

Summary
PknB is an essential serine/threonine kinase of Mycobacterium tuberculosis with possible roles in a number of signalling pathways involved in cell division and metabolism. We screened a library of >50,000 compounds for inhibitors of the in vitro phosphorylation of GarA (Rv1827) by PknB and identified a number of inhibitors. A program of synthetic medicinal chemistry was subsequently conducted around one class of inhibitors and was successful in generating ATP competitive inhibitors with potency in the nanomolar range. Compounds in this class showed cross-reactivity with the related M. tuberculosis kinase, PknF, but not with PknG in an in vitro autophosphorylation assay. These synthesised inhibitors were able to prevent the growth of M. tuberculosis in an Alamar blue assay and in an intracellular model of infection, but only in the micromolar range. We attempted to determine if cell wall permeability was an explanation for the discrepancy between the potent in vitro compared with relatively poor in vivo activity, but found no evidence that the activity of the inhibitors could be improved by weakening the cell wall. Despite a number of drug discovery efforts attempting to develop inhibitors against PknB, it is yet to be reported that any such inhibitors prevent mycobacterial growth at submicromolar concentrations.
Keywords: Mycobacterium tuberculosis, Kinases, PknB, Inhibitors
1. Introduction
Mycobacterium tuberculosis remains one of the world’s most devastating pathogens, with more than 13 million people suffering from an active tuberculosis infection and 1.8 million resulting deaths in 2008 alone.1 The emergence of multi-drug and extensively drug resistant strains has highlighted the need for new drugs to treat tuberculosis. Recent studies have focused on finding new pathways vulnerable to inhibition by small molecules and previously unexploited by drug discovery efforts. The inhibition of signalling pathways both in M. tuberculosis and the host may yield new classes of drug targets and a large amount of recent work has focused on developing this further.
Target based drug discovery, in which there is in vitro high throughput screening of a large number of small molecules against a validated target, has been used on a number of occasions to search for new anti-tuberculosis agents. We sought to find inhibitors of an essential M. tuberculosis serine/threonine protein kinase, PknB. Kinases are attractive as drug targets due to the range of crucial cellular processes in which they are involved. There has been much interest in developing ATP competitive kinase inhibitors for the treatment of cancer, a hallmark of which is often aberrant kinase activity. A large number of small molecule kinase inhibitors have been developed as potential anti-cancer drugs and there is a huge amount of interest in developing kinase inhibitors to treat a range of conditions.2 Kinase-focused libraries of small molecule inhibitors have been built-up as a result of these studies and a large amount of knowledge has been gained on the action of kinase inhibitors. The early success stories from the development of eukaryotic kinase inhibitors suggested that similar drugs could be developed to treat bacterial infections. The M. tuberculosis serine/threonine protein kinases (STPKs) are attractive targets partly because of the inferred importance of serine/threonine phosphorylation in M. tuberculosis: M. tuberculosis is unique within the bacterial world in having a much higher number of STPKs compared to the more common two-component signalling systems.3–6 Mycobacterium leprae, during extensive reductive evolution, has lost all but four of these kinases: PknA, PknB, PknG and PknL.7 PknA and PknB are essential in M. tuberculosis,8–10 while PknG has been reported to be involved in survival in the macrophage, either as a result of a role in glutamate metabolism11 or via modulation of host cell trafficking pathways.12
Previous studies have focused on the potential of PknB and PknG as drug targets in M. tuberculosis, although the majority have reported significantly less impressive activity against whole cells than against the purified protein in vitro.8,12–14 Despite this, there can be no doubt, from a number of lines of evidence, that PknB carries out function(s) essential to M. tuberculosis, and possesses a number of properties that suggest it is suitable for development of novel drugs: a crystal structure has also been determined for PknB15,16 and the kinase domain exhibits less than 30% similarity with eukaryotic kinases, which has positive implications for developing a drug specific for the bacterial kinases and not those of the host. The pknB gene (Rv0014c) is part of an operon highly conserved among the actinomycetes and also encoding pknA, the other essential kinase; pstP, a phosphoserine/threonine phosphatase; rodA, a cell division protein; pbpA; a protein involved in peptidoglycan biosynthesis; and two genes (fhaB and fhaA) coding for forkhead-associated proteins. Over-expression or depletion of this kinase results in gross changes in cell morphology, likely a result of the role of PknB in the phosphorylation-mediated regulation of a number of proteins involved in cell wall biosynthesis.10,17,18 In addition, both PknB and PknG are known to phosphorylate a protein involved in the regulation of glutamate metabolism,19–21 suggesting that an inhibitor of one or both of these kinases could also impact on the ability of M. tuberculosis to adequately regulate its central metabolic processes. Targeting of these bacterial kinases would therefore be a way of inhibiting evolutionarily-conserved steps in central metabolic processes.
We screened for small molecule inhibitors of PknB and, as a result of a medicinal chemistry program (manuscript in preparation), our lead compounds were able to inhibit PknB activity in vitro in the nanomolar range. However, the potency of our compounds against whole cells in culture or in a macrophage model of infection was two orders of magnitude lower than expected from the in vitro potency. An often suggested explanation for low anti-tuberculosis activity is the problem of cell wall permeability. Since the M. tuberculosis cell wall presents an extremely hydrophobic barrier which can impede the entry of drugs into the cell, we sought to determine if cell wall permeability might explain the difficulties in improving the potency of our PknB inhibitors. In addition, we investigated the role of efflux pumps, protein binding in the assay media and inhibitor specificity as alternative explanations.
2. Materials and methods
2.1. Compounds
Compounds for the high throughput screening included the MRCT compound collection comprising 45,000 diverse templates from commercially available collections, as well as 6400 kinase-focused templates (Biofocus DPI, Cambridge, UK) selected on the basis of bio-informatics provided by the crystal structure of PknB and other serine/threonine kinases. Focused libraries, based around compounds identified from the initial screen, were subsequently generated by synthetic medicinal chemistry.
2.2. Protein expression and purification
GarA was expressed in Escherichia coli and purified as described.22 The 279 residue kinase domain of PknB and the 292 residue kinase domain of PknF were expressed as 3C-protease cleavable GST-fusions in E. coli Rosetta 2 (DE3) pLysS cells. The proteins were purified with glutathione Sepharose 4B resin (GE Healthcare) and the GST-tag cleaved from the protein prior to elution using 3C protease (GE Healthcare).
2.3. High throughput protein kinase assays
A non-radioactive PknB kinase assay was developed to follow ATP depletion during the PknB-catalyzed phosphorylation of GarA (Rv1827). The Promega ‘Kinase Glo’ assay kit was used to measure levels of phosphorylation via detection of the remaining ATP after the completion of the kinase reaction. Briefly, luciferase and beetle luciferin were introduced to the reaction mix and, in the presence of Mg2+, ATP and oxygen, the oxygenation of luciferin generates one photon of light per turnover. PknB activity is inversely proportional to the intensity of the luminescence signal. Using this assay format the Km of ATP was determined to be 1.5 μM and ATP was used at this concentration for screening.
The following reaction mix was prepared: 0.15 μM PknB and 5 μM Rv1827 in reaction buffer (50 mM Tris–HCl, 150 mM NaCl, 1 mM DTT and 0.01% Triton X-100). The assay was carried out in 22 μl volumes in 384-well white plates (Matrix, 4316). A Beckman Biomek Fx liquid handling robot was used to dispense premixed reaction mix into the wells and the 2 μl of the compound/DMSO added to a final concentration of 10 μM/1% DMSO. The plates were incubated for 30 min and the reaction initiated with the addition of 1 mM MnCl2/1.5 μM ATP using the Perkin Elmer Flexdrop non-contact liquid dispenser. The reaction was stopped after 20 min with the addition of 22 μl Kinase Glo reagent and luminescence read with a BMG Polarstar reader. Control wells with and without PknB were included on every plate in columns 1, 2 and 23, 24 and were used to calculate Z′ values.23 Data from any plates with Z′<0.5 were rejected and were repeated.
2.4. Strains and growth conditions
M. tuberculosis H37Rv, Mycobacterium smegmatis mc2155, M. smegmatis ΔfbpA (gift from C. Thompson), and M. tuberculosis ΔompA (gift from P. Draper) cultures were grown at 37 °C in Dubos broth supplemented with 0.05% (vol/vol) Tween 80, 0.2% (vol/vol) glycerol, and 4% (vol/vol) Dubos medium albumin (Becton Dickinson). M. smegmatis strains were grown either in 5 ml volumes in 20 ml universals or in 50 ml volumes in 250 ml flasks, incubated in an orbital shaker at 150 rpm at 37 °C. M. tuberculosis was grown either in 100 ml volumes in a Bellco roll-in incubator (2 rpm) or in 10 ml volumes in static universals. Kanamycin was used at a final concentration of 25 μg ml−1. M. tuberculosis was grown on 7H11 supplemented with 10% OADC enrichment (Middlebrook) and 0.5% (vol/vol) glycerol.
2.5. M. tuberculosis minimum inhibitory concentration (MIC) determination
MICs were determined for M. tuberculosis using a 96-well plate method with Alamar blue, as described previously.24,25 A modified version of the assay used M. smegmatis with a 3 day incubation. The cell wall weakening and efflux pump inhibition assays were modifications of the Alamar blue assay, as described in the results. Z′ values were used as a measure of assay quality, using the following formula: Z′ = 1–3 × SSD/R, where SSD is the sum of the Standard Deviation of the negative controls and Standard Deviation of the positive controls, and R is the mean of the maximum signal control minus the mean of the negative signal control.23
2.6. Intracellular activity of PknB inhibitors in a macrophage infection by M. tuberculosis
Intracellular inhibition was tested in murine bone marrow-derived macrophages at a range of concentrations as previously described.25 Briefly, monocytes were isolated from Balb/c bone marrow and differentiated with L-cell supernatant for 5–7 days. The macrophages were infected with M. tuberculosis with an MOI of 0.5 bacteria: 1 macrophage for 4 h, and the infection incubated in the presence of various concentrations of the inhibitors for 5 days. At this point, the macrophages were lysed with dH20 + 0.05% Tween 80 and plated for CFU enumeration. Prior to the infection, the compounds were tested to rule out any effect of toxicity against the macrophages. Alamar blue was used to determine macrophage viability after incubation with the inhibitors (not shown).
2.7. Kinase autophosphorylation assay
In vitro autophosphorylation of PknA, PknB, PknG and PknF (1 μg) was carried out in 20 μl reaction mixture containing 2.5 μl buffer P (25 mM Tris–HCl, pH 7.0, 1 mM dithiothreitol, 5 mM MgCl2, 1 mM EDTA). The reaction was initiated with the additional of 0.08 μCi [γ-32P] ATP and incubated at 37 °C for 30 min. The reaction was terminated with the addition of SDS-PAGE sample buffer and heating the mixture at 100 °C for 5 min. The reaction mixtures were analyzed by SDS-PAGE. The gels were soaked in 20% tricholoroacetic acid for 10 min at 90 °C and dried. Radioactive proteins were visualized by autoradiography and quantified by Phosphorimage analysis using a STORM 840 Optical Scanner System and ImageQuant (version 5.2, Molecular Dynamics, Sunnyvale, CA). Images were adjusted for contrast and brightness using Adobe Photoshop.
2.8. Radioactive competition assay
Reactions were set up in a volume of 17 μl kinase buffer (50 mM Tris–HCl pH7.2, 1 mM DTT, 0.01% TX-100) containing 100 nM purified PknB and 3 μM purified Rv1827 and increasing amounts of each inhibitor diluted in neat DMSO (from 0.03 μM to 100 μM) in polypropylene U-bottomed 96 well plates. The reactions were started by the addition of 5 μl of solutions containing MnCl2 and ATP at Km or 50xKm, to achieve final concentrations of 1 mM MnCl2 with 1.5 μM ATP and 0.1 μCi 33P or 75 μM ATP and 1 μCi 33P.
After incubating for 70 min at room temperature, reactions were terminated by the addition of 20 μl 50% orthophosphoric acid. Reactions were then transferred to a 96 well glass filter capture plate (GF/C) and unbound 33P was washed away with 200 μl 1.5% orthophosphoric acid, using a Tomtec Harvester 9600. After drying overnight at room temperature, 25 μl scintillant was added to each well and quantification of Rv1827 phosphorylation achieved by detecting incorporated 33P with the PE Topcount.
2.9. Kinase profiling against a panel of mammalian kinases
This was carried out using a 33P-ATP filter-binding assay26 on a panel of 70 mammalian kinases at the National Centre for Protein Kinase Profiling in the MRC Protein Phosphorylation Unit at the University of Dundee.
2.10. Docking studies with PknB inhibitors
The mitoxantrone bound crystal structure (PDB:2FUM) was selected for docking studies owing to its greater suitability in terms of ligand-induced binding site architecture as compared to the ATP bound structure (PDB:1MRU). Protein preparation, grid generation and docking (GlideSP) were all carried out using the Schrodinger molecular modelling suite (Schrodinger LLC: New York. http://www.schrodinger.com).
3. Results
3.1. High throughput screening for PknB inhibitors
PknB has been previously validated as a suitable target for drug discovery and we sought to identify small molecule inhibitors targeted against this kinase. A number of potential substrates for PknB were tested, with GarA (Rv1827) being chosen as the most strongly phosphorylated protein in our assay. GarA has been described as an in vivo target for both PknB and PknG19,20 and interaction with this target is known in some detail.22,27 A non-radioactive PknB kinase assay was developed to follow ATP depletion during the PknB-catalyzed phosphorylation of GarA. Substrate Kms were determined for both ATP (1.5 μM) and Rv1827 (>20 μM). The Km ATP concentrations and an excess of peptide (5 μM) were selected to achieve a robust assay window >0.5 within the linear phase of the reaction, maximizing sensitivity to ATP competitive inhibitors.
In total ∼54,000 compounds were screened against PknB. The compound collection was sourced by MRCT from commercial suppliers. Drug-like filters to assess both physicochemical properties and known toxicophores were applied to the selection process. The MRCT library comprised ∼44,000 diverse compound collection and ∼9000 kinase-focused collection (targeting ATP binding site). In addition, ∼1000 natural products (Phytoquest) were screened. The average Z′ was 0.76 and a confirmed hit rate of 0.14% was observed. In total, 76 compounds confirmed IC50 in the range 1–200 μM.
Limitations of the Kinase glo assay format did not permit detailed enzyme kinetics to be performed. However using a radioactive assay, a number of compounds were assessed for ATP competitiveness by measuring IC50 at a high (75 μM) and a low (1.5 μM, Km) ATP concentration. From the Cheng–Prussoff equation28 we would predict that for an ATP competitive inhibitor, a 25-fold shift in IC50 would be seen between ATP at Km (1.5 μM) and 50XKm (75 μM). Five compounds from the series, including the original screen hit, with a range of potencies 50–1000 nM in the primary assay were selected. Fold shifts in the range 15.5–32 were observed for all compounds, consistent with an ATP competitive mechanism. It was expected that, due to the higher ATP concentration in the cells compared to the in vitro assay, a shift in MIC compared to IC50 enzyme activity would be observed. However, a consistent shift was not observed.
Docking studies (Figure 1) indicate that the compounds should be ATP competitive. They bind to the hinge region via two H-bonds between the ligand core and the peptide backbone of Val95 along with an additional H-bond via the ligand pyrazole NH to the peptide carbonyl of Glu93. Numerous Van Der Waals contacts contribute to binding, most notably between the ligand cyclopropyl group in the hydrophobic pocket towards the Met92 gatekeeper and also via extensive contact along the glycine rich loop at the entrance to the binding site.
Figure 1.

Docking of MRCT67127 into PknB active site. Green ribbon represents PknB (Code: 1MRU) with ATP co-crystallised (shown as green structure). Grey ribbon represents PknB (Code: 2FUM) with mitoxantrone co-crystallised. The mitoxantrone structure was removed and docking studies conducted with this PknB conformation: owing to the flat nature of the docked compounds (making them similar to mitoxantrone), this was considered a better way to obtain reliable results. MRT67127 is shown (grey structure) docked into PknB.
3.2. Anti-tuberculosis activity of lead compounds
Compounds that showed potent activity against PknB in vitro, no cytotoxicity and good selectivity against a panel of mammalian kinases, were tested in an M. tuberculosis Alamar blue MIC assay. Briefly, 2-fold dilutions of the inhibitors were incubated at 37 °C with approximately 1 × 105 cells per well in 96-well microplates for 7 days. Untreated M. tuberculosis and media only controls were included on every plate, and the control drugs isoniazid, streptomycin and ethambutol were tested in parallel with each batch of PknB inhibitors to ensure that the assay was reproducible. The Alamar blue reagent was added on the final day of the incubation, and the fluorescence measured at the completion of the experiment. A 90% inhibition of fluorescence compared to the untreated control was chosen as the cutoff for the determination of inhibitory concentrations. The statistical parameter, Z′-factor, was used as a measure of assay quality, with a score greater than 0.75 taken as an indication of good data.
MICs were determined for more than 100 compounds, with a range of MICs being observed varying between inactive at 250 μM to inhibitory at 8–16 μM. The compounds were also tested against M. smegmatis (data not shown) with a generally good agreement between the two assays. This confirms previously published experiments (e.g.29) which have suggested that testing compounds against M. smegmatis is a good model for TB drug discovery. There was a poor correlation between IC50 against the purified recombinant protein and anti-M. tuberculosis MIC, with the majority of compounds proving to be inactive against the whole cells. The lack of correlation between protein and whole cell activity is illustrated in Figure 2. Despite extensive rounds of chemistry, unfortunately it was not possible to reduce the M. tuberculosis MICs below the 10 μM level.
Figure 2.

Compounds inactive against M. tuberculosis had a range of IC50s against the purified protein. A similar spread was observed for the compounds able to inhibit M. tuberculosis. Starred section indicated the area of the graph where compounds highly active against both the purified protein and whole cells would appear.
3.3. Intracellular activity of PknB inhibitors against M. tuberculosis in a bone marrow derived macrophage model of infection
A subset of compounds was chosen for further investigation. The key criteria in this selection included reasonable activity against M. tuberculosis in vitro, a low MIC to IC50 ratio as a possible indication of good cell wall permeability, good selectivity against a panel of human kinases and good physicochemical parameters, including moderate logD and low hydrogen bond donor count, both of which are requirements for good membrane permeability. The intracellular activity of these compounds against M. tuberculosis in a bone marrow derived macrophage model of infection was tested.
The compounds showed a dose dependant inhibition of intracellular M. tuberculosis, although a number of the compounds were not able to inhibit 99% growth compared to the untreated control (chosen as the cutoff for determining intracellular inhibition). Figure 3 shows the inhibition of intracellular growth by the 12 PknB inhibitors chosen for further investigation. At the lowest concentration tested, 2.5 μM, none of the compounds were able to inhibit growth by more than 99% of the control. A number of the compounds were active at the higher concentrations of 10 and 20 μM. While concentrations up to 100 μM were tested, these are not shown due to a number of the compounds proving to have toxic effects against the macrophages, possibly due to inhibition of host kinases.
Figure 3.

Intracellular activity of PknB inhibitors in a bone marrow derived macrophage model. Macrophages were infected with an MOI of 0.5:1 and incubated in the presence of a dilution series of the inhibitors. After 5 days, the macrophages were lysed and the intracellular bacteria enumerated by CFU platings. Data is expressed as percentage survival compared to the untreated control. Inhibitor structures and corresponding in vitro activity against purified protein are shown in Table 1.
A summary of the data is shown in Table 1. Little correlation between intracellular activity and whole cell inhibition was found.
Table 1.
Structures and inhibitor activity against purified PknB and M. tuberculosis in an Alamar blue and an intracellular assay. IC50 is shown as the mean of three experimental repeats, Alamar blue and intracellular MIC is representative of three or more repeats.
| Inhibitor | Structure | IC50 PknB μM | MIC M. tuberculosis μM∗ | MIC Intracellular μM† |
|---|---|---|---|---|
| 1.MRT67127 | 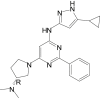 |
0.053 | 32 | 5 |
| 2.MRT67153 | 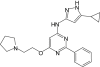 |
0.056 | 32 | 20 |
| 3.MRT68667 | 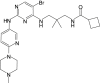 |
0.09 | 64 | 20 |
| 4.MRT68606 | 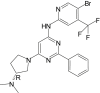 |
0.519 | 16 | >20 |
| 5.MRT68572 | 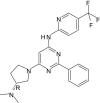 |
16.554 | 16 | >20 |
| 6.MRT68634 | 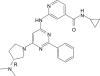 |
1.499 | 64 | >20 |
| 7.MRT67131 | 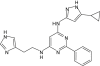 |
0.345 | 125 | >20 |
| 8.MRT67150 | 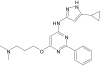 |
0.055 | 32 | 5 |
| 9.MRT67155 | 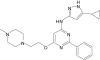 |
0.096 | 64 | 10 |
| 10.MRT67156 | 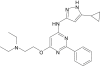 |
0.092 | 16 | 5 |
| 11.MRT67319 | 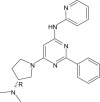 |
6.194 | 32 | 10 |
| 12.MRT68664 | 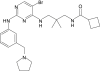 |
0.065 | 64 | 5 |
90% inhibition fluorescence compared to untreated control.
99% inhibition compared to untreated control.
3.4. Role of cell wall permeability and efflux on inhibitor MIC
Poor anti-tuberculosis activity by compounds that are potent in the in vitro protein assay is often attributed to impermeability of the M. tuberculosis cell wall. M. tuberculosis has a thick and ‘waxy’ cell wall, which limits the ability of compounds to penetrate and reach the cytoplasm.30 Much of the impermeability stems from the unique mycolic acid layer. While little is known about the specific properties of small molecules that enable them to diffuse across the M. tuberculosis cell wall, it is possible to modify certain functional groups of the compounds in ways that will hopefully improve entry into the cytoplasm. For example, modifications were made to our more active compounds in an attempt to vary the compounds’ LogP values and increase their hydrophobicity (to be discussed in a separate medicinal chemistry paper). In one case, it was possible to improve the MIC/IC50 ratio using this approach. However, the resulting activity of the compounds against M. tuberculosis was still poor. Before embarking on further chemistry with the hope of improving cell penetration, we attempted to determine whether cell wall permeability truly was a major contribution to low MIC values through the use of several cell wall-weakened models with the subset of inhibitors shown in Figure 3.
A number of chemicals and antibiotics were tested, including Triton X-100, glycine, lysozyme, isoniazid and ethambutol. There have been previous reports of sub-lethal concentrations of antibiotics targeted to the cell wall being able to enhance permeability of certain compounds.31,32 In particular, ethambutol has been demonstrated to enhance the activity of rifampicin, streptomycin and isoniazid against drug resistant strains.33 In our experiments, the cell wall weakening agent was used at 0.5 MIC in conjunction with the PknB inhibitors to determine whether the inhibitors were better able to inhibit M. tuberculosis in the presence of a weakened cell wall. As a control, we demonstrated that the MIC of rifampicin was reduced by 4-fold in the presence of sub-inhibitory concentrations of ethambutol, confirming published data that reported that accumulation of and sensitivity to rifampicin is enhanced by ethambutol.34 We tested a number of compounds, including the subset of 12 active compounds and a number that were previously inactive against M. tuberculosis. Several compounds were able to inhibit M. tuberculosis in the presence of ethambutol when they had been previously inactive. This implied that cell wall permeability was, indeed, a problem for some structural families. However, the MICs of the most efficacious compounds were not improved by ethambutol treatment.
In addition to the ethambutol cell-wall weakened model, we wanted to use a more defined model. An M. smegmatis fbpA mutant has been shown to possess increased sensitivity to a number of antibiotics due to alterations in its cell wall.35 FbpA is involved in mycolic acid biosynthesis and the mutant has a loss of cell wall hydrophobicity compared with the wild type. We tested the sensitivity of this mutant to the PknB inhibitors and, in agreement with the ethambutol data, there was no improvement in inhibition with a weakened cell wall. In comparison, vancomycin MIC was improved by 16-fold and erythromycin by 8-fold. This effect was specific to certain antibiotics as the MICs and streptomycin and ethambutol were not improved in the assay.
After testing the role of the mycolic acid layer in drug permeability, we sought also to investigate other possible paths of entry into the cell. Antibiotics can either diffuse across the cell wall, a possibility investigated in the experiments detailed above, or alternatively may enter through hydrophilic channels. Porins in the mycobacterial outer membrane are required for the diffusion of hydrophilic solutes and are the route of entry for small and hydrophilic antibiotics such as isoniazid and ethambutol.36 It has been demonstrated that an mspA porin deletion mutant of M. smegmatis has a multi-drug resistant phenotype to antibiotics including ampicillin.37 Although M. tuberculosis does not encode Msp-like proteins, expression of M. smegmatis MspA in M. tuberculosis enhances antibiotic sensitivity38 and an M. tuberculosis outer membrane channel-forming protein has been demonstrated to restore antibiotic sensitivity to a M. smegmatis mspA mutant.39 One of the best understood porins in M. tuberculosis is OmpA, that has been linked to the diffusion of pyrazinamide across the cell wall.40 M. tuberculosis ompA mutants have not been reported to have an antibiotic resistant phenotype similar to M. smegmatis mspA. Therefore, we tested the sensitivity of the mutant to a number of antibiotics compared to the wild type strain. Unlike M. smegmatis mspA, M. tuberculosis ompA did not have increased sensitivity to the small, hydrophilic antibiotics tested. However, it did show a slightly increased resistance to the large antibiotics vancomycin and erythromycin. This result was also observed for M. smegmatis mspA, despite both of these antibiotics being too large to enter the cell via porins. It is likely that alterations in the cell wall have a knock-on effect on entry of large antibiotics into the cell. This experiment does not provide any evidence that small antibiotics enter M. tuberculosis via OmpA channels, but we cannot rule out that OmpA is involved in the uptake of other antibiotics not tested. We also tested the ompA mutant with the PknB inhibitors and observed that the majority of them were equally active against the wild type and mutant.
The activity of efflux pump inhibitors has been shown to play a role in the acquired drug resistance of M. tuberculosis for a number of antibiotics,41–44 due to the upregulation of efflux pumps in drug resistant strains. It has been suggested that the use of efflux pump inhibitors in conjunction with anti-tuberculosis therapy could be used to reverse the efflux pump-mediated drug resistance in some infections.45 There is also some evidence that efflux plays a role in the intrinsic drug resistance of mycobacteria.46–48 Therefore, we tested whether the addition of sub-inhibitory concentrations of the efflux pump inhibitors reserpine and verapamil to our assay would improve the PknB inhibitor MICS. No change was observed in MIC at either 20 or 40 μg/ml of either efflux pump inhibitor, while the MIC of the ethidium bromide control was decreased by 4-fold. This is perhaps not surprising due to the observation that the effect of efflux inhibitors is most dramatic when there is overexpression or upregulation of efflux pumps, rather than against wild type levels.34,45
3.5. Specificity of PknB inhibitors
Attempts to select resistant mutants of the 12 most potent PknB inhibitors were unsuccessful, possibly due to either the high MICs of the compounds, or non-specific activity of the inhibitors. We plated 5 × 109 M. tuberculosis directly on to media containing 2, 4 and 8×MIC inhibitors as well as pre-growing the cells on 0.5 and 1×MIC before replating on the above concentrations. Neither method generated any resistant colonies. This, combined with the lack of correlation between MIC and activity in the in vitro protein assay led us to consider whether the inhibitors are truly targeting PknB. It is possible that some of the anti-tuberculosis activity of the inhibitors is due to off-target activity, meaning that further PknB-focused chemistry based around these structural families is unlikely to lead to an improvement in MIC. Alternatively, despite good selectivity over mammalian kinases, the inhibitors could be binding to a number of the other more closely related M. tuberculosis kinases, and it is possible that the inhibitor is being sequestered away from PknB by its activity against any of the eight non-essential kinases in M. tuberculosis.
We used an in vitro radioactivity based screen to measure the ability of the compounds to inhibit autophosphorylation of some of the other M. tuberculosis kinases. PknA, the other essential kinase, was tested but, due to high levels of stable phosphorylation of this kinase following purification, it was not sufficiently active in the assay. Due to the essential nature of PknA, we were not concerned about possible cross-reactivity of the PknB inhibitors with this kinase. In fact, this could be advantageous for the development of a TB drug. We also tested the ability of the inhibitors to inhibit PknF and PknG autophosphorylation. PknG is a soluble kinase while PknF is membrane-associated like PknB; both are non-essential.
It was observed that there is a high degree of cross-reactivity between the PknB inhibitors and PknF, the other membrane-associated kinase tested. However, PknG was less strongly inhibited at the concentrations tested (Figure 4). A possible explanation for this is the 39.5% sequence identity shared between PknB and PknF compared to only 27.3% between PknB and PknG.
Figure 4.

In vitro activity of the PknB inhibitors against PknF and PknG. The inhibitors were tested at 1 μM (not shown) and 10 μM for their ability to inhibit PknB, PknF and PknG in an autophosphorylation assay. There was good agreement between the anti-PknB activity in this assay and the non-radioactive assay used in the high throughput screen. The graph was created using the averaged band intensities from three independent autoradiographs.
4. Discussion
PknB has been shown to be an essential protein in M. tuberculosis,8 and is proposed to be involved in a number of vital pathways. Initially, it was implicated as having a role in cell division due to its chromosomal location within an operon containing genes known to be important for these processes. Depletion or overexpression of PknB results in gross changes in cell morphology, and some of its identified substrates are known to be important for cell wall and division-related functions.10 The cell wall is known to be a strong target for anti-tuberculosis drugs, due to its importance for the survival of the pathogen and its uniqueness. PknB is most strongly expressed during exponential growth10 and upon infection of a THP-1 macrophage cell line,49 suggesting that the kinase is involved in general growth and important during an infection. More recently, it has been seen that one of PknB’s substrates, GarA, has a role in intermediary metabolic processes. GarA negatively regulates two proteins involved in metabolism, 2-hydroxy-3-oxoadipate synthase and glutamate dehydrogenase and is itself negatively regulated via phosphorylation by PknB.20–22 Interestingly, GarA is also known to be phosphorylated in vivo by PknG on a neighbouring threonine to that phosphorylated by PknB.20
The known functions of PknB detailed above do suggest that this kinase would be a strong candidate for an anti-tuberculosis drug. We found a number of inhibitors able to inhibit PknB in vitro with IC50s in the 20 nM range. The MICs of these inhibitors against M. tuberculosis were found to be in the 16 μM to inactive range, despite several rounds of compound optimization. Some shift in MIC compared to IC50 was expected due to higher intracellular concentrations of ATP leading to competition. However, a consistent shift was not observed across the series, suggesting additional mechanisms are involved in the poor activity against whole cells. It should also be noted that the intracellular concentration of ATP in M. tuberculosis is in the micromolar range50 (A. Chang, unpublished data), which is significantly lower than that the accepted millimolar concentrations for mammalian cells. The poor MIC to IC50 ratios appear to be a similar result to the published data of other groups who have also attempted to develop PknB or PknG inhibitors.8,12–14 This poor activity was evident even in an intracellular assay.
We investigated the role of cell wall permeability, either diffusion through the hydrophobic cell wall or diffusion through hydrophilic channels, and found no evidence that this was a limiting step in the action of the inhibitors. We also investigated the role of efflux and observed that inhibitor activity was not improved in an efflux pump inhibited model.
A possibility is that the more potent inhibitors, which were chosen for compound optimization, were not targeting PknB in the cells. A similar problem was described by Payne et al.51 in their review of GlaxoSmithKline’s 7 years of antibacterial high throughput screen (HTS) research. They attributed the antibacterial activity of some of their lead compounds to non-specific membrane interactions that led to cell lysis. We did attempt to determine the in vivo target of the inhibitors by generating resistant mutants, however this proved unsuccessful. In a previous study, Fernandez et al. used M. smegmatis overexpressing PknB to demonstrate that an increase in PknB levels was able to increase resistance to their PknB inhibitors.8 However, they were only able to see a modest 2-fold increase in resistance compared to the control strain. Our attempts to perform a similar experiment in M. tuberculosis proved difficult, perhaps because M. tuberculosis is more sensitive to overexpression of PknB than M. smegmatis. Even a modest increase in PknB expression was sufficient to slow growth and alter cell morphology, and the effect of overexpression on inhibitor sensitivity was not significant (K.E.A. Lougheed, unpublished data).
It is also possible that PknB may not be bound by the inhibitors when it is in a physiologically relevant configuration. In other drug discovery efforts, it has been shown that small molecule inhibitors are ineffective when their targets are involved in protein–protein interactions.52 Such signalling complexes may behave differently in cells than in solution perhaps because the binding site is masked by interacting proteins or because of differences in the in vivo protein conformation compared to the purified protein against which the inhibitors were screened.
Although pknB is an essential gene, very low levels of activity may be sufficient for survival, especially under the conditions we used to test our compounds. The M. tuberculosis kinases do display some redundancy, at least in the in vitro screens which have been used to investigate the substrates of the kinases. It is also possible that the cross-reactivity of the inhibitors against other kinases or the binding of alternative proteins results in a sequestering of the available inhibitor away from PknB. In fact, our results have demonstrated that the majority of the inhibitors tested are also able to inhibit PknF in vitro, although PknG was not as strongly inhibited. Ideally, a more specific inhibitor would need to be developed that could completely inhibit PknB in vivo without its effects being diluted by off-target binding.
Our work appears to have highlighted one of the difficulties associated with target-based drug discovery: namely, that screening a relatively small library of inhibitors in vitro regularly fails to yield compounds with potent in vivo activity. However, the suitability of PknB as a drug target cannot yet be refuted. Our discovery of inhibitors with good in vitro activity and modest in vivo activity suggests that further screening on a larger scale may yet yield potent inhibitors able to completely inhibit the activity of PknB in vivo. To take into account the possibility of redundancy among the kinases, we believe further attempts to develop M. tuberculosis kinase inhibitors might usefully target a broad specificity inhibitor targeting multiple kinases as suggested by Hegymegi-Barakonyi et al.13
Funding
This work was supported by MRC Technology through a grant from the MRCT Development Gap Fund (A853-0058) and by the Medical Research Council (U117585867 and U117584228).
Competing interests
None declared.
Ethical approval
Not required.
Acknowledgements
We thank Kristian Birchall for providing Figure 1, Charles Thompson for the gift of the fbpA mutant, Philip Draper for the ompA mutant, and Elaine Davis for helpful discussions.
Contributor Information
Kathryn E.A. Lougheed, Email: k.lougheed@imperial.ac.uk.
Simon A. Osborne, Email: simon.osborne@tech.mrc.ac.uk.
Barbara Saxty, Email: barbara.saxty@tech.mrc.ac.uk.
David Whalley, Email: david.whalley@tech.mrc.ac.uk.
Tim Chapman, Email: tim.chapman@tech.mrc.ac.uk.
Nathalie Bouloc, Email: nathalie.bouloc@tech.mrc.ac.uk.
Jasveen Chugh, Email: jasveenchugh@gmail.com.
Timothy J. Nott, Email: nott@lunenfeld.ca.
Dony Patel, Email: dony.patel@googlemail.com.
Vicky L. Spivey, Email: vspivey@nimr.mrc.ac.uk.
Catherine A. Kettleborough, Email: catherine.kettleborough@tech.mrc.ac.uk.
Justin S. Bryans, Email: justin.bryans@tech.mrc.ac.uk.
Debra L. Taylor, Email: debbie.taylor@tech.mrc.ac.uk.
Stephen J. Smerdon, Email: ssmerdo@nimr.mrc.ac.uk.
Roger S. Buxton, Email: rbuxton@nimr.mrc.ac.uk.
References
- 1.Global Alliance for TB Drug Development Tuberculosis. Scientific blueprint for tuberculosis drug development. Tuberculosis (Edinb) 2001;81(Suppl. 1):1–52. doi: 10.1054/tube.2001.0288. [DOI] [PubMed] [Google Scholar]
- 2.Lapenna S., Giordano A. Cell cycle kinases as therapeutic targets for cancer. Nat Rev Drug Discov. 2009;8:547–566. doi: 10.1038/nrd2907. [DOI] [PubMed] [Google Scholar]
- 3.Cole S.T., Brosch R., Parkhill J., Garnier T., Churcher C., Harris D. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 4.Av-Gay Y., Everett M. The eukaryotic-like Ser/Thr protein kinases of Mycobacterium tuberculosis. Trends Microbiol. 2000;8:238–244. doi: 10.1016/s0966-842x(00)01734-0. [DOI] [PubMed] [Google Scholar]
- 5.Narayan A., Sachdeva P., Sharma K., Saini A.K., Tyagi A.K., Singh Y. Serine threonine protein kinases of mycobacterial genus: phylogeny to function. Physiol Genomics. 2007;29:66–75. doi: 10.1152/physiolgenomics.00221.2006. [DOI] [PubMed] [Google Scholar]
- 6.Molle V., Kremer L. Division and cell envelope regulation by Ser/Thr phosphorylation: Mycobacterium shows the way. Mol Microbiol. 2010;75:1064–1077. doi: 10.1111/j.1365-2958.2009.07041.x. [DOI] [PubMed] [Google Scholar]
- 7.Eiglmeier K., Parkhill J., Honore N., Garnier T., Tekaia F., Telenti A. The decaying genome of Mycobacterium leprae. Lepr Rev. 2001;72:387–398. [PubMed] [Google Scholar]
- 8.Fernandez P., Saint-Joanis B., Barilone N., Jackson M., Gicquel B., Cole S.T. The Ser/Thr protein kinase PknB is essential for sustaining mycobacterial growth. J Bacteriol. 2006;188:7778–7784. doi: 10.1128/JB.00963-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sassetti C.M., Boyd D.H., Rubin E.J. Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol. 2003;48:77–84. doi: 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- 10.Kang C.M., Abbott D.W., Park S.T., Dascher C.C., Cantley L.C., Husson R.N. The Mycobacterium tuberculosis serine/threonine kinases PknA and PknB: substrate identification and regulation of cell shape. Genes Dev. 2005;19:1692–1704. doi: 10.1101/gad.1311105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cowley S., Ko M., Pick N., Chow R., Downing K.J., Gordhan B.G. The Mycobacterium tuberculosis protein serine/threonine kinase PknG is linked to cellular glutamate/glutamine levels and is important for growth in vivo. Mol Microbiol. 2004;52:1691–1702. doi: 10.1111/j.1365-2958.2004.04085.x. [DOI] [PubMed] [Google Scholar]
- 12.Walburger A., Koul A., Ferrari G., Nguyen L., Prescianotto-Baschong C., Huygen K. Protein kinase G from pathogenic mycobacteria promotes survival within macrophages. Science. 2004;304:1800–1804. doi: 10.1126/science.1099384. [DOI] [PubMed] [Google Scholar]
- 13.Hegymegi-Barakonyi B., Szekely R., Varga Z., Kiss R., Borbely G., Nemeth G. Signalling inhibitors against Mycobacterium tuberculosis - early days of a new therapeutic concept in tuberculosis. Curr Med Chem. 2008;15:2760–2770. doi: 10.2174/092986708786242886. [DOI] [PubMed] [Google Scholar]
- 14.Szekely R., Waczek F., Szabadkai I., Nemeth G., Hegymegi-Barakonyi B., Eros D. A novel drug discovery concept for tuberculosis: inhibition of bacterial and host cell signalling. Immunol Lett. 2008;116:225–231. doi: 10.1016/j.imlet.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 15.Young T.A., Delagoutte B., Endrizzi J.A., Falick A.M., Alber T. Structure of Mycobacterium tuberculosis PknB supports a universal activation mechanism for Ser/Thr protein kinases. Nat Struct Biol. 2003;10:168–174. doi: 10.1038/nsb897. [DOI] [PubMed] [Google Scholar]
- 16.Ortiz-Lombardia M., Pompeo F., Boitel B., Alzari P.M. Crystal structure of the catalytic domain of the PknB serine/threonine kinase from Mycobacterium tuberculosis. J Biol Chem. 2003;278:13094–13100. doi: 10.1074/jbc.M300660200. [DOI] [PubMed] [Google Scholar]
- 17.Veyron-Churlet R., Zanella-Cleon I., Cohen-Gonsaud M., Molle V., Kremer L. Phosphorylation of the Mycobacterium tuberculosis beta-ketoacyl-acyl carrier protein reductase MabA regulates mycolic acid biosynthesis. J Biol Chem. 2010;285:12714–12725. doi: 10.1074/jbc.M110.105189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Parikh A., Verma S.K., Khan S., Prakash B., Nandicoori V.K. PknB-mediated phosphorylation of a novel substrate, N-acetylglucosamine-1-phosphate uridyltransferase, modulates its acetyltransferase activity. J Mol Biol. 2009;386:451–464. doi: 10.1016/j.jmb.2008.12.031. [DOI] [PubMed] [Google Scholar]
- 19.Villarino A., Duran R., Wehenkel A., Fernandez P., England P., Brodin P. Proteomic identification of M. tuberculosis protein kinase substrates: PknB recruits GarA, a FHA domain-containing protein, through activation loop-mediated interactions. J Mol Biol. 2005;350:953–963. doi: 10.1016/j.jmb.2005.05.049. [DOI] [PubMed] [Google Scholar]
- 20.O’Hare H.M., Duran R., Cervenansky C., Bellinzoni M., Wehenkel A.M., Pritsch O. Regulation of glutamate metabolism by protein kinases in mycobacteria. Mol Microbiol. 2008;70:1408–1423. doi: 10.1111/j.1365-2958.2008.06489.x. [DOI] [PubMed] [Google Scholar]
- 21.de Carvalho L.P., Zhao H., Dickinson C.E., Arango N.M., Lima C.D., Fischer S.M. Activity-based metabolomic profiling of enzymatic function: identification of Rv1248c as a mycobacterial 2-hydroxy-3-oxoadipate synthase. Chem Biol. 2010;17:323–332. doi: 10.1016/j.chembiol.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nott T.J., Kelly G., Stach L., Li J., Westcott S., Patel D. An intramolecular switch regulates phosphoindependent FHA domain interactions in Mycobacterium tuberculosis. Sci Signal. 2009;2 doi: 10.1126/scisignal.2000212. ra12. [DOI] [PubMed] [Google Scholar]
- 23.Zhang J.H., Chung T.D., Oldenburg K.R. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 24.Collins L., Franzblau S.G. Microplate alamar blue assay versus BACTEC 460 system for high-throughput screening of compounds against Mycobacterium tuberculosis and Mycobacterium avium. Antimicrob Agents Chemother. 1997;41:1004–1009. doi: 10.1128/aac.41.5.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lougheed K.E., Taylor D.L., Osborne S.A., Bryans J.S., Buxton R.S. New anti-tuberculosis agents amongst known drugs. Tuberculosis (Edinb) 2009;89:364–370. doi: 10.1016/j.tube.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bain J., Plater L., Elliott M., Shpiro N., Hastie C.J., McLauchlan H. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.England P., Wehenkel A., Martins S., Hoos S., Andre-Leroux G., Villarino A. The FHA-containing protein GarA acts as a phosphorylation-dependent molecular switch in mycobacterial signaling. FEBS Lett. 2009;583:301–307. doi: 10.1016/j.febslet.2008.12.036. [DOI] [PubMed] [Google Scholar]
- 28.Cheng Y., Prusoff W.H. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 29.Andries K., Verhasselt P., Guillemont J., Gohlmann H.W., Neefs J.M., Winkler H. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science. 2005;307:223–227. doi: 10.1126/science.1106753. [DOI] [PubMed] [Google Scholar]
- 30.Brennan P.J., Nikaido H. The envelope of mycobacteria. Annu Rev Biochem. 1995;64:29–63. doi: 10.1146/annurev.bi.64.070195.000333. [DOI] [PubMed] [Google Scholar]
- 31.Malaviya A., Gomes J. Enhanced biotransformation of sitosterol to androstenedione by Mycobacterium sp. using cell wall permeabilizing antibiotics. J Ind Microbiol Biotechnol. 2008;35:1235–1239. doi: 10.1007/s10295-008-0419-5. [DOI] [PubMed] [Google Scholar]
- 32.Rastogi N., Goh K.S., David H.L. Enhancement of drug susceptibility of Mycobacterium avium by inhibitors of cell envelope synthesis. Antimicrob Agents Chemother. 1990;34:759–764. doi: 10.1128/aac.34.5.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jagannath C., Reddy V.M., Gangadharam P.R. Enhancement of drug susceptibility of multi-drug resistant strains of Mycobacterium tuberculosis by ethambutol and dimethyl sulphoxide. J Antimicrob Chemother. 1995;35:381–390. doi: 10.1093/jac/35.3.381. [DOI] [PubMed] [Google Scholar]
- 34.Piddock L.J., Williams K.J., Ricci V. Accumulation of rifampicin by Mycobacterium aurum, Mycobacterium smegmatis and Mycobacterium tuberculosis. J Antimicrob Chemother. 2000;45:159–165. doi: 10.1093/jac/45.2.159. [DOI] [PubMed] [Google Scholar]
- 35.Nguyen L., Chinnapapagari S., Thompson C.J. FbpA-Dependent biosynthesis of trehalose dimycolate is required for the intrinsic multidrug resistance, cell wall structure, and colonial morphology of Mycobacterium smegmatis. J Bacteriol. 2005;187:6603–6611. doi: 10.1128/JB.187.19.6603-6611.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lambert P.A. Cellular impermeability and uptake of biocides and antibiotics in Gram-positive bacteria and mycobacteria. J Appl Microbiol. 2002;92(Suppl.):46S–54S. [PubMed] [Google Scholar]
- 37.Stephan J., Mailaender C., Etienne G., Daffe M., Niederweis M. Multidrug resistance of a porin deletion mutant of Mycobacterium smegmatis. Antimicrob Agents Chemother. 2004;48:4163–4170. doi: 10.1128/AAC.48.11.4163-4170.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mailaender C., Reiling N., Engelhardt H., Bossmann S., Ehlers S., Niederweis M. The MspA porin promotes growth and increases antibiotic susceptibility of both Mycobacterium bovis BCG and Mycobacterium tuberculosis. Microbiology. 2004;150:853–864. doi: 10.1099/mic.0.26902-0. [DOI] [PubMed] [Google Scholar]
- 39.Siroy A., Mailaender C., Harder D., Koerber S., Wolschendorf F., Danilchanka O. Rv1698 of Mycobacterium tuberculosis represents a new class of channel-forming outer membrane proteins. J Biol Chem. 2008;283:17827–17837. doi: 10.1074/jbc.M800866200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Raynaud C., Laneelle M.A., Senaratne R.H., Draper P., Laneelle G., Daffe M. Mechanisms of pyrazinamide resistance in mycobacteria: importance of lack of uptake in addition to lack of pyrazinamidase activity. Microbiology. 1999;145:1359–1367. doi: 10.1099/13500872-145-6-1359. [DOI] [PubMed] [Google Scholar]
- 41.Takiff H.E., Cimino M., Musso M.C., Weisbrod T., Martinez R., Delgado M.B. Efflux pump of the proton antiporter family confers low-level fluoroquinolone resistance in Mycobacterium smegmatis. Proc Natl Acad Sci USA. 1996;93:362–366. doi: 10.1073/pnas.93.1.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gupta A.K., Chauhan D.S., Srivastava K., Das R., Batra S., Mittal M. Estimation of efflux mediated multi-drug resistance and its correlation with expression levels of two major efflux pumps in mycobacteria. J Commun Dis. 2006;38:246–254. [PubMed] [Google Scholar]
- 43.Escribano I., Rodriguez J.C., Llorca B., Garcia-Pachon E., Ruiz M., Royo G. Importance of the efflux pump systems in the resistance of Mycobacterium tuberculosis to fluoroquinolones and linezolid. Chemotherapy. 2007;53:397–401. doi: 10.1159/000109769. [DOI] [PubMed] [Google Scholar]
- 44.Srivastava S., Musuka S., Sherman C., Meek C., Leff R., Gumbo T. Efflux-pump-derived multiple drug resistance to ethambutol monotherapy in Mycobacterium tuberculosis and the pharmacokinetics and pharmacodynamics of ethambutol. J Infect Dis. 2010;201:1225–1231. doi: 10.1086/651377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Amaral L., Martins M., Viveiros M. Enhanced killing of intracellular multidrug-resistant Mycobacterium tuberculosis by compounds that affect the activity of efflux pumps. J Antimicrob Chemother. 2007;59:1237–1246. doi: 10.1093/jac/dkl500. [DOI] [PubMed] [Google Scholar]
- 46.Ramon-Garcia S., Martin C., De Rossi E., Ainsa J.A. Contribution of the Rv2333c efflux pump (the Stp protein) from Mycobacterium tuberculosis to intrinsic antibiotic resistance in Mycobacterium bovis BCG. J Antimicrob Chemother. 2007;59:544–547. doi: 10.1093/jac/dkl510. [DOI] [PubMed] [Google Scholar]
- 47.Ramon-Garcia S., Martin C., Thompson C.J., Ainsa J.A. Role of the Mycobacterium tuberculosis P55 efflux pump in intrinsic drug resistance, oxidative stress responses, and growth. Antimicrob Agents Chemother. 2009;53:3675–3682. doi: 10.1128/AAC.00550-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li X.Z., Zhang L., Nikaido H. Efflux pump-mediated intrinsic drug resistance in Mycobacterium smegmatis. Antimicrob Agents Chemother. 2004;48:2415–2423. doi: 10.1128/AAC.48.7.2415-2423.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Singh A., Singh Y., Pine R., Shi L., Chandra R., Drlica K. Protein kinase I of Mycobacterium tuberculosis: cellular localization and expression during infection of macrophage-like cells. Tuberculosis (Edinb) 2006;86:28–33. doi: 10.1016/j.tube.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 50.Koul A., Vranckx L., Dendouga N., Balemans W., Van den Wyngaert I., Vergauwen K. Diarylquinolines are bactericidal for dormant mycobacteria as a result of disturbed ATP homeostasis. J Biol Chem. 2008;283:25273–25280. doi: 10.1074/jbc.M803899200. [DOI] [PubMed] [Google Scholar]
- 51.Payne D.J., Gwynn M.N., Holmes D.J., Pompliano D.L. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat Rev Drug Discov. 2007;6:29–40. doi: 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- 52.Hoshi N., Langeberg L.K., Gould C.M., Newton A.C., Scott J.D. Interaction with AKAP79 modifies the cellular pharmacology of PKC. Mol Cell. 2010;37:541–550. doi: 10.1016/j.molcel.2010.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]