

Abstract
Postsynaptic density 95 (PSD-95), the major scaffold at excitatory synapses, is critical for synapse maturation and learning. In rodents, eye opening, the onset of pattern vision, triggers a rapid movement of PSD-95 from visual neuron somata to synapses. We showed previously that the PI3 kinase–Akt pathway downstream of BDNF/TrkB signaling stimulates synaptic delivery of PSD-95 via vesicular transport. However, vesicular transport requires PSD-95 palmitoylation to attach it to a lipid membrane. Also, PSD-95 insertion at synapses is known to require this lipid modification. Here, we show that BDNF/TrkB signaling is also necessary for PSD-95 palmitoylation and its transport to synapses in mouse visual cortical layer 2/3 neurons. However, palmitoylation of PSD-95 requires the activation of another pathway downstream of BDNF/TrkB, namely, signaling through phospholipase Cγ and the brain-specific PKC variant protein kinase M ζ (PKMζ). We find that PKMζ selectively regulates phosphorylation of the palmitoylation enzyme ZDHHC8. Inhibition of PKMζ results in a reduction of synaptic PSD-95 accumulation in vivo, which can be rescued by overexpressing ZDHHC8. Therefore, TrkB and PKMζ, two critical regulators of synaptic plasticity, facilitate PSD-95 targeting to synapses. These results also indicate that palmitoylation can be regulated by a trophic factor. Our findings have implications for neurodevelopmental disorders as well as aging brains.
Introduction
Postsynaptic density 95 (PSD-95) is the major PSD scaffolding protein for ionotropic glutamate receptors and the signaling molecules through which these receptors function (Kim and Sheng, 2004). Overexpression of PSD-95 increases spine number, size, and synaptic efficacy (El-Husseini et al., 2000a). In the visual pathway of rodents, rapid increases in synaptic PSD-95 precede increases in synaptic strength and refinement that occur during the period encompassing eye opening (EO) (Yoshii et al., 2003; Lu and Constantine-Paton, 2004). Hippocampal slices lacking PSD-95 show prolonged NMDA receptor (NMDAR) mediated Ca2+ currents (Beique et al., 2006) and, in cultured slices, a reduced developmental increase of AMPA receptors (AMPARs) (Ehrlich et al., 2007). PSD-95 knock-out mice show disrupted spatial learning and no long-term depression (LTD) but have enhanced long-term potentiation (LTP) (Migaud et al., 1998). Conversely, overexpression of PSD-95 occludes LTP while enhancing LTD (Beique and Andrade, 2003; Stein et al., 2003; Ehrlich and Malinow, 2004). These findings suggest that PSD-95 levels are critical for activity-dependent regulation of NMDARs and AMPARs by setting the range of upregulation or downregulation of these receptors.
BDNF and its receptor TrkB participate in many aspects of neural circuit formation (McAllister et al., 1999; Poo, 2001; Huang and Reichardt, 2003; Minichiello, 2009; Yoshii and Constantine-Paton, 2010). In the central visual pathway, EO and the onset of pattern vision increase BDNF/TrkB signaling (Castren et al., 1992; Viegi et al., 2002), the formation of mature dendritic spines (Chakravarthy et al., 2006), and visual cortical plasticity (Cabelli et al., 1995). BDNF also facilitates the maturation of inhibition implicated in closure of the critical period for ocular dominance plasticity (Huang et al., 1999). TrkB colocalizes with NMDA receptors (Gomes et al., 2006), and interacts with PSD-95 in an activity-dependent manner (Ji et al., 2005; Yoshii and Constantine-Paton, 2007). Furthermore, both BDNF/TrkB signaling and a brain-specific protein kinase C variant, protein kinase M ζ (PKMζ), are involved in late-LTP (Kang and Schuman, 1995; Korte et al., 1995; Figurov et al., 1996; Patterson et al., 1996; Poo, 2001; Kovalchuk et al., 2002; Pastalkova et al., 2006; Minichiello, 2009), and maintenance of long-term memory (Alonso et al., 2005; Pastalkova et al., 2006; Shema et al., 2007). BDNF increases the expression and activity of PKMζ (Adasme et al., 2011).
We showed previously that BDNF/TrkB signaling through the PI3 kinase–Akt pathway facilitates PSD-95 transport to synapses via vesicular transport (Ji et al., 2005; Yoshii and Constantine-Paton, 2007). Here, we employ a genetically modified mouse strain, TrkBF616A, to specifically block the receptor and show that TrkB activation is also necessary for trafficking PSD-95 to visual synapses after eye opening (AEO) in vivo. We also report that BDNF/TrkB signaling regulates palmitoylation of PSD-95. To our surprise, this process is mediated by PLC and PKMζ. Inhibition of PKMζ also suppresses the synaptic redistribution of PSD-95 in visual cortical layer 2/3 pyramidal neurons. Furthermore, the phosphorylation of palmitoyl acyltransferase (PAT) ZDHHC8 is regulated by PKMζ. Finally, we demonstrate that ZDHHC8 overexpression rescues the suppression of synaptic transport of PSD-95 by a PKMζ blocking peptide in vivo.
Materials and Methods
Animals.
TrkBF616A mice were kindly provided by Dr. David Ginty (Johns Hopkins University, Baltimore, MD). Wild-type (WT) C57BL6, mice were from Charles River Laboratories. Animals were caged and maintained in a 12 h light/dark cycle. Both sexes were used for experiments. All manipulations were performed in accord with the guidelines of the Massachusetts Institute of Technology (MIT) Institutional Animal Care and Use Committee.
Chemicals.
1-(Tert-butyl)-3-(naphthalen-1-ylmethyl)-1H-pyrazolo [3,4-d]pyrimidin-4-amine (1NM-PP1) and 3-([1,1′-biphenyl]-4-yl)-1-(tert-butyl)-1H-pyrazolo[3,4-d] pyrimidin-4-amine (BPh-PP1) were prepared using procedures described previously (Bishop et al., 1999). Their chemical structures are shown in Figure 1A. These were applied chronically using the slow release plastic Elvax. U73122 (PLC inhibitor), chelerythrine, PD98059 (mitogen-activated protein kinase inhibitor), and wortmannin were purchased from Sigma-Aldrich. The PKMζ inhibitory pseudosubstrate (myr-SIYRRGARRWRKL-OH) and scrambled peptide (myr-RLYRKRIWRSAGROH) (Pastalkova et al., 2006) were made in the MIT Biopolymers Laboratory.
Figure 1.
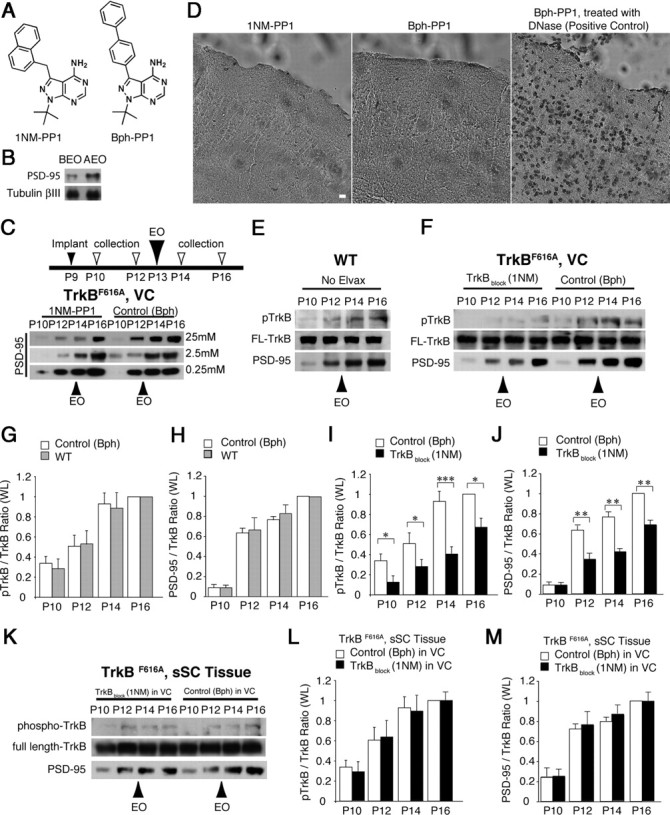
TrkB blockade suppresses developmental increase of PSD-95 synthesis. A, Chemical structure of 1NM-PP1 (left) and Bph-PP1 (right). B, PSD-95 level significantly increased 6 h AEO compared to BEO at P13 in synaptosomal fractions from mouse visual cortex. C, Among three different concentrations, Elvax containing 1NM-PP1 at 25 mm showed the highest suppression of PSD-95 at P12, P14, and P16 compared to Bph-PP1 containing Elvax. The inset diagram (top) in this figure and those in the following figures illustrate the sequence of procedures for the experiments. D, The TUNEL assay showed no apoptosis in VC under the 1NM-PP1-Elvax (left) or Bph-PP1-Elvax (middle). As a positive control, slices were treated with DNase (right). Scale bar, 10 μm. E, During the period bracketing eye opening, in whole lysates of WT mouse VC, PSD-95 and pTrkB increased, whereas full-length TrkB (FL-TrkB) levels remained constant. F, Both pTrkB and PSD-95 levels were suppressed in whole lysates of VC when TrkB was blocked with 1NM-PP1. FL-TrkB levels in the same lanes are used to normalize because their levels remained constant. G, H, Both pTrkB (G) and PSD-95 (H) levels were similar between WT and Bph-PP1-treated control VC. I, J, Both pTrkB (I) and PSD-95 (J) levels were suppressed in whole lysates of VC when TrkB was blocked with 1NM-PP1. K–M, When TrkB was blocked in VC over the interval encompassing the eye opening (P10–P16), both phosphorylated TrkB (K, L) and PSD-95 (K, M) increased normally in the same animals' superficial visual layers of the superior colliculus (sSC), indicating that the blocking effect was restricted to cortex. The blots in C–F and K were first probed for pTrkB, then stripped and probed for PSD-95 and FL-TrkB. *p < 0.05; **p < 0.01; ***p < 0.001. Error bars represent SEM.
Elvax preparation.
Elvax was made as described previously (Simon et al., 1992). 1NM-PP1 or Bph-PP1 (25 mm) was prepared in a 10% Elvax solution in methylene chloride. The solvent was evaporated at 20°C, and the resulting plugs were lyophilized overnight, then sliced at 90 μm on a cryostat. Slices were cut to trapezoid-shape (parallel sides, 2–3 × 4–5 mm; height, 4–5 mm). Following craniotomy under isoflurane anesthesia, pieces of Elvax were placed on the surface of both occipital cortices.
TUNEL assay.
The FragEL DNA Fragmentaion Detection Kit (Calbiochem) was used for the TUNEL assay. The procedures were performed according to the manufacturer's instructions.
Antibodies.
For immunoblotting, mouse monoclonal anti-PSD-95 (catalog #K28/43; NeuroMab), anti-TrkB antibody (catalog #T16020; BD Biosciences), anti-phospho-TrkB (catalog #ab51187; Abcam), anti-SAP102 (synapse-associated protein 102, kindly provided by Dr. Johannes Hell, UC Davis, Davis, CA), anti-HA tag (catalog #3724; Cell Signaling Technology), and anti-phospho-(Ser) PKC substrate (catalog #2261; Cell Signaling Technology) antibodies were used. For immunohistochemistry, rabbit anti-PSD-95 (catalog #2507; Cell Signaling Technology) was used.
Plasmid construction.
The 2A cleavage sequence from Thosea asigna virus together with the nucleotide sequences encoding PKMζ pseudosubstrate or its scrambled peptide were attached by PCR to the TagRFP-T sequence using the following primers (Szymczak et al., 2004; Trichas et al., 2008): universal forward primer, cgatcaattgatggtgtctaagggcgaagagctgattaag; first PCR reverse primer, gttagcagacttcctctgccctctccactgcccttatacagctcgtccatgccattaag; second PCR reverse primer, cttgggccaggattctcctcgacgtcaccgcatgttagcagacttcctctgccctctccac; third PCR reverse primer for pseudo-PKM ζ substrate, gttatgtacatcacagcttcctccacctcctggcgcccctcctgtagatgcttgggccaggattctcctcgacg; reverse primer for scramble peptide, gttatgtacatcacctgccggcgctcctccagatcctcttcctatacagccttgggccaggattctcctcgacg.
The modified TagRFP-T sequences were inserted under the CAG promoter of the pCAGIG plasmid by replacing the IRES and GFP sequences. TagRFP-T was a gift from the laboratory of R. Tsien (UCSD, San Diego CA). pCAGIG was originally from the laboratory of the C. Cepko (Harvard Medical School, Boston, MA).
Synaptosome fractionation, immunoblotting, immunoprecipitation.
Identification of visual cortex (VC) was based on the Paxinos atlas (Paxinos and Franklin, 2001). The VC tissues were collected by cutting the occipital one-third of the cortex along the anteroposterior axis and 3 mm from the midline along mediolateral axis. Whole lysates, synaptosome preparation, and immunoblotting were performed as described previously (Yoshii et al., 2003). Protein concentration of each sample was measured using the Pierce BCA Protein Assay Kit (Thermo Scientific), and 10 μg of sample was loaded in each lane for SDS-PAGE separation. Following immunoblotting, films were scanned and band densities were quantified using NIH ImageJ. Full-length TrkB bands were constant between postnatal day 10 (P10) and P16; thus, phosphorylated TrkB (pTrkB) and PSD-95 band densities were normalized to the TrkB band. Control samples at P16 showed highest levels of PSD-95 and pTrkB; thus, fold changes at each time point were calculated relative to the protein level at P16 control on the same gels (Fig. 1G–J,L,M). Results are reported as mean ± SEM.
All immunoblot analyses used three sets of protein samples from three different litters. Two gels were run for each sample and blotted (N = 6).
Primary neuronal culture and lipofection.
Primary neuronal culture and lipofection were performed as described previously (Ji et al., 2005; Yoshii and Constantine-Paton, 2007).
Metabolic labeling with 3H palmitic acid and immunoprecipitation.
Primary VC cultures were prepared from embryonic day 16 (E16) mouse brains. Cultured visual cortical neurons (6 × 106 cells/plate; 21–28 d in vivo) were preincubated for 30 min in serum-free Neurobasal medium with fatty acid-free bovine serum albumin (10 mg/ml; Sigma), and were then labeled for 4 h in neurobasal media containing 2 mCi/ml 3H palmitic acid (PerkinElmer) with or without BDNF and/or various blockers. Labeled neurons were washed with PBS. BDNF and/or inhibitors were added from the preincubation period through the end of the 4 h incubation period.
To observe PSD-95 palmitoylation, we scraped labeled neurons using 0.5 ml of lysis buffer A (20 mm Tris-HCl, pH 7.4, 1 mm EDTA, 100 mm NaCl, and 1% SDS), and after 5 min extraction, 1% Triton X-100 was added to a final volume of 1 ml. The suspensions were centrifuged at 20,000 × g for 10 min, and supernatants were incubated with anti-PSD-95 antibody for 1 h and then incubated for 1 h with 30 μl of protein A-Sepharose (Pharmacia) slurry at 4°C. Immunoprecipitates were washed three times with buffer containing 20 mm Tris-HCl, pH 7.4, 1 mm EDTA, 100 mm NaCl, and 1% Triton X-100, and were resuspended in 33 μl of SDS-PAGE sample buffer. We used 30 μl for autoradiography and the remaining 3 μl for immunoblotting. The ratios of palmitoylated PSD-95 densities in autoradiographies to total PSD-95 densities in corresponding immunoblots were calculated. Each experiment was repeated four times.
Immunohistochemistry.
After 4% paraformaldehyde perfusion and vibratome sectioning at 50 μm, coronal brain sections were permeabilized using pepsin (Gene Tex; catalog #GTX28194) at 37°C for 10 min and were rinsed twice with PBS. The sections were reacted overnight at room temperature with mouse anti-PSD-95 in PBS containing 4% donkey serum and were rinsed in three changes of PBS (15 min each). Subsequently, they were incubated in Alexa-488-tagged antibody (Invitrogen) and then washed with PBS three times (15 min each). Images were captured using a 40× objective lens under identical settings with a Nikon PCM 2000 confocal microscope. The visual cortex was identified on coronal sections using the Paxinos atlas. The Z-series of optical sections taken at intervals of 0.5 μm through 20 μm of tissue were reconstructed.
In utero electroporation.
Following laparotomy on timed pregnant mice, the uterus was externalized. Approximately 0.5–0.6 μl of solution containing PSD-95-GFP construct (2 μg/μl) was injected into the forebrain ventricle at E15.5 through the uterine wall. In later experiments, constructs encoding red fluorescent protein (RFP)-tagged pseudosubstrate and/or HA-tagged ZDHHC8 were added at 0.01 μg/μl. Using an ECM 830 apparatus (Harvard Apparatus), brains were electroporated with five pulses of 28 V for 50 ms at intervals of 950 ms. After recovery, pregnancies continued, and pups were delivered and nursed normally.
Cortical injection.
The PKMζ inhibitory pseudosubstrate and scrambled peptide were dissolved in PBS to a concentration of 10 nmol/ml. Under isoflurane anesthesia, the skin was opened. The skull was thinned with a drill. A pulled capillary pipette connected to a Nanoject II (Drummond Scientific) could then be stereotactically advanced to 350 μm below the skull surface to target visual cortical layer 2/3. One hundred and eighty nanoliters of a PKMζ pseudosubstrate peptide solution containing Cholera-toxin B conjugated with Alexa-647 (Invitrogen) were injected over 1 min. In each animal, injections were made in four different sites of the visual cortex. The eyes were opened immediately after the surgery at P13. For each experiment described, data were acquired from three experimental animals and three controls.
Imaging analysis.
Sections containing PSD-95-GFP-expressing neurons were imaged at successive depths of 0.5 μm using a Nikon PCM 2000 confocal microscope. In each experiment, data were acquired from three experimental animals and three controls. The same laser intensity was used between treatment and control groups, and all analyzed neurons included the cell's nucleus indicated by a “halo” pattern of GFP signal in somata. Neurons with a soma showing fluorescence either saturated (intensity value of 255 throughout the nucleus in ImageJ) or dim (intensity value <150 in ImageJ) were excluded from the analyses. Total pixel intensities were analyzed with ImageJ. Secondary branches of apical dendrites located between 60–150 μm from the soma were selected with 40 × 20 μm rectangles. For each cell analyzed, a threshold was set by eye during the initial evaluation so as to represent PSD-95-GFP puncta and exclude diffuse labels in dendritic shafts (see Fig. 3C). The same threshold was applied to all neurons in the same experiment. Total pixel intensity was calculated as the sum of each pixel number multiplied by its intensity as measured above threshold for each cell. This value was then averaged across all dendritic segments sampled in the experiment.
Figure 3.
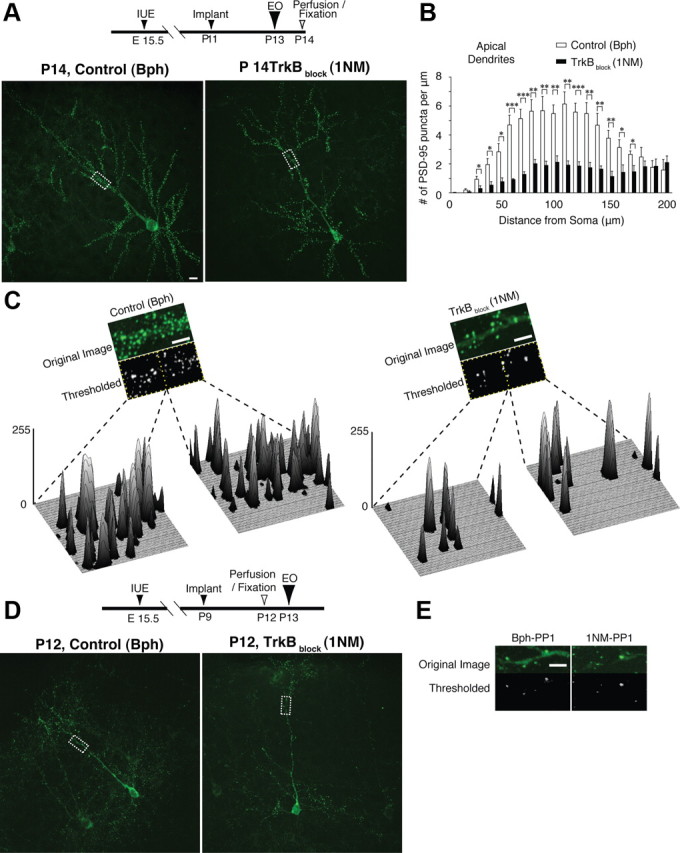
TrkB blockade suppresses increases of PSD-95 synaptic puncta after eye opening. A, PSD-95-GFP-expressing neurons in layer 2/3 of VC were treated in vivo with 1NM-PP1 (TrkB block) or Bph-PP1 (control) over the eye-opening interval. Dashed rectangles are magnified in C. B, Using a Sholl analysis, numbers of PSD-95 puncta on apical dendrites were significantly reduced at many distances from the soma TrkB-blocked neurons after eye opening. The same analysis for basal dendrites showed the same trend, but only 2 Sholl intervals of 13 showed significance at p < 0.05. C, PSD-95-GFP puncta intensity in secondary branches of apical dendrites was reduced in TrkB-blocked neurons. Three-dimensional graphs show intensities of puncta after thresholding that eliminate GFP signal in dendritic shafts and allow visualization of the PSD-95 distribution and intensity along the dendritic segment. D, E, Before EO, treatment with Bph-PP1 or 1NM-PP1 from P9 to P12 revealed that PSD-95-GFP puncta intensities in secondary branches of apical dendrites were unchanged between neurons treated for the same duration but not over the eye-opening interval. Dashed rectangles are magnified in E. *p < 0.05; **p < 0.01; ***p < 0.001. Error bars represent SEM. Scale bars: 10 μm.
Neurolucida software (MBF Bioscience) was used for quantitative analyses of neuronal morphology, including PSD-95 puncta number, branching, and largest cell body area.
Statistics.
Student's t tests was used for comparison of two groups. One-way ANOVA with a post hoc Tukey's test was used for comparisons of more than two groups.
Results
Developmental increases in PSD-95 are accelerated by EO and regulated by TrkB
We reported previously that rapid activity-dependent transport of PSD-95 to synapses was triggered by EO in the rat central visual system (Yoshii et al., 2003), and we have confirmed a similar significant increase in synaptosomal PSD-95 AEO compared to before eye opening (BEO) in WT mouse visual cortex (Fig. 1B) (PSD-95AEO/PSD-95BEO = 2.1 ± 0.2; p < 0.01; N = 6). TrkB phosphorylation was also increased AEO as compared to BEO (pTrkBAEO/pTrkBBEO = 1.8 ± 0.3; p < 0.01; N = 6).
To test TrkB effects on PSD-95 in visual neurons in vivo, we used a genetically modified mouse strain, TrkBF616A, with a mutation in the ATP-binding pocket of TrkB that allows an adenosine analog (1NM-PP1) to specifically block kinase activity of the receptor (Chen et al., 2005). We surgically implanted thin slices of the slow-release polymer Elvax containing different concentrations of 1NM-PP1 or a newly developed control compound Bph-PP1 (Fig. 1A) on the surface of VC at P9. We found a dose-dependent decrease in phospho-TrkB (data not shown) and synaptic PSD-95 in 1NM-PP1-treated VC (Fig. 1C), and subsequently used Elvax with the most effective concentration (25 mm) of 1NM-PP1 or the control molecule Bph-PP1 for all in vivo experiments. No cell death was detected after these implants (Fig. 1D).
VC lysate changes in TrkBF616A mice
After normalizing to full-length TrkB in the same lane, both WT mice and TrkBF616A control mice (Bph-PP1-Elvax on VC) showed similar increases in TrkB phosphorylation (Fig. 1E–G) and in PSD-95 levels (Fig. 1E,F,H) over the EO interval (P12–P14). The different patterns of increase in Figure 1, G and H, suggest that in VC whole lysates, more TrkB is activated after EO when the cortex becomes driven by high-contrast vision (Heimel et al., 2010), whereas PSD-95 levels increase from before EO (P12) through at least 3 d afterward.
As expected, ratios of phospho-TrkB to total TrkB in these lysates were significantly reduced in TrkB-suppressed (1NM-PP1-treated) VCs compared to control (Bph-PP1-treated) VCs (Fig. 1F,I). Ratios of PSD-95 to total TrkB levels were also suppressed in TrkB-blocked VCs from P12 through P16 (Fig. 1F,J).
In contrast to the VC, PSD-95 levels were not perturbed in the visual layers of the superior colliculus or the dorsal thalamus, indicating that 1NM-PP1 from cortically placed Elvax was not affecting lower levels of the visual pathway (Fig. 1K–M).
Synaptic analyses in TrkBF616A mice
We found previously in rat that PSD-95 increases in synaptic fractions immediately after EO were not seen in whole lysates and did not require protein synthesis (Yoshii et al., 2003), suggesting that EO and pattern vision onset acutely trigger trafficking of PSD-95 to the synapse with no new protein synthesis of the scaffold during this interval.
In mouse VC treated with Bph-PP1-Elvax (control), synaptosome PSD-95 levels increased after EO (Fig. 2A) (synaptosome PSD-95 level change at P14 relative to P12, 1.35 ± 0.05; N = 6). However, 1NM-PP1-Elvax (TrkB blocking) placed on VC suppressed the increase of PSD-95 in the synaptosome compared to the Bph-PP1 after EO (synaptosome PSD-95 level change at P14 relative to P12, 1.01 ± 0.05; N = 6; control vs 1NM-PP1, p < 0.05). While synaptosome PSD-95 in control was enriched compared to levels in control whole lysates (Fig. 2A), TrkB-blockade with 1NM-PP1-Elvax also suppressed the PSD-95 synaptosome enrichment over whole lysates during the same 2 d interval (Fig. 2A) (synaptosome enrichment for PSD-95 level in synpatosome/PSD-95 level in whole lysate: 1.78 ± 0.06 in control, 1.15 ± 0.05 in TrkBblock; p < 0.01; N = 6).
Figure 2.

TrkB blockade suppresses the synaptosome increases and endogenous dendritic PSD-95 after eye opening. A, When TrkB signaling was suppressed, synaptosomal PSD-95 level as compared to control was neither increased after EO nor enriched compared to whole lysates (WL). B, Immunohistochemistry of PSD-95 in layer 2/3 VC from two different animals implanted with Bph-PP1 Elvax to the left hemisphere and 1NM-PP1 Elvax to the right. After eye opening, the punctate distribution of PSD-95 seen in the TrkB-blocked cortices (1NM) compared to the contralateral Bph-treated cortices show reductions in endogenous, immunostained, PSD-95 puncta but enhanced staining in somata. Dashed lines indicate the pial surface. A dashed square shows the size of the quantified area for total pixel intensity of PSD-95-positive immunolabels. Scale bar, 10 μm.
We previously documented BDNF/TrkB-driven PSD-95 transport in dendrites of cultured neurons from mouse VC (Yoshii and Constantine-Paton, 2007). As a first step in determining whether TrkB-mediated transport from somata and proximal dendrites to synapses occurred in mouse cortical neurons in vivo, we used immunocytochemistry to examine changes in endogenous PSD-95 (Fig. 2B). Mice with control Bph-Elvax on one VC and 1NM-PP1 Elvax on the other were maintained over the EO interval, and, after animals were killed and tissue and slices were processed, the local distribution of endogenous PSD-95 was examined in VC layer 2/3 neurons of both cortices. Suppression of TrkB signaling caused a marked reduction in the number and intensity of PSD-95 puncta among the dendrites of these neurons, and layer 2/3 cell bodies were filled by diffuse anti-PSD-95 immunoreactivity. Pixel intensities of PSD-95-positive immunolabels in 20 × 20 μm areas (Fig. 2B, dashed square) were sampled and quantified (50,100 ± 1700 A.U.; N = 20). Control neurons in the other VCs of the same mice showed the converse distribution: high densities of PSD-95 throughout the superficial dendrites with PSD-95 immunoreactivity barely visible in layer 2/3 cell bodies (83,900 ± 3600 A.U.; N = 20; p < 0.001).
To quantify TrkB effects on the distribution of PSD-95 in single neurons, PSD-95-GFP constructs were electroporated into newly generated 2/3 VC neurons of TrkBF616A mice at embryonic day 15.5. At either P9 or P11, 1NM-PP1-Elvax or Bph-PP1-Elvax was placed on their VCs. All animals survived 3 d of Bph-PP1 or 1NM-PP1 treatment. Pups implanted at P9 were killed for cortical analyses at P12 before EO. Eyes of pups implanted on P11 were opened on P13, and pups were killed on P14. We analyzed PSD-95-GFP puncta in littermates after EO (P14) and before EO (P12).
At P14 (after P11 Elvax implantations), PSD-95-GFP-expressing control neurons had many protrusions supporting PSD-95 puncta, whereas neurons treated with 1NM-PP1 had fewer PSD-95 puncta (Fig. 3A). This difference in numbers of PSD-95-positive protrusions was much more significant for apical dendrite puncta (Fig. 3B) than basal dendrite puncta (data not shown; p < 0.05 in only 2 of 13 segments; N = 17 neurons).
Consequently, we focused on apical dendrites and estimated the density of PSD-95-GFP in synaptic puncta by selecting fixed lengths of dendrite at the point of secondary branching (Fig. 3A, boxed areas) and thresholding the images (Fig. 3C) to best represent synaptic PSD-95-GFP. All pixel intensities in the thresholded segments from each neuron in the control and TrkB-blocked treatment groups were summed separately to give the total pixel intensity for each group (Yoshii and Constantine-Paton, 2007). The pooled pixel intensity in apical dendrites with TrkB blocked (8540 ± 900 A.U.) was highly significantly reduced relative to the apical dendrite pixel intensities (30,100 ± 3900 A.U.) in control neurons (N = 34 for each group; p < 0.001).
A Sholl analysis examining differences in numbers of PSD-95 puncta at different distances from somata between Bph-PP1 and 1NM-PP1 treatments of apical dendrite anatomical puncta is shown in Figure 3B and shows significant differences across the apical dendritic tree (N = 17 neurons). Total PSD-95-GFP pixel intensities across 34 basal dendrites of these same neurons were also reduced in 1NM-PP1- compared to Bph-PP1-treated cells; however, the significance was less robust (24,700 ± 5900 A.U. in control vs 6500 ± 1000 A.U. in 1NM-PP1; p < 0.05). Thus, an effect of TrkB blockade on dendritic protrusions is consistent with the previous report using a dominant-negative TrkB mutant mouse (Chakravarthy et al., 2006).
In the Sholl analysis, the total dendritic branching was marginally different between TrkB-blocked neurons and controls (p < 0.05 in only 1 of 15 segments at 70 μm from the somata; N = 17). Cell body sizes were also only modestly altered (165 ± 15 μm2 in control vs 140 ± 5 μm2 in 1NM-PP1; p < 0.05; N = 17). These findings are in contrast with data from forebrain-specific TrkB knock-out mice (Xu et al., 2000). The short-duration block of TrkB in our experiments compared to prolonged absence of the receptor in the knock-out mice could explain these differences.
To determine whether the effect on apical puncta change was due to EO and not simply due to the presence of Elvax on cortex, we performed a similar experiment on the layer 2/3 VC neurons that had also been exposed to 1NM-PP1 for the same duration but not over the EO interval. Before EO, similar low-density distributions of PSD-95-GFP puncta were present in both 1NM-PP1- and Bph-PP1-treated neurons (Fig. 3D,E). Total pixel intensities at P12 were 6980 ± 750 A.U. in control VC neurons treated with Bph-PP1 (N = 20) and 6850 ± 1050 A.U. in VC neurons treated with 1NM-PP1 (N = 20; p = 0.33).
TrkB-PLCγ-PKMζ regulates PSD-95 palmitoylation
Palmitoylation of PSD-95 can be triggered by glutamate (El-Husseini Ael et al., 2002) and is crucial for this scaffold's trafficking and insertion at postsynaptic sites (El-Husseini et al., 2000b). Our previous work suggested that in young cortical neurons, NMDA receptor currents activate BDNF/TrkB signaling and drive movement of PSD-95 from the ER to the Golgi apparatus in cell bodies before dendritic transport (Yoshii and Constantine-Paton, 2007). However, PSD-95 is synthesized by cytoplasmic ribosomes, implying that it must be palmitoylated to allow association with ER and Golgi membranes. Therefore, we hypothesized that BDNF/TrkB signaling might also trigger palmitoylation, and this might be a trigger for PSD-95 palmitoylation to enable the scaffold's membrane association and microtubule-based vesicular transport to synapses. Despite recent progress in purification of palmitoylation using chemical probing, metabolic labeling with a radioisotope is the standard procedure for identification of palmitoylated proteins (Fukata and Fukata, 2010), but radioactive labeling of whole mouse pups was not feasible. Therefore, we dissociated cortical neurons from TrkBF616A mouse embryos and labeled them with 3H-palmitic acid.
Subsequently, we immunopreciptated PSD-95 from lysates of cultures with no treatment or with either BDNF or 1NM-PP1 added. Palmitoylation of PSD-95 from cultures treated with BDNF was higher than without treatment (palmitoylated PSD-95 ratio relative to control, 1.25 ± 0.05: p < 0.01: N = 4), and palmitoylation of PSD-95 from cultures treated with 1NM-PP1 was the lowest (palmitoylated PSD-95 ratio relative to control, 0.45 ± 0.11; p < 0.001; N = 4) (Fig. 4A). In addition, immunopreciptation of PSD-95 from the lysates revealed that levels of palmitoylated PSD-95 were significantly greater in cultures treated with Bph-PP1- compared to 1NM-PP1-treated cultures (palmitoylated PSD-95 ratio relative to Bph-PP1 treatment, 0.37 ± 0.11; p < 0.001; N = 4) (Fig. 4B). Neuronal cultures were subsequently exposed to antagonists of the three pathways downstream of TrkB: mitogen-activated protein kinase (MAPK), phosphoinositide 3 kinase (PI3K), and phospholipase Cγ (PLCγ) (Huang and Reichardt, 2003). The PLC blocker U73122 significantly reduced PSD-95 palmitoylation (palmitoylated PSD-95 ratio relative to control, 0.35 ± 0.17; p < 0.001; N = 4), while blockers of MAPK (PD98059; palmitoylated PSD-95 ratio relative to control, 1.15 ± 0.21; p = 0.38; N = 4) and PI3K (wortmannin; palmitoylated PSD-95 ratio relative to control, 1.03 ± 0.1; p = 0.21; N = 4) did not (Fig. 4C). PLCγ is known to regulate PKC. Consequently, we applied chelerythrine, a broad PKC blocker. This treatment also suppressed PSD-95 palmitoylation (palmitoylated PSD-95 ratio relative to control, 0.39 ± 0.08; p < 0.001; N = 4) (Fig. 4D). PKMζ is a brain-specific variant of PKC. It is an important mediator of late, protein synthesis-dependent LTP in hippocampus (Sajikumar et al., 2005; Pastalkova et al., 2006) and is critical for maintenance of long-term memory (Shema et al., 2007, 2011). PKMζ also regulates experience-induced LTP in primary visual cortex (Cooke and Bear, 2010), and BDNF activates PKMζ (Adasme et al., 2011). Furthermore, blockade of PKMζ has recently been shown to reduce PSD-95 in Xenopus tadpole tectal neurons (Liu et al., 2009). PKMζ can be specifically inhibited by a pseudosubstrate peptide that is myristoylated and thus permeable to cell membranes (Pastalkova et al., 2006). Therefore, we treated another set of cultures with BDNF and the myristoylated PKMζ pseudosubstrate. These showed a significant reduction in PSD-95 palmitoylation (palmitoylated PSD-95 ratio relative to control, 0.48 ± 0.18; p < 0.001; N = 4) compared to cultures treated with BDNF and the scrambled peptide (palmitoylated PSD-95 ratio relative to control, 0.94 ± 0.06; p = 0.2; N = 4) or BDNF alone (Fig. 4E). Future experiments using in vivo deletions of PLCγ and PKMζ will be necessary to confirm this current, single-dose, in vitro pharmacological blockade.
Figure 4.

Immunoprecipitation experiments demonstrate that BDNF/TrkB signaling regulates PSD-95 palmitoylation through PLCγ and PKMζ in VC cultures. A, Palmitoylation of PSD-95 was increased in cultured VC neurons by BDNF (50 ng ml−1) and decreased by TrkB block (1NM-PP1, 3 μm; N = 4). B, VC cultures with TrkB block (1NM-PP1, 3 μm) in the presence of BDNF (50 ng ml−1) showed a reduction in PSD-95 palmitoylation ration compared to control (Bph-PP1, 3 μm; N = 4). C, VC cultures treated with blockers of the PLC pathway (U73122, 1 μm), but not the MAPK (PD98059, 50 μm) or PI3K (wortmannin, 100 nm) pathways, in the presence of BDNF (50 ng ml−1) showed a decrease in PSD-95 palmitoylation (N = 4). D, VC cultures treated with the broad PKC blocker (chelerythrine, 2.5 μm) in the presence of BDNF (50 ng ml−1) showed decreased palmitoylation (N = 4). E, VC cultures treated with PKMζ inhibitory peptide (a pseudosubstrate, 1 μm) or the scrambled peptide (1 μm) in the presence of BDNF (50 ng ml−1) showed a decrease only with the pseudosubstrate (N = 4). Cultures in B–E were pretreated with BDNF (50 ng ml−1). The signaling blocked is indicated for each lane below the blots. All immunoprecipitates (B–E) were resuspended in 33 μl of SDS-PAGE sample buffer; 30 μl were used for autoradiography [PSD-95 (3H Palm)], and the remaining 3 μl for immunoblotting [PSD-95 (IB)]. **P < 0.01; ***p < 0.001. Error bars represent SEM.
PKMζ blockade causes reduction in PSD-95 puncta
We next examined the contribution of PKMζ to synaptic localization of PSD-95 in vivo. At P13, before EO, we injected either the PKMζ pseudosubstrate or the scrambled pseudosubstrate into the VC of animals expressing PSD-95-GFP in layer 2/3 neurons. We added Alexa-647-conjugated cholera toxin B (ACTB), a non-toxic and broad neuronal tracer, to the injected peptide solutions. Eyes were opened immediately after the peptide/ACTB injection, and 24 h later the animals were killed, and VC layer 2/3 neurons were examined for PSD-95 puncta distribution and intensity (Fig. 5A,B). VC neurons expressing ACTB and presumably PKMζ inhibitory pseudosubstrate showed significantly lower total pixel intensity (9200 ± 1300) than neurons containing the scrambled peptides (28,400 ± 4200; N = 16 dendrites in 8 neurons for each group; p < 0.001).
Figure 5.

Cortical injection of PKMζ inhibitory pseudosubstrate suppresses synaptic increase of PSD-95 in vivo after eye opening. A, Overlays depict blue-stained cell bodies of neurons that absorbed either a scrambled or pseudosubstrate peptide solution containing an ACTB, a cell-tracking agent. B, Visual cortical neurons treated with PKMζ inhibitory pseudosubstrate had a reduction in PSD-95 total puncta intensity compared with neurons treated with the scrambled peptide.
We ensured that the pseudosubstrate was the active agent in the cells analyzed by building pseudosubstrate and scrambled peptide DNA constructs tagged with RFP (RFP-2A-pseudo and RFP-2A-scr, respectively) DNA (Fig. 6A). First, we confirmed that endogenous PSD-95 puncta were reduced in cultured cortical neurons expressing the pseudosubstrate-RFP (N = 22 dendrites in 9 cells; mean, 7.10 ± 1.0 puncta per 100 μm dendrite) compared to the scrambled-peptide-RFP (N = 24 dendrites in 9 cells; mean, 10.2 ± 0.9 puncta per 100 μm of dendrite; peudosubstrate-RFP vs scrambled-peptide-RFP, p < 0.05) (Fig. 6B). To better discern the PSD-95 puncta changes, we next either co-electroporated the pseudosubstrate-RFP or the scrambled-peptide-RFP construct with PSD-95-GFP into separate cortices at E15.5 and examined PSD-95 puncta in VC layer 2/3 24 h after EO (Fig. 6C,D). Neurons expressing the pseudosubstrate-RFP showed significantly lower total puncta intensity (33,900 ± 3500) than neurons expressing the scrambled-peptide-RFP (24,500 ± 2800; N = 18 for each group; p < 0.01).
Figure 6.
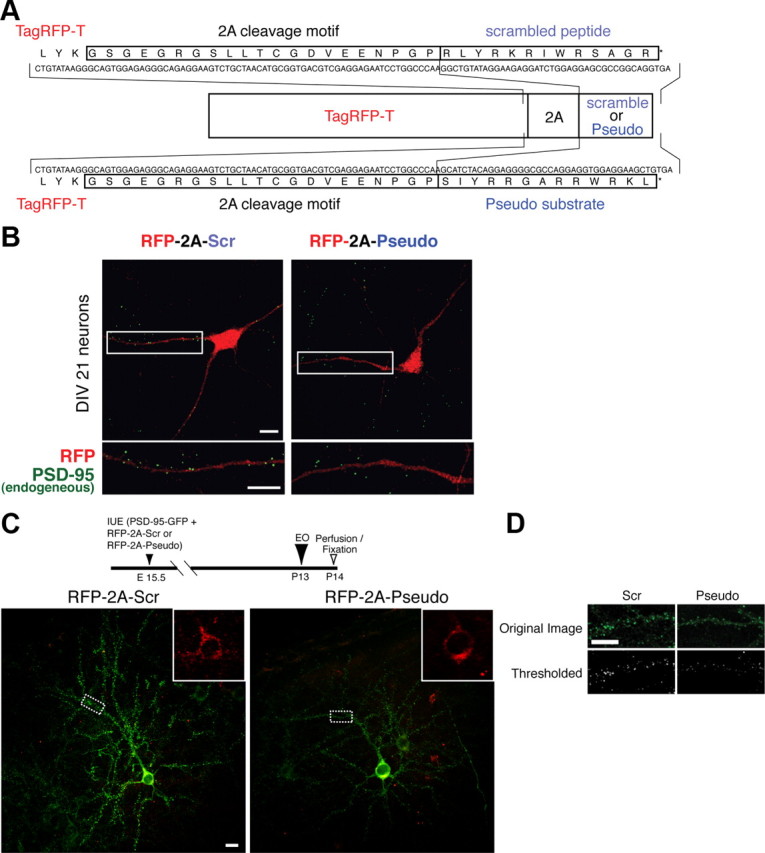
Genetically encoded PKMζ inhibitory pseudosubstrate suppresses synaptic increase of PSD-95 in vivo after eye opening. A, Nucleotide and amino acid sequences of the 2A cleavage motif followed by either the scrambled peptide or the PKMζ pseudosubstrate peptide in the expression plasmids of TagRFP-T. B, Endogenous dendritic PSD-95 puncta are decreased in neurons expressing the pseudosubstrate but not in neurons expressing scrambled peptide or untransfected neurons (data not shown). C, Visual cortical neurons electroporated at E15.5 with constructs coding for either the pseudosubstrate-RFP or the scrambled-peptide-RFP plus PSD-95-GFP. Insets depict pseudosubstrate-RFP or scrambled-peptide-RFP. The diagram (top) illustrates the sequence of in vivo experiments. D, Visual cortical neurons expressing PKMζ inhibitory pseudosubstrate showed a significant reduction in PSD-95 puncta intensity compared with neurons expressing the scrambled peptide. Scale bars: 10 μm.
Finally, we asked whether any palmitoylation enzyme(s) were regulated by PKMζ. Protein palmitoylation is accomplished by the ZDHHC family of PATs. However, there are over 20 known PATs (Fukata and Fukata, 2010). We selected six ZDHHCs based on known synaptic function and proven PSD-95 palmitoylation ability (Fukata et al., 2004; Huang et al., 2004; Fukata and Fukata, 2010). We reasoned that the putative PAT(s) should be phosphorylated by PKMζ. We transfected HA-tagged ZDHHCs 2, 3, 7, 8, 15, and 17 (gifts from M. Fukata, National Institute for Physiological Sciences, Okazaki, Aichi, Japan) in sets of primary cortical neuronal cultures, and, for each PAT, cultures were treated with either scrambled peptide or pseudosubstrate. Subsequently, we pulled down the PATs from culture lysates using an anti-HA antibody and immunoblotted to visualize HA-tagged ZDHHCs (Fig. 7A). As indicated by asterisks in Figure 7A, amounts of each PAT were equal between the two treatment groups (ZDHHC2pseudo/ZDHHC2scr = 1.02 ± 0.02; ZDHHC3pseudo/ZDHHC3scr = 1.05 ± 0.08; ZDHHC7pseudo/ZDHHC7scr = 1.02 ± 0.03; ZDHHC8pseudo/ZDHHC8scr = 0.98 ± 0.03; ZDHHC15pseudo/ZDHHC15scr = 1.0 ± 0.02; ZDHHC17pseudo/ZDHHC17scr = 0.95 ± 0.08). We then reprobed the blotting membranes with an antibody against phosphorylated-serine PKC family substrates and found that only ZDHHC8 showed a pronounced phosphorylation band in the scrambled pseudosubsrate-treated control cultures, whereas the phosphorylation band was absent when cultures were treated with the PKMζ pseudosubstrate (Fig. 7B).
Figure 7.
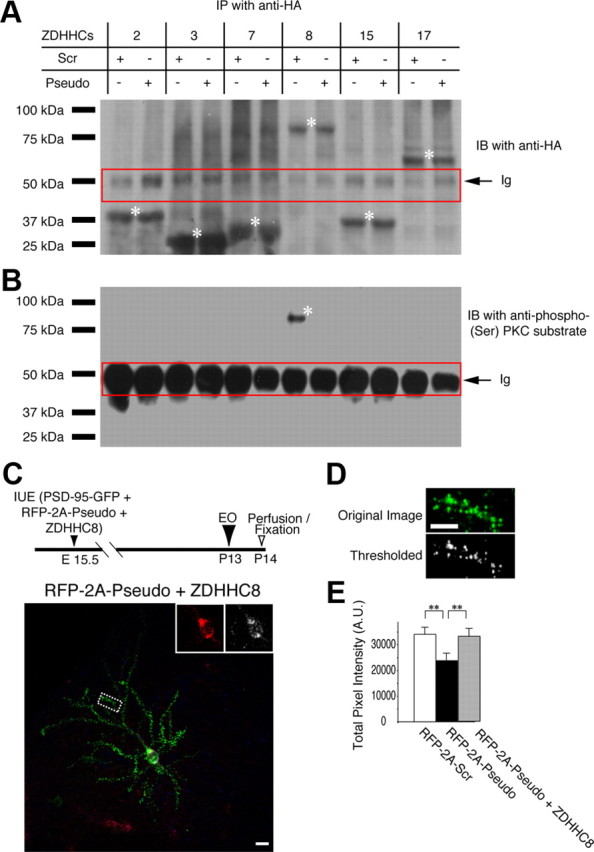
ZDHHC8 is responsible for PKMζ-regulated synaptic increase of PSD-95 in vivo after eye opening. A, HA-tagged ZDHHCs were expressed in neurons with either a scrambled peptide or pseudosubstrate. Immunoprecipitated ZDHHCs were visualized using an anti-HA tag antibody (N = 3). The red rectangle encloses the Igs. Asterisks indicate the HA-tagged PATs. B, Only ZDHHC8 with scrambled peptide treatment shows a band visualized by anti-phospho-(Ser) PKC substrate antibody. The band is absent in a sample treated by the pseudosubstrate (N = 3). The red rectangle encloses the Igs. The asterisk indicates phospho-ZDHHC8. C, Visual cortical neurons electroporated at E15.5 with constructs coding for pseudosubstrate-RFP, HA-tagged ZDHHC8, and PSD-95-GFP. Insets depict coexpression of pseudosubstrate-RFP (red) and HA-tagged ZDHHC8 stained with anti-HA tag antibody (pseudocolored in white for better visibility). The diagram illustrates the sequence of the in vivo experiments. D, E, An overexpression of ZDHHC8 rescues suppression of PSD-95 puncta intensity by the PKMζ inhibitory pseudosubstrate. Two branches from nine neurons (N = 18 total) were analyzed for each group. **p < 0.01. Error bars represent SEM. Scale bars: 10 μm.
These findings motivated us to test whether overexpression of ZDHHC8 could rescue the in vivo suppression of PSD-95 synaptic redistribution by PKMζ blockade in layer 2/3 VC neurons after EO. We electroporated constructs for PSD-95-GFP, RFP-tagged pseudosubstrate, and HA-tagged ZDHHC8 in utero and, 1 d after EO, killed the animals and stained the cortical slices with an anti-HA tag antibody followed by Alexa-647-conjugated secondary antibody. With this procedure, we determined whether a GFP- and RFP-containing neuron also overexpressed ZDHHC8 (Fig. 7C). We found that the addition of the WT ZDHHC8 gene completely restored total pixel intensity of PSD-95-GFP puncta, despite the pseudosubstrate inhibition of PKMζ (Fig. 7D,E) (33,000 ± 3600; N = 18; p < 0.01 between RFP-2A-pseudo and RFP-2A-pseudo + ZDHHC8). In the same slice, some neurons expressed PSD-95-GFP and RFP-tagged pseudosubstrate, but no detectable ZDHHC8. In these neurons, total pixel intensity of PSD-95-GFP puncta was suppressed, similar to neurons in Figure 6C (right).
Discussion
Transport of PSD-95 to synapses is dependent on pattern vision and mediated by activation of TrkB and PKMζ in mouse visual cortex
Previously, we reported that redistribution of PSD-95 to synapses is triggered by EO and the onset of the patterned vision in young rats (Yoshii et al., 2003). AMPAR increases at synapses follow within the next 24 h (Lu and Constantine-Paton, 2004). During this interval, more TrkB is associated with PSD-95 (Yoshii and Constantine-Paton, 2007). The current study confirms that the same activity-dependent transport occurs in mouse visual cortex and is dependent on the BDNF/TrkB signaling pathway. We show that EO induces increases of synaptosome PSD-95 levels, while PSD-95 levels in whole lysates remain stable between P12 and P14 (Fig. 2A). The punctate distribution of PSD-95 in control visual cortex also increases after EO (total pixel intensity, ∼30,000) compared with that before EO (total pixel intensity, ∼7000) in visual cortex (Fig. 3C,D). In addition, TrkB phosphorylation gradually increases during development (Fig. 1G), over a period encompassing EO. The increase in TrkB phosphorylation is consistent with an increase in BDNF mRNA during this period (Castren et al., 1992).
Our data also indicate that specific TrkB blockade using a chemical genetic approach suppresses increases in PSD-95 synthesis in whole lysates during early development (Fig. 1F,J) and enrichment of PSD-95 at visual cortical synapses triggered by EO (Fig. 2A). Suppression of TrkB signaling also causes a reduction of endogenous PSD-95 puncta and increases endogenous PSD-95 immunolabeling in cell bodies, suggesting a decrease in PSD-95 transport to synapses (Fig. 2B). These results indicate that, in intact mouse visual cortex, TrkB signaling regulates the developmental increase in PSD-95 and that EO, the onset of pattern vision, facilitates TrkB activation as well as the synaptic redistribution of PSD-95.
Chemical genetic inactivation of TrkB signaling in the intact TrkBF616A mouse has been shown to block the reinstatement of deprived eye synapses after brief monocular deprivation (Kaneko et al., 2008). In light of the present work, this is likely due to failure to deliver PSD-95 to the synapses driven by the newly reactivated eye's cortical inputs, because reduction or elimination of PSD-95 is known to decrease synapse number on young neurons (Beique et al., 2006; Elias et al., 2006; Yoshii and Constantine-Paton, 2007, 2010), and we have shown previously that 4 d of eye reclosure after EO causes PSD-95 levels in visual neuron synaptic fractions to decrease to pre-EO levels with a rapid recovery after eye reopening (Yoshii et al., 2003).
TrkB regulates excitatory synapse maturation and synaptic localization of PSD-95
The role of BDNF/TrkB signaling on inhibitory neurons in visual cortex has been well studied (Huang et al., 1999). Evidence for BDNF/TrkB functions in pyramidal neurons (Bracken and Turrigiano, 2009) and their developing spines is also abundant (Tyler and Pozzo-Miller, 2003; Chakravarthy et al., 2006; Tanaka et al., 2008). Furthermore, it has been established that PSD-95 at synapses is crucial to spine maturation (Kim and Sheng, 2004; van Zundert et al., 2004) and that BDNF/TrkB signaling plays a central role in facilitating glutamate synapse stabilization/maturation through the PSD-95 scaffold (Ji et al., 2005; Yoshii and Constantine-Paton, 2007, 2010). Our current results provide further evidence that BDNF/TrkB signaling regulates transport of PSD-95 to synapses in layer 2/3 pyramidal neurons, thus contributing to activity-dependent maturation of excitatory visual synapses.
PSD-95 palmitoylation is regulated by a trophic factor signaling pathway
Palmitoylation has been shown to be required for PSD-95 transport and synaptic insertion (Topinka and Bredt, 1998; El-Husseini et al., 2000b), and PSD-95 palmitoylation is known to require synaptic activity (El-Husseini Ael et al., 2002; Noritake et al., 2009). Considerably less is known about the signaling pathway that regulates this palmitoylation. Here, we document that the TrkB pathway regulates palmitoylation of PSD-95 through PLC and the PKC/PKMζ pathway (Fig. 4–7). We also show, for the first time, that PKMζ regulates phosphorylation of only ZDHHC8 out of the six palmitoyl acyltransferases known to act on PSD-95 (Fig. 7A,B), and overexpression of ZDHHC8 rescues significant amounts of PSD-95 transport to synapses in the presence of the PKMζ pseudosubstrate, which reduces, but probably does not completely block, PKMζ activity when electroporated in vivo (Fig. 7C–E). One possibility is that phosphorylation of ZDHHC8 directly regulates its enzymatic function of palmitoylation through a mechanism such as conformational change. However, phosphorylation may also regulate intracellular localization or stability of ZDHHC8.
Several reports indicate that one mechanism through which PKMζ might work to maintain LTP and memory is to block the endocytosis of the AMPAR subunit GluR2 by interfering with N-ethylmaleimide-sensitive factor-mediated GluR2 endocytosis (Yao et al., 2008; Migues et al., 2010). GluR2 is also palmitoylated by GODZ (Golgi apparatus-specific protein with a DHHC zinc finger domain; or ZDHHC3) (Hayashi et al., 2005). However, palmitoylation of GluR2 was unchanged between control and TrkB-blocked samples (data not shown), indicating differential regulations of these two palmitoylated synaptic proteins.
Palmitoylation and vesicular transport: two critical components of PSD-95 trafficking
Neurotrophins and protein palmitoylation interact in key ways with diverse membrane-associated proteins and regulate their subcellular localizations and functions throughout the brain. ZDHHC8 has been shown to be localized in the Golgi apparatus and to palmitoylate PSD-95 (Mukai et al., 2008).
We previously used fluorescent recovery after photobleaching and in vitro immunocytochemistry to show that the PI3K–Akt pathway is required for the NMDAR driven, BDNF-induced trafficking of PSD-95 from visual cortical neuron somata to dendrites. The same report showed that TrkB signaling facilitates PSD-95 transport from the ER to the Golgi apparatus (Yoshii and Constantine-Paton, 2007). Our current work shows that EO-induced increase in visual activity, also operating through BDNF/TrkB signaling, upregulates PLCγ signaling to induce ZDHHC8-dependent palmitoylation of PSD-95. Thus BDNF/TrkB signaling drives two synergistic pathways in parallel: (1) the PI3K–Akt pathway triggers PSD-95 vesicular transport systems in cell bodies and dendrites to bring the scaffold to the synapse; (2) the PLC pathway drives palmitoylation of PSD-95, allowing the scaffold synthesized in cytoplasm to attach to plasma membranes and travel via the dendritic microtubule transport system as we have shown previously (Yoshii and Constantine-Paton, 2007). TrkB-induced increases in PSD-95 at synapses (Yoshii et al., 2003; Ji et al., 2005; Yoshii and Constantine-Paton, 2007) and the subsequent synaptic signaling mediated through this scaffold (Ehrlich et al., 2007; Yoshii and Constantine-Paton, 2007) are known to increase stabilization of young synapses, a precondition for many of the events involved in neural circuit maturation (McAllister et al., 1999; Poo, 2001; Huang and Reichardt, 2003; Minichiello, 2009; Yoshii and Constantine-Paton, 2010).
Synaptic trafficking of PSD-95 is regulated by both TrkB signaling and PKMζ
PLCγ-binding sites on TrkB have been shown to be crucial for maintaining late LTP (Minichiello et al., 2002) and also limbic epileptogenesis (He et al., 2010). It is widely accepted that PLCγ activates PKC and that PKMζ is a brain-specific PKC variant. In fact, BDNF has been shown to upregulate the expression and, because PKMζ is a constitutively active enzyme, the activity of PKMζ (Adasme et al., 2011). Furthermore, both TrkB and PKMζ are crucial for the maintenance of late-phase LTP, consolidation of long-term memory (Pastalkova et al., 2006; Minichiello, 2009), synaptic tagging (Barco et al., 2005; Sajikumar et al., 2005), and synaptic localization of PSD-95 (Yoshii and Constantine-Paton, 2007; Liu et al., 2009).
Conclusion
We have documented a new mechanism for trafficking synaptic molecules in vivo that is triggered by the developmental milestone of eye opening; namely, TrkB and PKMζ play significant, if not critical roles in the transport of PSD-95 to synapses after eye opening in layer 2/3 pyramidal neurons of mouse visual cortex. This mechanism involves the PSD-95 palmitoylation enzyme ZDHHC8, which is thus a prime candidate for effecting the critical palmitoylation known to bring PSD-95 to synapses. The same or a similar mechanism is likely to play a role in various forms of learning and memory as well as the developmental tuning of circuitry as a result of environmentally induced activity.
Perhaps most significantly, the knowledge of a neurotrophin-pathway-dependent palmitoylation of PSD-95 can provide insights and eventual treatments for devastating developmental and psychiatric diseases such as autism spectrum disorders and schizophrenia, to name a few (Mukai et al., 2008; Fukata and Fukata, 2010; Yoshii and Constantine-Paton, 2010). This TrkB-triggered pathway is also involved in learning and memory, and also may be critical to the maintenance of memory and cognition in the aging brain (Shema et al., 2011).
Footnotes
The authors declare no competing financial interests.
This work was supported by NIH Grant R01EY006039-27 (M.C.P.), Department of Defense Grant W81XWH-09-1-0088 (A.Y.), and NIH Grant R01EB001987 (K.M.S., C.Z.). We thank Dr. David D. Ginty (Johns Hopkins University, Baltimore) for alerting us to and providing the TrkBF616A mice; Drs. Masaki and Yuko Fukata (National Institute for Physiological Sciences, Okazaki, Aichi, Japan) for providing HA-tagged ZDHHC constructs; Dr. Miguel Bosch (Massachusetts Institute of Technology, Cambridge, MA) for providing the CAG promoter-driven PSD-95-GFP construct; and Dr. Johannes Hell (University of California, Davis, Davis, CA) for providing the anti-SAP102 antibody. We thank Dr. Charles Jennings (McGovern Institute, Massachusetts Institute of Technology) for constructive feedback concerning this manuscript and Dr. H. Robert Horvitz for proofreading.
References
- Adasme T, Haeger P, Paula-Lima AC, Espinoza I, Casas-Alarcon MM, Carrasco MA, Hidalgo C. Involvement of ryanodine receptors in neurotrophin-induced hippocampal synaptic plasticity and spatial memory formation. Proc Natl Acad Sci U S A. 2011;108:3029–3034. doi: 10.1073/pnas.1013580108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso M, Bekinschtein P, Cammarota M, Vianna MR, Izquierdo I, Medina JH. Endogenous BDNF is required for long-term memory formation in the rat parietal cortex. Learn Mem. 2005;12:504–510. doi: 10.1101/lm.27305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barco A, Patterson S, Alarcon JM, Gromova P, Mata-Roig M, Morozov A, Kandel ER. Gene expression profiling of facilitated L-LTP in VP16-CREB mice reveals that BDNF is critical for the maintenance of LTP and its synaptic capture. Neuron. 2005;48:123–137. doi: 10.1016/j.neuron.2005.09.005. [DOI] [PubMed] [Google Scholar]
- Beique JC, Andrade R. PSD-95 regulates synaptic transmission and plasticity in rat cerebral cortex. J Physiol. 2003;546:859–867. doi: 10.1113/jphysiol.2002.031369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beique JC, Lin DT, Kang MG, Aizawa H, Takamiya K, Huganir RL. Synapse-specific regulation of AMPA receptor function by PSD-95. Proc Natl Acad Sci U S A. 2006;103:19535–19540. doi: 10.1073/pnas.0608492103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop AC, Kung CY, Shah K, Witucki L, Shokat KM, Liu Y. Generation of monospecific nanomolar tyrosine kinase inhibitors via a chemical genetic approach. J Am Chem Soc. 1999;121:627–631. [Google Scholar]
- Bracken BK, Turrigiano GG. Experience-dependent regulation of TrkB isoforms in rodent visual cortex. Dev Neurobiol. 2009;69:267–278. doi: 10.1002/dneu.20701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabelli RJ, Hohn A, Shatz CJ. Inhibition of ocular dominance column formation by infusion of NT-4/5 or BDNF. Science. 1995;267:1662–1666. doi: 10.1126/science.7886458. [DOI] [PubMed] [Google Scholar]
- Castren E, Zafra F, Thoenen H, Lindholm D. Light regulates expression of brain-derived neurotrophic factor mRNA in rat visual cortex. Proc Natl Acad Sci U S A. 1992;89:9444–9448. doi: 10.1073/pnas.89.20.9444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarthy S, Saiepour MH, Bence M, Perry S, Hartman R, Couey JJ, Mansvelder HD, Levelt CN. Postsynaptic TrkB signaling has distinct roles in spine maintenance in adult visual cortex and hippocampus. Proc Natl Acad Sci U S A. 2006;103:1071–1076. doi: 10.1073/pnas.0506305103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Ye H, Kuruvilla R, Ramanan N, Scangos KW, Zhang C, Johnson NM, England PM, Shokat KM, Ginty DD. A chemical-genetic approach to studying neurotrophin signaling. Neuron. 2005;46:13–21. doi: 10.1016/j.neuron.2005.03.009. [DOI] [PubMed] [Google Scholar]
- Cooke SF, Bear MF. Visual experience induces long-term potentiation in the primary visual cortex. J Neurosci. 2010;30:16304–16313. doi: 10.1523/JNEUROSCI.4333-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich I, Malinow R. Postsynaptic density 95 controls AMPA receptor incorporation during long-term potentiation and experience-driven synaptic plasticity. J Neurosci. 2004;24:916–927. doi: 10.1523/JNEUROSCI.4733-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich I, Klein M, Rumpel S, Malinow R. PSD-95 is required for activity-driven synapse stabilization. Proc Natl Acad Sci U S A. 2007;104:4176–4181. doi: 10.1073/pnas.0609307104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Husseini AE, Schnell E, Chetkovich DM, Nicoll RA, Bredt DS. PSD-95 involvement in maturation of excitatory synapses. Science. 2000a;290:1364–1368. [PubMed] [Google Scholar]
- El-Husseini AE, Craven SE, Chetkovich DM, Firestein BL, Schnell E, Aoki C, Bredt DS. Dual palmitoylation of PSD-95 mediates its vesiculotubular sorting, postsynaptic targeting, and ion channel clustering. J Cell Biol. 2000b;148:159–172. doi: 10.1083/jcb.148.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Husseini AE, Schnell E, Dakoji S, Sweeney N, Zhou Q, Prange O, Gauthier-Campbell C, Aguilera-Moreno A, Nicoll RA, Bredt DS. Synaptic strength regulated by palmitate cycling on PSD-95. Cell. 2002;108:849–863. doi: 10.1016/s0092-8674(02)00683-9. [DOI] [PubMed] [Google Scholar]
- Elias GM, Funke L, Stein V, Grant SG, Bredt DS, Nicoll RA. Synapse-specific and developmentally regulated targeting of AMPA receptors by a family of MAGUK scaffolding proteins. Neuron. 2006;52:307–320. doi: 10.1016/j.neuron.2006.09.012. [DOI] [PubMed] [Google Scholar]
- Figurov A, Pozzo-Miller LD, Olafsson P, Wang T, Lu B. Regulation of synaptic responses to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature. 1996;381:706–709. doi: 10.1038/381706a0. [DOI] [PubMed] [Google Scholar]
- Fukata M, Fukata Y, Adesnik H, Nicoll RA, Bredt DS. Identification of PSD-95 palmitoylating enzymes. Neuron. 2004;44:987–996. doi: 10.1016/j.neuron.2004.12.005. [DOI] [PubMed] [Google Scholar]
- Fukata Y, Fukata M. Protein palmitoylation in neuronal development and synaptic plasticity. Nat Rev Neurosci. 2010;11:161–175. doi: 10.1038/nrn2788. [DOI] [PubMed] [Google Scholar]
- Gomes RA, Hampton C, El-Sabeawy F, Sabo SL, McAllister AK. The dynamic distribution of TrkB receptors before, during, and after synapse formation between cortical neurons. J Neurosci. 2006;26:11487–11500. doi: 10.1523/JNEUROSCI.2364-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Rumbaugh G, Huganir RL. Differential regulation of AMPA receptor subunit trafficking by palmitoylation of two distinct sites. Neuron. 2005;47:709–723. doi: 10.1016/j.neuron.2005.06.035. [DOI] [PubMed] [Google Scholar]
- He XP, Pan E, Sciarretta C, Minichiello L, McNamara JO. Disruption of TrkB-mediated phospholipase Cgamma signaling inhibits limbic epileptogenesis. J Neurosci. 2010;30:6188–6196. doi: 10.1523/JNEUROSCI.5821-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heimel JA, Saiepour MH, Chakravarthy S, Hermans JM, Levelt CN. Contrast gain control and cortical TrkB signaling shape visual acuity. Nat Neurosci. 2010;13:642–648. doi: 10.1038/nn.2534. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem. 2003;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- Huang K, Yanai A, Kang R, Arstikaitis P, Singaraja RR, Metzler M, Mullard A, Haigh B, Gauthier-Campbell C, Gutekunst CA, Hayden MR, El-Husseini A. Huntingtin-interacting protein HIP14 is a palmitoyl transferase involved in palmitoylation and trafficking of multiple neuronal proteins. Neuron. 2004;44:977–986. doi: 10.1016/j.neuron.2004.11.027. [DOI] [PubMed] [Google Scholar]
- Huang ZJ, Kirkwood A, Pizzorusso T, Porciatti V, Morales B, Bear MF, Maffei L, Tonegawa S. BDNF regulates the maturation of inhibition and the critical period of plasticity in mouse visual cortex. Cell. 1999;98:739–755. doi: 10.1016/s0092-8674(00)81509-3. [DOI] [PubMed] [Google Scholar]
- Ji Y, Pang PT, Feng L, Lu B. Cyclic AMP controls BDNF-induced TrkB phosphorylation and dendritic spine formation in mature hippocampal neurons. Nat Neurosci. 2005;8:164–172. doi: 10.1038/nn1381. [DOI] [PubMed] [Google Scholar]
- Kaneko M, Hanover JL, England PM, Stryker MP. TrkB kinase is required for recovery, but not loss, of cortical responses following monocular deprivation. Nat Neurosci. 2008;11:497–504. doi: 10.1038/nn2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H, Schuman EM. Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science. 1995;267:1658–1662. doi: 10.1126/science.7886457. [DOI] [PubMed] [Google Scholar]
- Kim E, Sheng M. PDZ domain proteins of synapses. Nat Rev Neurosci. 2004;5:771–781. doi: 10.1038/nrn1517. [DOI] [PubMed] [Google Scholar]
- Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Bonhoeffer T. Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc Natl Acad Sci U S A. 1995;92:8856–8860. doi: 10.1073/pnas.92.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovalchuk Y, Hanse E, Kafitz KW, Konnerth A. Postsynaptic induction of BDNF-mediated long-term potentiation. Science. 2002;295:1729–1734. doi: 10.1126/science.1067766. [DOI] [PubMed] [Google Scholar]
- Liu XF, Tari PK, Haas K. PKM zeta restricts dendritic arbor growth by filopodial and branch stabilization within the intact and awake developing brain. J Neurosci. 2009;29:12229–12235. doi: 10.1523/JNEUROSCI.2842-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Constantine-Paton M. Eye opening rapidly induces synaptic potentiation and refinement. Neuron. 2004;43:237–249. doi: 10.1016/j.neuron.2004.06.031. [DOI] [PubMed] [Google Scholar]
- McAllister AK, Katz LC, Lo DC. Neurotrophins and synaptic plasticity. Annu Rev Neurosci. 1999;22:295–318. doi: 10.1146/annurev.neuro.22.1.295. [DOI] [PubMed] [Google Scholar]
- Migaud M, Charlesworth P, Dempster M, Webster LC, Watabe AM, Makhinson M, He Y, Ramsay MF, Morris RG, Morrison JH, O'Dell TJ, Grant SG. Enhanced long-term potentiation and impaired learning in mice with mutant postsynaptic density-95 protein. Nature. 1998;396:433–439. doi: 10.1038/24790. [DOI] [PubMed] [Google Scholar]
- Migues PV, Hardt O, Wu DC, Gamache K, Sacktor TC, Wang YT, Nader K. PKMzeta maintains memories by regulating GluR2-dependent AMPA receptor trafficking. Nat Neurosci. 2010;13:630–634. doi: 10.1038/nn.2531. [DOI] [PubMed] [Google Scholar]
- Minichiello L. TrkB signalling pathways in LTP and learning. Nat Rev Neurosci. 2009;10:850–860. doi: 10.1038/nrn2738. [DOI] [PubMed] [Google Scholar]
- Minichiello L, Calella AM, Medina DL, Bonhoeffer T, Klein R, Korte M. Mechanism of TrkB-mediated hippocampal long-term potentiation. Neuron. 2002;36:121–137. doi: 10.1016/s0896-6273(02)00942-x. [DOI] [PubMed] [Google Scholar]
- Mukai J, Dhilla A, Drew LJ, Stark KL, Cao L, MacDermott AB, Karayiorgou M, Gogos JA. Palmitoylation-dependent neurodevelopmental deficits in a mouse model of 22q11 microdeletion. Nat Neurosci. 2008;11:1302–1310. doi: 10.1038/nn.2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noritake J, Fukata Y, Iwanaga T, Hosomi N, Tsutsumi R, Matsuda N, Tani H, Iwanari H, Mochizuki Y, Kodama T, Matsuura Y, Bredt DS, Hamakubo T, Fukata M. Mobile DHHC palmitoylating enzyme mediates activity-sensitive synaptic targeting of PSD-95. J Cell Biol. 2009;186:147–160. doi: 10.1083/jcb.200903101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastalkova E, Serrano P, Pinkhasova D, Wallace E, Fenton AA, Sacktor TC. Storage of spatial information by the maintenance mechanism of LTP. Science. 2006;313:1141–1144. doi: 10.1126/science.1128657. [DOI] [PubMed] [Google Scholar]
- Patterson SL, Abel T, Deuel TA, Martin KC, Rose JC, Kandel ER. Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron. 1996;16:1137–1145. doi: 10.1016/s0896-6273(00)80140-3. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. San Diego: Academic; 2001. The mouse brain in stereotaxic coordinates. [Google Scholar]
- Poo MM. Neurotrophins as synaptic modulators. Nat Rev Neurosci. 2001;2:24–32. doi: 10.1038/35049004. [DOI] [PubMed] [Google Scholar]
- Sajikumar S, Navakkode S, Sacktor TC, Frey JU. Synaptic tagging and cross-tagging: the role of protein kinase Mzeta in maintaining long-term potentiation but not long-term depression. J Neurosci. 2005;25:5750–5756. doi: 10.1523/JNEUROSCI.1104-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shema R, Sacktor TC, Dudai Y. Rapid erasure of long-term memory associations in the cortex by an inhibitor of PKM zeta. Science. 2007;317:951–953. doi: 10.1126/science.1144334. [DOI] [PubMed] [Google Scholar]
- Shema R, Haramati S, Ron S, Hazvi S, Chen A, Sacktor TC, Dudai Y. Enhancement of consolidated long-term memory by overexpression of protein kinase Mzeta in the neocortex. Science. 2011;331:1207–1210. doi: 10.1126/science.1200215. [DOI] [PubMed] [Google Scholar]
- Simon DK, Prusky GT, O'Leary DD, Constantine-Paton M. N-methyl-D-aspartate receptor antagonists disrupt the formation of a mammalian neural map. Proc Natl Acad Sci U S A. 1992;15:10593–10597. doi: 10.1073/pnas.89.22.10593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein V, House DR, Bredt DS, Nicoll RA. Postsynaptic density-95 mimics and occludes hippocampal long-term potentiation and enhances long-term depression. J Neurosci. 2003;23:5503–5506. doi: 10.1523/JNEUROSCI.23-13-05503.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szymczak AL, Workman CJ, Wang Y, Vignali KM, Dilioglou S, Vanin EF, Vignali DA. Correction of multi-gene deficiency in vivo using a single “self-cleaving” 2A peptide-based retroviral vector. Nat Biotechnol. 2004;22:589–594. doi: 10.1038/nbt957. [DOI] [PubMed] [Google Scholar]
- Tanaka J, Horiike Y, Matsuzaki M, Miyazaki T, Ellis-Davies GC, Kasai H. Protein synthesis and neurotrophin-dependent structural plasticity of single dendritic spines. Science. 2008;319:1683–1687. doi: 10.1126/science.1152864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topinka JR, Bredt DS. N-terminal palmitoylation of PSD-95 regulates association with cell membranes and interaction with K+ channel Kv1.4. Neuron. 1998;20:125–134. doi: 10.1016/s0896-6273(00)80440-7. [DOI] [PubMed] [Google Scholar]
- Trichas G, Begbie J, Srinivas S. Use of the viral 2A peptide for bicistronic expression in transgenic mice. BMC Biol. 2008;6:40. doi: 10.1186/1741-7007-6-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler WJ, Pozzo-Miller L. Miniature synaptic transmission and BDNF modulate dendritic spine growth and form in rat CA1 neurones. J Physiol. 2003;553:497–509. doi: 10.1113/jphysiol.2003.052639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Zundert B, Yoshii A, Constantine-Paton M. Receptor compartmentalization and trafficking at glutamate synapses: a developmental proposal. Trends Neurosci. 2004;27:428–437. doi: 10.1016/j.tins.2004.05.010. [DOI] [PubMed] [Google Scholar]
- Viegi A, Cotrufo T, Berardi N, Mascia L, Maffei L. Effects of dark rearing on phosphorylation of neurotrophin Trk receptors. Eur J Neurosci. 2002;16:1925–1930. doi: 10.1046/j.1460-9568.2002.02270.x. [DOI] [PubMed] [Google Scholar]
- Xu B, Zang K, Ruff NL, Zhang YA, McConnell SK, Stryker MP, Reichardt LF. Cortical degeneration in the absence of neurotrophin signaling: dendritic retraction and neuronal loss after removal of the receptor TrkB. Neuron. 2000;26:233–245. doi: 10.1016/s0896-6273(00)81153-8. [DOI] [PubMed] [Google Scholar]
- Yao Y, Kelly MT, Sajikumar S, Serrano P, Tian D, Bergold PJ, Frey JU, Sacktor TC. PKM zeta maintains late long-term potentiation by N-ethylmaleimide-sensitive factor/GluR2-dependent trafficking of postsynaptic AMPA receptors. J Neurosci. 2008;28:7820–7827. doi: 10.1523/JNEUROSCI.0223-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshii A, Constantine-Paton M. BDNF induces transport of PSD-95 to dendrites through PI3K-AKT signaling after NMDA receptor activation. Nat Neurosci. 2007;10:702–711. doi: 10.1038/nn1903. [DOI] [PubMed] [Google Scholar]
- Yoshii A, Constantine-Paton M. Postsynaptic BDNF-TrkB signaling in synapse maturation, plasticity, and disease. Dev Neurobiol. 2010;70:737–750. doi: 10.1002/dneu.20765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshii A, Sheng MH, Constantine-Paton M. Eye opening induces a rapid dendritic localization of PSD-95 in central visual neurons. Proc Natl Acad Sci U S A. 2003;100:1334–1339. doi: 10.1073/pnas.0335785100. [DOI] [PMC free article] [PubMed] [Google Scholar]