41 truncation mutant. Mapping these conserved residues onto the MamAΔ41 structures revealed that Arg50 and Asp79 form a salt bridge which stabilizes the NTD hook-like shape by connecting TPR1 and TPR2 (Fig. 6A). To disrupt the Arg50-Asp79 salt bridge, it is sufficient to mutate a single residue of the pair. As such, we introduced the R50E mutation into MamAΔ41, purified the mutated protein and determined its structure (3ASD, see Table S1). The MamAΔ41 R50E structure revealed major conformational changes, in which the NTD was rotated by almost 30°, relative to the wild type CTD (Fig. 6A), leading to destruction of the concave binding surface, as well as to the deformation of the overall protein hook-like shape. The origin of this NTD movement emerged from the same hinge region as in the wild type protein (Asp112), providing additional support to our two-domain observation. Because the Arg50-Asp79 salt bridge interconnects H1 to H3, we suspected that its disruption would enhance H1 flexibility and affect stability of the entire TPR1 motif. TPR1 motif destabilization was verified in the R50E structure by the absence of electron density for the TPR1 motif, indicating its high flexibility. Additional evidence of the disorder and flexible properties of TPR1 was provided by SDS-PAGE analysis of isolated and melted R50E crystals, which revealed no protein degradation (Fig. S9). Although the R50E P4322 space group contains a single monomer, crystal packing revealed that the symmetry-related CTDs form a typical super-helix-like shape, common in other sequential TPR-containing proteins. Three symmetry monomers create a complete superhelical turn, whereas adjacent NTDs are perpendicular to the superhelical main vertical axis (Fig. 6B). MamA complex formation was disrupted as a result of the R50E mutation (Fig. 6D), while the in vivo subcellular localization of MamA R50E–GFP revealed a largely diffuse pattern (80% of all cells; n = 300 from independent experiments) (Fig. 2). Given that disruption of the salt bridge alters NTD shape and its orientation, relative to the CTD, we suggest that the concave binding pocket can no longer recognize its partner, leading to alterations in MamA homooligomerization and in vivo localization.
Fig. 6.

Effect of conserved MamA residue mutation. (A) Superposition of native MamAΔ41 (green) and MamAΔ41 R50E (NTD- yellow, CTD- red) reveals a large rotation of the mutant NTD. Highly conserved residues Arg50 and Asp 159 form a salt bridge connecting TPR1 and TPR2 (light pink) in the wild type structure. (B) Side and top views of representative symmetry-related monomers demonstrate the superhelical crystal packing of MamAΔ41 R50E. The continuous superhelix is formed by the CTD (light gray) of each monomer, while adjacent NTDs (dark gray) lie perpendicular to the superhelix long axis. A single representative monomer in this packing is highlighted (NTD- yellow, CTD- red) (C) Superposition of native MamAΔ41 (brown) and MamAΔ41 D159K (light pink) reveals no major structural differences caused by this point mutation. (D) Size exclusion (Superdex 200) chromatograms of wild type MamA and mutants demonstrating alterations in the MamA oligomeric state. Full-length wild type M. magneticum MamA, M. gryphiswaldense MamA and MamA D159K (blue, pink, and orange respectively) eluted at a volume corresponding to ∼500 kDa. Full-length MamA R50E mutant eluted at volumes appropriate for the dimer and monomer. The MamAΔ41 mutant (red) eluted at a volume appropriate for a monomer.
In contrast, D159K mutation had no effect on the overall structure of MamAΔ41 (3ASH and 3ASG, see Table S1), on MamA complex formation (Fig. 6 B and D), or on in vivo subcellular localization (Fig. 2). However, we cannot exclude the possibility that this mutation altered other protein functions without causing any structural effect or protein mislocalization.
Discussion
Organelle formation and proper function require the assembly of multiprotein complexes. In magnetosomes, a bacterial organelle involved in magnetic-sensing, MamA is predicted to play a role in protein assembly and activation of biomineralization (9). This function is supported by our M. magneticum AMB-1 and M. gryphiswaldense MSR-1 MamA structures that adopt a TPR fold, known to serve as a platform for multiprotein complexes. In contrast to previously described TPR-containing proteins, MamA self-assembles into large and stable homooligomer complexes. Originally, these complexes were regarded as an artifact of protein aggregation (11) but have recently been shown to exist in vivo (10). We were able to biochemically validate that MamA complexes are not protein aggregates, as they adopt a globular shape with narrow size distributions, and by their ability to undergo partial dissociation under conditions of extreme pH or in the presence of Tween-20 but not under many other reagents used to dissociate aggregates. We, therefore, speculate that previous reports of MamA dissociation from magnetosome chains under similar complex dissociation conditions (8, 34) reflected complex disassembly and a loss of magnetosome-binding surfaces.
One can ask how a sequential TPR protein is able to homooligomerize into a highly stable complex. The first hint at a strategy came from examination of our truncation mutants. MamAΔ26, in which a single helix of a putative TPR motif is missing, showed compromised homooligomeric complex formation, while in MamAΔ41, lacking a full putative TPR, these complexes were completely abolished, indicative of the putative TPR as participating in complex formation. Because we showed that the first helix of the putative TPR motifs binds to MamAΔ41, additional sites of interactions on MamAΔ41 are needed to create a MamA complex. These can be deduced by comparison to previously determined TPR proteins. A detailed structural comparison of MamA to other TPR-containing proteins revealed several key differences. Although MamA is a sequential TPR, it adopts a hook-like fold with separation into two domains that can undergo conformational change, rather than a superhelix fold. The concave and convex sites of TPR-containing proteins were previously described as being able to bind proteins or peptides (30–33). Two of our MamA structures demonstrated such ability, in which a residual cleavage sequence (H11) adopted a helical conformation and associated with the concave and convex sites in complete agreement with the structure of previously determined peptide-bound TPR complexes.
At least three protein–protein interaction sites are required to create a protein assembly framework on top of a large homooligomer. Based on our data, it seems that MamA contains at least three such binding sites, namely a putative TPR sequence, a concave binding site and a convex binding site. We speculate that the putative TPR motif binds at the concave site to assemble into a homooligomer, although we cannot rule out the possibility that the convex site binds the putative TPR (Fig. 7). Our assumption is, however, supported by the truncation mutants, as well as by the R50E mutation which deformed the concave site. Both the truncation and the point mutations led to destabilization of the MamA complex. It is important to note that the concave site can bind a TPR protein, as was observed previously (32), in agreement with the H11-binding site. We also propose that once MamA forms a large homooligomer, the convex permeable binding site (or sites), predicted by the surface properties and protein shape (Fig. S10), is responsible for protein–protein interactions with other magnetosome-associated proteins.
Fig. 7.

A schematic simplified model for MamA homooligomerization and assembly with other magnetosome-associated proteins leading to the formation of a multiprotein complex. The putative TPR motif of the MamA monomer binds at the concave surface of another monomer in a repetitive manner to create a highly stable homooligomer. The MamA convex sites bind other magnetosome-associated proteins to support proper activation of magnetite biomineralization throughout the chain.
Notably, four other magnetosome-associated proteins with molecular mass of 26.8, 31.6, 54.0, and 63.5 kDa were found to bind MamA in vivo (10). The fact that several magnetosome-associated proteins can simultaneously bind to MamA and that variations in the MamA homooligomerization status can cause differences in MamA complex outer surface curvature, support our model in which surfaces presented by MamA complexes serve as a multiprotein assembly sites. This assumption is also supported by the mislocalization of MamA truncation mutants in vivo. A monomer with a limited binding surface will not be able to support the binding affinities needed for magnetosome binding, as normally achieved by a large MamA homooligomer. Furthermore, the large binding surface created could aid in increasing the local concentrations of other magnetosome-associated proteins to allow for proper biomineralization. Although this is an appealing model, we cannot rule out that the four proteins in question are assembled into a large complex, whereas only one of the four binds MamA. In addition, these four proteins could each act as independent partners that all compete for the same site on MamA. Finally, the four magnetosome-associated proteins could represent single protein degradation products.
Overall, our structural and biochemical data shed new light onto the assembly of TPR motif-containing proteins into a large scaffold that may support protein–protein interactions. This unique large homooligomeric TPR-based scaffold may provide additional mechanic support or serve as an interaction platform which allows for proper functioning of a prokaryotic organelle.
Methods
Protein Expression and Purification.
The full length and truncation mutant mamA genes were amplified from genomic DNA of two magnetotactic bacteria species, Magnetospirillum magneticum AMB-1 and Magnetospirillum gryphiswaldense MSR-1 (German Collection of Microorganisms and Cell Cultures - DMSZ, 6361) using the polymerase chain reaction (PCR). In the amplified DNA fragments, a NcoI site was introduced at the initiation codon, ATG, followed by insertion of a glycine-encoding codon (GGA) to maintain the reading frame. The termination codon was replaced with a ScoI site. The fragments were digested with NcoI and SacI and cloned into the respective sites of the pET52(b) expression vector (Novagen) which encodes a C-terminal histidine10 tag, giving rise to plasmid p52MamA. Expression and purification of full-length proteins and MamAΔ26 truncation mutants were similarly achieved using previously published MamAΔ41 expression and purification protocols, with small alternations (36). These alterations included changing the NaCl concentration in buffer C to 1 mM and in buffer E to 5 mM for purifying full-length MamA. Likewise, the NaCl concentration of buffer C was altered to 1 M and to 100 mM in buffer E for MamAΔ26 purification.
Site-Directed Mutagenesis by PCR.
MamA mutants were generated using QuickChange site-directed mutagenesis (Stratagene). Coding and antisense primers containing a single mutagenic site were used for PCR amplification.
Negative Staining and TEM Imagining.
A 5 μL drop of protein sample (0.01 mg mL-1) was applied onto a glow-discharged, carbon-coated copper grid for 1 min and then negatively stained with 1–2% uranyl acetate. Images were collected with a 120 kV JEOL JEM-1230 transmission electron microscope at a magnification of × 30,000.
Peptide Synthesis.
Peptide synthesis was carried out using 9-fluorenylmethoxycarbonyl (Fmoc) solid phase peptide synthesis on Rink amide resin (0.2 mmol/g). Peptide synthesis was performed on an automated peptide synthesizer in the presence of four equivalent (eq) of amino acids, eight eq of N,N-diisopropylethylamine, and four eq of O-Benzotriazole-N,N,N’,N’-tetramethyl-uronium-hexafluoro-phosphate (HBTU)/ N-Hydroxybenzotriazole (HOBt) to the initial loading of the resin. Fmoc-deprotection was achieved using 20% piperidine in dimethylformamide (DMF). To cleave the peptides from the resin, a mixture of TFA, triisopropylsilane, and water (95∶2.5∶2.5) was added to the dried peptide-resin. Analytical HPLC was performed on a thermo-instrument (Spectra System p4000) using an analytical column (Jupiter 5 μm, C4, 300A 150 × 4.6 mm) and a flow rate of 1.2 mL/ min. Preparative HPLC was performed on a Waters instrument using a preparative column (Jupiter 10 μm, C4, 300A, 250 × 10 mm) and a flow rate of 25 mL/ min (Fig. S11). Mass spectrometry analysis was carried out using LQC Fleet Trap (Thermo Scientific). All reactions were carried out at room temperature. Resins, protected amino acids, and HBTU were purchased from Novabiochem. DMF was of biotech grade.
Isothermal Titration Calorimetry.
Titration calorimetry measurements were performed with an ITC200 calorimeter (Microcal) at 25 °C. Both protein and peptides solutions were diluted to the same final buffer concentration of 10 mM Tris•HCl, pH 8.0, 150 mM NaCl and 10% dimethyl sulfoxide (DMSO). Aliquots (2 μL) of the 0.1 mM peptide solution were added by means of a 40 μL rotating stirrer-syringe to the reaction cell, containing 200 μL of the 0.01 mM protein solution. The heat of dilution was determined to be negligible in separate titrations of the peptide into the buffer solution. The data were fitted using ORIGIN 7.0 software (Origin Lab).
Fluorescence Microscopy.
Localization of GFP-tagged proteins was carried out using a Nikon Eclipse 80i microscope equipped with a Qimaging RETIGA 2000R Fast 1394 camera. Images were obtained with the 100× oil objective and analyzed with QCapturePro 5.1 software. M. magneticum AMB-1 ΔmamA cells transformed with the pAK20 plasmids were grown from single colonies to stationery phase in MG medium [described previously (2)] (supplemented with 30 μM Fe-malate) in the presence of 10 μg mL-1 kanamycin in microcentrifuge tubes (1.7 mL tubes filled with 1.6 mL medium). Cultures were then diluted 1∶100 into 10 mL of fresh media and incubated in a microaerobic chamber at 30 °C, where the oxygen concentration was maintained below 10% for 3 d prior to imaging. In the fluorescence microscopy experiments, all strains were mounted onto 1% agarose pads and imaged on the same day. Exposure time was 5 s.
Crystallization and Structure Determination.
Purified MamAΔ41 and the mutants crystallized at different conditions at 13 °C (Table S1). Crystals were harvested without addition of cryoprotecting solution and flash-cooled in liquid nitrogen. Data collection was performed at beamline ID14-2 and ID29 at the European Synchrotron Radiation Facility (ESRF), Grenoble, France. Data were reduced and scaled using the HKL2000 (40) suite. For phasing, similar crystals to 3AS5 (Table S2) were soaked in the same reservoir condition with 0.6 M NaI for 30 s. Heavy atom data was collected at the home source using an image plate detector system (MAR 345 mm) (X-Ray Research) and RU-H3RHB (Rigaku) rotating anode generator. Data collection was performed at 100 °K and a total of 378 frames (189°) were collected with an oscillation range of 0.5°. The exposure time was 7 min per image and the crystal-to-detector distance was 200 mm. Phases obtained by SAD technique and by initial auto-building in Phenix (41). 2FQ7 was overlaid onto the partial model obtained from Phenix and the combined model was converted to polyalanine. This model was used for the molecular replacement protocol in Phaser (42) against high resolution native data (Table S1), followed by solvent flattening in Solomon (43). The output map and protein sequence were entered into the auto-build cycle in Phenix and the resulting model was manually edited according to unit cell symmetry. ARP/WARP (44) used the correct model and high resolution data to obtain an interpretable high resolution map. The final model was built by COOT and refined in REFMAC (45). For Rfree calculation, 5% of the data was excluded. Phases for all other structures were obtained by Phaser molecular replacement using the 3AS5 chain A. Structural figures were prepared with PyMOL (38).
Least-Squares Overlaps.
R.M.S. calculations were performed with SwissPDB viewer (37) using the domain alternate fit to align structures on the basis of the conserved domain and to define the conformational changes of the structural homologues.
Electrostatic Potential Calculations.
Electrostatic calculations were done in PyMOL (38), using the Adaptive Poisson-Boltzmann Solver (APBS) plug-in (39).
Coordinates.
Structures have been submitted to the Protein Data Bank (accession codes 3AS4, 3AS5, 3AS8, 3ASD, 3ASF, 3ASH, and 3ASG).
Supplementary Material
Acknowledgments.
We thank ESRF (Grenoble, France) beamlines ID 14-2, ID 23-1, and ID 29 for assistance during data collection. We also thank Dr. Anat Ben-Zvi and Dr. Amir Aharoni for fruitful discussion, Rina Jeger for her assistance in TEM imaging and Dr. Simy Weil for MALDI-TOF sample preparation.
Footnotes
This article is a PNAS Direct Submission.
The authors declare no conflict of interest.
See Author Summary on page 13369.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 3AS4, 3AS5, 3AS8, 3ASD, 3ASF, 3ASH, and 3ASG).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1103367108/-/DCSupplemental.
References
- 1.Faivre D, Schuler D. Magnetotactic bacteria and magnetosomes. Chem Rev. 2008;108:4875–4898. doi: 10.1021/cr078258w. [DOI] [PubMed] [Google Scholar]
- 2.Murat D, Quinlan A, Vali H, Komeili A. Comprehensive genetic dissection of the magnetosome gene island reveals the step-wise assembly of a prokaryotic organelle. Proc Natl Acad Sci USA. 2010;107:5593–5598. doi: 10.1073/pnas.0914439107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schuler D. Genetics and cell biology of magnetosome formation in magnetotactic bacteria. FEMS Microbiol Rev. 2008;32:654–672. doi: 10.1111/j.1574-6976.2008.00116.x. [DOI] [PubMed] [Google Scholar]
- 4.Komeili A. Molecular mechanisms of magnetosome formation. Annu Rev Biochem. 2007;76:351–366. doi: 10.1146/annurev.biochem.74.082803.133444. [DOI] [PubMed] [Google Scholar]
- 5.Bazylinski DA, Frankel RB. Magnetosome formation in prokaryotes. Nat Rev Microbiol. 2004;2:217–230. doi: 10.1038/nrmicro842. [DOI] [PubMed] [Google Scholar]
- 6.Jogler C, et al. Comparative analysis of magnetosome gene clusters in magnetotactic bacteria provides further evidence for horizontal gene transfer. Environ Microbiol. 2009;11:1267–1277. doi: 10.1111/j.1462-2920.2009.01854.x. [DOI] [PubMed] [Google Scholar]
- 7.Matsunaga T, Okamura Y. Genes and proteins involved in bacterial magnetic particle formation. Trends Microbiol. 2003;11:536–541. doi: 10.1016/j.tim.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 8.Taoka A, et al. Spatial localizations of Mam22 and Mam12 in the magnetosomes of Magnetospirillum magnetotacticum. J Bacteriol. 2006;188:3805–3812. doi: 10.1128/JB.00020-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Komeili A, Vali H, Beveridge TJ, Newman DK. Magnetosome vesicles are present before magnetite formation, and MamA is required for their activation. Proc Natl Acad Sci USA. 2004;101:3839–3844. doi: 10.1073/pnas.0400391101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamamoto D, et al. Visualization and structural analysis of the bacterial magnetic organelle magnetosome using atomic force microscopy. Proc Natl Acad Sci USA. 2010;107:9382–9387. doi: 10.1073/pnas.1001870107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Okuda Y, Fukumori Y. Expression and characterization of a magnetosome-associated protein, TPR-containing MAM22, in Escherichia coli. FEBS Lett. 2001;491:169–173. doi: 10.1016/s0014-5793(01)02178-0. [DOI] [PubMed] [Google Scholar]
- 12.Young JC, Barral JM, Ulrich Hartl F. More than folding: localized functions of cytosolic chaperones. Trends Biochem Sci. 2003;28:541–547. doi: 10.1016/j.tibs.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 13.Fransen M, et al. Comparison of the PTS1- and Rab8b-binding properties of Pex5p and Pex5Rp/TRIP8b. (Translated from eng) Biochim Biophys Acta. 2008;1783:864–873. doi: 10.1016/j.bbamcr.2008.02.013. [DOI] [PubMed] [Google Scholar]
- 14.Brocard C, Hartig A. Peroxisome targeting signal 1: is it really a simple tripeptide? Biochim Biophys Acta. 2006;1763:1565–1573. doi: 10.1016/j.bbamcr.2006.08.022. [DOI] [PubMed] [Google Scholar]
- 15.Mirus O, Bionda T, von Haeseler A, Schleiff E. Evolutionarily evolved discriminators in the 3-TPR domain of the Toc64 family involved in protein translocation at the outer membrane of chloroplasts and mitochondria. J Mol Model. 2009;15:971–982. doi: 10.1007/s00894-008-0449-y. [DOI] [PubMed] [Google Scholar]
- 16.Baker MJ, Frazier AE, Gulbis JM, Ryan MT. Mitochondrial protein-import machinery: correlating structure with function. Trends Cell Biol. 2007;17:456–464. doi: 10.1016/j.tcb.2007.07.010. [DOI] [PubMed] [Google Scholar]
- 17.D’Andrea LD, Regan L. TPR proteins: the versatile helix. Trends Biochem Sci. 2003;28:655–662. doi: 10.1016/j.tibs.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 18.Gatsos X, et al. Protein secretion and outer membrane assembly in Alphaproteobacteria. FEMS Microbiol Rev. 2008;32:995–1009. doi: 10.1111/j.1574-6976.2008.00130.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tiwari D, et al. Key residues in Mycobacterium tuberculosis protein kinase G play a role in regulating kinase activity and survival in the host. J Biol Chem. 2009;284:27467–27479. doi: 10.1074/jbc.M109.036095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Edqvist PJ, et al. Tetratricopeptide repeats in the type III secretion chaperone, LcrH: their role in substrate binding and secretion. Mol Microbiol. 2006;59:31–44. doi: 10.1111/j.1365-2958.2005.04923.x. [DOI] [PubMed] [Google Scholar]
- 21.Grunberg K, et al. Biochemical and proteomic analysis of the magnetosome membrane in Magnetospirillum gryphiswaldense. Appl Environ Microbiol. 2004;70:1040–1050. doi: 10.1128/AEM.70.2.1040-1050.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zeytuni N, Zarivach R. Crystallization and preliminary crystallographic analysis of the Magnetospirillum magneticum AMB-1 and M. gryphiswaldense MSR-1 magnetosome-associated proteins MamA. Acta Crystallogr F . 2010;66:824–827. doi: 10.1107/S1744309110018300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holm L, Kaariainen S, Rosenstrom P, Schenkel A. Searching protein structure databases with DaliLite v.3. Bioinformatics. 2008;24:2780–2781. doi: 10.1093/bioinformatics/btn507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Han D, Oh J, Kim K, Lim H, Kim Y. Crystal structure of YrrB: a TPR protein with an unusual peptide-binding site. Biochem Biophys Res Commun. 2007;360:784–790. doi: 10.1016/j.bbrc.2007.06.129. [DOI] [PubMed] [Google Scholar]
- 25.Kajander T, Cortajarena AL, Mochrie S, Regan L. Structure and stability of designed TPR protein superhelices: unusual crystal packing and implications for natural TPR proteins. Acta Crystallogr D. 2007;63:800–811. doi: 10.1107/S0907444907024353. [DOI] [PubMed] [Google Scholar]
- 26.Jinek M, et al. The superhelical TPR-repeat domain of O-linked GlcNAc transferase exhibits structural similarities to importin alpha. Nat Struct Mol Biol. 2004;11:1001–1007. doi: 10.1038/nsmb833. [DOI] [PubMed] [Google Scholar]
- 27.Kim K, et al. Crystal structure of PilF: functional implication in the type 4 pilus biogenesis in Pseudomonas aeruginosa. Biochem Biophys Res Commun. 2006;340:1028–1038. doi: 10.1016/j.bbrc.2005.12.108. [DOI] [PubMed] [Google Scholar]
- 28.Lim H, Kim K, Han D, Oh J, Kim Y. Crystal structure of TTC0263, a thermophilic TPR protein from Thermus thermophilus HB27. Mol Cells. 2007;24:27–36. [PubMed] [Google Scholar]
- 29.Trindade MB, Job V, Contreras-Martel C, Pelicic V, Dessen A. Structure of a widely conserved type IV pilus biogenesis factor that affects the stability of secretin multimers. J Mol Biol. 2008;378:1031–1039. doi: 10.1016/j.jmb.2008.03.028. [DOI] [PubMed] [Google Scholar]
- 30.Scheufler C, et al. Structure of TPR domain-peptide complexes: critical elements in the assembly of the Hsp70-Hsp90 multichaperone machine. Cell. 2000;101:199–210. doi: 10.1016/S0092-8674(00)80830-2. [DOI] [PubMed] [Google Scholar]
- 31.Scherr N, et al. Structural basis for the specific inhibition of protein kinase G, a virulence factor of Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 2007;104:12151–12156. doi: 10.1073/pnas.0702842104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sampathkumar P, Roach C, Michels PA, Hol WG. Structural insights into the recognition of peroxisomal targeting signal 1 by Trypanosoma brucei peroxin 5. J Mol Biol. 2008;381:867–880. doi: 10.1016/j.jmb.2008.05.089. [DOI] [PubMed] [Google Scholar]
- 33.Lunelli M, Lokareddy RK, Zychlinsky A, Kolbe M. IpaB-IpgC interaction defines binding motif for type III secretion translocator. Proc Natl Acad Sci USA. 2009;106:9661–9666. doi: 10.1073/pnas.0812900106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schuler D. Molecular analysis of a subcellular compartment: the magnetosome membrane in Magnetospirillum gryphiswaldense. Arch Microbiol. 2004;181:1–7. doi: 10.1007/s00203-003-0631-7. [DOI] [PubMed] [Google Scholar]
- 35.Wallace AC, Laskowski RA, Thornton JM. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995;8:127–134. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
- 36.Zeytuni N, Zarivach R. Purification of the M. magneticum strain AMB-1 magnetosome associated protein MamADelta41. J Vis Exp. 2010;37 doi: 10.3791/1844. doi: 10.3791/1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 38.DeLano WL. San Carlos: DeLano Scientific; 2002. The PyMOL Molecular Graphics System; p. 0.99. [Google Scholar]
- 39.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Macromolecular Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 41.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D . 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McCoy AJ, et al. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Abrahams JP, Leslie AG. Methods used in the structure determination of bovine mitochondrial F1 ATPase. Acta Crystallogr D. 1996;52:30–42. doi: 10.1107/S0907444995008754. [DOI] [PubMed] [Google Scholar]
- 44.Morris RJ, Perrakis A, Lamzin VS. ARP/wARP’s model-building algorithms. I. The main chain. Acta Crystallogr D. 2002;58:968–975. doi: 10.1107/s0907444902005462. [DOI] [PubMed] [Google Scholar]
- 45.Vagin AA, et al. REFMAC5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr D. 2004;60:2184–2195. doi: 10.1107/S0907444904023510. [DOI] [PubMed] [Google Scholar]
