

Abstract
Parvalbumin-positive (PV+) fast-spiking basket cells are thought to play key roles in network functions related to precise time keeping during behaviorally relevant hippocampal synchronous oscillations. Although they express relatively few receptors for neuromodulators, the highly abundant and functionally important neuropeptide cholecystokinin (CCK) is able to selectively depolarize PV+ basket cells, making these cells sensitive biosensors for CCK. However, the molecular mechanisms underlying the CCK-induced selective and powerful excitation of PV+ basket cells are not understood. We used single and paired patch-clamp recordings in acute rat hippocampal slices, in combination with post hoc identification of the recorded interneurons, to demonstrate that CCK acts via G-protein-coupled CCK2 receptors to engage sharply divergent intracellular pathways to exert its cell-type-selective effects. In contrast to CCK2 receptors on pyramidal cells that signal through the canonical Gq–PLC pathway to trigger endocannabinoid-mediated signaling events, CCK2 receptors on neighboring PV+ basket cells couple to an unusual, pertussis-toxin-sensitive pathway. The latter pathway involves ryanodine receptors on intracellular calcium stores that ultimately activate a nonselective cationic conductance to depolarize PV+ basket cells. CCK has highly cell-type-selective effects even within the PV+ cell population, as the PV+ dendrite-targeting bistratified cells do not respond to CCK. Together, these results demonstrate that an abundant ligand such as CCK can signal through the same receptor in different neurons to use cell-type-selective signaling pathways to provide divergence and specificity to its effects.
Introduction
Cholecystokinin (CCK), a peptide initially discovered in the gastrointestinal tract, is one of the most widely expressed neuropeptides in the brain that is present at especially high levels in the hippocampus (Beinfeld et al., 1981; Crawley, 1985). Two classes of G-protein-coupled receptors mediate the actions of CCK, with CCK2 receptors representing the predominant subtype found in the brain and CCK1 receptors being expressed mainly in the gut and a few discrete brain regions (Innis and Snyder, 1980; Sankaran et al., 1980; Honda et al., 1993). In general agreement with their abundance and wide expression, CCK and CCK2 receptors have been linked to a variety of functions, including learning and memory and feeding and nociception, and they have also been implicated in several neurological and psychiatric disorders including anxiety, panic attacks, schizophrenia, and epilepsy (Noble and Roques, 2006; Lee and Soltesz, 2011a).
The cellular targets and molecular mechanisms underlying the CCK effects are beginning to be identified, including the CCK-mediated control of the perisomatically targeting GABAergic basket cells in the hippocampus. The basket cells, characterized by their strategic innervation of the somata and proximal dendrites of postsynaptic neurons, play a critical role in regulating the output and synchrony of large neuronal populations (Freund, 2003; Freund and Katona, 2007). Basket cells can be divided into two functionally distinct groups, the CCK-expressing (CCK+) and the parvalbumin-expressing (PV+) basket cells. PV+ basket cells are fast-spiking and nonaccommodating in their spiking properties, have fast membrane time constants, and release GABA synchronously, making them highly suitable for generating precisely timed network oscillations. In contrast, CCK+ basket cells fire at lower frequencies, are accommodating in their firing, show slower membrane time constants and asynchronous GABA release, and are influenced by a variety of neuromodulators. Thus, basket cells contribute distinct, cell-type-specific functions related to the timing and plasticity of network activity (Hefft and Jonas, 2005; Glickfeld and Scanziani, 2006; Soltesz, 2006).
CCK has been shown to act as a critical, cell-type-selective modulator of these two basket cell classes. In particular, CCK suppresses GABA transmission from CCK+ basket cells onto CA1 pyramidal cells, but it strongly increases the output of PV+ basket cells through a powerful depolarizing action (Földy et al., 2007). The mechanism underlying the actions of CCK on CCK+ basket cell to pyramidal cell transmission has been shown to involve retrograde cannabinoid signaling and presynaptic cannabinoid type 1 receptors (CB1Rs) (Földy et al., 2007; Lee and Soltesz, 2011b), but the mechanisms responsible for the potent CCK-mediated depolarization of PV+ basket cells are not known. Our data in this study show, for the first time, that although the same type of CCK receptor (the CCK2 receptor) is involved in the opposing actions of CCK on the two basket cell types, the CCK receptor is differentially coupled to distinct G-protein-mediated signaling pathways to exert its contrasting, cell-type-selective effects on CCK+ versus PV+ basket cell functions. These results demonstrate a novel mechanism that enables an abundant neuropeptide to achieve the seemingly contradictory dual goals of diversity and specificity of its actions within the hippocampal neuronal network.
Materials and Methods
All protocols were approved by the Institutional Animal Care and Use Committee of the University of California, Irvine.
Electrophysiology.
Transverse entorhinal–hippocampal slices (350 μm) were prepared from P16–P22 Sprague–Dawley rats of either sex. Slices were incubated for 1 h at 33°C in sucrose-containing artificial cerebrospinal fluid (ACSF; 85 mm NaCl, 75 mm sucrose, 2.5 mm KCl, 25 mm glucose, 1.25 mm NaH2PO4, 4 mm MgCl2, 0.5 mm CaCl2, and 24 mm NaHCO3), then transferred to an oxygenated standard ACSF solution (126 mm NaCl, 2.5 mm KCl, 26 mm NaHCO3, 2 mm CaCl2, 2 mm MgCl2, 1.25 mm NaH2PO4, and 10 mm glucose) for electrophysiological recordings performed at 33°C. Slices were visualized with an upright microscope (BX-50, Olympus) with infrared differential interference contrast (IR-DIC) optics. Recordings were made using MultiClamp700A amplifier (Molecular Devices). Signals were filtered at 4 kHz using a Bessel filter and digitized at 10 kHz with a Digidata 1320A analog–digital interface (Molecular Devices). Series resistances were carefully monitored and the recordings were discarded if the series resistance changed significantly or reached 20 MΩ. Drugs were purchased from either Tocris Bioscience or Sigma and were dissolved according to manufacturers' instructions. CCK-8s, the sulfated eight-amino acid C-terminal fragment of CCK, was used in all experiments, as it is the predominant form of CCK in the brain (Dockray, 1980).
PV+ basket cells or PV+ bistratified cells under voltage clamp at −60 mV were recorded with the following internal solution: 40 mm CsCl, 90 mm K-gluconate, 1.8 mm NaCl, 1.7 mm MgCl2, 3.5 mm KCl, 0.05 mm EGTA, 10 mm HEPES, 2 mm MgATP, 0.4 mm Na2GTP, 10 mm phosphocreatine, and 0.2% biocytin (pH 7.2, 270–290 mOsm). In experiments with GDP-β-S, the Na2GTP was omitted from the internal solution (note that control experiments were performed to ensure that the longer recording time alone did not affect the CCK-induced current; data not shown). Summary graphs are presented with the plots normalized to the baseline current before CCK application, and drug effects are compared with the current plots aligned to the onset of the peak holding current. The I/V curve of the CCK-induced current was measured with a ramp protocol ranging from −100 mV to +20 mV applied at a rate of 0.05 mV/ms. Experiments for the I/V curve were conducted in the presence of TTX (1 μm) and cadmium (100 μ M) to reduce any confounding effects from the opening of voltage-gated sodium and calcium channels. The CCK-induced current was isolated by subtracting the I/V curve under control conditions from the peak current seen in the presence of CCK.
For paired recordings, whole-cell recordings were obtained from presynaptic CCK+ basket cells with internal solution containing 126 mm K-gluconate, 4 mm KCl, 10 mm HEPES, 4 mm MgATP, 0.3 mm GTP-Na, 10 mm phosphocreatine, and 0.2% biocytin (pH 7.2, 270–290 mOsm). The synaptically coupled postsynaptic cells were recorded under voltage clamp, with internal solution containing 40 mm CsCl, 90 mm K-gluconate, 1.8 mm NaCl, 1.7 mm MgCl2, 3.5 mm KCl, 0.05 mm EGTA, 10 mm HEPES, 2 mm MgATP, 0.4 mm Na2GTP, 10 mm phosphocreatine, and 0.2% biocytin (pH 7.2, 270–290 mOsm). Presynaptic interneurons were held under current clamp at −60 mV and stimulated at 10 Hz, as this frequency resulted in stable responses over time and allowed for a sufficient number of events for reliable analysis (see Földy et al., 2006). Effective unitary IPSCs (euIPSCs; events including both successes and failures) were individually inspected and included in analysis based on their onset latency (for details, see Neu et al., 2007) after the presynaptic action potential. Because CCK actions on GABAergic transmission are powerful but transient (Deng and Lei, 2006; Földy et al., 2007) (note that the transiency is most likely due to receptor desensitization that was not studied here), quantification of CCK effects during the paired recordings was performed at three distinct time points (as in Földy et al., 2007; Lee and Soltesz, 2011b): (1) immediately before CCK application (referred to as “control” in the text and Figs. 2 and 3); (2) at the time of the maximal effect during CCK application (“peak”; typically observed ∼3–4 min after the starting of the CCK application, with the peak effect lasting up to 1 min); and (3) >6 min after the start of the CCK application, past the initial “peak” CCK effects (“postpeak”).
Figure 2.
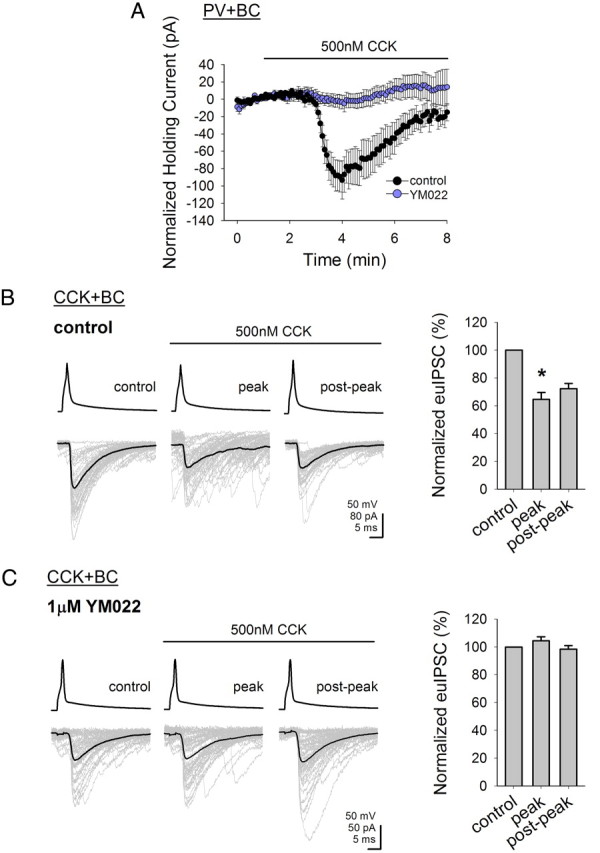
Effects of CCK on both PV+ basket cells and on the CCK+ basket cell to pyramidal cell transmission are mediated by CCK2 receptors. A, Summary graph showing the effect of CCK (500 nm) on the holding current of PV+ basket cells; the CCK-induced shift in holding current can be blocked by application of YM022 (1 μm), a selective CCK2 receptor antagonist (Vholding = −60 mV). B, CCK (500 nm) significantly suppressed the amplitudes of the euIPSC in the pyramidal cell evoked by the presynaptic CCK+ basket cell. Individual IPSCs are shown in gray and the average is shown in black; summary data are shown on the right. C, CCK-induced suppression of the evoked euIPSCs is blocked by YM022; summary data are shown on the right. Error bars represent SEM, and the asterisks denote significance at p < 0.05.
Figure 3.
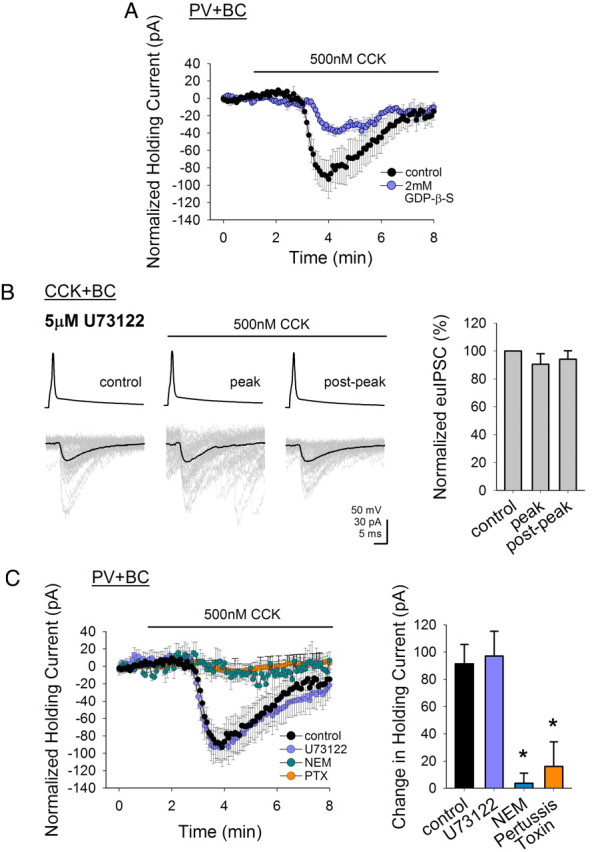
CCK2 receptors couple to different signaling pathways depending on cell type. A, GDP-β-S included in the recording pipette significantly reduces the CCK-induced shift in the holding current of PV+ basket cells. B, Blockade of phospholipase C by U73122 abolishes the CCK-induced reduction of the euIPSCs by the presynaptic CCK+ basket cell. Individual IPSCs are shown in gray and the average is shown in black; summary data are shown on the right. C, U73122 does not abolish the CCK-induced effects in the holding current of PV+ basket cells. N-ethylmaleimide (NEM) or Pertussis toxin (PTX), both blockers of Gi/o-protein-coupled receptors, prevents the CCK-mediated shift in holding current in PV+ basket cells (Vholding = −60 mV). Error bars represent SEM, and the asterisks denote significance at p < 0.05.
Recordings were analyzed using Clampfit software (Molecular Devices), MiniAnalysis (Synaptosoft), and SigmaPlot (Systat Software). Data are presented as mean ± SEM, and statistical significance of data was determined using a one-way ANOVA followed by a post hoc Holm–Sidak test, with a level of significance of p < 0.05.
Calcium imaging.
Simultaneous whole-cell voltage-clamp recording (as described above) and intracellular calcium imaging were performed on PV+ basket cells, using an electrophysiological setup equipped with an Olympus BX61WI microscope and a spinning disk confocal system (CSU 22, Yokogawa) coupled with an electron multiplying charge-coupled device camera (iXon+, Andor Technology). For intracellular calcium imaging, 50 μm Oregon Green 488 BAPTA-1 (OGB; Invitrogen) was added to the recording internal solution and the dye was excited at 488 nm. Four frames were averaged during acquisition, resulting in a final frame rate of ∼25 Hz. Images were analyzed in ImageJ (NIH; Abramoff et al., 2004). ΔF/F represents the increase in fluorescence at the time of the peak CCK effect (as determined with the simultaneous electrophysiological recording) over baseline intensity.
Immunochemistry and neuroanatomy.
Only cells fulfilling both immunocytochemical and morphological criteria were used for data analysis. After recording, slices were transferred into a fixative solution containing 4% paraformaldehyde and 0.2% picric acid in 0.1 m phosphate buffer. Slices were resectioned into 50-μm-thin sections and immunoreactivity for PV was tested with a rabbit polyclonal antibody (PV-28, Swant; diluted 1:1000 in Tris-buffered saline containing 2% normal goat serum and 0.5% Triton-X); immunoreactivity for CCK was revealed with a mouse monoclonal antibody (mAb 9303, generously provided by the CURE Digestive Diseases Research Center, Antibody RIA Core, Los Angeles; NIH Grant DK41301; diluted 1:1000). The reactions were visualized with a goat anti-rabbit IgG conjugated to Alexa 488 (diluted 1:500 in Tris-buffered saline containing 2% normal goat serum; Invitrogen) and a goat anti-mouse IgG conjugated to Alexa 594 (diluted 1:500), streptavidin conjugated to Alexa-350 for biocytin (diluted 1:500). The sections were then mounted in Vectashield (Vector Laboratories) and analyzed with a fluorescent microscope. To reveal the axonal and dendritic arbors of the interneurons in detail, the biocytin-filled cells were subsequently visualized with 3,3′-diaminobenzidine tetrahydrochloride (0.015%) using a standard ABC kit (Vector) (as described by Szabadics et al., 2010).
Results
Data for this study were obtained using whole-cell recordings from PV+ basket cells, PV+ bistratified cells, and paired recordings between synaptically coupled CCK+ basket cells and pyramidal cells (see Figs. 1, 6A). The distinct responses to hyperpolarizing and depolarizing current pulses were used as an initial guide to identify the interneuronal subtype. The identity of every interneuron included in this study was confirmed by both immunocytochemistry (expression of PV vs CCK) and axonal morphology (axonal arborization restricted mainly to the stratum pyramidale for PV+ and CCK+ basket cells and axons ramifying predominantly in the stratum oriens and stratum radiatum for the PV+ bistratified cells).
Figure 1.

Identification of recorded perisomatically targeting basket cells in the CA1 region of the hippocampus. A, B, Camera lucida drawings of a representative PV+ (A) and CCK+ (B) basket cell used in this study; the dense axonal arborization is restricted mainly to the stratum pyramidale. Photomicrographs show post hoc immunocytochemistry for the biocytin-filled recorded cell, indicating expression of PV (A) or CCK (B). The current-clamp recording traces demonstrate the distinct responses of the two types of cells in response to hyperpolarizing and depolarizing current steps; note the fast-spiking firing pattern of the PV+ cell versus the spike frequency adaptation in the CCK+ cells. Str. Ori., Stratum oriens; Str. Pyr., stratum pyramidale; Str. Rad., stratum radiatum; Str. L.-M., stratum lacunosum-moleculare.
Figure 6.

CCK does not activate PV+ dendrite-targeting bistratified cells. A, Camera lucida drawings of a representative PV+ dendrite-targeting bistratified cell used in this study; note that the axonal arbor are not restricted to the stratum pyramidale as seen in Figure 1. Instead, the axons sparsely traverse through the stratum pyramidale and extend primarily in the stratum radiatum and stratum oriens. Photomicrographs show post hoc immunocytochemistry for the biocytin-filled recorded cell, indicating expression of PV. The current-clamp recording traces demonstrate the response of the recorded cell in response to hyperpolarizing and depolarizing current steps; the dendrite-targeting cells also show a fast-spiking pattern and demonstrate a slight sag seen during the hyperpolarizing pulse. B, Summary graph demonstrating that application of CCK (500 nm) does not elicit any response in PV+ dendrite-targeting bistratified cells. Error bars represent SEM, and the asterisks denote significance at p < 0.05. Str. Ori, Stratum oriens; Str. Pyr., stratum pyramidale; Str. Rad., stratum radiatum; Str. L.-M., stratum lacunosum-moleculare.
Throughout this study, CCK was applied at one, high concentration of 500 nm to evoke maximal responses to CCK. This also ensured that any differences in signaling pathways would not simply be due to variations in effective agonist concentration, as it has been observed with other G-protein-coupled receptors (GPCRs; see Discussion). Importantly, the opposing effects of CCK on the PV+ basket cells (excitation) and on the CCK+ basket cell to pyramidal cell transmission (inhibition) previously have been shown to be present even at low nanomolar concentrations of CCK (Földy et al., 2007).
CCK acts via the CCK2 receptor on both the PV+ basket cells and the CCK+ basket cell to pyramidal cell transmission
We first sought to definitively identify the CCK receptor subtype responsible for the actions of CCK in both basket cell subtypes. We have previously shown using current-clamp recordings that CCK directly depolarizes PV+ basket cells (Földy et al., 2007). To determine the mechanism responsible for this depolarization, we conducted voltage-clamp recordings of PV+ basket cells at a holding potential of −60 mV. Application of 500 nm CCK induced an inward shift in the holding current in the PV+ basket cells (−91.4 ± 17.4 pA; n = 5) (Fig. 2A). As CCK2 receptors are the prevalent form of CCK receptors in the brain (Innis and Snyder, 1980; Honda et al., 1993), we tested the role of CCK2 receptors in this current with the selective CCK2 receptor antagonist YM022 (Nishida et al., 1994) and found that application of 1 μm YM022 was able to abolish the CCK-induced current (−2.7 ± 7.8 pA, n = 5) (Fig. 2A).
It has been reported that CCK acts in an opposing (compared with the excitation of the PV+ basket cells) manner on CCK+ basket cell to pyramidal cell transmission by depressing GABA release from these cells (Földy et al., 2007). We first confirmed the latter finding with paired recordings between synaptically coupled CCK+ basket cells and pyramidal cells, showing that CCK (500 nm) attenuates the amplitude of the euIPSCs (peak reduction by CCK: −34.7 ± 7.8% compared with the pre-CCK control period; control: −142 ± 7.0 pA, peak: −91.8 ± 7.0 pA, postpeak: −102.7 ± 5.4 pA; n = 3) (Fig. 2B). Next, the same paired recording experiment was conducted in the presence of the CCK2 receptor antagonist YM022 (1 μm), and the data showed that YM022 was also able to block the CCK-induced depression of GABA release from CCK+ basket cells (peak reduction by CCK in YM022: 0% compared with the pre-CCK control period; control: −37.5 ± 11.3 pA, peak: −39.2 ± 10.9 pA, postpeak: −36.9 ± 9.2 pA; n = 5) (Fig. 2C).
These results indicate that the CCK-induced excitatory, depolarizing current in PV+ basket cells and the CCK-induced inhibition of the CCK+ basket cell to pyramidal cell transmission both require the activation of CCK2 receptors.
CCK2 receptor signaling diverges in the control of PV+ versus CCK+ basket cell output
Next, we characterized the downstream pathway following CCK2 receptor activation. CCK2 receptors are classified as GPCRs (Noble and Roques, 1999; Dufresne et al., 2006), and blockade of GPCR activity with guanosine 5′-O-(2-thiodiphosphate) (GDP-β-S), a nonhydrolyzable analog of GDP that prevents GPCR activation, inhibits the CCK-induced depression of GABA release from CCK+ basket cells (Földy et al., 2007). To examine whether the CCK-induced excitation of PV+ basket cells also uses G-protein-dependent signaling, we conducted whole-cell recordings with GDP-β-S (2 mm) dialyzed (15–20 min) into the intracellular pipette. GDP-β-S significantly reduced the CCK-induced inward current in PV+ basket cells compared with control (to −38.2 ± 3.8 pA; n = 5) (Fig. 3A), demonstrating that the depolarizing action of CCK on PV+ basket cells also involves a G-protein-coupled mechanism.
CCK2 receptors are classically thought to be linked to the Gq/11-protein–phospholipase C (PLC) pathway, largely based on studies using expression of CCK2 receptors in various cell lines or immunoblocking studies using anti-PLC or anti-Gq antibodies (Noble and Roques, 1999; Dufresne et al., 2006). The actions of CCK on CCK+ basket cell transmission involve a CB1R-mediated mechanism (Földy et al., 2007), and activation of Gq/11-linked receptors and PLC enhances the synthesis of endocannabinoids (Piomelli, 2003; Hashimotodani et al., 2005; Kano et al., 2009). Therefore, to examine whether activation of PLC played a downstream role after CCK2 receptor activation, we preincubated the hippocampal slices for >1 h with U73122 (5 μm), a PLC inhibitor, to block PLC activity. As expected, U73122 was able to significantly reduce the CCK-induced depression of GABA release from CCK+ basket cells (peak reduction by CCK in U73122: 9.6 ± 7.6% compared with the pre-CCK control period; control: −53.5 ± 16.7 pA, peak: −48.7 ± 14.2 pA, postpeak: −48.1 ± 12.9 pA; n = 4) (Fig. 3B).
Next, we tested whether PLC was also involved in the downstream signaling leading to the CCK-induced effects on PV+ basket cells. Surprisingly, we did not find any significant change in the CCK-induced inward current after blocking PLC, compared with control (in U73122: −97.1 ± 18.4 pA; n = 7) (Fig. 3C), indicating that in the PV+ basket cells, the Gq/11–PLC pathway does not seem to play a role in the CCK-induced effects. Note that in the gray traces shown in the second panel (“peak”) of Figure 3B, application of CCK after U73122 incubation increased the frequency of spontaneous IPSCs in the postsynaptic pyramidal cell. As described by Földy et al. (2007), the CCK-induced increase in the spontaneous IPSCs in the pyramidal cells is due to the CCK-induced increase in the firing of presynaptic firing PV+ basket cells. Therefore, the finding that the CCK-induced increases in spontaneous IPSCs persist in the presence of U73122 while U73122 blocks the CCK-induced effects on the CCK+ basket cell to pyramidal cell transmission is in agreement with the hypothesis that the mechanisms underlying CCK actions involving CCK2 receptors diverge in PV+ basket cells and pyramidal cells.
Agonist-binding and behavioral studies (Scemama et al., 1988; Knapp et al., 1990; Derrien et al., 1994; Lodge et al., 2003) have suggested that there can actually be a heterogeneity in CCK2 receptor signaling. More recent studies have confirmed these initial observations, showing that the CCK2 receptors can also couple to Gi/o proteins, at least in expression systems and in the spinal cord (Pommier et al., 1999, 2003; Oz et al., 2007). Therefore, we tested the possibility that in the PV+ basket cells, CCK2 receptors may in fact be coupling with the less canonical G-protein signaling pathway. Hippocampal slices were preincubated (10–20 min) with 250 μm N-ethylmaleimide (NEM), which alkylates the cysteine residue on Gi/o proteins and thus uncouples the Gi/o proteins from their associated receptors (Shapiro et al., 1994). The presence of NEM significantly reduced the CCK-induced current (in NEM: −3.6 ± 7.5 pA; n = 4) (Fig. 3C), suggesting that Gi/o proteins are involved in the CCK-mediated effects on PV+ basket cells. As NEM can have other nonspecific effects, we also used pertussis toxin (PTX), a more specific blocker of Gi/o proteins that acts by catalyzing the ADP-ribosylation of the α subunit of the G protein (Katada et al., 1982). Preincubation with PTX (>12 h) also significantly reduced the CCK-induced current (in PTX: −16.0 ± 18.1 pA; n = 3) (Fig. 3C), further confirming that in contrast to CCK+ basket cells, a Gi/o-mediated pathway plays a critical role in CCK2 signaling in PV+ basket cells [control experiments were performed to ensure that the long incubation time (required for PTX activity) alone did not affect the CCK-induced current; data not shown]. Thus, these findings show that although CCK activates the CCK2 receptors to exert its effects on both types of basket cells, the downstream intracellular pathways begin to diverge sharply depending on cell type. To depress CCK+ basket cell transmission, CCK2 receptors couple to a Gq/11-protein–PLC to trigger cannabinoid-mediated depression of GABA release, whereas in PV+ basket cells, CCK2 receptors do not use PLC, but instead couple to a pertussis-toxin-sensitive signaling pathway to depolarize the cells.
Intracellular mechanism mediating the CCK-induced depolarization of PV+ basket cells
After establishing the initial steps in the actions of CCK in PV+ basket cells, we then examined the effector ion channel mediating the CCK-induced current. Neither tetrodotoxin (TTX, 1 μm) nor extracellular cesium (2 mm) had any significant effect on the CCK-induced current (CCK current in TTX: −84.6 ± 19.8 pA, n = 3; in cesium: −85.5 ± 7.1 pA, n = 4) (Fig. 4A), indicating that most likely neither voltage-gated sodium channels, h-channels, nor potassium channels are involved.
Figure 4.

Mechanism underlying the depolarizing actions of CCK in PV+ basket cells. A, Blockers of voltage-gated sodium channels, potassium, or h-channels with tetrodotoxin (TTX) or cesium (Cs+) do not block the CCK-induced current in PV+ basket cells. B, Low extracellular calcium greatly potentiates the CCK-induced current, and low extracellular sodium abolishes the current. C, Blockers of TRP channels significantly reduce the CCK-induced current (Vholding = −60 mV). Error bars represent SEM, and the asterisks denote significance at p < 0.05.
To determine which ion was contributing to the CCK-induced inward current, we performed experiments in which Na+ and Ca2+ were substituted or reduced. In the first condition, extracellular Na+ was reduced to 10% of the Na+ concentration (to 15.3 mm) in the control ACSF and substituted with an equimolar amount of choline chloride. In the presence of this low extracellular Na+ concentration, the CCK-induced inward current was abolished to 5.2 ± 7.2 pA (n = 5) (Fig. 4B). Similar results were seen when the extracellular Na+ was substituted with an equimolar amount of N-methyl-d-glucamine, as the CCK-induced current was reduced to −4.9 ± 3.7 pA (n = 5) compared with pre-CCK control. Next, to assess the role of calcium in this current, we used a low (0–0.2 mm) extracellular Ca2+ recording medium by substituting Ca2+ with an equimolar amount of Mg2+ to maintain the divalent cation concentration. We found that the CCK-induced current was greatly increased under these conditions (to −307.2 ± 95.7 pA; n = 4) (Fig. 4B). These ionic properties are most reminiscent of a nonselective cationic channel, where the inward current mediated by extracellular sodium is increased when the larger extracellular Ca2+ ions are removed. A likely candidate for such a channel is the nonselective cation-conducting transient receptor potential (TRP) channels, and indeed, the latter phenomenon involving enhanced responses after the removal of extracellular Ca2+ ions has been observed for CCK-mediated effects in other regions of the brain (Tsujino et al., 2005; Meis et al., 2007; Chung and Moore, 2009).
TRP channels were first identified in Drosophila mutants carrying the trp mutation in photoreceptors that showed transient responses to continuous light (Hardie and Minke, 1992; Niemeyer et al., 1996). TRP channels are also widely expressed in mammalian systems and respond to various stimuli from sensory inputs, hormones, growth factors, and neuromodulators. They play a critical role in regulating Na+ and Ca+ levels in both excitable and nonexcitable cells, and they exhibit a wide variety of activation mechanisms (for review, see Clapham, 2003; Abramowitz and Birnbaumer, 2009; Kiselyov and Patterson, 2009). Although TRP channels generally have been thought to associate with Gq/11-coupled pathways, recent findings (Jeon et al., 2008; Otsuguro et al., 2008; Xiao et al., 2010) indicate that TRP channels can in fact also be activated via pertussis-toxin-sensitive Gi/o-protein pathways.
To assess the involvement of TRP channels in the CCK-induced current, various pharmacological agents were applied during the recordings. The TRP channel blockers used here have nonspecific effects, but they have all blocked TRP-channel-mediated effects in numerous studies looking at TRP channels and are currently the best pharmacological tools available to assess TRP channel activity (Zhang et al., 2008; Qiu et al., 2010). Bath application of 2-APB (100 μm) or flufenamic acid (FFA) was able to significantly attenuate the CCK-induced current (in 2-APB: −17.7 ± 5.1 pA; n = 4; in FFA: −26.3 ± 11.4 pA; n = 4; note that 2-APB can also block IP3 receptors, but, as shown below, IP3 receptors do not appear to be involved in the CCK-induced current) (Fig. 4C).
To further describe this current, we also looked at its I/V characteristics using a ramp protocol ranging from −100 mV to +20 mV (0.05 mV/ms). The CCK-mediated current was isolated by subtracting the currents recorded before from the current recorded during the time of the peak CCK effect. The current showed a characteristic I/V shape, similar to what has been seen with TRPC1/4 and TRPC1/5 subunit heteromers (Strübing et al., 2001) (Fig. 5A), and a clear reversal at −22.1 ± 3.2 mV (n = 5), indicating a mixed-cation conductance.
Figure 5.

Intracellular calcium stores are involved in the induction of the CCK-mediated nonselective cationic current in PV+ basket cells. A, I/V relationship of the CCK-induced current. Inset shows schematic outline of ramp protocol (from −100 mV to 20 mV at 0.05 mV/ms). B, IP3 receptor blockade with heparin has no effect on the CCK-induced current, whereas blocking ryanodine receptors with ryanodine or 8-NH2-cADPR significantly attenuates the CCK-induced current. C, Simultaneous electrophysiological recording and intracellular calcium imaging of a PV+ basket cell filled with the calcium indicator Oregon Green BAPTA (OGB, 50 μm). CCK-induced current (top trace, black) takes place in conjunction with an increase in intracellular calcium, as indicated by the increased fluorescence of the OGB (bottom plot, blue) (Vholding = −60 mV). Error bars represent SEM, and the asterisks denote significance at p < 0.05.
As TRP channel activation has been (albeit somewhat controversially) linked to intracellular calcium store release (Minke and Cook, 2002; Salido et al., 2009), we next investigated the role of the IP3 and ryanodine receptors on the calcium stores. Addition of 2 mg/ml heparin, an IP3 receptor blocker, into the intracellular pipette had no significant effect on the CCK-induced current (in heparin: −83.6 ± 18.7 pA; n = 6) (Fig. 5B), indicating that IP3 receptors are most likely not involved in the CCK-induced current. This further suggests that the inhibitory effect of 2-APB on the CCK-induced current (Fig. 4C) is indeed through the blockade of TRP channels rather than by blocking IP3 receptors. It is also interesting to note that immunocytochemical studies in the rat hippocampus and striatum have shown that IP3 receptors do not colocalize with parvalbumin (Martone et al., 1997; Hertle and Yeckel, 2007).
Ryanodine receptors are also located on the endoplasmic reticulum and are involved in CCK-mediated signaling for the generation of Ca2+ oscillations (Cancela and Petersen, 1998). To test the possibility that it may be the ryanodine receptors playing a role, we tested two blockers of the ryanodine receptors. First, we preincubated (>30 min) the hippocampal slices in 10 μm ryanodine to block ryanodine receptors and found that ryanodine significantly reduced the CCK-induced current (to −26.3 ± 11.9 pA; n = 3) (Fig. 5B). As cyclic ADP-ribose, synthesized by ADP-ribose cyclase, is an endogenous agonist for ryanodine receptor (Galione et al., 1991; Morikawa et al., 2003), we also tested the effect of applying 8-NH2-cADPR, a synthetic analog of cyclic ADP-ribose that acts an antagonist, into the pipette. The latter manipulation also significantly reduced the CCK-induced current (to −28.7 ± 11.6 pA; n = 4) (Fig. 5B). These experiments strongly suggest that perturbation of intracellular calcium stores, specifically through ryanodine receptors, can play a critical role in mediating the CCK-induced current.
To further confirm this hypothesis, we used calcium imaging to directly visualize whether CCK would induce any Ca2+ release from calcium stores in the PV+ basket cells. We added 50 μm of the calcium indicator OGB into the recording pipette and conducted simultaneous voltage-clamp recording and calcium imaging of PV+ basket cells. Application of CCK induced a rise in intracellular calcium (ΔF/F: 11.5 ± 4.2%, n = 4; see Materials and Methods) (Fig. 5C) in the PV+ basket cells, indicating that CCK2 receptor signaling involves the opening of intracellular calcium stores. Together, these experiments identify the downstream intracellular messengers involved in the CCK2-receptor-coupled signaling pathway that mediate the depolarizing effects of CCK in PV+ basket cells.
PV+ dendrite-targeting bistratified cells do not respond to CCK
In addition to the more widely known PV+ basket cells that innervate the perisomatic regions of postsynaptic neurons, the PV+ cell population also contains a subtype of cells whose axons target the dendritic regions of principal cells. This subtype, named the bistratified cells (as its axons mainly project into the stratum radiatum and oriens; Buhl et al., 1994), is also fast spiking and GABAergic, but they display important functional differences as compared with the PV+ basket cells. The bistratified cells, compared with PV+ basket cells, spike at different phases during various network oscillations (Klausberger et al., 2004; Tukker et al., 2007) and show different forms of long-term plasticity (Nissen et al., 2010), indicating their highly distinct contributions to the modulation of network activity.
To test whether the PV+ bistratified cells were also affected by CCK, we performed voltage-clamp recordings from post hoc identified PV+ bistratified cells (Fig. 6A). In contrast to the PV+ basket cells, the PV+ bistratified cells showed minimal response to CCK application (−6.4 ± 9.5 pA; n = 5) (Fig. 6B). Note that CCK did increase spontaneous IPSC frequency onto the recorded PV+ bistratified cells, acting as a positive control for CCK (control: 8.5 ± 2.0 Hz, peak CCK effect: 26 ± 2.4 Hz). These data obtained from the bistratified cells suggest that even within a cell population, CCK can have specialized actions by activating one subgroup but not the other.
Discussion
CCK, a neuropeptide expressed at unusually high levels in the brain, plays key roles in several types of normal and pathological conditions. Despite its abundance, CCK demonstrates exceptionally precise specificity in its effects at the cellular level, most notably in its control of the GABAergic basket cells of the hippocampus, where CCK directly depolarizes the PV+ basket cells while indirectly suppressing GABA release from CCK+ basket cells (Földy et al., 2007). Basket cells innervate the perisomatic region of principal cells, playing a crucial role in shaping the output of principal cells to perform key operations within the network. By its precise, opposing regulation of basket cell activity, CCK acts as a molecular switch to gate one source of functionally distinct inhibition over the other.
However, the mechanism that enables CCK to exert its dual actions in regulating basket cell output has been unclear. The predominant form of CCK receptors in the brain are the G-protein-coupled CCK2 receptors, with the G-protein-coupled CCK1 receptors expressed in select few regions as well (Innis and Snyder, 1980; Sankaran et al., 1980; Van Dijk et al., 1984; Hill et al., 1987; Honda et al., 1993). In the present study, we first show that both the CCK-induced depolarizing current in PV+ basket cells and the CCK-induced suppression of transmission from CCK+ basket cells require CCK2 receptors, and then demonstrate that, surprisingly, CCK2 receptors couple to different intracellular messengers in different cells, inducing the activation of differing pathways to produce divergent effects (summarized in Fig. 7).
Figure 7.

Schematic representation summarizing the cell-type-dependent selectivity of CCK2 receptor signaling. A, CCK signals through the same CCK2 receptor to use divergent signaling pathways and exert opposing effects on hippocampal basket cells, as described in B and C. B, In PV+ basket cells, CCK2 receptors couple to an unusual, pertussis-toxin-sensitive pathway and ryanodine receptors on calcium stores that ultimately activate a nonselective cationic conductance to depolarize the cells. C, CCK2 receptors signal through the more canonical Gq–PLC pathway in pyramidal cells in conjunction with CB1 receptor signaling to regulate CCK+ basket cell transmission.
A known source of complexity of ligand–GPCR signaling is the expression of different receptors for a common ligand within a given cell. In hippocampal oriens/alveus interneurons, for example, application of DHPG can activate both metabotropic glutamate receptor type 1a and type 5 to generate distinct Ca2+-signaling pathways and plasticity mechanisms (Topolnik et al., 2006), and in CA1 pyramidal cells, vasoactive intestinal peptide (VIP) can activate different VIP receptors to regulate synaptic transmission (Yang et al., 2009). However, as our results show that the same CCK receptor subtype mediates the effects both on the PV+ basket cells and on the CCK+ basket cell to pyramidal cell transmission, a different mechanism is likely to be involved that does not evoke the need for the expression of different receptors for a common ligand in these two cell types.
The early traditional views on GPCR signaling have proposed a relatively linear, narrow cascade, where one GPCR would interact with one particular G protein (or G proteins within one family) to exert its effects. CCK2 receptors, for example, generally have been thought to be coupled to a Gq/11-protein-mediated pathway (Noble and Roques, 1999; Dufresne et al., 2006). However, it is increasingly being recognized that a GPCR can, in fact, couple through different G proteins and pathways (Hermans, 2003; Baker and Hill, 2007; Maudsley et al., 2007) to introduce greater functional versatility in their responses. For example, GPCRs can couple to two different pathways depending on the concentration of agonist used, as seen with the gonadotropin-releasing hormone receptor, where it is thought that certain conformations of the receptor, sensitive to different agonist concentrations, couple to multiple signaling pathways to influence GABAergic transmission (Krsmanovic et al., 2003; Chen and Moenter, 2009), and with the V1a vasopressin receptor, where different concentrations of vasopressin can trigger diverse pathways to affect vasoconstriction (Henderson and Byron, 2007). However, because it has been previously shown that even low nanomolar concentrations of CCK can induce the same opposing effects in PV+ basket cells and CCK+ basket cell transmission (Földy et al., 2007), and we have used the same high-saturating concentration of CCK in all of the present experiments, differences in agonist concentration alone cannot explain the cell-type-specific CCK signaling in our study.
Instead, the data in the present paper indicate that CCK2 receptor signaling is segregated according to cell type. The CCK-mediated suppression of CCK+ basket cell transmission involves a CB1R-mediated mechanism (Földy et al., 2007; Lee and Soltesz, 2011b), and, as seen with other GPCR-mediated cannabinoid signaling (Piomelli, 2003; Kano et al., 2009), we show here that this CCK effect also involves the traditional Gq/11–PLC pathway. As the mechanism for the CCK-induced activation of PV+ basket cells was not known, we set to define the signaling pathway underlying the depolarization of the PV+ basket cells. Surprisingly, our data demonstrated that in PV+ basket cells, CCK2 signaling was not coupled to the Gq/11–PLC pathway, but instead, to a pertussis-toxin-sensitive Gi/o-protein pathway. This pathway has been alluded to in earlier studies (Knapp et al., 1990; Pommier et al., 1999, 2003), and recently in spinal cord motorneurons (Oz et al., 2007), but it has never been identified in cortical regions.
Additional experiments aimed at further defining the intracellular cascade indicated that the CCK-induced depolarizing current in PV+ basket cells was most likely mediated by a TRP-like cationic conductance, based on our results with ion substitution experiments, TRP channel blockers, and analysis of the current reversal potential. The activation of the TRP channel was then linked to the involvement of intracellular stores, specifically through the ryanodine receptors, in the PV+ basket cells. Due to the lack of subtype-specific pharmacology, future studies will be necessary for the precise identification of the exact subtype(s) of TRP channels involved in the CCK-induced excitation of PV+ basket cells.
Thus, the present results indicate that CCK2 receptor signaling may help define a new criterion for GPCR signaling selectivity at the cell-type-specific level, where a highly abundant ligand such as CCK can use the same GPCR subtype to distinguish different intracellular pathways, depending on the specific cell type. In light of the fact that CCK is expressed at such high levels (up to microgram quantities) in the brain (Crawley, 1985), this presents an important mechanism for CCK to provide precise regulation of its actions within the network. Interestingly, cell-type specificity has been reported for the CCK-induced enhancement of glutamate release as well (Deng et al., 2010), suggesting that highly specific spatiotemporal patterns of CCK actions may underlie the diversity of behavioral effects of CCK on learning and memory, anxiety, satiety, and nociception (Noble and Roques, 2006; Lee and Soltesz, 2011a).
In addition to PV+ basket cells, PV+ interneurons also consist of a subgroup that targets the dendritic regions of principal cells (Buhl et al., 1994). Therefore, we set out to investigate the role of CCK in modulating PV+ dendrite-targeting bistratified cells as well. Unexpectedly, we found that the PV+ bistratified cells, in fact, showed minimal response to CCK. In vivo data have revealed sharp differences between the spike timing of PV+ basket versus bistratified cells during behaviorally relevant neuronal oscillations (Klausberger et al., 2003, 2004; Tukker et al., 2007), and recent studies also have shown that PV+ basket cells and bistratified cells express different forms of long-term plasticity (Nissen et al., 2010). Therefore, the current data on the differential effects of CCK on the two PV+ cell populations are in general agreement with the latter in vivo and plasticity studies (Klausberger et al., 2003, 2004; Tukker et al., 2007; Nissen et al., 2010) indicating the existence of significant differences in the factors involved in the modulation of perisomatic versus dendritic inhibition provided by fast-spiking cells.
The strict requirement for the identification of cell type by its firing pattern, its immunopositivity for PV or CCK, and further confirmation of its axonal morphology have enabled us to unequivocally distinguish between the different cell-type-specific pathways. The effects of CCK, CCK2 receptors, and CB1 receptors in anxiety and other mood disorders are well known; however, attempts to bring a CCK2 receptor antagonist to therapeutic use have not been successful (Herranz, 2003; Wang et al., 2005; Harro, 2006; Lee and Soltesz, 2011a). PV+ interneurons have been shown to be involved in cognitive functions, deletion of the PV+ interneurons within the network can cause deficits in spatial working memory, and they are damaged in schizophrenia and by various drugs of abuse (Lewis et al., 2005; Behrens et al., 2007; Fuchs et al., 2007; Murray et al., 2011). Therefore, the present results on the mechanisms underlying the excitatory effects of CCK on PV+ cells suggest that selective manipulation of the differing CCK2 receptor signaling pathways may provide a more targeted potential future therapeutic approach for cognitive, mood, and neurological disorders.
Footnotes
This work was supported by National Institutes of Health Grant NS38580 (to I.S.), the American Epilepsy Society (to S.Y.L.), and the George E. Hewitt Foundation for Medical Research (to J.S.). We thank Rose Zhu for excellent technical assistance, and Drs. Lutz Hilgenberg and Martin Smith for discussions.
References
- Abramoff MD, Magelhaes PJ, Ram SJ. Image processing with ImageJ. Biophotonics International. 2004;11:36–42. [Google Scholar]
- Abramowitz J, Birnbaumer L. Physiology and pathophysiology of canonical transient receptor potential channels. FASEB J. 2009;23:297–328. doi: 10.1096/fj.08-119495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker JG, Hill SJ. Multiple GPCR conformations and signalling pathways: implications for antagonist affinity estimates. Trends Pharmacol Sci. 2007;28:374–381. doi: 10.1016/j.tips.2007.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens MM, Ali SS, Dao DN, Lucero J, Shekhtman G, Quick KL, Dugan LL. Ketamine-induced loss of phenotype of fast-spiking interneurons is mediated by NADPH-oxidase. Science. 2007;318:1645–1647. doi: 10.1126/science.1148045. [DOI] [PubMed] [Google Scholar]
- Beinfeld MC, Meyer DK, Eskay RL, Jensen RT, Brownstein MJ. The distribution of cholecystokinin immunoreactivity in the central nervous system of the rat as determined by radioimmunoassay. Brain Res. 1981;212:51–57. doi: 10.1016/0006-8993(81)90031-7. [DOI] [PubMed] [Google Scholar]
- Buhl EH, Halasy K, Somogyi P. Diverse sources of hippocampal unitary inhibitory postsynaptic potentials and the number of synaptic release sites. Nature. 1994;368:823–828. doi: 10.1038/368823a0. [DOI] [PubMed] [Google Scholar]
- Cancela JM, Petersen OH. The cyclic ADP ribose antagonist 8-NH2-cADP-ribose blocks cholecystokinin-evoked cytosolic Ca2+ spiking in pancreatic acinar cells. Pflugers Arch. 1998;435:746–748. doi: 10.1007/s004240050578. [DOI] [PubMed] [Google Scholar]
- Chen P, Moenter SM. GABAergic transmission to gonadotropin-releasing hormone (GnRH) neurons is regulated by GnRH in a concentration-dependent manner engaging multiple signaling pathways. J Neurosci. 2009;29:9809–9818. doi: 10.1523/JNEUROSCI.2509-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung L, Moore SD. Cholecystokinin excites interneurons in rat basolateral amygdala. J Neurophysiol. 2009;102:272–284. doi: 10.1152/jn.90769.2008. [DOI] [PubMed] [Google Scholar]
- Clapham DE. TRP channels as cellular sensors. Nature. 2003;426:517–524. doi: 10.1038/nature02196. [DOI] [PubMed] [Google Scholar]
- Crawley JN. Comparative distribution of cholecystokinin and other neuropeptides. Why is this peptide different from all other peptides? Ann NY Acad Sci. 1985;448:1–8. doi: 10.1111/j.1749-6632.1985.tb29900.x. [DOI] [PubMed] [Google Scholar]
- Deng PY, Lei S. Bidirectional modulation of GABAergic transmission by cholecystokinin in hippocampal dentate gyrus granule cells of juvenile rats. J Physiol. 2006;572:425–442. doi: 10.1113/jphysiol.2005.104463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng PY, Xiao Z, Jha A, Ramonet D, Matsui T, Leitges M, Shin HS, Porter JE, Geiger JD, Lei S. Cholecystokinin facilitates glutamate release by increasing the number of readily releasable vesicles and releasing probability. J Neurosci. 2010;30:5136–5148. doi: 10.1523/JNEUROSCI.5711-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derrien M, McCort-Tranchepain I, Ducos B, Roques BP, Durieux C. Heterogeneity of CCK-B receptors involved in animal models of anxiety. Pharmacol Biochem Behav. 1994;49:133–141. doi: 10.1016/0091-3057(94)90467-7. [DOI] [PubMed] [Google Scholar]
- Dockray GJ. Cholecystokinins in rat cerebral cortex: identification, purification and characterization by immunochemical methods. Brain Res. 1980;188:155–165. doi: 10.1016/0006-8993(80)90564-8. [DOI] [PubMed] [Google Scholar]
- Dufresne M, Seva C, Fourmy D. Cholecystokinin and gastrin receptors. Physiol Rev. 2006;86:805–847. doi: 10.1152/physrev.00014.2005. [DOI] [PubMed] [Google Scholar]
- Földy C, Neu A, Jones MV, Soltesz I. Presynaptic, activity-dependent modulation of cannabinoid type 1 receptor-mediated inhibition of GABA release. J Neurosci. 2006;26:1465–1469. doi: 10.1523/JNEUROSCI.4587-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Földy C, Lee SY, Szabadics J, Neu A, Soltesz I. Cell type-specific gating of perisomatic inhibition by cholecystokinin. Nat Neurosci. 2007;10:1128–1130. doi: 10.1038/nn1952. [DOI] [PubMed] [Google Scholar]
- Freund TF. Interneuron diversity series: rhythm and mood in perisomatic inhibition. Trends Neurosci. 2003;26:489–495. doi: 10.1016/S0166-2236(03)00227-3. [DOI] [PubMed] [Google Scholar]
- Freund TF, Katona I. Perisomatic inhibition. Neuron. 2007;56:33–42. doi: 10.1016/j.neuron.2007.09.012. [DOI] [PubMed] [Google Scholar]
- Fuchs EC, Zivkovic AR, Cunningham MO, Middleton S, Lebeau FE, Bannerman DM, Rozov A, Whittington MA, Traub RD, Rawlins JN, Monyer H. Recruitment of parvalbumin-positive interneurons determines hippocampal function and associated behavior. Neuron. 2007;53:591–604. doi: 10.1016/j.neuron.2007.01.031. [DOI] [PubMed] [Google Scholar]
- Galione A, Lee HC, Busa WB. Ca(2+)-induced Ca2+ release in sea urchin egg homogenates: modulation by cyclic ADP-ribose. Science. 1991;253:1143–1146. doi: 10.1126/science.1909457. [DOI] [PubMed] [Google Scholar]
- Glickfeld LL, Scanziani M. Distinct timing in the activity of cannabinoid-sensitive and cannabinoid-insensitive basket cells. Nat Neurosci. 2006;9:807–815. doi: 10.1038/nn1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie RC, Minke B. The trp gene is essential for a light-activated Ca2+ channel in Drosophila photoreceptors. Neuron. 1992;8:643–651. doi: 10.1016/0896-6273(92)90086-s. [DOI] [PubMed] [Google Scholar]
- Harro J. CCK and NPY as anti-anxiety treatment targets: promises, pitfalls, and strategies. Amino Acids. 2006;31:215–230. doi: 10.1007/s00726-006-0334-x. [DOI] [PubMed] [Google Scholar]
- Hashimotodani Y, Ohno-Shosaku T, Tsubokawa H, Ogata H, Emoto K, Maejima T, Araishi K, Shin HS, Kano M. Phospholipase C beta serves as a coincidence detector through its Ca2+ dependency for triggering retrograde endocannabinoid signal. Neuron. 2005;45:257–268. doi: 10.1016/j.neuron.2005.01.004. [DOI] [PubMed] [Google Scholar]
- Hefft S, Jonas P. Asynchronous GABA release generates long-lasting inhibition at a hippocampal interneuron–principal neuron synapse. Nat Neurosci. 2005;8:1319–1328. doi: 10.1038/nn1542. [DOI] [PubMed] [Google Scholar]
- Henderson KK, Byron KL. Vasopressin-induced vasoconstriction: two concentration-dependent signaling pathways. J Appl Physiol. 2007;102:1402–1409. doi: 10.1152/japplphysiol.00825.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermans E. Biochemical and pharmacological control of the multiplicity of coupling at G-protein-coupled receptors. Pharmacol Ther. 2003;99:25–44. doi: 10.1016/s0163-7258(03)00051-2. [DOI] [PubMed] [Google Scholar]
- Herranz R. Cholecystokinin antagonists: pharmacological and therapeutic potential. Med Res Rev. 2003;23:559–605. doi: 10.1002/med.10042. [DOI] [PubMed] [Google Scholar]
- Hertle DN, Yeckel MF. Distribution of inositol-1,4,5-trisphosphate receptor isotypes and ryanodine receptor isotypes during maturation of the rat hippocampus. Neuroscience. 2007;150:625–638. doi: 10.1016/j.neuroscience.2007.09.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill DR, Campbell NJ, Shaw TM, Woodruff GN. Autoradiographic localization and biochemical characterization of peripheral type CCK receptors in rat CNS using highly selective nonpeptide CCK antagonists. J Neurosci. 1987;7:2967–2976. doi: 10.1523/JNEUROSCI.07-09-02967.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda T, Wada E, Battey JF, Wank SA. Differential gene expression of CCK(A) and CCK(B) receptors in the rat brain. Mol Cell Neurosci. 1993;4:143–154. doi: 10.1006/mcne.1993.1018. [DOI] [PubMed] [Google Scholar]
- Innis RB, Snyder SH. Distinct cholecystokinin receptors in brain and pancreas. Proc Natl Acad Sci U S A. 1980;77:6917–6921. doi: 10.1073/pnas.77.11.6917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon JP, Lee KP, Park EJ, Sung TS, Kim BJ, Jeon JH, So I. The specific activation of TRPC4 by Gi protein subtype. Biochem Biophys Res Commun. 2008;377:538–543. doi: 10.1016/j.bbrc.2008.10.012. [DOI] [PubMed] [Google Scholar]
- Kano M, Ohno-Shosaku T, Hashimotodani Y, Uchigashima M, Watanabe M. Endocannabinoid-mediated control of synaptic transmission. Physiol Rev. 2009;89:309–380. doi: 10.1152/physrev.00019.2008. [DOI] [PubMed] [Google Scholar]
- Katada T, Amano T, Ui M. Modulation by islet-activating protein of adenylate cyclase activity in C6 glioma cells. J Biol Chem. 1982;257:3739–3746. [PubMed] [Google Scholar]
- Kiselyov K, Patterson RL. The integrative function of TRPC channels. Front Biosci. 2009;14:45–58. doi: 10.2741/3230. [DOI] [PubMed] [Google Scholar]
- Klausberger T, Magill PJ, Márton LF, Roberts JD, Cobden PM, Buzsáki G, Somogyi P. Brain-state- and cell-type-specific firing of hippocampal interneurons in vivo. Nature. 2003;421:844–848. doi: 10.1038/nature01374. [DOI] [PubMed] [Google Scholar]
- Klausberger T, Márton LF, Baude A, Roberts JD, Magill PJ, Somogyi P. Spike timing of dendrite-targeting bistratified cells during hippocampal network oscillations in vivo. Nat Neurosci. 2004;7:41–47. doi: 10.1038/nn1159. [DOI] [PubMed] [Google Scholar]
- Knapp RJ, Vaughn LK, Fang SN, Bogert CL, Yamamura MS, Hruby VJ, Yamamura HI. A new, highly selective CCK-B receptor radioligand ([3H][N-methyl-Nle28,31]CCK26–33): evidence for CCK-B receptor heterogeneity. J Pharmacol Exp Ther. 1990;255:1278–1286. [PubMed] [Google Scholar]
- Krsmanovic LZ, Mores N, Navarro CE, Arora KK, Catt KJ. An agonist-induced switch in G protein coupling of the gonadotropin-releasing hormone receptor regulates pulsatile neuropeptide secretion. Proc Natl Acad Sci U S A. 2003;100:2969–2974. doi: 10.1073/pnas.0535708100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SY, Soltesz I. Cholecystokinin: a multi-functional molecular switch of neuronal circuits. Dev Neurobiol. 2011a;71:83–91. doi: 10.1002/dneu.20815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Soltesz I. Requirement for CB1 but not GABAB receptors in the cholecystokinin mediated inhibition of GABA release from cholecystokinin expressing basket cells. J Physiol. 2011b;589:891–902. doi: 10.1113/jphysiol.2010.198499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis DA, Hashimoto T, Volk DW. Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci. 2005;6:312–324. doi: 10.1038/nrn1648. [DOI] [PubMed] [Google Scholar]
- Lodge DJ, Roques BP, Lawrence AJ. Atypical behavioural responses to CCK-B receptor ligands in Fawn-Hooded rats. Life Sci. 2003;74:1–12. doi: 10.1016/j.lfs.2003.03.003. [DOI] [PubMed] [Google Scholar]
- Martone ME, Alba SA, Edelman VM, Airey JA, Ellisman MH. Distribution of inositol-1,4,5-trisphosphate and ryanodine receptors in rat neostriatum. Brain Res. 1997;756:9–21. doi: 10.1016/s0006-8993(96)01430-8. [DOI] [PubMed] [Google Scholar]
- Maudsley S, Martin B, Luttrell LM. G protein-coupled receptor signaling complexity in neuronal tissue: implications for novel therapeutics. Curr Alzheimer Res. 2007;4:3–19. doi: 10.2174/156720507779939850. [DOI] [PubMed] [Google Scholar]
- Meis S, Munsch T, Sosulina L, Pape HC. Postsynaptic mechanisms underlying responsiveness of amygdaloid neurons to cholecystokinin are mediated by a transient receptor potential-like current. Mol Cell Neurosci. 2007;35:356–367. doi: 10.1016/j.mcn.2007.03.012. [DOI] [PubMed] [Google Scholar]
- Minke B, Cook B. TRP channel proteins and signal transduction. Physiol Rev. 2002;82:429–472. doi: 10.1152/physrev.00001.2002. [DOI] [PubMed] [Google Scholar]
- Morikawa H, Khodakhah K, Williams JT. Two intracellular pathways mediate metabotropic glutamate receptor-induced Ca2+ mobilization in dopamine neurons. J Neurosci. 2003;23:149–157. doi: 10.1523/JNEUROSCI.23-01-00149.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray AJ, Sauer JF, Riedel G, McClure C, Ansel L, Cheyne L, Bartos M, Wisden W, Wulff P. Parvalbumin-positive CA1 interneurons are required for spatial working but not for reference memory. Nat Neurosci. 2011;14:297–299. doi: 10.1038/nn.2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neu A, Földy C, Soltesz I. Postsynaptic origin of CB1-dependent tonic inhibition of GABA release at cholecystokinin-positive basket cell to pyramidal cell synapses in the CA1 region of the rat hippocampus. J Physiol. 2007;578:233–247. doi: 10.1113/jphysiol.2006.115691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemeyer BA, Suzuki E, Scott K, Jalink K, Zuker CS. The Drosophila light-activated conductance is composed of the two channels TRP and TRPL. Cell. 1996;85:651–659. doi: 10.1016/s0092-8674(00)81232-5. [DOI] [PubMed] [Google Scholar]
- Nishida A, Miyata K, Tsutsumi R, Yuki H, Akuzawa S, Kobayashi A, Kamato T, Ito H, Yamano M, Katuyama Y. Pharmacological profile of (R)-1-[2,3-dihydro-1-(2′-methylphenacyl)-2-oxo-5-phenyl-1H-1,4-benzodiazepin-3-yl]-3-(3-methylphenyl)urea (YM022), a new potent and selective gastrin/cholecystokinin-B receptor antagonist, in vitro and in vivo. J Pharmacol Exp Ther. 1994;269:725–731. [PubMed] [Google Scholar]
- Nissen W, Szabo A, Somogyi J, Somogyi P, Lamsa KP. Cell type-specific long-term plasticity at glutamatergic synapses onto hippocampal interneurons expressing either parvalbumin or CB1 cannabinoid receptor. J Neurosci. 2010;30:1337–1347. doi: 10.1523/JNEUROSCI.3481-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble F, Roques BP. CCK-B receptor: chemistry, molecular biology, biochemistry and pharmacology. Prog Neurobiol. 1999;58:349–379. doi: 10.1016/s0301-0082(98)00090-2. [DOI] [PubMed] [Google Scholar]
- Noble F, Roques BP. Cholecystokinin peptides in brain function. In: Lajtha A, Lim R, editors. Handbook of neurochemistry and molecular neurobiology. New York: Springer; 2006. pp. 545–571. [Google Scholar]
- Otsuguro K, Tang J, Tang Y, Xiao R, Freichel M, Tsvilovskyy V, Ito S, Flockerzi V, Zhu MX, Zholos AV. Isoform-specific inhibition of TRPC4 channel by phosphatidylinositol 4,5-bisphosphate. J Biol Chem. 2008;283:10026–10036. doi: 10.1074/jbc.M707306200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oz M, Yang KH, Shippenberg TS, Renaud LP, O'Donovan MJ. Cholecystokinin B-type receptors mediate a G-protein-dependent depolarizing action of sulphated cholecystokinin octapeptide (CCK-8s) on rodent neonatal spinal ventral horn neurons. J Neurophysiol. 2007;98:1108–1114. doi: 10.1152/jn.00148.2007. [DOI] [PubMed] [Google Scholar]
- Piomelli D. The molecular logic of endocannabinoid signalling. Nat Rev Neurosci. 2003;4:873–884. doi: 10.1038/nrn1247. [DOI] [PubMed] [Google Scholar]
- Pommier B, Da Nascimento S, Dumont S, Bellier B, Million E, Garbay C, Roques BP, Noble F. The cholecystokinin B receptor is coupled to two effector pathways through pertussis toxin-sensitive and -insensitive G proteins. J Neurochem. 1999;73:281–288. doi: 10.1046/j.1471-4159.1999.0730281.x. [DOI] [PubMed] [Google Scholar]
- Pommier B, Marie-Claire C, Da Nascimento S, Wang HL, Roques BP, Noble F. Further evidence that the CCK2 receptor is coupled to two transduction pathways using site-directed mutagenesis. J Neurochem. 2003;85:454–461. doi: 10.1046/j.1471-4159.2003.01690.x. [DOI] [PubMed] [Google Scholar]
- Qiu J, Fang Y, Rønnekleiv OK, Kelly MJ. Leptin excites proopiomelanocortin neurons via activation of TRPC channels. J Neurosci. 2010;30:1560–1565. doi: 10.1523/JNEUROSCI.4816-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salido GM, Sage SO, Rosado JA. Biochemical and functional properties of the store-operated Ca2+ channels. Cell Signal. 2009;21:457–461. doi: 10.1016/j.cellsig.2008.11.005. [DOI] [PubMed] [Google Scholar]
- Sankaran H, Goldfine ID, Deveney CW, Wong KY, Williams JA. Binding of cholecystokinin to high affinity receptors on isolated rat pancreatic acini. J Biol Chem. 1980;255:1849–1853. [PubMed] [Google Scholar]
- Scemama JL, Robberecht P, Waelbroeck M, De Neef P, Pradayrol L, Vaysse N, Christophe J. CCK and gastrin inhibit adenylate cyclase activity through a pertussis toxin-sensitive mechanism in the tumoral rat pancreatic acinar cell line AR 4–2J. FEBS Lett. 1988;242:61–64. doi: 10.1016/0014-5793(88)80985-2. [DOI] [PubMed] [Google Scholar]
- Shapiro MS, Wollmuth LP, Hille B. Modulation of Ca2+ channels by PTX-sensitive G-proteins is blocked by N-ethylmaleimide in rat sympathetic neurons. J Neurosci. 1994;14:7109–7116. doi: 10.1523/JNEUROSCI.14-11-07109.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltesz I. Diversity in the neuronal machine: order and variability in interneuronal microcircuits. New York: Oxford UP; 2006. [Google Scholar]
- Strübing C, Krapivinsky G, Krapivinsky L, Clapham DE. TRPC1 and TRPC5 form a novel cation channel in mammalian brain. Neuron. 2001;29:645–655. doi: 10.1016/s0896-6273(01)00240-9. [DOI] [PubMed] [Google Scholar]
- Szabadics J, Varga C, Brunner J, Chen K, Soltesz I. Granule cells in the CA3 area. J Neurosci. 2010;30:8296–8307. doi: 10.1523/JNEUROSCI.5602-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topolnik L, Azzi M, Morin F, Kougioumoutzakis A, Lacaille JC. mGluR1/5 subtype-specific calcium signalling and induction of long-term potentiation in rat hippocampal oriens/alveus interneurones. J Physiol. 2006;575:115–131. doi: 10.1113/jphysiol.2006.112896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujino N, Yamanaka A, Ichiki K, Muraki Y, Kilduff TS, Yagami K, Takahashi S, Goto K, Sakurai T. Cholecystokinin activates orexin/hypocretin neurons through the cholecystokinin A receptor. J Neurosci. 2005;25:7459–7469. doi: 10.1523/JNEUROSCI.1193-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tukker JJ, Fuentealba P, Hartwich K, Somogyi P, Klausberger T. Cell type-specific tuning of hippocampal interneuron firing during gamma oscillations in vivo. J Neurosci. 2007;27:8184–8189. doi: 10.1523/JNEUROSCI.1685-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dijk A, Richards JG, Trzeciak A, Gillessen D, Möhler H. Cholecystokinin receptors: biochemical demonstration and autoradiographical localization in rat brain and pancreas using [3H] cholecystokinin8 as radioligand. J Neurosci. 1984;4:1021–1033. doi: 10.1523/JNEUROSCI.04-04-01021.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Wong PT, Spiess J, Zhu YZ. Cholecystokinin-2 (CCK2) receptor-mediated anxiety-like behaviors in rats. Neurosci Biobehav Rev. 2005;29:1361–1373. doi: 10.1016/j.neubiorev.2005.05.008. [DOI] [PubMed] [Google Scholar]
- Xiao R, Tian J, Tang J, Zholos AV, Zhu Michael X. Critical role of pertussis toxin sensitive G proteins in the activation of TRPC4 and TRPC5 channels. Biophysical J. 2010;98:326a–327a. [Google Scholar]
- Yang K, Trepanier CH, Li H, Beazely MA, Lerner EA, Jackson MF, MacDonald JF. Vasoactive intestinal peptide acts via multiple signal pathways to regulate hippocampal NMDA receptors and synaptic transmission. Hippocampus. 2009;19:779–789. doi: 10.1002/hipo.20559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Roepke TA, Kelly MJ, Rønnekleiv OK. Kisspeptin depolarizes gonadotropin-releasing hormone neurons through activation of TRPC-like cationic channels. J Neurosci. 2008;28:4423–4434. doi: 10.1523/JNEUROSCI.5352-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]