

Abstract
Many signals have risen and fallen in the tide of investigation into mechanisms of myocardial hypertrophy and heart failure (HF). In our opinion, the multifunctional Ca and calmodulin-dependent protein kinase II (CaMKII) has emerged as a molecule to watch, in part because a solid body of accumulated data essentially satisfy Koch's postulates, showing that the CaMKII pathway is a core mechanism for promoting myocardial hypertrophy and heart failure. Multiple groups have now confirmed the following: (1) that CaMKII activity is increased in hypertrophied and failing myocardium from animal models and patients; (2) CaMKII overexpression causes myocardial hypertrophy and HF and (3) CaMKII inhibition (by drugs, inhibitory peptides and gene deletion) improves myocardial hypertrophy and HF. Patients with myocardial disease die in equal proportion from HF and arrhythmias, and a major therapeutic obstacle is that drugs designed to enhance myocardial contraction promote arrhythmias. In contrast, inhibiting the CaMKII pathway appears to reduce arrhythmias and improve myocardial responses to pathological stimuli. This brief paper will introduce the molecular physiology of CaMKII and discuss the impact of CaMKII on ion channels, Ca handling proteins and transcription in myocardium. This article is part of a Special Issue entitled “Key Signaling Molecules Special Issue”.
Keywords: Calmodulin kinase II, Cell signaling, Oxidation, Hypertrophy, Heart failure, Arrhythmias
1. Molecular physiology of CaMKII
CaMKII is a serine–threonine kinase that exists as an elaborate holoenzyme complex consisting of a pair of hexameric stacked rings (Fig. 1). There are four CaMKII gene products (α, β, γ, δ). These CaMKII isoforms have different tissue distribution and may have subtle differences in Ca/CaM sensitivity and activation kinetics, but, at present, understanding of the potentially specific roles for various CaMKII isoforms is incomplete. The predominant, though not exclusive, form in myocardium appears to be CaMKIIδ. Two major splice variants of CaMKIIδ are expressed in the adult heart, CaMKIIδB [1,2] and CaMKIIδC [2–4], the former containing an 11 amino acid nuclear localization sequence [5]. It appears that CaMKIIδ is functionally significant for myocardial pathology, as it was recently shown that targeted deletion of CaMKIIδ is sufficient to prevent adverse consequences of transaortic banding, a surgical model of pathological afterload augmentation [6,7] (Table 1). Each of the dozen CaMKII monomers that compose the holoenzyme consists of three domains (Fig. 1). Under basal conditions CaMKII activity is submaximal because the N-terminus catalytic domain is constrained by the pseudosubstrate region within the regulatory domain. CaMKII indirectly senses increases in intracellular Ca by binding calcified calmodulin (Ca/CaM) at the CaM-binding region in the regulatory domain, which is adjacent to the pseudosubstrate region. Ca/CaM binding reorders CaMKII so that the catalytic domain is not constrained by the pseudosubstrate domain. During brief, low frequency increases in intracellular [Ca], CaMKII deactivates after Ca/CaM unbinds from the regulatory domain.
Fig. 1.
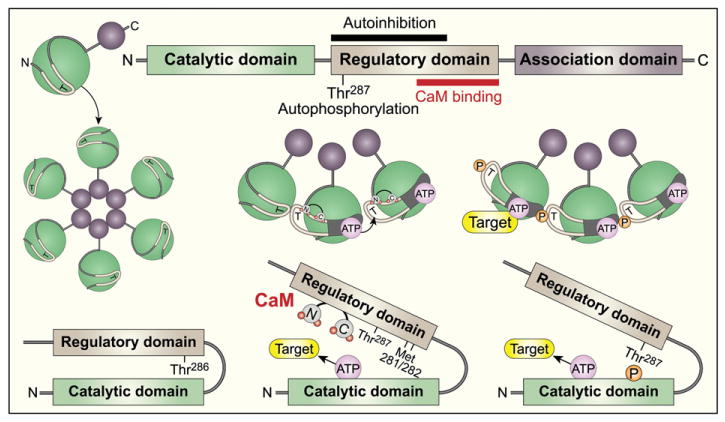
CaMKII structural domains and regulation. CaMKII monomers consist of an N terminal catalytic domain and a C terminal association domain that bound a regulatory domain (top). The association domains (maroon circles) are required for assembly of the CaMKII monomers into the holoenzyme (middle panels). Under resting conditions the catalytic domain is constrained by the regulatory domain (left middle and bottom panels). After intracellular Ca2+ rises and complexes with calmodulin (CaM) the Ca2+/CaM binds to the C terminal portion of the CaMKII regulatory domain (mid portion of the top, middle and bottom panels) to prevent autoinhibition of the regulatory domain on the catalytic domain, activating CaMKII. With sustained Ca2+/CaM or increased oxidation, CaMKII transitions into a Ca2+/CaM-autonomous active enzyme after autophosphorylation (at Thr 287) or oxidation (at Met281/282) of amino acids in the regulatory domain.
Table 1.
| Genotype/description | Phenotype |
|---|---|
| AC3-I/AC3-C α–MHC promoter transgenic [9,11] | Cardiomyopathy resistance to MI, isoproterenol infusion and angiotensin II. |
| CaMKIIN α–MHC promoter transgenic [80] | Reduced myocardial NF-κB signaling |
| CaMKIIδB α–MHC promoter transgenic [1,2] | Hypertrophy and secondary cardiomyopathy |
| CaMKIIδC α–MHC promoter transgenic [2–4] | Spontaneous cardiomyopathy, arrhythmias, and sudden death |
| CaMKIV α–MHC promoter transgenic [16] | Elevated myocardial CaMKII, hypertrophy and proarrhythmia |
| CaMKIIδ−/− Global knockout [6,7] | Resistance to aortic banding induced cardiomyopathy |
| CaMKIIγ−/− Global knockout [12] | Reduced macrophage and endothelial cell apoptosis in response to ER stress |
2. CaMKII activity becomes Ca/CaM independent by autophosphorylation and oxidation
If Ca/CaM elevations are prolonged or occur at high frequency, the CaMKII monomers catalyze intersubunit phosphorylations at an autophosphorylation site (Thr 286/287, the precise numbering varies according to isoform) in the regulatory domain [8]. Thr 287 autophosphorylation reduces the likelihood of Ca/CaM unbinding by increasing the affinity by a factor of 105 [9], but also confers residual Ca/CaM-independent activity after Ca/CaM dissociation [10]. Ca/CaM-autonomous activity also emerges under conditions favoring oxidation of a pair of Met residues (281/282) [11] which are present on the CaMKII isoforms most relevant to myocardial biology (CaMKIIδ and γ [12]). CaMKII activation by oxidation requires initial Ca/CaM binding and does not promote the increase in Ca/CaM affinity (so-called CaM trapping) seen with autophosphorylation, presumably because CaM trapping is prevented by oxidation of a Met residue embedded in the CaM binding region (Met 308). Increased oxidation can shift the Ca dependence for CaMKII activation to low levels that may favor CaMKII activation even under ambient intracellular Ca activity [13]. CaMKII Met oxidation is reversed by methionine sulfoxide reductase A (MsrA) [11] a finding that provides potential insight into deleterious consequences of MsrA gene deletion [11,14] and the benefits of MsrA overexpression [15]. It appears that Thr autophosphorylation and Met oxidation are interactive processes, because Thr autophosphorylation is increased under circumstances of enhanced intracellular reactive oxygen species (ROS) [11]. ROS inactivates many phosphatases, so ROS could favor Thr autophosphorylation by reducing the capacity to dephosphorylate Thr 286/287. Increased Met oxidation may also lead to improved accessibility of Thr 286/287 for autophosphorylation, even in the absence of elevated Ca/CaM.
3. Ca/CaM-autonomous CaMKII activity is implicated in heart disease
The ability of CaMKII to transition between Ca/CaM-dependence and independence has important implications for physiology and disease. On one hand, CaMKII activity appears to be important for Ca regulated/mediated physiological activities such as excitation-contraction coupling (ECC), excitation-transcription coupling (ETC) and ‘fight or flight’ heart rate increases. On the other hand, Ca/CaM-autonomous CaMKII activity appears to be increased in myocardial disease where it may contribute to apoptosis, arrhythmias [16], defective ECC and ETC favoring pathological hypertrophy.
4. CaMKII effects on cardiac ion channels and Ca handling proteins
CaMKII can phosphorylate numerous ion channels and Ca transport proteins and the consequent alterations in cellular electrophysiology and ECC can contribute to arrhythmogenesis and contractile dysfunction seen in HF (Fig. 2) [17]. The effects of CaMKII signaling to ion channels and the integrated impact of these effects on action potential properties (such as configuration, duration, restitution, afterdepolarizations) are complex [18,19]. For example, CaMKII tends to increase inward Ca and late Na currents which tends to prolong action potential duration, but also increases certain K currents that tend to shorten action potential duration (see more details below). While this is an over simplification, it is easy to appreciate that these CaMKII-dependent changes can impact regional action potential characteristics and contribute to arrhythmogenesis. Let us consider how certain individual currents are modified by CaMKII.
Fig. 2.
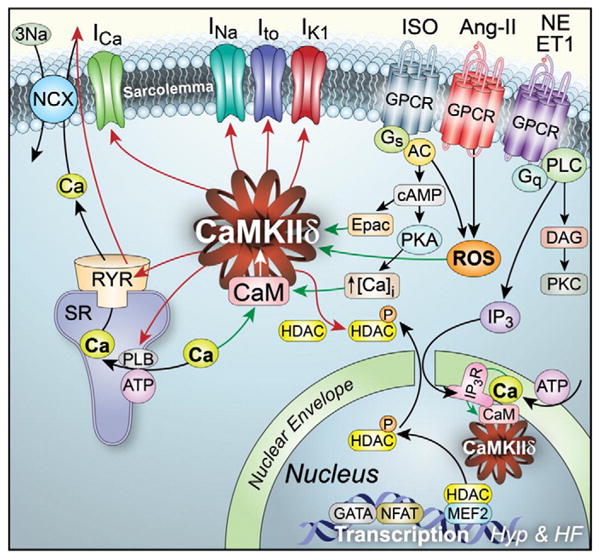
The relationship of CaMKII to upstream activators and target proteins in cardiomyocytes. CaMKII is present in cardiomyocytes where it is activated by Ca/CaM, ROS and Epac. Activation of β-adrenergic receptors (β-AR) can activate CaMKII by way of adenylyl cyclase (AC) that leads to increased Epac and increased protein kinase A (PKA). Other G-protein coupled receptor (GPCR) agonists (angiotensin II, AngII; norepinephrine, NE and endothelin-1, ET1) can increase cytoplasmic ROS by activating NAPDH oxidase, and increase Ca release from inositol triphosphate receptors (IP3R) in the nuclear envelope that activate nuclear CaMKII to increase prehypertrophic signaling by type II histone deacetylase (HDAC) derepression of myocyte enhancer factor 2 (MEF2). Activated CaMKIIδ (the predominant myocardial isoform) induces stimulatory actions by phosphorylating major Ca homeostatic proteins to increase L-type Ca current (ICa), phospholamban (PLB) to increase cytosolic Ca uptake by the sarcoplasmic reticulum (SR), and ryanodine receptor (RyR) to increase SR Ca release, which activates inward Na current due to the Na/Ca exchanger (NCX). CaMKII catalyzes phosphorylation of voltage-gated ion channels responsible for Na current (INa), transient outward K current (Ito) and inward rectifier K current (IK1).
5. Voltage-gated Ca channels
With regard to L-type Ca channels, Ca/CaM participates in Ca-dependent inactivation [20], which is important in limiting Ca entry into cardiac myocytes. Ca current (ICa,L) is also activated by CaMKII (ICa facilitation), seen functionally as a positive staircase of ICaL, where amplitude increases and inactivation slows over a series of several pulses [21–23]. Evidence has been presented that CaMKII-dependent phosphorylation at both the carboxy terminal of the α subunit and Thr498 in the β2a subunit of the channel mediate this effect [24,25]. CaMKII-induced changes in ICa,L may contribute to triggered arrhythmias, especially those attributed to early afterdepolarizations (EADs), which are thought to be mediated by inappropriate reactivation of ICa,L during long action potentials (Fig. 2) [16].
T-type Ca current (ICaT), while not present in adult ventricular myocytes (except during hypertrophy or HF) can also be enhanced by CaMKII-dependent phosphorylation [26–28]. The functional effect of CaMKII signaling on cardiac ICaT has not yet been fully elucidated.
6. Voltage-gated Na channels
Cardiac sodium current (INa) gating can also be modulated by CaMKII, [29,30] and CaMKII coimmunoprecipitates with and phosphorylates NaV1.5 channels. Either acute or chronic overexpression of CaMKIIδC shifts INa availability to more negative membrane potentials, enhances accumulation of Na channels in the intermediate inactivated state and slows recovery from inactivation. These loss-of-function INa effects would tend to produce Brugada Syndrome like effects, similar to that seen with certain monogenic Na channel mutations in humans. CaMKII also enhances late non-inactivating INa, which phenocopies INa alterations seen during ischemia and HF and with human mutations associated with long QT syndrome 3 (LQT3). Intriguingly, the multiple CaMKII-induced changes in INa gating are rate dependent [31] and almost identical to those seen in a human genetic mutation in NaV1.5 (1795InsD) that is associated with arrhythmogenic LQT3 and Brugada syndrome in the same patients. Since CaMKII is upregulated and more active in HF [32], CaMKII-dependent INa regulation in HF may be an important form of acquired arrhythmia. CaMKII is targeted to NaV1.5 by βIV spectrin in cardiomyocytes and neurons, a localization mechanism that is required for phosphorylation of a residue on the I–II intracellular domain (S571) which contributes to the gating effects on INa described above [33].
7. Voltage-gated K channels
Transient outward K current (Ito) which contributes to early cardiac action potential repolarization, is also modulated by CaMKII [18,34–36]. CaMKII can slow Ito inactivation and accelerate recovery from inactivation. Both effects would shorten action potential duration and refractory period, potentially causing proarrhythmic heterogeneity in action potential duration. These acute kinetic effects may apply to both Ito,fast and Ito,slow, which are attributed to KV4.2/KV4.3 and KV1.4 genes. Chronic CaMKII overexpression recapitulates the downregulation of Ito,fast (and KV4.3 protein) and upregulation of Ito,slow (and KV1.4 protein), as seen in HF [18]. CaMKII has also been reported to bind specifically to KV4.3 channel in myocytes, providing a recruitable reservoir of CaMKII which may participate in the modulation of other channels (e.g. Ca channels) [37]. This combination of effects makes the net effects on action potential duration and arrhythmogenesis complicated to anticipate, especially when combined with the CaMKII effects on other cardiac ion channels. In this context computational modeling has emerged as an important tool to understand the possible overall consequences of CaMKII on cardiac electrophysiology and arrhythmogenesis [19,38].
The inwardly rectifying IK1 is important in stabilizing the resting membrane potential, and reductions in IK1 (as in HF) can increase excitability. Short-term or chronic CaMKII overexpression causes downregulation of IK1 (and Kir2.1 protein expression that underlies IK1) [18], whereas in chronic CaMKII inhibition IK1 density is increased [39]. CaMKII may also acutely regulate channel gating by increasing baseline IK1 amplitude (based on CaMKII inhibitor effects) [18]. Overall, many sarcolemmal ion channels are subject to CaMKII-dependent regulation (Fig. 2).
8. CaMKII regulates SR Ca uptake and release
On one hand, the SR Ca-ATPase (SERCA2) had been reported to be a CaMKII target with apparent activation [40], but follow-up studies have indicated that this may not be the case [41]. On the other hand, CaMKII is well known to phosphorylate phospholamban (PLB), the small SR transmembrane protein that binds to and inhibits SERCA2 in cardiac myocytes. PLB phosphorylation relieves this inhibition and enhances SR Ca-ATPase activity. Thus CaMKII-dependent PLN phosphorylation at Thr17 (like PKA-dependent phosphorylation of Ser16) can contribute to the lusitropic (prorelaxant) effect and enhanced SR Ca uptake and release seen with increased heart rate and sympathetic stimulation.
The SR Ca release channel (ryanodine receptor, RyR) associates with and is directly phosphorylated by CaMKII. Not all results agree, but the prevailing view is that CaMKII phosphorylates RyR at Ser 2814 (at least), which activates RyR gating during both diastole and ECC [17,42–46]. In HF, where CaMKII expression and activation are increased, RyR phosphorylation and diastolic SR Ca leak are increased [32]; this diastolic SR Ca leak can initiate delayed afterdepolarizations (DADs) where the depolarizing current is inward Na/Ca exchange current. The CaMKII-induced RyR diastolic leak appears to mimic and indeed is synergistic with the human RyR mutations responsible for catecholaminergic polymorphic ventricular tachycardia (CPVT) [47]. As with INa above, these studies in genetically modified animal models provide proof of concept that this sort of CaMKII-modified RyR behavior may be a major arrhythmogenic mediator in HF and atrial fibrillation [48,49].
9. CaMKII activation near the nuclear envelope is important for transcription
The 1,4,5-trisphosphate receptor (IP3R) is another, more ubiquitous intracellular Ca release channel that is typically activated by G-protein coupled receptor and phospholipase C induced local increases in IP3. Adult myocytes express mainly type 2 IP3R, which are relatively enriched at the nuclear envelope in ventricle, but are also in the SR in atria [50]. IP3Rs may be involved in hypertrophic signaling (see below). IP3R can also initiate arrhythmogenic SR Ca release events and DADs by recruiting diastolic RyR-dependent release during neurohumoral activation [51]. CaMKII phosphorylates the IP3R2, inhibiting channel opening, and thus providing a negative feedback mechanism for IP3R channel gating [52]. This particular aspect of CaMKII might be antiarrhythmic and antihypertrophic.
10. Neurohumoral agonist pathways are ‘upstream’ activators of CaMKII
CaMKII is activated in response to signals generated through G-protein coupled receptors (GPCRs, Fig. 2). Norepinephrine (NE) and phenylephrine (PE), ligands for the α1-adrenergic receptor, and endothelin (ET-1), which acts on the endothelin receptor, induce GPCR coupling to the Gq protein and thus increase phospholipase C activity and InsP3 formation. In cardiomyocytes these agonists activate CaMKII and increase its autophosphorylation at Thr 286/287 [53–56] Mechanistically this appears to occur through Ca release from nuclear stores sensitive to InsP3 [54]. Isoproterenol (ISO) stimulates β-adrenergic receptors (β-AR) which couple to Gs and cyclic AMP formation. This activates protein kinase A (PKA), which increases intracellular Ca, in part through phosphorylation of the L-type Ca channel. ISO also activates and increases the autophosphorylation of CaMKII in cardiomyocytes [57–59]. How this occurs is not clear because there is evidence both for and against the expected PKA and Ca channel dependent pathway [60]. Another proposed mechanism involves the guanine nucleotide exchange factor Epac (Exchange protein directly activated by cAMP) [58,61] which has been shown to associate with CaMKII and β-arrestin at the β1AR [59]. Angiotensin (AngII), another GPCR agonist, also activates CaMKII, but here the proposed mechanism involves ROS generation and CaMKII oxidation as well as phosphorylation [11,13].
11. CaMKII connects neurohumoral agonist stimulation to hypertrophic gene programs
The Gq/PLC/InsP3 signaling pathway activated by NE, PE or ET-1 has been extensively linked to hypertrophic growth and gene expression. Neonatal rat ventricular myocytes (NRVMs) provided the initial model used to elucidate basic cellular mechanisms underlying hypertrophic changes in cell size, myofilament organization, protein synthesis and fetal gene expression. When NRVMs are cultured with PE, NE or ET-1 the expression of atrial natriuretic peptide (ANF), brain natriuretic peptide (BNP), myosin light chain-2 (MLC-2), and the α- and β-myosin heavy chain is affected. Pharmacological inhibition of CaMKII prevents these responses and forced expression of CaMKII in NRVMs elicits this same program of fetal gene expression [53,62,63]. Of note, CaMKIIδB, the splice variant that includes a nuclear localization sequence, is most effective at inducing hypertrophy and ANF expression in NRVMs [62]. ISO treatment has also been shown to induce fetal gene expression in NRVMs through a pathway that is not dependent on protein kinase A but rather on CaMKII [64]. Leukemia inhibitory factor (LIF), which acts through a gp130 linked receptor, is another agonist that induces hypertrophy in NRVMs through CaMKII mediated signaling [65]. The findings suggesting involvement of CaMKII in cardiac hypertrophy have been further substantiated using in vivo models. In response to transverse aortic constriction (TAC), an intervention that induces in vivo hypertrophy through GPCR activation, there are rapid and sustained increases in CaMKII phosphorylation and expression [3,66]. In addition cardiac specific transgenic expression of CaMKII leads to cardiomyocyte hypertrophy, associated with increases in cardiomyocyte size and induction of the fetal gene program [1–3] (Table 1).
12. CaMKII in cardiac transcriptional regulation
What are the mechanisms by which CaMKII alters cardiomyocyte growth and hypertrophic gene expression? The best characterized pathway is through CaMKII mediated phosphorylation of class II HDACs, in particular HDAC4 and HDAC5 [2,54,67,68]. relieving and derepressing MEF-2 mediated gene expression [2,69,70]. The involvement of HDACs and MEF2 signaling in development of cardiac hypertrophy is evidenced by enhanced responsiveness to TAC induced hypertrophy in mice in which class II HDACs are mutated or genetically deleted [69]. Recent studies also suggest a role for CaMKII in phosphorylation-induced transcriptional regulation of calcineurin (CaN), a regulator of the NFAT transcription factor involved in cardiac hypertrophy [71,72]. The sodium/calcium exchanger, NCX, is upregulated during hypertrophy, and induction of NCX expression by ISO was demonstrated to require CaMKII mediated effects on the transcription factor AP- 1 [73]. As mentioned above, CaMKII may also mediate transcriptional alterations in K+ channel protein expression.
13. The role of CaMKII in cardiomyopathy and cardiomyocyte survival
Prolonged TAC, myocardial infarction (MI) or high dose ISO administration induce dilated cardiomyopathy, HF, and ischemic damage. Recently mice in which the CaMKIIδ isoform was genetically deleted were generated. Loss of CaMKIIδ was shown to significantly attenuate the adverse cardiac remodeling, including ventricular dilation and dysfunction, induced by long term TAC [6,7]. In vivo administration of AngII and/or ISO also induces hypertrophy and resultant pathological remodeling, which is prevented in mice that express a CaMKII inhibitory peptide [9] (Table 1).
Cardiac pathophysiological responses are associated with apoptotic cell death and inflammation. βAR receptor induced apoptosis has been shown to require CaMKII in studies using pharmacological inhibitors and transgenic expression of the CaMKII inhibitory AC3-I or AIP [9,11,57,74,75]. Cardiomyocyte apoptosis appears to be largely mediated through the CaMKIIδC splice variant via a mitochondrial death pathway [57,75]. This could result from the profound effect of CaMKIIδC on RyR mediated SR Ca leak and mitochondrial Ca [76] or through its effect on expression of p53 and the pro-apoptotic BAX protein. [77]. A collection of mouse models with genetic alteration in CaMKII ativity is shown in Table 1. Remarkably, CaMKIIδB appears to suppress cardiomyocyte apoptosis. This has been suggested to occur through phosphorylation of the transcription factor HSF1 and subsequent induction of HSP70 [78] or through effects on GATA-4 mediated co-activation and induction of the antiapoptotic protein Bcl-2 [79]. There is also compelling evidence for effects of CaMKII on transcriptional regulation of proinflammatory gene expression, in particular that of complement factor B, through modulation of NFκB signaling pathways [80].
14. The Ying and Yang of CaMKII in cardiomyopathy and cardiomyocyte survival
CaMKII activation following TAC, oxidative stress or neurohumoral input can potentially induce (or down regulate) myriad genes and the proteins they encode. At present it appears that many of the genes regulated by CaMKII are maladaptive and would thus be targets for therapeutic CaMKII inhibition. It is likely, however, that CaMKII serves physiological as well as more pathophysiological roles, even at the level of regulated gene expression. Whether there are truly differential effects of CaMKIIδB and -δC on cardiac gene transcription and cardiomyocyte survival remains to be elucidated, but further understanding could provide caveats and insights into the use of CaMKII inhibitors in heart disease.
15. Conclusion
Taken together, abundant evidence indicates that CaMKII may be directly involved in the increased apoptosis, ineffective ECC and pathological hypertrophy that are major underlying causes for HF and sudden death in common forms of structural heart disease. CaMKII also appears to promote proinflammatory signaling [80] that characterizes ischemia, infarction and HF. Understanding the roles of this extensively regulated and pluripotent signaling molecule is an important goal for cardiovascular science and potentially for developing new and improved therapies for patients with myocardial hypertrophy and HF who are at high risk for arrhythmias and sudden death. Although CaMKIIδ knockout mice appear to survive normally, it will be important to learn if CaMKII inhibitory drugs, likely lacking isoform selectivity, will cause untoward side effects due to off target actions or undesirable effects of CaMKII inhibition in extra-cardiac tissue. While many biological questions remain, the next step to evaluate the therapeutic potential of CaMKII inhibitors, for arrhythmia and heart failure, awaits development of suitable drugs for clinical studies.
Acknowledgments
We thank Ms. Lorene Bender for secretarial assistance and Mr. Shawn Roach for graphic design.
Funding: Fondation Leducq (M.E.A., D.M.B.) and NIH R37-HL30077 (D.M.B.), P01-HL80101 (J.H.B., D.M.B.), R01 HL028143 (J.H.B.); R01 HL 079031, R01 HL 096652, and R01 HL 070250 (M.E.A.).
Contributor Information
Mark E. Anderson, Email: Mark-e-anderson@uiowa.edu.
Joan Heller Brown, Email: Jhbrown@ucsd.edu.
Donald M. Bers, Email: dmbers@ucdavis.edu.
References
- 1.Zhang T, Johnson EN, Gu Y, Morissette MR, Sah VP, Gigena M, et al. The cardiac-specific nuclear δB isoform of Ca2+/calmodulin-dependent protein kinase II induces hypertrophy and dilated cardiomyopathy associated with increased PP2A activity. J Biol Chem. 2002;277(2):1261–7. doi: 10.1074/jbc.M108525200. [DOI] [PubMed] [Google Scholar]
- 2.Zhang T, Kohlhaas M, Backs J, Mishra S, Phillips W, Dybkova N, et al. CaMKIIδ isoforms differentially affect calcium handling but similarly regulate HDAC/MEF2 transcriptional responses. J Biol Chem. 2007;282(48):35078–87. doi: 10.1074/jbc.M707083200. [DOI] [PubMed] [Google Scholar]
- 3.Zhang T, Maier LS, Dalton ND, Miyamoto S, Ross J, Jr, Bers DM, et al. The deltaC isoform of CaMKII is activated in cardiac hypertrophy and induces dilated cardiomyopathy and heart failure. Circ Res. 2003;92(8):912–9. doi: 10.1161/01.RES.0000069686.31472.C5. [DOI] [PubMed] [Google Scholar]
- 4.Sag CM, Wadsack DP, Khabbazzadeh S, Abesser M, Grefe C, Neumann K, et al. Calcium/calmodulin-dependent protein kinase II contributes to cardiac arrhythmogenesis in heart failure. Circ Heart Fail. 2009;2(6):664–75. doi: 10.1161/CIRCHEARTFAILURE.109.865279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Edman CF, Schulman H. Identification and characterization of delta B-CaM kinase and delta C-CaM kinase from rat heart, two new multifunctional Ca2+/calmodulin-dependent protein kinase isoforms. Biochem Biophys Acta. 1994;1221(1):89–101. doi: 10.1016/0167-4889(94)90221-6. [DOI] [PubMed] [Google Scholar]
- 6.Backs J, Backs T, Neef S, et al. The delta isoform of CaM kinase II is required for pathological cardiac hypertrophy and remodeling after pressure overload. Proc Natl Acad Sci USA. 2009;106(7):2342–7. doi: 10.1073/pnas.0813013106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ling H, Zhang T, Pereira L, et al. Requirement for Ca2+/calmodulin-dependent kinase II in the transition from pressure overload-induced cardiac hypertrophy to heart failure in mice. J Clin Invest. 2009;119(5):1230–40. doi: 10.1172/JCI38022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Koninck P, Schulman H. Sensitivity of CaM kinase II to the frequency of Ca oscillations. Science. 1998;279(5348):227–30. doi: 10.1126/science.279.5348.227. [DOI] [PubMed] [Google Scholar]
- 9.Zhang R, Khoo MS, Wu Y, Yang Y, Grueter CE, Ni G, et al. Calmodulin kinase II inhibition protects against structural heart disease. Nat Med. 2005;11(4):409–17. doi: 10.1038/nm1215. [DOI] [PubMed] [Google Scholar]
- 10.Meyer T, Hanson PI, Stryer L, Schulman H. Calmodulin trapping by calcium-calmodulin-dependent protein kinase. Science. 1992;256(5060):1199–202. doi: 10.1126/science.256.5060.1199. [DOI] [PubMed] [Google Scholar]
- 11.Erickson JR, Joiner ML, Guan X, et al. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell. 2008;133(3):462–74. doi: 10.1016/j.cell.2008.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Timmins JM, Ozcan L, Seimon TA, et al. Calcium/calmodulin-dependent protein kinase II links ER stress with Fas and mitochondrial apoptosis pathways. J Clin Invest. 2009 October;119(10):2925–41. doi: 10.1172/JCI38857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Palomeque J, Rueda OV, Sapia L, et al. Angiotensin II-induced oxidative stress resets the Ca dependence of Ca-calmodulin protein kinase II and promotes a death pathway conserved across different species. Circ Res. 2009;105(12):1204–12. doi: 10.1161/CIRCRESAHA.109.204172. [DOI] [PubMed] [Google Scholar]
- 14.Moskovitz J, Bar-Noy S, Williams WM, Requena J, Berlett BS, Stadtman ER. Methionine sulfoxide reductase (MsrA) is a regulator of antioxidant defense and lifespan in mammals 36. Proc Natl Acad Sci USA. 2001;98(23):12920–5. doi: 10.1073/pnas.231472998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ruan H, Tang XD, Chen ML, et al. High-quality life extension by the enzyme peptide methionine sulfoxide reductase. Proc Natl Acad Sci USA. 2002;99(5):2748–53. doi: 10.1073/pnas.032671199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu Y, Temple J, Zhang R, et al. Calmodulin kinase II and arrhythmias in a mouse model of cardiac hypertrophy. Circ. 2002;106(10):1288–93. doi: 10.1161/01.cir.0000027583.73268.e7. [DOI] [PubMed] [Google Scholar]
- 17.Bers DM, Grandi E. CaMKII regulation of cardiac ion channels. J Cadriovasc Res. 2009;54:180–7. doi: 10.1097/FJC.0b013e3181a25078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wagner S, Hacker E, Grandi E, Weber SL, Dybkova N, Sossalla S, et al. Ca/calmodulin kinase II differentially modulates potassium currents. Circ Arrhythm Electrophysiol. 2009;2(3):285–94. doi: 10.1161/CIRCEP.108.842799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grandi E, Puglisi JL, Wagner S, Maier LS, Severi S, Bers DM. Simulation of Ca-calmodulin-dependent protein kinase II on rabbit ventricular myocyte ion currents and action potentials. Biophys J. 2007;93(11):3835–47. doi: 10.1529/biophysj.107.114868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pitt GS, Zuhlke RD, Hudmon A, Schulman H, Reuter H, Tsien RW. Molecular basis of calmodulin tethering and Ca-dependent inactivation of L-type Ca channels. J Biol Chem. 2001;276(33):30794–802. doi: 10.1074/jbc.M104959200. [DOI] [PubMed] [Google Scholar]
- 21.Anderson ME, Braun AP, Schulman H, Premack BA. Multifunctional Ca/calmodulin-dependent protein kinase mediates Ca-induced enhancement of the L-type Ca current in rabbit ventricular myocytes. Circ Res. 1994;75(5):854–61. doi: 10.1161/01.res.75.5.854. [DOI] [PubMed] [Google Scholar]
- 22.Yuan W, Bers DM. Ca-dependent facilitation of cardiac Ca current is due to Ca-calmodulin-dependent protein kinase. Am J Physiol Heart Circ Physiol. 1994;267(3):H982–93. doi: 10.1152/ajpheart.1994.267.3.H982. [DOI] [PubMed] [Google Scholar]
- 23.Xiao RP, Cheng H, Lederer WJ, Suzuki T, Lakatta EG. Dual regulation of Ca/calmodulin-dependent kinase II activity by membrane voltage and by calcium influx. Proc Natl Acad Sci USA. 1994;91(20):9659–63. doi: 10.1073/pnas.91.20.9659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hudmon A, Schulman H, Kim J, Maltez JM, Tsien RW, Pitt GS. CaMKII tethers to L-type Ca channels, establishing a local and dedicated integrator of Ca signals for facilitation. J Cell Biol. 2005;171(3):537–47. doi: 10.1083/jcb.200505155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grueter CE, Abiria SA, Dzhura I, et al. L-type Ca(2+) Channel facilitation mediated by phosphorylation of the beta subunit by CaMKII. Mol Cell. 2006;23(5):641–50. doi: 10.1016/j.molcel.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 26.Izumi T, Kihara Y, Sarai N, Yoneda T, Iwanaga Y, Inagaki K, et al. Reinduction of T-type calcium channels by endothelin-1 in failing hearts in vivo and in adult rat ventricular myocytes in vitro. Circulation. 2003;108(20):2530–5. doi: 10.1161/01.CIR.0000096484.03318.AB. [DOI] [PubMed] [Google Scholar]
- 27.Barrett PQ, Lu HK, Colbran R, Czernik A, Pancrazio JJ. Stimulation of unitary T-type Ca channel currents by calmodulin-dependent protein kinase II. Am J Physiol Cell Physiol. 2000;279(6):C1694–703. doi: 10.1152/ajpcell.2000.279.6.C1694. [DOI] [PubMed] [Google Scholar]
- 28.Welsby PJ, Wang H, Wolfe JT, Colbran RJ, Johnson ML, Barrett PQ. A mechanism for the direct regulation of T-type calcium channels by Ca/calmodulin-dependent kinase II. J Neurosci. 2003;23(31):10116–21. doi: 10.1523/JNEUROSCI.23-31-10116.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wagner S, Dybkova N, Rasenack ECL, Jacobshagen C, Fabritz L, Kirchhof P, et al. Ca/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J Clin Invest. 2006;116(12):3127–38. doi: 10.1172/JCI26620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maltsev VA, Reznikov V, Undrovinas NA, Sabbah HN, Undrovinas A. Modulation of late sodium current by Ca, calmodulin, and CaMKII in normal and failing dog cardiomyocytes: similarities and differences. Am J Physiol Heart Circ Physiol. 2008;294(4):H1597–608. doi: 10.1152/ajpheart.00484.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Veldkamp MW, Viswanathan PC, Bezzina C, Baartscheer A, Wilde AAM, Balser JR. Two distinct congenital arrhythmias evoked by a multidysfunctional Na+ channel. Circ Res. 2000;86(9):91e–7e. doi: 10.1161/01.res.86.9.e91. [DOI] [PubMed] [Google Scholar]
- 32.Ai X, Curran JW, Shannon TR, Bers DM, Pogwizd SM. Ca/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca leak in heart failure. Circ Res. 2005;97(12):1314–22. doi: 10.1161/01.RES.0000194329.41863.89. [DOI] [PubMed] [Google Scholar]
- 33.Hund TJ, Koval OM, Li J, Wright PJ, Qian L, Snyder JS, et al. A βIV-spectrin/CaMKII signaling complex is essential for membrane excitability in mice. J Clin Invest. 2010;120(10):3508–19. doi: 10.1172/JCI43621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tessier S, Karczewski P, Krause EG, Pansard Y, Acar C, Lang-Lazdunski M, et al. Regulation of the transient outward K+ current by Ca/calmodulin-dependent protein kinases II in human atrial myocytes. Circ Res. 1999;85(9):810–9. doi: 10.1161/01.res.85.9.810. [DOI] [PubMed] [Google Scholar]
- 35.Colinas O, Gallego M, Setien R, Lopez-Lopez JR, Perez-Garcia MT, Casis O. Differential modulation of Kv4.2 and Kv4.3 channels by calmodulin-dependent protein kinase II in rat cardiac myocytes. Am J Physiol Heart Circ Physiol. 2006;291(4):H1978–87. doi: 10.1152/ajpheart.01373.2005. [DOI] [PubMed] [Google Scholar]
- 36.Sergeant GP, Ohya S, Reihill JA, Perrino BA, Amberg GC, Imaizumi Y, et al. Regulation of Kv4.3 currents by Ca/calmodulin-dependent protein kinase II. Am J Physiol Cell Physiol. 2005;288(2):C304–13. doi: 10.1152/ajpcell.00293.2004. [DOI] [PubMed] [Google Scholar]
- 37.Wang Y, Cheng J, Tandan S, Jiang M, McCloskey DT, Hill JA. Transient-outward K+ channel inhibition facilitates L-type Ca current in heart. J Cardiovasc Electrophysiol. 2006;17(3):298–304. doi: 10.1111/j.1540-8167.2006.00362.x. [DOI] [PubMed] [Google Scholar]
- 38.Hund TJ, Rudy Y. Rate dependence and regulation of action potential and calcium transient in a canine cardiac ventricular cell model. Circulation. 2004;110(20):3168–74. doi: 10.1161/01.CIR.0000147231.69595.D3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li J, Marionneau C, Zhang R, Shah V, Hell JW, Nerbonne JM, et al. Calmodulin kinase II inhibition shortens action potential duration by upregulation of K+ currents. Circ Res. 2006;99(10):1092–9. doi: 10.1161/01.RES.0000249369.71709.5c. [DOI] [PubMed] [Google Scholar]
- 40.Toyofuku T, Kurzydlowski K, Narayanan N, MacLennan DH. Identification of Ser38 as the site in cardiac sarcoplasmic reticulum Ca/calmodulin-dependent protein kinase. J Biol Chem. 1994;269:26492–6. [PubMed] [Google Scholar]
- 41.Odermatt A, Kurzydlowski K, MacLennan DH. The Vmax of the Ca-ATPase of cardiac sarcoplasmic reticulum (SERCA2a) is not altered by Ca/calmodulin-dependent phosphorylation or by interaction with phospholamban. J Biol Chem. 1996;271:14206–13. doi: 10.1074/jbc.271.24.14206. [DOI] [PubMed] [Google Scholar]
- 42.Wehrens XHT, Lehnart SE, Reiken SR, Marks AR. Ca/calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circ Res. 2004;94(6):e61–70. doi: 10.1161/01.RES.0000125626.33738.E2. [DOI] [PubMed] [Google Scholar]
- 43.Rodriguez P, Bhogal MS, Colyer J. Stoichiometric phosphorylation of cardiac ryanodine receptor on serine 2809 by calmodulin-dependent kinase II and protein kinase A. J Biol Chem. 2003;278(40):38593–600. doi: 10.1074/jbc.C301180200. [DOI] [PubMed] [Google Scholar]
- 44.Li L, Satoh H, Ginsburg KS, Bers DM. The effect of Ca-calmodulin-dependent protein kinase II on cardiac excitation–contraction coupling in ferret ventricular myocytes. J Physiol. 1997;501(Pt 1):17–31. doi: 10.1111/j.1469-7793.1997.017bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guo T, Zhang T, Mestril R, Bers DM. Ca/calmodulin-dependent protein kinase II phosphorylation of ryanodine receptor does affect calcium sparks in mouse ventricular myocytes. Circ Res. 2006;99(4):398–406. doi: 10.1161/01.RES.0000236756.06252.13. [DOI] [PubMed] [Google Scholar]
- 46.Yang D, Zhu WZ, Xiao B, Brochet DXP, Chen SRW, Lakatta EG, et al. Ca/calmodulin kinase II-dependent phosphorylation of ryanodine receptors suppresses Ca sparks and Ca waves in cardiac myocytes. Circ Res. 2007;100(3):399–407. doi: 10.1161/01.RES.0000258022.13090.55. [DOI] [PubMed] [Google Scholar]
- 47.Dybkova N, Sedej S, Napolitano C, Neef S, Rokita AG, Hunlich M, et al. Overexpression of CaMKIIδc in RyR2R4496C+/− knock-in mice leads to altered intracellular Ca2+−handling and increased mortality. J Am Coll Cardiol. 2011;57(4):469–79. doi: 10.1016/j.jacc.2010.08.639. [DOI] [PubMed] [Google Scholar]
- 48.van Oort RJ, McCauley MD, Dixit SS, Pereira L, Yang Y, Respress JL, et al. Ryanodine receptor phosphorylation by calcium/calmodulin-dependent protein kinase II promotes life-threatening ventricular arrhythmias in mice with heart failure. Circulation. 2010;122(25):2669–79. doi: 10.1161/CIRCULATIONAHA.110.982298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chelu M, Sarma S, Sood S, Wang S, Oort V, Jeroen R, et al. Calmodulin kinase II mediated sarcoplasmic reticulum calcium leak promotes atrial fibrillation. J Clin Invest. 2009;119(7):1940–51. doi: 10.1172/JCI37059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Perez PJ, Ramos-Franco J, Fill M, Mignery GA. Identification and functional reconstitution of the type 2 inositol 1, 4, 5-trisphosphate receptor from ventricular cardiac myocytes. J Biol Chem. 1997;272(38):23961–9. doi: 10.1074/jbc.272.38.23961. [DOI] [PubMed] [Google Scholar]
- 51.Li X, Zima AV, Sheikh F, Blatter LA, Chen J. Endothelin-1-induced arrhythmogenic Ca signaling is abolished in atrial myocytes of inositol-1, 4, 5-trisphosphate(IP3)-receptor type 2-deficient mice. Circ Res. 2005;96(12):1274–81. doi: 10.1161/01.RES.0000172556.05576.4c. [DOI] [PubMed] [Google Scholar]
- 52.Bare DJ, Kettlun CS, Liang M, Bers DM, Mignery GA. Cardiac type 2 inositol 1, 4, 5-trisphosphate receptor: interaction and modulation by calcium/calmodulin-dependent protein kinase II. J Biol Chem. 2005;280(16):15912–20. doi: 10.1074/jbc.M414212200. [DOI] [PubMed] [Google Scholar]
- 53.Zhu W, Zou Y, Shiojima I, Kudoh S, Aikawa R, Hayashi D, et al. Ca/calmodulin-dependent kinase II and calcineurin play critical roles in endothelin-1-induced cardiomyocyte hypertrophy. J Biol Chem. 2000;275(20):15239–45. doi: 10.1074/jbc.275.20.15239. [DOI] [PubMed] [Google Scholar]
- 54.Wu X, Zhang T, Bossuyt J, Li X, McKinsey TA, Dedman JR, et al. Local InsP3-dependent perinuclear Ca signaling in cardiac myocyte excitation–transcription coupling. J Clin Invest. 2006;116(3):675–82. doi: 10.1172/JCI27374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.O-Uchi J, Komukai K, Kusakari Y, Obata T, Hongo K, Sasaki H, et al. Alpha1-adrenoceptor stimulation potentiates L-type Ca current through Ca/calmodulin-dependent PK II (CaMKII) activation in rat ventricular myocytes. Proc Natl Acad Sci USA. 2005;102(26):9400–5. doi: 10.1073/pnas.0503569102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ago T, Yang Y, Zhai P, Sadoshima J. Nifedipine inhibits cardiac hypertrophy and left ventricular dysfunction in response to pressure overload. J Cardiovasc Transpl Res. 2010;4:304–13. doi: 10.1007/s12265-010-9182-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhu WZ, Wang SQ, Chakir K, et al. Linkage of beta(1)-adrenergic stimulation to apoptotic heart cell death through protein kinase A-independent activation of Ca/calmodulin kinase II. J Clin Invest. 2003;111(5):617–25. doi: 10.1172/JCI16326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pereira L, Metrich M, Fernandez-Velasco M, Lucas A, Leroy J, Perrier R, et al. The cAMP binding protein Epac modulates Ca sparks by a Ca/calmodulin kinase signalling pathway in rat cardiac myocytes. J Physiol. 2007;583(Pt 2):685–94. doi: 10.1113/jphysiol.2007.133066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mangmool S, Shukla AK, Rockman HA. Beta-arrestin-dependent activation of Ca (2+)/calmodulin kinase II after beta(1)-adrenergic receptor stimulation. J Cell Biol. 2010;189(3):573–87. doi: 10.1083/jcb.200911047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Grimm M, Brown JH. Beta-adrenergic receptor signaling in the heart: role of CaMKII. J Mol Cell Cardiol. 2010;48(2):322–30. doi: 10.1016/j.yjmcc.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Oestreich EA, Malik S, Goonasekera SA, Blaxall BC, Kelley GG, Dirksen RT, et al. Epac and phospholipase Cepsilon regulate Ca release in the heart by activation of protein kinase Cepsilon and calcium-calmodulin kinase II. J Biol Chem. 2009;284(3):1514–22. doi: 10.1074/jbc.M806994200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ramirez MT, Zhao XL, Schulman H, Brown JH. The nuclear deltaB isoform of Ca/calmodulin-dependent protein kinase II regulates atrial natriuretic factor gene expression in ventricular myocytes. J Biol Chem. 1997;272(49):31203–8. doi: 10.1074/jbc.272.49.31203. [DOI] [PubMed] [Google Scholar]
- 63.Sei CA, Irons CE, Sprenkle AB, McDonough PM, Brown JH, Glembotski CC. The alpha-adrenergic stimulation of atrial natriuretic factor expression in cardiac myocytes requires calcium influx, protein kinase C, and calmodulin-regulated pathways. J Biol Chem. 1991;266(24):15910–6. [PubMed] [Google Scholar]
- 64.Sucharov CC, Mariner PD, Nunley KR, Long C, Leinwand L, Bristow MR. A beta1-adrenergic receptor CaM kinase II-dependent pathway mediates cardiac myocyte fetal gene induction. Am J Physiol Heart Circ Physiol. 2006;291(3):H1299–308. doi: 10.1152/ajpheart.00017.2006. [DOI] [PubMed] [Google Scholar]
- 65.Kato T, Sano M, Miyoshi S, Sato T, Hakuno D, Ishida H, et al. Calmodulin kinases II and IV and calcineurin are involved in leukemia inhibitory factor-induced cardiac hypertrophy in rats. Circ Res. 2000;87(10):937–45. doi: 10.1161/01.res.87.10.937. [DOI] [PubMed] [Google Scholar]
- 66.Colomer JM, Mao L, Rockman HA, Means AR. Pressure overload selectively up-regulates Ca/calmodulin-dependent protein kinase II in vivo. Mol Endocrinol. 2003;17(2):183–92. doi: 10.1210/me.2002-0350. [DOI] [PubMed] [Google Scholar]
- 67.Backs J, Song K, Bezprozvannaya S, Chang S, Olson EN. CaM kinase II selectively signals to histone deacetylase 4 during cardiomyocyte hypertrophy. J Clin Invest. 2006;116(7):1853–64. doi: 10.1172/JCI27438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bossuyt J, Helmstadter K, Wu X, Clements-Jewery H, Haworth RS, Avkiran M, et al. Ca2+/calmodulin-dependent protein kinase IIdelta and protein kinase D overexpression reinforce the histone deacetylase 5 redistribution in heart failure. Circ Res. 2008;102(6):695–702. doi: 10.1161/CIRCRESAHA.107.169755. [DOI] [PubMed] [Google Scholar]
- 69.Zhang CL, McKinsey TA, Chang S, Antos CL, Hill JA, Olson EN. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell. 2002;110(4):479–88. doi: 10.1016/s0092-8674(02)00861-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Passier R, Zeng H, Frey N, Naya FJ, Nicol RL, McKinsey TA, et al. CaM kinase signaling induces cardiac hypertrophy and activates the MEF2 transcription factor in vivo. J Clin Invest. 2000;105(10):1395–406. doi: 10.1172/JCI8551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.MacDonnell SM, Weisser-Thomas J, Kubo H, Hanscome M, Liu Q, Jaleel N, et al. CaMKII negatively regulates calcineurin-NFAT signaling in cardiac myocytes. Circ Res. 2009;105(4):316–25. doi: 10.1161/CIRCRESAHA.109.194035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lu YM, Shioda N, Yamamoto Y, Han F, Fukunaga K. Transcriptional upregulation of calcineurin Abeta by endothelin-1 is partially mediated by calcium/calmodulin-dependent protein kinase IIdelta3 in rat cardiomyocytes. Biochim Biophys Acta. 2010;1799(5–6):429–41. doi: 10.1016/j.bbagrm.2010.02.004. [DOI] [PubMed] [Google Scholar]
- 73.Mani SK, Egan EA, Addy BK, Grimm M, Kasiganesan H, Thiyagarajan T, et al. Beta-adrenergic receptor stimulated Ncx1 upregulation is mediated via a CaMKII/AP-1 signaling pathway in adult cardiomyocytes. J Mol Cell Cardiol. 2010;48(2):342–51. doi: 10.1016/j.yjmcc.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yang Y, Zhu WZ, Joiner ML, Zhang R, Oddis CV, Hou Y, et al. Calmodulin kinase II inhibition protects against myocardial cell apoptosis in vivo. Am J Physiol Heart Circ Physiol. 2006;291(6):H3065–75. doi: 10.1152/ajpheart.00353.2006. [DOI] [PubMed] [Google Scholar]
- 75.Zhu W, Woo AY, Yang D, Cheng H, Crow MT, Xiao RP. Activation of CaMKIIdeltaC is a common intermediate of diverse death stimuli-induced heart muscle cell apoptosis. J Biol Chem. 2007;282(14):10833–9. doi: 10.1074/jbc.M611507200. [DOI] [PubMed] [Google Scholar]
- 76.Zhang T, Guo T, Mishra S, Dalton ND, Kranias EG, Peterson KL, et al. Phospholamban ablation rescues sarcoplasmic reticulum Ca(2+) handling but exacerbates cardiac dysfunction in CaMKIIdelta(C) transgenic mice. Circ Res. 2010;106(2):354–62. doi: 10.1161/CIRCRESAHA.109.207423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Toko H, Takahashi H, Kayama Y, Oka T, Minamino T, Okada S, et al. Ca/calmodulin-dependent kinase II{delta} causes heart failure by accumulation of p53 in dilated cardiomyopathy. Circulation. 2010;122(9):891–9. doi: 10.1161/CIRCULATIONAHA.109.935296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Peng W, Zhang Y, Zheng M, Cheng H, Zhu W, Cao CM, et al. Cardioprotection by CaMKII-deltaB is mediated by phosphorylation of heat shock factor 1 and subsequent expression of inducible heat shock protein 70. Circ Res. 2010;106(1):102–10. doi: 10.1161/CIRCRESAHA.109.210914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Little GH, Saw A, Bai Y, Dow J, Marjoram P, Simkhovich B, et al. Critical role of nuclear calcium/calmodulin-dependent protein kinase IIdeltaB in cardiomyocyte survival in cardiomyopathy. J Biol Chem. 2009;284(37):24857–68. doi: 10.1074/jbc.M109.003186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Singh MV, Kapoun A, Higgins L, et al. Ca/calmodulin-dependent kinase II triggers cell membrane injury by inducing complement factor B gene expression in the mouse heart. J Clin Invest. 2009;119(4):986–96. doi: 10.1172/JCI35814. [DOI] [PMC free article] [PubMed] [Google Scholar]