

Abstract
Context
Primary mitochondrial dysfunction is one of the most common causes of inherited disorders predominantly involving the neuromuscular system. Advances in the molecular study of mitochondrial DNA have changed our vision and our approach to primary mitochondrial disorders. Many of the mitochondrial disorders are caused by mutations in nuclear genes and are inherited in an autosomal recessive pattern. Among the autosomal inherited mitochondrial disorders, those related to DNA polymerase γ dysfunction are the most common and the best studied. Understanding the molecular mechanisms and being familiar with the recent advances in laboratory diagnosis of this group of mitochondrial disorders are essential for pathologists to interpret abnormal histopathology and laboratory results and to suggest further studies for a definitive diagnosis.
Objectives
To help pathologists better understand the common clinical syndromes originating from mutations in DNA polymerase γ and its associated proteins and use the stepwise approach of clinical, laboratory, and pathologic diagnosis of these syndromes.
Data Sources
Review of pertinent published literature and relevant Internet databases.
Conclusions
Mitochondrial disorders are now better recognized with the development of molecular tests for clinical diagnosis. A cooperative effort among primary physicians, diagnostic pathologists, geneticists, and molecular biologists with expertise in mitochondrial disorders is required to reach a definitive diagnosis.
Mitochondrial disorders are a group of diseases originating primarily from mitochondrial dysfunction. The accurate incidence and prevalence of mitochondrial disorders are not known because of high childhood mortality and evident underdiagnosis; however, they have traditionally been regarded as rare and obscure. The complete sequencing of the human mitochondrial genome (mtDNA) and the identification of pathogenic mtDNA mutations have facilitated recognizing and diagnosing mitochondrial disorders.1,2 Recent epidemiology studies suggest that mitochondrial disorders are far more common than generally accepted. A survey of the population of northeast England found that 9.2 in 100 000 people have diseases with primary mutations in the mtDNA, and a further 16.5 in 100 000 are at risk for developing mtDNA-related diseases.3 These estimated data indicate that primary mitochondrial dysfunction is one of the common causes of inherited disorders predominantly involving the neuromuscular system. It is estimated that the minimal lifetime risk of developing a mitochondrial disorder is 1 in 5000 live births. However, a recent study revealed that the prevalence could be even greater because pathogenic mtDNA mutations were detected in more than 1 in 200 live births.3
The mitochondrion is a eukaryotic cell organelle that is involved in multiple cellular activities, including multiple steps of metabolism, such as the Krebs cycle, energy conversion through electron transport, the urea cycle, heme synthesis, and the internal signaling pathway of programmed cell death mediated by the release of cytochrome c. The mitochondrion’s most important role in the cell is in converting energy harnessed from oxidative metabolism to adenosine triphosphate, the major universal energy source for most cell activities. Diseases generally included in mitochondrial disorders are the clinical syndromes caused primarily by disruption of energy production through oxidative phosphorylation (OXPHOS). Traditionally, mutations in mtDNA and dysfunction of the proteins encoded by mtDNA were regarded as classic mechanisms causing mitochondrial disorders. As the knowledge of genetic defects and molecular mechanisms grows, it has become obvious that most mitochondrial disease mutations are located in nuclear genes encoding mitochondrial proteins that are transported into the mitochondria. These disorders are mostly inherited in an autosomal recessive pattern. Among the autosomal inherited mitochondrial disorders, those related to DNA polymerase γ (pol γ) dysfunction are the most common and best studied.4
Mitochondrial disorders usually have a heterogeneous presentation with multiorgan involvement. Although there is no specific treatment for mitochondrial disorders, correct diagnosis is important for symptomatic control and avoidance of potential mistakes in management. Furthermore, prognostic and genetic counseling also requires accurate understanding of the molecular basis in each case. Pathologists need to know the characteristic clinical findings of these diseases to correctly interpret histologic changes and abnormal laboratory results, as well as to suggest further studies for definitive diagnosis. The purpose of this review is to help pathologists better understand the most common clinical syndromes originating primarily from mutations of pol γ and associated proteins and use the stepwise approach of clinical, laboratory, and pathologic diagnosis of these clinical syndromes. A clear understanding of the mechanism in each clinical case, along with the molecular studies, is important in determining a definitive final diagnosis.
MITOCHONDRIAL DNA AND INHERITANCE OF MITOCHONDRIAL DISORDERS
Mitochondria originated from free-living, aerobic bacteria that were engulfed by an ancestral eukaryotic cell and evolved in symbiosis with the host cell and its progeny.5,6 The symbiotic partnership further developed by transferring most of the bacterial DNA to the host chromosome to reduce the workload of mitochondria. As a result, many proteins unique to the mitochondria are now encoded in eukaryotic nuclear DNA. These proteins are translated by the cytoplasmic machinery and subsequently transported into the mitochondria. Human mtDNA is circular and double-stranded, with 16 569 base pairs encoding only 13 polypeptides that are absolutely crucial for the OXPHOS complexes (Figure 1), in addition to 22 transfer RNAs and 2 ribosomal RNAs.7 Because of this dual genetic origin of mitochondrial proteins, mitochondrial disorders can have a complicated inheritance.
Figure 1.
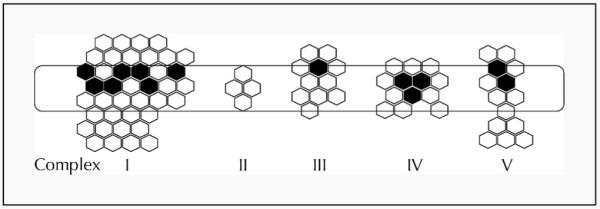
Protein complexes in the mitochondrial respiratory chain and oxidative phosphorylation system. Each hexagon represents a polypeptide product of a single gene; solid black hexagons represent the polypeptides that are encoded in mitochondrial DNA. Adapted from Schapira AHV,7 Lancet 2006;368(9527):71, with permission.
Mitochondria are dynamic organelles that constantly undergo fission and fusion. In the process of cell mitosis or meiosis, mtDNA is randomly distributed. Because mtDNA molecules are not divided evenly, like nuclear DNA, their inheritance is not Mendelian; instead, it is a “cytoplasmic inheritance.” In the process of fertilization, spermatozoa contribute very little, if any, mitochondria to the cytoplasm of a fertilized egg. Human mtDNA inheritance thus follows a “maternal inheritance” mechanism. However, this inheritance is complicated by one cell containing multiple copies of the mtDNA, and wild type mtDNA can coexist with mutant mtDNA; a phenomenon known as heteroplasmy. During oogenesis, wild type and mutant mtDNA are distributed randomly with mitochondria into progeny oocytes, which will then contain different proportions of mutant mtDNA.8 In the process of embryogenesis, mitochondria are randomly or nonrandomly distributed into somatic cells.9,10 This “mitotic segregation” of mtDNA causes significantly variable levels of mutant mtDNA in different cells, tissues, and organs. The overall mitochondrial function will be significantly affected only when the level of mutant mtDNA in a cell reaches a phenotypic threshold.11 The level of heteroplasmy in different cells and tissues can usually determine the phenotype of mitochondrial disorders. On the other hand, the energy dependence of the tissue or cell type also contributes significantly to the phenotype, based on different levels of tolerance to mitochondrial dysfunction. Furthermore, mitochondria in specific tissues may express tissue-specific proteins.6 Nuclear DNA background and other poorly understood factors may add to the complexity of mitochondrial inheritance.6,11 In clinical presentation, the same mutation can have multiple phenotypes and the same phenotype may result from different mutations.
POL Γ AND MITOCHONDRIAL DNA REPLICATION
Consistent with the symbiotic origin of the mitochondrion, replication of mtDNA differs from nuclear DNA replication. The replisome, a complex composed of multiple proteins needed for DNA replication, is much simpler for mtDNA than for nuclear DNA. In humans, there is only one mtDNA polymerase, pol γ, which was confirmed in 1976.12 Its role in human mtDNA replication was confirmed, and the gene of the human pol γ catalytic subunit was subsequently cloned.13-15 In vitro experiments showed a minimal mtDNA replisome of a heterotrimeric DNA polymerase, which consists of a catalytic subunit (pol γA) and a homodimer of the accessory subunit (pol γB), a helicase (Twinkle, encoded by PEO1 [C10ORF2]) and a mitochondrial single-stranded DNA-binding protein (mtSSB).16 Other accessory proteins are likely to engage in the replication machinery in vivo (reviewed in detail by Graziewicz et al17). Because pol γ is the only DNA polymerase in human mitochondria, it is not only responsible for mtDNA replication but also is involved in mtDNA repair. Understandably, its mutation and dysfunction will result in significant disruption of healthy mitochondrial function, with marked clinical effects. A detailed study on the functional domains of both pol γA and pol γB is required to understand the molecular mechanism of mitochondrial disorders caused by pol γ dysfunction.
Human pol γA is a 140 kDa polypeptide with 1239 amino acids,18 encoded by POLG (GenBank Accession AF497906.1 [or NG_008218.1]) in band 15q25. It is a family-A DNA polymerase, with both DNA polymerase activity and exonuclease proofreading activity. The polymerase domain is at the C-terminal portion, and the 3′ to 5′ exonuclease domain is located near the N-terminus. Between the polymerase and exonuclease domains is the linker region, which is known to be involved in binding the accessory subunit.19,20 A mitochondrial targeting sequence is present at the N-terminus, followed by a polyglutamine segment that is encoded by a CAG repeat in exon 2 of the POLG gene (Figure 2). The 3′ to 5′ exonuclease activity serves as a proofreading mechanism to excise the mispaired nucleotide at 3′ termini, ensuring high fidelity in DNA replication. The relationship between the exonuclease activity and the fidelity of mtDNA replication was confirmed by in vitro experiments in yeast.21 One critical amino acid mutation in the exonuclease domain that markedly decreases the mismatch-specific 3′ to 5′ exonuclease activity resulted in a several 100-fold increase in the frequency of spontaneous mutations of the mtDNA.22 Interestingly, mice expressing an exonuclease-deficient pol γ develop a mtDNA mutator phenotype and instead of developing pol γ–type mitochondrial disease, they develop a premature aging phenotype.23,24 A 5′-deoxyribose phosphate lyase activity was found to be intrinsic to the human pol γA subunit.25 This activity is involved in base excision repair,17 which is important for mtDNA maintenance. The efficiency of the lyase activity is also enhanced by pol γB.26 Pol γ also contains a reverse transcriptase activity with a catalytic rate greater than that of human immunodeficiency virus 1 (HIV-1) reverse transcriptase18,27; however, the significance of this activity in vivo is not clear.28 Recently, the crystal structure of the pol γ holoenzyme was solved.20 This model can be used to predict the functional defects that may result from the mutations identified in clinical patients.
Figure 2.

Human DNA polymerase γ catalytic subunit (pol γA) gene (POLG), functional domains, mutations, and clinical syndromes presumably related to the mutational changes. Exon numbers are marked in the gene diagram. Amino acids are labeled with one letter code. The color of the box labeling the mutation is the same with the color of the clinical syndrome box on the bottom. Abbreviations: MIRAS, mitochondrial recessive ataxia syndrome; NRTI, nucleoside reverse transcriptase inhibitor; SANDO, sensory ataxic neuropathy, dysarthria, and ophthalmoparesis; SCAE, spinocerebellar ataxia-epilepsy syndrome. Adapted from http://tools.niehs.nih.gov/polg, with permission.
The 55 kDa accessory subunit pol γB is a 474 amino acid polypeptide encoded by the POLG2 gene on band 17q23-24 (GenBank Accession AF142992).29 The 3-dimensional structure of the mouse pol γB was determined to be a crystallized dimer in 2001.30 The human pol γB structure was subsequently solved by Fan et al.31 These models help to determine the functionally important domains and critical amino acid residues that may be important in mtDNA replication, as well as in disease. Several domains of pol γB interact with pol γA as shown by electron microscopy and the recent pol γ holoenzyme crystal structure.20,32 Of note, pol γB promotes extension of mismatched termini, thus lowering the fidelity of DNA replication. The homopolymeric runs in mtDNA that are prone to frame shift mutation in vivo are most sensitive to this.33
The mtDNA helicase, Twinkle, is encoded by PEO1 (C10ORF2), which is located on band 10q24 (GenBank Accession AF292004 [or NG_013029.1]).34 The full-length protein is 684 amino acids, with a molecular weight of 77 kDa. It is homologous to bacteriophage T7 gene 4 primase/helicase and functions as a 5′ to 3′ DNA helicase. However, Twinkle lacks primase activity. Recently, it was shown that the Twinkle helicase can form both hexamers and heptamers in vitro.35
The gene encoding the human mtSSB, SSBP1, is located at band 7q34. As an essential protein in the mtDNA replisome, it greatly enhances pol γ activity.16,36 An in vitro mutational study revealed that Drosophila mtSSB defective in DNA binding was not efficient in stimulating pol γ activity, resulting in mtDNA depletion.37 The mtSSB protein has been cloned and the crystal structure resolved38; however, there has been no report of mitochondrial disorders resulting from mtSSB mutation to date.
DISEASE-RELATED MUTATIONS IN PROTEINS INVOLVED IN MITOCHONDRIAL DNA INTEGRITY
Mutations in pol γA, pol γB, and the Twinkle helicase have been linked to mitochondrial dysfunction, and account for most mutations in autosomal inherited mitochondrial disorders.17,39,40 Pol γ with diminished DNA polymerase activity and/or reduced exonuclease activity with weakened proofreading can lead to a significant decrease in mtDNA copy number, or multiple deletions in mtDNA, which in turn translates to defects in essential OXPHOS proteins, the functional consequences of which can be related to clinical outcome.17,39,40
DNA sequencing has becomes an integral part of clinical diagnoses, and for mitochondrial disorders, in particular, it has become essential because POLG is a major disease locus. The Human DNA Polymerase γ Web site (http://tools.niehs.nih.gov/polg/. Accessed November 10, 2010) currently shows more than 180 disease mutations for the POLG gene (Figure 2). However, only a few of these mutations have been characterized biochemically to determine their functional defects at the molecular level. These mutations were recently reviewed in detail by Chan and Copeland,4 and since then, new mutations have been characterized by Kasiviswanathan et al.41 The most common POLG mutation is the A467T mutation within the linker region of pol γ, which is found in more than one-third of all individuals who have a pol γ–associated mitochondrial disorder.4 The 2 next mostcommon disease mutations, W748S39 and G848S,41 in addition to A467T, comprise the mutations in most affected individuals. A467T impairs the interaction between the 2 subunits of pol γ and reduces polymerase activity.19 Additionally, biochemical analysis of a point mutation in POLG2 (G451E) revealed that this mutation impaired subunit interaction with pol γA, reducing the functional capacity of the holoenzyme because the mutant pol γB failed to enhance DNA binding and processivity of the holoenzyme. The G451E mutation was found in a patient with autosomal dominant progressive external ophthalmoplegia (PEO) (PEOA4).42 Mixing studies using both wild type and the G451E pol γB did not reveal a dominant negative effect, and the authors42 suggested that the disease phenotype may arise through haplotype insufficiency or through heterodimerization of the wild type and the G451E pol γB proteins. This may cause stalling at the DNA replication fork and promote mtDNA deletions. G416A mutation in POLG2 was found in conjunction with a Y582C OPA1 mutation in a patient with PEO.43 However, biochemical analysis of the recombinant G416A pol γB did not reveal any defects, suggesting that the OPA1 mutation may explain the PEO in that patient. 43 A novel heteroallelic 24–base pair insertion (c.1207_1208ins24) was also found in POLG2 and may cause dysfunction because of missplicing.44
The PEO1 (C10ORF2) mutations are associated with autosomal dominant progressive external ophthalmoplegia (PEO) (PEOA3), parkinsonism, as well as infantileonset spinocerebellar ataxia.40,45,46 Biochemical analyses of purified mutant PEO1 proteins have been performed, but the results have been mixed.16,47,48 The functional defects of mutant Twinkle proteins have also been studied in the ortholog in Drosophila.49 Transgenic mice carrying a point mutation at amino acid residue 360 (A360T), or a short repeat of amino acids 353 to 365 reproduced histopathology and histochemical changes resulting from mitochondrial respiratory chain dysfunction. Furthermore, mtDNA depletion and multiple mtDNA deletions were confirmed in the muscle and brain tissue of the transgenic mice, which were further linked to respiratory chain dysfunction by single muscle fiber polymerase chain reaction analysis.50
Several proteins involved in mitochondrial nucleotide transportation and metabolism are essential for providing building material for mtDNA. Depletion and multiple deletions of mtDNA caused by mutational change in adenine nucleotide translocator (ANT1 [SLC25A4]), thymidine phosphorylase (ECGF1 [TYMP]), mitochondrial thymidine kinase (TK2), deoxyguanosine kinase (DGUOK), succinyl-coenzyme A ligase (adenosine 5′-diphosphate–forming) subunit β (SUCLA2), succinyl-CoA ligase (GDP-forming) subunit alpha (SUCLG1), optic atrophy 1 protein (OPA1), p53-inducible ribonucleotide reductase (RRM2B), and the mitochondrial inner membrane protein MPV17 have been reported in similar clinical syndromes.4,40,51
CLINICAL SYNDROMES CAUSED BY POL γ MUTATIONS
Progressive External Ophthalmoplegia:(Online Mendelian Inheritance in Man (OMIM) Database Numbers: Autosomal Dominant PEOA1:157640, PEOA3:609286, and PEOA4:610131 and Autosomal Recessive PEOB: 9258450)
Progressive external ophthalmoplegia is a descriptive term that refers to a heterogeneous group of diseases characterized by chronic, progressive, bilateral, and usually symmetric ocular motility deficit and ptosis. It was recognized as a syndrome in the late 19th century.52 Histology and ultrastructural changes found in muscle biopsies suggested a mitochondrial defect.53,54 Respiratory chain dysfunction was subsequently identified, confirming the primary mitochondrial dysfunction as the cause of the syndrome in these cases.55 In 1989, deletions of mtDNA isolated from muscle biopsies were demonstrated in Italian families with the heritable autosomal dominant PEO, linking the original defect further to a nuclear protein.56 Since then, many chromosomal genetic mutations have been related to PEO, most of which are inherited in an autosomal recessive pattern.40
The characteristic clinical presentation of bilateral ptosis in PEO typically starts in early adulthood. Ptosis can also be asymmetric, present as strabismus and diplopia, or diplopia when fixating at near objects. Some patients may progressively develop pareses of all extraocular muscles, resulting in an almost complete ophthalmoplegia. Variable severity of retinal pigmentary changes that may result in visual loss can occur. Optic nerve atrophy, a typical sign in Leber hereditary optic neuropathy, can occasionally be present in some patients with PEO.57
Like other mitochondrial disorders, PEO can include multisystem involvement. Muscle involvement results in proximal muscle weakness, exercise intolerance, wasting, dysarthria, and dysphagia. Degenerative changes, such as hypogonadism, cataracts, and hearing loss, are also frequent presentations.58 Other neurologic symptoms can occur,59 and cardiac abnormalities with arrhythmias, which are sometimes fatal, are significant in the late stage of some patients.60 The disease of patients with multiple organ involvement is sometimes given other names, such as “ophthalmoplegia plus syndrome” and “Kearns-Sayre syndrome” to differentiate the disease from classic PEO.57,58 Because PEO is a descriptive term, the clinical picture may present in many mitochondrial disorders as well as other neuromuscular disorders. The most important differential diagnosis in mitochondrial PEO is myasthenia gravis, which is far more common and is usually present with ophthalmoplegia in the early stage. No specific therapy for PEO has yet been developed. Surgical correction of ptosis was sometimes attempted but is usually unhelpful because of the progressive nature of the disease.61 Some patients with cardiac conduction disorders have benefited from pacemakers.
Infantile Hepatocerebral Syndrome (Alpers Syndrome) (OMIM 203700 and 251880)
Infantile diffuse cerebral degeneration was reported by Bernard Alpers in 193162 and was further clarified in 1960 as a clinical syndrome,63 which was later referred to as Alpers syndrome. However, the link between cerebral neuropathy and liver failure was later recognized by Huttenlocher et al in 197664; therefore, it is sometimes also referred to as Alpers-Huttenlocher syndrome. The POLG mutations were first identified in Alpers syndrome in 1999.65 Since then, several mutations have been related to Alpers syndrome, which is now well established as an autosomal recessive clinical syndrome of primary mitochondrial dysfunction.66
Alpers syndrome is characterized by psychomotor retardation, intractable epilepsy, and liver failure in infants and young children.67 Symptoms usually begin between birth and 3 years. Hypotonia, hemiparesis, and ataxia are frequent presentation; sensory neuropathy is rarely reported. In older patients, severe developmental delay and cortical blindness are noticed. Neurophysiologic examination reveals a loss of visual evoked potentials, slow wave activity with superimposed polyspikes, and frequent numbers and duration of the bursts of suppression of the cortical electric activity. Hepatic failure may not be obvious in the early stage but usually advances progressively. In patients with epilepsy, acute liver failure after exposure to valproic acid or sodium divalproate is a characteristic finding.68-70 Most patients died within months of the onset.64,71 Alpers syndrome is a rare disease, and the characteristic presentation with both cerebral degeneration and liver failure is not usually obvious in the early stage. Therefore, the differential diagnosis is broad. Before molecular studies on the mtDNA, mitochondrial proteins, and the nuclear proteins involved in mtDNA replication were available, definitive diagnosis of Alpers syndrome was rarely made without autopsy. An early suspicion of Alpers syndrome is important, in order to avoid using valproic acid and sodium divalproate to control epilepsy, which is an invariable neurology presentation in the early stages of the disease.72
Ataxia-Neuropathy Syndrome (OMIM 607459)
Patients with sensory ataxic neuropathy, dysarthria, and ophthalmoparesis (SANDO) were first reported in 1997 for a possible novel mitochondrial disease associated with multiple mtDNA deletions. Van Goethem73 later reported more cases and considered them a variant form of autosomal recessive PEO. Although all patients eventually developed PEO, neuromuscular involvement preceded ptosis in most cases. Reported cases are either autosomal recessive or sporadic, and the age of onset is between 16 and 30 years. Several different terms have been used to name this syndrome, for example, mitochondrial recessive ataxia syndrome (MIRAS) and spinocerebellar ataxia-epilepsy syndrome (SCAE). The most prominent features are a sensory ataxic neuropathy with loss of kinesthetic and vibratory sensation, ataxic gait, positive Romberg sign, and areflexia. The presence of ophthalmoplegia and dysarthria is required for the diagnosis of SANDO. The disease usually progresses slowly, but severe and early onset myopathy, muscle atrophy, and severe dysphagia are seen in some patients.
Other Clinical Syndromes
Leigh syndrome (OMIM 256000)74 is the most common childhood-onset clinical syndrome of the mitochondrial respiratory chain dysfunction.75 However, it is not commonly seen in patients with pol γ–related mutations. Other clinical syndromes rarely related to mutations in the pol γ complex are mitochondrial neurogastrointestinal encephalopathy syndrome (MNGIE, OMIM 603041),76,77 Charcot-Marie-Tooth disease,78-80 male infertility, testicular cancer, idiopathic parkinsonism, premature ovarian failure and nucleoside reverse transcriptase inhibitors toxicity in patients infected with human immunodeficiency virus.4,81,82
DIAGNOSIS OF MITOCHONDRIAL DISORDERS CAUSED BY POL γ–RELATED MUTATIONS
Although some clinical presentations, such as progressive external ophthalmoplegia sensory ataxic neuropathy, dysarthria, and ophthalmoparesis; or a combination of cerebral degeneration and liver failure in Alpers syndrome, may be quite characteristic, most patients with mitochondrial disorders have variable and nonspecific presentation, especially in the early stages of disease. Furthermore, there are no specific laboratory or image studies to reliably screen mitochondrial disorders. Recognition and diagnosis of mitochondrial disorders are challenging for both clinicians and pathologists. A high alertness on the part of the primary physician and cooperation between clinicians, pathologists, and medical geneticists are crucial to reach a definitive diagnosis. The guidelines for initial diagnosis and in-depth evaluation of mitochondrial disorders have recently been reviewed by Haas et al83 and the Mitochondrial Medicine Society’s Committee on Diagnosis.84 Issues most pertinent to anatomic and clinical pathologists are discussed here.
Red Flag Clinical Presentations of Mitochondrial Disorders
As described previously, in the “Clinical Syndromes Caused by Pol γ Mutations,” mitochondrial disorders usually have multiorgan disturbance, with the highly energy-demanding organs most commonly involved. Familiarity with the clinical presentation of mitochondrial disorders will help pathologists to correctly interpret histologic changes and laboratory findings, as well as to suggest further studies for definitive diagnosis. Clinical presentations suggestive of mitochondrial disorders usually involve the neurologic, muscular (including myocardial), ophthalmologic, and gastrointestinal (especially hepatic) systems. The common, red-flag clinical findings that should raise suspicion for mitochondrial disorders were reviewed in detail by Haas et al.83 Although none of the clinical presentations is specific for a mitochondrial disorder, some findings are more commonly associated with mitochondrial dysfunction. For example, epilepsy, either partial or myoclonus, is a common neurologic presentation related to mitochondrial dysfunction. Other neurologic impairment patterns, such as cerebral and basal ganglia neuron injury or degeneration, are described in the “Clinical Syndromes Caused by Pol γ Mutations.” Sustained insufficiency of energy supply from mitochondria induces cardiomyopathy with rhythm or conduction disturbance. Ophthalmoplegia, with paresis and ptosis, is not only the classic presentation of PEO but also the most common presentation of other mitochondrial syndromes; it may also be accompanied by visual defects and optic neuropathy. In the gastrointestinal system, unexplained or valproate-induced liver failure is a frequent first finding of mitochondrial dysfunction. In the early years of child-hood, well-defined symptoms or signs may not be obtained, but unexplained nonspecific presentations associated with metabolic disturbances, such as failure to thrive, lactic acidosis, and neuromuscular problems disproportionate to activity, are often seen in the early stage of mitochondrial disorders. When clinical presentation involves 3 or more organs/systems, pathologic and laboratory evaluations on mitochondrial function are strongly indicated.
Radiology Findings
Magnetic resonance imaging is a useful tool in the diagnosis of mitochondrial disorders, especially in Leigh syndrome and mitochondrial encephalopathy with lactic acidosis and strokelike episodes (MELAS).85 However, its diagnostic value in pol γ–related clinical syndromes is limited. Cerebral atrophy or edema is a frequent finding in Alpers syndrome. Rarely, signs of leukoencephalopathy and calcifications of the basal ganglia can be present in magnetic resonance imaging and computed tomography imaging57 of patients with PEO. However, these are only nonspecific findings. Magnetic resonance spectroscopy is a new neuroimaging technique that can reveal abnormal changes in some metabolic products related to respiratory chain dysfunction by shift of chemical peaks.86-88 The metabolic products most commonly studied using magnetic resonance spectroscopy are lactate, N-acetyl-l-aspartate, succinate, total creatine, choline, and myoinositol. The change in lactate level is most helpful for the diagnosis of mitochondrial disorders. Evaluation of brain tissue lactate level by magnetic resonance spectroscopy can be more sensitive than blood lactate level in detecting respiratory chain dysfunction, especially when a patient is in the acute phase of disease.88 Deficient respiratory chain activity can cause a significant increase in succinate concentration, which can be detected by magnetic resonance spectroscopy. Obviously, these changes are not specific for any particular cause of mitochondrial dysfunction.
Anatomic Pathology
As described previously, different tissues have significantly different tolerance levels to mitochondrial dysfunction. It is crucial to biopsy the most profoundly affected tissue to identify specific morphology consistent with mitochondrial disorders. Skin fibroblasts and muscle tissue are most commonly submitted for anatomic pathology study. Liver biopsy could be very helpful when it is the only available tissue with detectable morphologic abnormalities, such as in Alpers syndrome. On the other hand, cardiac muscle biopsy itself is rarely helpful because of the high incidence of mitochondrial changes secondary to commonly seen cardiomyopathy and chronic heart failure.84,89 Of note, although skin may not be a good tissue source for morphology diagnosis, cultured fibroblasts from a relatively simple skin biopsy procedure could be very useful for further study to identify a specific enzymatic or molecular defect.
Mitochondria respond to functional deficiency by proliferation. At the early stage of an OXPHOS disorder, the only morphologic change that might be noted in a muscle biopsy is accumulation of mitochondria in the subsarcolemmal region. As the disease advances, accumulated mitochondria build up, giving rise to the well-known ragged-red fibers, clearly identifiable with Gomori trichrome stain as a characteristic change of mitochondrial disorders. However, ragged-red fiber is not frequently seen in infants who are not old enough to accumulate mitochondria as red granular deposits. Furthermore, this finding is not specific for mitochondrial disorders. Several enzyme histochemical stains, such as nicotinamide adenine dinucleotide (reduced form) dehydrogenase, succinate dehydrogenase (complex II), and cytochrome c oxidase (COX, complex IV) stains, are useful for identifying dysfunction in respiratory chain activity. However, they may also be positive in other myopathic or degenerative muscular diseases. Therefore, they may be no more specific than ragged-red fibers. Three of the 13 subunits of COX complex are coded by mtDNA (Figure 1). A deficiency in COX activity may reflect a low level of wild type mtDNA. In contrast, all subunits of complex II are coded by nuclear genes. Therefore, sequential or simultaneous staining of COX and succinate dehydrogenase provides useful information on the differential diagnoses of mitochondrial disorders originating from different genetic backgrounds. In the presence of healthy or strong succinate dehydrogenase activity, a low COX activity together with ragged-red fibers usually points to problems with mtDNA integrity, resulting in markedly impaired intrinsic mitochondrial protein synthesis.90 As would be expected, low COX activity in the background of healthy succinate dehydrogenase activity is a frequent finding in disorders resulting from pol γ–related mutations. In tissues with heteroplasmic mtDNA, if the mutations in the mtDNA involve the COX complex proteins, the COX stain will show a mosaic pattern, reflecting different levels of mutant mtDNA. Respiratory chain enzyme deficiency can also be confirmed with a liver biopsy, although that is not routinely studied.91
Some characteristic histopathology findings are very helpful in diagnosing Alpers syndrome at autopsy. Cerebral degeneration, including neuronal loss, is usually identified in the gray matter and is especially significant in the olivary nuclei, and spongiform degeneration and associated gliosis may develop from focal to diffuse. The liver shows massive hepatocyte necrosis with bile duct proliferation and fibrosis that often rapidly progresses to cirrhosis.67
The diagnostic value of electron microscopy is limited and controversial. Although electron microscopy is well known to recognize quantitative and morphologic changes in mitochondria, none of the ultrastructural findings are specific for primary mitochondrial disorders. However, in patients who are suspected of having a primary mitochondrial disorder, electron microscopy could be more sensitive than histochemical stains in finding abnormal mitochondria, leading to further diagnostic workup.
Laboratory Diagnosis
Before molecular tests became routine clinical practice, diagnosis of mitochondrial disorders was heavily dependent on the evaluation of the metabolic product that might increase because of the dysfunction of oxidative phosphorylation. These tests use blood, urine, or cerebral spinal fluid and thus require less-invasive sampling. The lactate to pyruvate ratio indirectly reflects the average cytoplasmic nicotinamide adenine dinucleotide (reduced form) to nicotinamide adenine dinucleotide+ ratio92 and, therefore, is an easy screening method for metabolic changes due to mitochondrial dysfunction. Evaluation of several amino acid levels, such as the alanine level, the alanine to lysine ratio, or the alanine to phenylalanine and tyrosine ratio, which reflects the relative alanine level, may also reflect overall mitochondrial function. Elevation of tyrosine in newborn screening is associated with Alpers syndrome.84 Impaired oxidative phosphorylation also causes accumulation of intermediates of the tricarboxylic acid cycle, ethylmalonic acid, and 3-methyl glutaconic acid. Evaluation of these organic acids may also provide evidence to mitochondrial dysfunction. Defects in fatty acid β-oxidation secondary to OXPHOS disorders may be reflected by elevations in blood carnitine and acylcarnitine levels. In the early stages or mild forms of mitochondrial disorders, abnormal levels of metabolites can sometimes be observed by provocative clinical testing, such as carbohydrate loading, fasting studies, or exercise testing. Overall, although metabolic tests are convenient and most can be performed in routine chemistry laboratories using high-performance liquid chromatography and/or tandem mass spectrometry, these tests are not sensitive enough to detect mild or early stages of the diseases. The usually localized tissue distribution of dysfunctional mitochondria also makes it difficult to generate detectable systemic metabolic changes. Furthermore, metabolic tests are rarely diagnostic of any particular mitochondrial disease.93 Many disease conditions, such as secondary enzyme deficiencies, metabolic disorders, nutritional deficiencies, and neurologic disorders, can have positive laboratory results.
A variety of methods have been used to assess the activity of respiratory chain enzymes in fresh or frozen biopsy tissue.84 Locating the defect at specific enzymes is very helpful in narrowing down the mechanisms of mitochondrial disorders and selecting appropriate molecular tests to confirm the diagnosis. Mutations related to pol γ complex result in multiple enzyme defects in complex I, III, IV, and V, reflecting dysfunction of most or all proteins encoded by mtDNA. These enzyme activity tests are more sensitive and specific than metabolic product tests with blood, urine, or cerebral spinal fluid; however, these tests are not practical for the routine clinical laboratory, and invasive procedures are required to obtain the tissue sample.
Molecular Studies
The development of genetic and molecular methods for clinical laboratory diagnosis has changed the approach toward definitive pathologic diagnosis of mitochondrial disorders. Previously obscure syndromes caused by primary mitochondrial dysfunction can now be diagnosed at the molecular level, helping clinicians to understand the fundamental disease mechanism. A syndrome with a full-characteristic clinical presentation, such as hepatocerebral degeneration (Alpers syndrome), may suggest a specific molecular test (POLG gene sequencing) for establishing a solid diagnosis, without requiring any invasive tissue biopsy or histopathology diagnosis.66 However, clinical presentations of many mitochondrial disorders are not fully developed or straightforward. The dual-genome origin, a broad range of mutations, and the diversified levels of heteroplasmy with variable tissue distribution make it impossible to set up a set of molecular tests to diagnose all primary mitochondrial disorders. A series of laboratory tests are required before a selective set of molecular studies can be considered. Mitochondrial disorders caused by pol γ dysfunction usually have a marked depletion of mtDNA or multiple mtDNA deletions and mutations, which may be considered as a general screening test. The diagnostic value of the test results depends on the relevance of the tissue from which the mtDNA is extracted, and a definitive diagnosis depends on finding mutational changes in the nuclear DNA that alter the function of pol γ and associated proteins, impairing mtDNA synthesis and maintenance (Figure 3).
Figure 3.

Mechanism and diagnostic approach of mitochondrial disorders from polymerase γ dysfunction. The unfilled (white) boxes from left to right depict the mechanism of that group of mitochondrial disorders from gene mutation to clinical presentation; genes in the gray boxes are less commonly related to those disorders with the same molecular mechanism. The black boxes specify applicable diagnostic tests at each step, usually from right to left. The bottom panel indicates the value of clinical evaluation and diagnostic tests. The functional protein products of the chromosomal genes are described in the text. Abbreviations: MRI, magnetic resonance imaging; MRS, magnetic resonance spectroscopy; mtDNA, mitochondrial DNA; OXPHOS, oxidative phosphorylation; PCR, polymerase chain reaction.
To detect mtDNA depletion, a traditional method is quantitative Southern blot, based on densitometry scanning of autoradiograph films. Southern blot is labor intensive and time consuming compared with recently developed quantitative polymerase chain reaction methods.94 Furthermore, through mutant primer design, polymerase chain reaction methods can also be used to quantitate specific mtDNA mutations, serving as a way to evaluate the heteroplasmic level of mtDNA. Southern blot analysis is still indispensible for identifying large-scale deletions and multiple deletions, which frequently occur with pol γ malfunction. Sanger sequencing is not commonly used for diagnosing heteroplasmic mtDNA mutational changes because that method does not provide quantitative information, and the level of mutant mtDNA may be significantly related to the severity of the functional defects.95 Recently developed microarray-based sequencing methods may overcome that disadvantage.96 Finally, to definitively confirm the pol γ–related mutations, nuclear DNA sequencing of POLG and related genes encoding associated proteins (eg, POLG2, PEO1 [C10ORF2], MPV17) is needed (Figure 3). Care should be taken when interpreting the functional relationship of the mutations to their clinical syndromes. Except for rare syndromes caused by dominant-type mutations (autosomal dominant PEO) or complicated mutations with other genes involved, most phenotypes require mutations on both chromosome alleles (trans) or compound mutations on the same allele (cis). Furthermore, additional mutations in POLG and related genes are continuously being reported (Figure 2); however, most mutations have not been functionally and biochemically characterized, although many are assumed to be functionally significant, as predicted from the structural model and/or the amino acid conservation. Therefore, a solid diagnosis is still heavily dependent on the clinical judgment and support from relevant laboratory data from metabolic studies in addition to the nuclear DNA and mtDNA mutations found in molecular studies.
CONCLUSIONS
Advances in the molecular study of mtDNA have changed our vision and our approach to primary mitochondrial disorders. We already have detailed knowledge of the pathogenesis of some of the autosomal inherited mitochondrial disorders caused by pol γ–related mutational changes, with in vitro biochemistry or in vivo studies confirming the molecular pathogenesis. However, further studies are needed to establish a clear link between the mutation and the clinical syndrome. The fundamental pathogenic pathway in this category of mitochondrial disorders is a defective pol γ causing depletion or deletion of mtDNA. Some characteristic combination of clinical presentations may point to molecular defects, but most clinical cases have multiple organ system involvement, and a definitive diagnosis may be elusive to clinicians and pathologists, especially at the early stages of disease. A cooperative effort between primary physicians, diagnostic pathologists, geneticists, and molecular biologists who have expertise in mitochondrial disorders is required to reach a final definitive diagnosis. Usually, characteristic histopathology, metabolic studies, and enzymatic analysis of the OXPHOS respiratory chain may point the direction to a specific set of molecular studies. Quantification and deletion analysis of MtDNA can be used for suspected mitochondrial disease, in addition to sequence analysis of nuclear genes encoding proteins involved in the maintenance of the mtDNA (Figure 3). New molecular studies will reduce the necessity of invasive procedures for diagnostic purposes, as well as lead to more cases that can be diagnosed without the range of tissue sampling and testing that is traditionally associated with the autopsy.
Acknowledgments
Sherine S. L. Chan, PhD, was supported by Career Development Award 5 R00 ES015555-03 from the National Institutes of Health.
The authors have no relevant financial interest in the products or companies described in this article.
Footnotes
Drs Zhang and Chan contributed equally to this review.
Contributor Information
Linsheng Zhang, Department of Pathology and Laboratory Medicine, Medical University of South Carolina.
Sherine S. L. Chan, Department of Pharmaceutical and Biomedical Sciences, South Carolina College of Pharmacy, Medical University of South Carolina.
Daynna J. Wolff, Department of Pathology and Laboratory Medicine, Medical University of South Carolina.
References
- 1.Anderson S, Bankier AT, Barrell BG, et al. Sequence and organization of the human mitochondrial genome. Nature. 1981;290(5806):457–465. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- 2.Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Elliott HR, Samuels DC, Eden JA, Relton CL, Chinnery PF. Pathogenic mitochondrial DNA mutations are common in the general population. Am J Hum Genet. 2008;83(2):254–260. doi: 10.1016/j.ajhg.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chan SSL, Copeland WC. DNA polymerase gamma and mitochondrial disease: understanding the consequence of POLG mutations. Biochim Biophys Acta. 2009;1787(5):312–319. doi: 10.1016/j.bbabio.2008.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martin W, Muller M. The hydrogen hypothesis for the first eukaryote. Nature. 1998;392(6671):37–41. doi: 10.1038/32096. [DOI] [PubMed] [Google Scholar]
- 6.Alberts B. Molecular Biology of the Cell. 4th ed. Garland Science; New York, NY: 2002. pp. xxxivpp. 808–821. [Google Scholar]
- 7.Schapira AH. Mitochondrial disease. Lancet. 2006;368(9529):70–82. doi: 10.1016/S0140-6736(06)68970-8. [DOI] [PubMed] [Google Scholar]
- 8.Chinnery PF, Howell N, Lightowlers RN, Turnbull DM. MELAS and MERRF: the relationship between maternal mutation load and the frequency of clinically affected offspring. Brain. 1998;121(10):1889–1894. doi: 10.1093/brain/121.10.1889. [DOI] [PubMed] [Google Scholar]
- 9.Chinnery PF, Zwijnenburg PJ, Walker M, et al. Nonrandom tissue distribution of mutant mtDNA. Am J Med Genet. 1999;85(5):498–501. [PubMed] [Google Scholar]
- 10.Frederiksen AL, Andersen PH, Kyvik KO, Jeppesen TD, Vissing J, Schwartz M. Tissue specific distribution of the 3243A→G mtDNA mutation. J Med Genet. 2006;43(8):671–677. doi: 10.1136/jmg.2005.039339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rossignol R, Faustin B, Rocher C, Malgat M, Mazat JP, Letellier T. Mitochondrial threshold effects. Biochem J. 2003;370(3):751–762. doi: 10.1042/BJ20021594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bolden A, Noy GP, Weissbach A. DNA polymerase of mitochondria is a gamma-polymerase. J Biol Chem. 1977;252(10):3351–3356. [PubMed] [Google Scholar]
- 13.Ropp PA, Copeland WC. Cloning and characterization of the human mitochondrial DNA polymerase, DNA polymerase gamma. Genomics. 1996;36(3):449–458. doi: 10.1006/geno.1996.0490. [DOI] [PubMed] [Google Scholar]
- 14.Davis AF, Ropp PA, Clayton DA, Copeland WC. Mitochondrial DNA polymerase gamma is expressed and translated in the absence of mitochondrial DNA maintenance and replication. Nucleic Acids Res. 1996;24(14):2753–2759. doi: 10.1093/nar/24.14.2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lestienne P. Evidence for a direct role of the DNA polymerase gamma in the replication of the human mitochondrial DNA in vitro. Biochem Biophys Res Commun. 1987;146(3):1146–1153. doi: 10.1016/0006-291x(87)90767-4. [DOI] [PubMed] [Google Scholar]
- 16.Korhonen JA, Pham XH, Pellegrini M, Falkenberg M. Reconstitution of a minimal mtDNA replisome in vitro. EMBO J. 2004;23(12):2423–2429. doi: 10.1038/sj.emboj.7600257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Graziewicz MA, Longley MJ, Copeland WC. DNA polymerase gamma in mitochondrial DNA replication and repair. Chem Rev. 2006;106(2):383–405. doi: 10.1021/cr040463d. [DOI] [PubMed] [Google Scholar]
- 18.Longley MJ, Ropp PA, Lim SE, Copeland WC. Characterization of the native and recombinant catalytic subunit of human DNA polymerase gamma: identification of residues critical for exonuclease activity and dideoxynucleotide sensitivity. Biochemistry. 1998;37(29):10529–10539. doi: 10.1021/bi980772w. [DOI] [PubMed] [Google Scholar]
- 19.Chan SSL, Longley MJ, Copeland WC. The common A467T mutation in the human mitochondrial DNA polymerase (POLG) compromises catalytic efficiency and interaction with the accessory subunit. J Biol Chem. 2005;280(36):31341–31346. doi: 10.1074/jbc.M506762200. [DOI] [PubMed] [Google Scholar]
- 20.Lee YS, Kennedy WD, Yin YW. Structural insight into processive human mitochondrial DNA synthesis and disease-related polymerase mutations. Cell. 2009;139(2):312–324. doi: 10.1016/j.cell.2009.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Foury F, Vanderstraeten S. Yeast mitochondrial DNA mutators with deficient proofreading exonucleolytic activity. EMBO J. 1992;11(7):2717–2726. doi: 10.1002/j.1460-2075.1992.tb05337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hu JP, Vanderstraeten S, Foury F. Isolation and characterization of ten mutator alleles of the mitochondrial DNA polymerase-encoding MIP1 gene from Saccharomyces cerevisiae. Gene. 1995;160(1):105–110. doi: 10.1016/0378-1119(95)00215-r. [DOI] [PubMed] [Google Scholar]
- 23.Trifunovic A, Wredenberg A, Falkenberg M, et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429(6990):417–423. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- 24.Kujoth GC, Hiona A, Pugh TD, et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309(5733):481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- 25.Longley MJ, Prasad R, Srivastava DK, Wilson SH, Copeland WC. Identification of 5′-deoxyribose phosphate lyase activity in human DNA polymerase gamma and its role in mitochondrial base excision repair in vitro. Proc Natl Acad Sci U S A. 1998;95(21):12244–12248. doi: 10.1073/pnas.95.21.12244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pinz KG, Bogenhagen DF. The influence of the DNA polymerase gamma accessory subunit on base excision repair by the catalytic subunit. DNA Repair (Amst) 2006;5(1):121–128. doi: 10.1016/j.dnarep.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 27.Murakami E, Feng JY, Lee H, Hanes J, Johnson KA, Anderson KS. Characterization of novel reverse transcriptase and other RNA-associated catalytic activities by human DNA polymerase gamma: importance in mitochondrial DNA replication. J Biol Chem. 2003;278(38):36403–36409. doi: 10.1074/jbc.M306236200. [DOI] [PubMed] [Google Scholar]
- 28.Lee HR, Johnson KA. Fidelity and processivity of reverse transcription by the human mitochondrial DNA polymerase. J Biol Chem. 2007;282(44):31982–31989. doi: 10.1074/jbc.M705392200. [DOI] [PubMed] [Google Scholar]
- 29.Lim SE, Longley MJ, Copeland WC. The mitochondrial p55 accessory subunit of human DNA polymerase gamma enhances DNA binding, promotes processive DNA synthesis, and confers N-ethylmaleimide resistance. J Biol Chem. 1999;274(53):38197–38203. doi: 10.1074/jbc.274.53.38197. [DOI] [PubMed] [Google Scholar]
- 30.Carrodeguas JA, Theis K, Bogenhagen DF, Kisker C. Crystal structure and deletion analysis show that the accessory subunit of mammalian DNA polymerase γ, PolyB, functions as a homodimer. Mol Cell. 2001;7(1):43–54. doi: 10.1016/s1097-2765(01)00153-8. [DOI] [PubMed] [Google Scholar]
- 31.Fan L, Kim S, Farr CL, et al. A novel processive mechanism for DNA synthesis revealed by structure, modeling and mutagenesis of the accessory subunit of human mitochondrial DNA polymerase. J Mol Biol. 2006;358(5):1229–1243. doi: 10.1016/j.jmb.2006.02.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yakubovskaya E, Lukin M, Chen Z, et al. The EM structure of human DNA polymerase γ reveals a localized contact between the catalytic and accessory subunits. EMBO J. 2007;26(19):4283–4291. doi: 10.1038/sj.emboj.7601843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Longley MJ, Nguyen D, Kunkel TA, Copeland WC. The fidelity of human DNA polymerase gamma with and without exonucleolytic proofreading and the p55 accessory subunit. J Biol Chem. 2001;276(42):38555–38562. doi: 10.1074/jbc.M105230200. [DOI] [PubMed] [Google Scholar]
- 34.Spelbrink JN, Li FY, Tiranti V, et al. Human mitochondrial DNA deletions associated with mutations in the gene encoding Twinkle, a phage T7 gene 4-like protein localized in mitochondria. Nat Genet. 2001;28(3):223–231. doi: 10.1038/90058. [DOI] [PubMed] [Google Scholar]
- 35.Ziebarth TD, Gonzalez-Soltero R, Makowska-Grzyska MM, Nunez-Ramirez R, Carazo JM, Kaguni LS. Dynamic effects of cofactors and DNA on the oligomeric state of human mitochondrial DNA helicase. J Biol Chem. 2010;285(19):14639–14647. doi: 10.1074/jbc.M109.099663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maier D, Farr CL, Poeck B, et al. Mitochondrial single-stranded DNA-binding protein is required for mitochondrial DNA replication and development in Drosophila melanogaster. Mol Biol Cell. 2001;12(4):821–830. doi: 10.1091/mbc.12.4.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Farr CL, Matsushima Y, Lagina AT, 3rd, Luo N, Kaguni LS. Physiological and biochemical defects in functional interactions of mitochondrial DNA polymerase and DNA-binding mutants of single-stranded DNA-binding protein. J Biol Chem. 2004;279(17):17047–17053. doi: 10.1074/jbc.M400283200. [DOI] [PubMed] [Google Scholar]
- 38.Yang C, Curth U, Urbanke C, Kang C. Crystal structure of human mitochondrial single-stranded DNA binding protein at 2.4 A resolution. Nat Struct Biol. 1997;4(2):153–157. doi: 10.1038/nsb0297-153. [DOI] [PubMed] [Google Scholar]
- 39.Chan SSL, Longley MJ, Copeland WC. Modulation of the W748S mutation in DNA polymerase γ by the E1143G polymorphism in mitochondrial disorders. Hum Mol Genet. 2006;15(23):3473–3483. doi: 10.1093/hmg/ddl424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Copeland WC. Inherited mitochondrial diseases of DNA replication. Annu Rev Med. 2008;59:131–146. doi: 10.1146/annurev.med.59.053006.104646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kasiviswanathan R, Longley MJ, Chan SS, Copeland WC. Disease mutations in the human mitochondrial DNA polymerase thumb subdomain impart severe defects in mitochondrial DNA replication. J Biol Chem. 2009;284(29):19501–19510. doi: 10.1074/jbc.M109.011940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Longley MJ, Clark S, Yu Wai Man C, et al. Mutant POLG2 disrupts DNA polymerase gamma subunits and causes progressive external ophthalmoplegia. Am J Hum Genet. 2006;78(6):1026–1034. doi: 10.1086/504303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ferraris S, Clark S, Garelli E, et al. Progressive external ophthalmoplegia and vision and hearing loss in a patient with mutations in POLG2 and OPA1. Arch Neurol. 2008;65(1):125–131. doi: 10.1001/archneurol.2007.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Walter MC, Czermin B, Muller-Ziermann S, et al. Late-onset ptosis and myopathy in a patient with a heterozygous insertion in POLG2. J Neurol. 2010;257(9):1517–1523. doi: 10.1007/s00415-010-5565-9. [DOI] [PubMed] [Google Scholar]
- 45.Vandenberghe W, Van Laere K, Debruyne F, Van Broeckhoven C, Van Goethem G. Neurodegenerative Parkinsonism and progressive external ophthalmoplegia with a Twinkle mutation. Mov Disord. 2009;24(2):308–309. doi: 10.1002/mds.22198. [DOI] [PubMed] [Google Scholar]
- 46.Hakonen AH, Goffart S, Marjavaara S, et al. Infantile-onset spinocerebellar ataxia and mitochondrial recessive ataxia syndrome are associated with neuronal complex I defect and mtDNA depletion. Hum Mol Genet. 2008;17(23):3822–3835. doi: 10.1093/hmg/ddn280. [DOI] [PubMed] [Google Scholar]
- 47.Holmlund T, Farge G, Pande V, Korhonen J, Nilsson L, Falkenberg M. Structure-function defects of the twinkle amino-terminal region in progressive external ophthalmoplegia. Biochim Biophys Acta. 2009;1792(2):132–139. doi: 10.1016/j.bbadis.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 48.Longley MJ, Humble MM, Sharief FS, Copeland WC. Disease variants of the human mitochondrial DNA helicase encoded by C10orf2 differentially alter protein stability, nucleotide hydrolysis and helicase activity. J Biol Chem. 2010;285(39):29690–29702. doi: 10.1074/jbc.M110.151795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Matsushima Y, Kaguni LS. Differential phenotypes of active site and human adPEO mutations in Drosophila mitochondrial DNA helicase expressed in Schneider cells. J Biol Chem. 2007;282(13):9436–9444. doi: 10.1074/jbc.M610550200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tyynismaa H, Mjosund KP, Wanrooij S, et al. Mutant mitochondrial helicase Twinkle causes multiple mtDNA deletions and a late-onset mitochondrial disease in mice. Proc Natl Acad Sci U S A. 2005;102(49):17687–17692. doi: 10.1073/pnas.0505551102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dimmock D, Tang LY, Schmitt ES, Wong LJ. Quantitative evaluation of the mitochondrial DNA depletion syndrome. Clin Chem. 2010;56(7):1119–1127. doi: 10.1373/clinchem.2009.141549. [DOI] [PubMed] [Google Scholar]
- 52.Langdon HM, Cadwalader WB. Chronic progressive external ophthalmoplegia: report of a case with necropsy. Trans Am Ophthalmol Soc. 1928;26:247–260. [PMC free article] [PubMed] [Google Scholar]
- 53.Zintz R, Villiger W. Electron microscopic findings in 3 cases of chronic progressive ocular muscular dystrophy [in German] Ophthalmologica. 1967;153(6):439–459. doi: 10.1159/000305086. [DOI] [PubMed] [Google Scholar]
- 54.Olson W, Engel WK, Walsh GO, Einaugler R. Oculocraniosomatic neuromuscular disease with “ragged-red” fibers. Arch Neurol. 1972;26(3):193–211. doi: 10.1001/archneur.1972.00490090019001. [DOI] [PubMed] [Google Scholar]
- 55.Lou HC, Reske-Nielsen E. Letter: Progressive external ophthalmoplegia: evidence for a disorder in pyruvate-lactate metabolism. Arch Neurol. 1976;33(6):455–456. doi: 10.1001/archneur.1976.00500060061014. [DOI] [PubMed] [Google Scholar]
- 56.Zeviani M, Servidei S, Gellera C, Bertini E, DiMauro S, DiDonato S. An autosomal dominant disorder with multiple deletions of mitochondrial DNA starting at the D-loop region. Nature. 1989;339(6222):309–311. doi: 10.1038/339309a0. [DOI] [PubMed] [Google Scholar]
- 57.Bau V, Zierz S. Update on chronic progressive external ophthalmoplegia. Strabismus. 2005;13(3):133–142. doi: 10.1080/09273970500216432. [DOI] [PubMed] [Google Scholar]
- 58.Edmond JC. Mitochondrial disorders. Int Ophthalmol Clin. 2009;49(3):27–33. [Google Scholar]
- 59.Suomalainen A, Majander A, Haltia M, et al. Multiple deletions of mitochondrial DNA in several tissues of a patient with severe retarded depression and familial progressive external ophthalmoplegia. J Clin Invest. 1992;90(1):61–66. doi: 10.1172/JCI115856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hong D, Bi H, Yao S, Wang Z, Yuan Y. Clinical phenotype of autosomal dominant progressive external ophthalmoplegia in a family with a novel mutation in the C10orf2 gene. Muscle Nerve. 2010;41(1):92–99. doi: 10.1002/mus.21439. [DOI] [PubMed] [Google Scholar]
- 61.Daut PM, Steinemann TL, Westfall CT. Chronic exposure keratopathy complicating surgical correction of ptosis in patients with chronic progressive external ophthalmoplegia. Am J Ophthalmol. 2000;130(4):519–521. doi: 10.1016/s0002-9394(00)00558-4. [DOI] [PubMed] [Google Scholar]
- 62.Alpers BJ. Diffuse progressive degeneration of the gray matter of the cerebrum. Arch Neurol Psychiatry. 1931;25(3):469–505. [Google Scholar]
- 63.Alpers BJ. Progressive cerebral degeneration of infancy. J Nerv Ment Dis. 1960;130(6):442–448. doi: 10.1097/00005053-196006000-00002. [DOI] [PubMed] [Google Scholar]
- 64.Huttenlocher PR, Solitare GB, Adams G. Infantile diffuse cerebral degeneration with hepatic cirrhosis. Arch Neurol. 1976;33(3):186–192. doi: 10.1001/archneur.1976.00500030042009. [DOI] [PubMed] [Google Scholar]
- 65.Naviaux RK, Nyhan WL, Barshop BA, et al. Mitochondrial DNA polymerase gamma deficiency and mtDNA depletion in a child with Alpers’ syndrome. Ann Neurol. 1999;45(1):54–58. doi: 10.1002/1531-8249(199901)45:1<54::aid-art10>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 66.Nguyen KV, Sharief F, Chan SSL, Copeland WC, Naviaux RK. Molecular diagnosis of Alpers syndrome. J Hepatology. 2006;45(1):108–116. doi: 10.1016/j.jhep.2005.12.026. [DOI] [PubMed] [Google Scholar]
- 67.Gordon N. Alpers syndrome: progressive neuronal degeneration of children with liver disease. Dev Med Child Neurol. 2006;48(12):1001–1003. doi: 10.1017/S0012162206002209. [DOI] [PubMed] [Google Scholar]
- 68.Delarue A, Paut O, Guys JM, et al. Inappropriate liver transplantation in a child with Alpers-Huttenlocher syndrome misdiagnosed as valproate-induced acute liver failure. Pediatr Transplant. 2000;4(1):67–71. doi: 10.1034/j.1399-3046.2000.00090.x. [DOI] [PubMed] [Google Scholar]
- 69.Kayihan N, Nennesmo I, Ericzon BG, Nemeth A. Fatal deterioration of neurological disease after orthotopic liver transplantation for valproic acid-induced liver damage. Pediatr Transplant. 2000;4(3):211–214. doi: 10.1034/j.1399-3046.2000.00115.x. [DOI] [PubMed] [Google Scholar]
- 70.Schwabe MJ, Dobyns WB, Burke B, Armstrong DL. Valproate-induced liver failure in one of two siblings with Alpers disease. Pediatr Neurol. 1997;16(4):337–343. doi: 10.1016/s0887-8994(97)00030-1. [DOI] [PubMed] [Google Scholar]
- 71.Wilson DC, McGibben D, Hicks EM, Allen IV. Progressive neuronal degeneration of childhood (Alpers syndrome) with hepatic cirrhosis. Eur J Pediatr. 1993;152(3):260–262. doi: 10.1007/BF01956158. [DOI] [PubMed] [Google Scholar]
- 72.Saneto RP, Lee IC, Koenig MK, et al. POLG DNA testing as an emerging standard of care before instituting valproic acid therapy for pediatric seizure disorders. Seizure. 2010;19(3):140–146. doi: 10.1016/j.seizure.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Van Goethem G, Martin JJ, Dermaut B, et al. Recessive POLG mutations presenting with sensory and ataxic neuropathy in compound heterozygote patients with progressive external ophthalmoplegia. Neuromuscul Disord. 2003;13(2):133–142. doi: 10.1016/s0960-8966(02)00216-x. [DOI] [PubMed] [Google Scholar]
- 74.Naess K, Freyer C, Bruhn H, et al. MtDNA mutations are a common cause of severe disease phenotypes in children with Leigh syndrome. Biochim Biophys Acta. 2009;1787(5):484–490. doi: 10.1016/j.bbabio.2008.11.014. [DOI] [PubMed] [Google Scholar]
- 75.Castro-Gago M, Blanco-Barca MO, Campos-Gonzalez Y, Arenas-Barbero J, Pintos-Martinez E, Eiris-Punal J. Epidemiology of pediatric mitochondrial respiratory chain disorders in northwest Spain. Pediatr Neurol. 2006;34(3):204–211. doi: 10.1016/j.pediatrneurol.2005.07.011. [DOI] [PubMed] [Google Scholar]
- 76.Van Goethem G, Schwartz M, Lofgren A, Dermaut B, Van Broeckhoven C, Vissing J. Novel POLG mutations in progressive external ophthalmoplegia mimicking mitochondrial neurogastrointestinal encephalomyopathy. Eur J Hum Genet. 2003;11(7):547–549. doi: 10.1038/sj.ejhg.5201002. [DOI] [PubMed] [Google Scholar]
- 77.Giordano C, Powell H, Leopizzi M, et al. Fatal congenital myopathy and gastrointestinal pseudo-obstruction due to POLG1 mutations. Neurology. 2009;72(12):1103–1105. doi: 10.1212/01.wnl.0000345002.47396.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Banchs I, Casasnovas C, Alberti A, et al. Diagnosis of Charcot-Marie-Tooth disease. J Biomed Biotechnol. 2009:10. doi: 10.1155/2009/985415. doi:10.1155/2009/985415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Szigeti K, Lupski JR. Charcot-Marie-Tooth disease. Eur J Hum Genet. 2009;17(6):703–710. doi: 10.1038/ejhg.2009.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Harrower T, Stewart JD, Hudson G, et al. POLG1 mutations manifesting as autosomal recessive axonal Charcot-Marie-Tooth disease. Arch Neurol. 2008;65(1):133–136. doi: 10.1001/archneurol.2007.4. [DOI] [PubMed] [Google Scholar]
- 81.Yamanaka H, Gatanaga H, Kosalaraksa P, et al. Novel mutation of human DNA polymerase γ associated with mitochondrial toxicity induced by anti-HIV treatment. J Infect Dis. 2007;195(10):1419–1425. doi: 10.1086/513872. [DOI] [PubMed] [Google Scholar]
- 82.Bailey CM, Kasiviswanathan R, Copeland WC, Anderson KS. R964C mutation of DNA polymerase γ imparts increased stavudine toxicity by decreasing nucleoside analog discrimination and impairing polymerase activity. Antimicrob Agents Chemother. 2009;53(6):2610–2612. doi: 10.1128/AAC.01659-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Haas RH, Parikh S, Falk MJ, et al. Mitochondrial disease: a practical approach for primary care physicians. Pediatrics. 2007;120(6):1326–1333. doi: 10.1542/peds.2007-0391. [DOI] [PubMed] [Google Scholar]
- 84.Haas RH, Parikh S, Falk MJ, et al. The in-depth evaluation of suspected mitochondrial disease. Mol Genet Metab. 2008;94(1):16–37. doi: 10.1016/j.ymgme.2007.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Valanne L, Ketonen L, Majander A, Suomalainen A, Pihko H. Neuroradiologic findings in children with mitochondrial disorders. AJNR Am J Neuroradiol. 1998;19(2):369–377. [PMC free article] [PubMed] [Google Scholar]
- 86.Bianchi MC, Tosetti M, Battini R, et al. Proton MR spectroscopy of mitochondrial diseases: analysis of brain metabolic abnormalities and their possible diagnostic relevance. AJNR Am J Neuroradiol. 2003;24(10):1958–1966. [PMC free article] [PubMed] [Google Scholar]
- 87.Lin DD, Crawford TO, Barker PB. Proton MR spectroscopy in the diagnostic evaluation of suspected mitochondrial disease. AJNR Am J Neuroradiol. 2003;24(1):33–41. [PMC free article] [PubMed] [Google Scholar]
- 88.Dinopoulos A, Cecil KM, Schapiro MB, et al. Brain MRI and proton MRS findings in infants and children with respiratory chain defects. Neuropediatrics. 2005;36(5):290–301. doi: 10.1055/s-2005-872807. [DOI] [PubMed] [Google Scholar]
- 89.Lesnefsky EJ, Moghaddas S, Tandler B, Kerner J, Hoppel CL. Mitochondrial dysfunction in cardiac disease: ischemia—reperfusion, aging, and heart failure. J Mol Cell Cardiol. 2001;33(6):1065–1089. doi: 10.1006/jmcc.2001.1378. [DOI] [PubMed] [Google Scholar]
- 90.Scaglia F, Towbin JA, Craigen WJ, et al. Clinical spectrum, morbidity, and mortality in 113 pediatric patients with mitochondrial disease. Pediatrics. 2004;114(4):925–931. doi: 10.1542/peds.2004-0718. [DOI] [PubMed] [Google Scholar]
- 91.Gauthier-Villars M, Landrieu P, Cormier-Daire V, et al. Respiratory chain deficiency in Alpers syndrome. Neuropediatrics. 2001;32(3):150–152. doi: 10.1055/s-2001-16614. [DOI] [PubMed] [Google Scholar]
- 92.Debray FG, Mitchell GA, Allard P, Robinson BH, Hanley JA, Lambert M. Diagnostic accuracy of blood lactate-to-pyruvate molar ratio in the differential diagnosis of congenital lactic acidosis. Clin Chem. 2007;53(5):916–921. doi: 10.1373/clinchem.2006.081166. [DOI] [PubMed] [Google Scholar]
- 93.Barshop BA. Metabolomic approaches to mitochondrial disease: correlation of urine organic acids. Mitochondrion. 2004;4(5–6):521–527. doi: 10.1016/j.mito.2004.07.010. [DOI] [PubMed] [Google Scholar]
- 94.Bai R-K, Wong L-JC. Detection and quantification of heteroplasmic mutant mitochondrial DNA by real-time amplification refractory mutation system quantitative PCR analysis: a single-step approach. Clin Chem. 2004;50(6):996–1001. doi: 10.1373/clinchem.2004.031153. [DOI] [PubMed] [Google Scholar]
- 95.Wong LJ. Molecular genetics of mitochondrial disorders. Dev Disabil Res Rev. 2010;16(2):154–162. doi: 10.1002/ddrr.104. [DOI] [PubMed] [Google Scholar]
- 96.Coon KD, Valla J, Szelinger S, et al. Quantitation of heteroplasmy of mtDNA sequence variants identified in a population of AD patients and controls by array-based resequencing. Mitochondrion. 2006;6(4):194–210. doi: 10.1016/j.mito.2006.07.002. [DOI] [PubMed] [Google Scholar]