

Abstract
Along with compounds from terrestrial microorganisms, the constituents of higher plants have provided a substantial number of the natural product-derived drugs used currently in western medicine. Interest in the elucidation of new structures of the secondary metabolite constituents of plants has remained high among the natural products community over the first decade of the 21st century, particularly of species that are used in systems of traditional medicine or are utilized as botanical dietary supplements. In this review, progress made in the senior author’s laboratory in research work on naturally occurring sweeteners and other taste-modifying substances and on potential anticancer agents from tropical plants will be described.
Introduction
As society enters the second decade of the 21st century, there remains a considerable interest in the use of organic molecules from organisms in drug discovery for a diverse range of disease states.1,2 Of the new small-molecule drugs of natural origin introduced to western medicine over the last ten years, the majority have been derived from terrestrial microbes, with others from higher plants, marine organisms, and terrestrial animals.3,4 The terms “natural product” and “secondary metabolite” now have a common meaning, with such compounds evolving to enhance the survival of the producing organism.5 Natural products are biosynthesized in the correct chiral form to exhibit biological activity in many in vitro and in vivo laboratory test systems, and are regarded as being complementary to compounds produced by synthesis and combinatorial chemistry for drug discovery programs.6,7 Although natural product drug discovery research is no longer regarded in such favorable terms as it once was by many large pharmaceutical companies, newly refined bioassay and other procedures have been developed for enhancing the purification of natural product samples leading, in part, to the more rapid elucidation of lead compound structures.2,8,9
Medicinal plants have been used for millennia as one of the main sources of therapeutic agents for mankind.10 Of the approximately 270,000 terrestrial plants that have been classified taxonomically,11 some 30,000 higher plants are used medicinally.12 Drug discovery from medicinal plants in the 19th century led to the purification of several early drugs inclusive of codeine, digitoxin, morphine, and quinine, which are all still widely used.13 The discovery of new drug leads from medicinal plants may be aided by ethnopharmacology, which is a mode of scientific investigation relating to the indigenous medicinal uses of a particular species.14 Fabricant and Farnsworth documented 122 plant natural products with therapeutic use in their pure form, of which over 70% were developed at least partially as a consequence of the availability of ethnopharmacological information.15 From 210 small-molecule therapeutic agents included in a recent World Health Organization Model List of Essential Medicines, 17 of these are of plant origin.16 Moreover, in a recently published textbook of pharmacognosy, most of the compounds included are derived from plants, rather than other types of organisms.17
Therefore, there remains much interest in the constituents of plants as therapeutic agents. This may be strongly supported by the relatively large number of plant-derived drugs that have been approved by the U.S. Food and Drug Administration (FDA) in the first decade of the 21st century (Table 1). It may be seen from this table that 15 pure substances are tabulated, with ten being new structural entities. Several compounds are well-known, and have been approved for the first time for the use(s) specified. Veregen®, a standardized mixture of the catechins of green tea [Camellia sinensis (L) Kuntze; Theaceae] is the first of a new class of botanical prescription drugs to be approved by the FDA, and, in order for this have occurred, stringent standards of both quality control procedures used and clinical testing carried out were met.18 Newly approved oncological applications for previously approved anticancer agents such as docetaxel and irinotecan are not included in this table.
Table 1.
Plant Natural Products and Derivatives Approved by the U.S. FDA from 2001–2010.a
| year approved | generic name | natural lead compound | trade name | indication |
|---|---|---|---|---|
| 2001 | galanthamine | galanthamine | Razadyne® | dementia associated with Alzheimer’s disease |
| 2002 | nitisinoneb | leptospermone | Orfadin® | hereditary tyrosinemia type 1 |
| 2003 | miglustatb | 1-deoxynojirimycin | Zavesca® | type 1 Gaucher disease |
| 2004 | tiotropiumb bromide | atropine | Spiriva® | COPD and exacerbation of COPD |
| 2004 | trospiumb chloride | atropine | Sanctura® | overactive bladder |
| 2004 | solifenacinb | quinine | VESIcare® | overactive bladder |
| 2005 | paclitaxel (protein-bound) | paclitaxel (taxol) | Abraxane® | breast cancer |
| 2006 | sinecatechinsc | green tea phenols | Veregen® | genital warts |
| 2006d | nabiloneb | Δ9- tetrahydrocannabinol | Cesamet® | chemotherapy-induced nausea |
| 2008 | methylnaltrexoneb bromide | morphine | Relistor® | opioid induced constipation |
| 2008 | tetrabenazineb | emetine | Xenazine® | Huntington’s-associated cholera |
| 2009 | artemetherb and lumefantrine | artemesinin | Coartem® | malaria |
| 2009 | colchicine | colchicine | Colcrys® | gout |
| 2010 | cabazitaxelb | paclitaxel (taxol) | Jevtana® | hormone-refractory metastatic prostate cancer |
| 2010 | dextromethorphan and quinidine | morphine and quinidine | Nuedexta™ | pseudobulbar affect |
| 2010 | capsaicin | Capsaicin | Qutenza® | postherpetic neuralgia |
Information taken from www.drugs.com.
New molecular entity.
Approved as a mixture of compounds as a “botanical drug”.
Originally approved in 1985, but only introduced to the market in May 2006 (www.valent.com).
In terms of the discovery of new natural products that might reach the drug market in the future, considerable progress is being made in terms of the development of new leads from terrestrial microbial,19,20 marine,20–22 and higher plant20,23 sources. However, on comparing the secondary metabolites from plants with those from terrestrial microbial and marine sources, it may be perceived that there is a comparable lack of structural novelty among the compounds under investigation. There are about 200,000 secondary metabolites that have been reported to date from plants,24 with isoprenoids, phenolic compounds, and alkaloids representing the major compound groups biosynthesized. Crude plant extracts tend to be very complex, which has the effect of making the work-up of biologically active constituents using solvent partition and chromatographic methods quite lengthy. Moreover, many plant extracts contain “vegetable tannins”, polyphenolic substances that interact non-specifically with protein biological targets due to hydrogen-bond interactions, which require special treatment for removal.25 Plant collections relying on access to biodiversity in developing countries require intellectual property (IP) negotiations that may be time-consuming to formulate, especially if ethnopharmacological observations are involved.26 According to Appendino and Pollastro, the necessity of dealing with IP considerations is a major reason why large pharmaceutical companies have tended to phase out drug discovery work on plants, in particular.26
In spite of the above-mentioned complications in terms of drug discovery, there is today a very great deal of interest among members of the global natural products community in investigating new plant constituents. To give some support for this, Figure 1 shows pie charts of new compounds published in the Journal of Natural Products in two single years at the beginning (2001) and end (2010) of the last decade. The compounds are grouped according to the major classes of organism of origin. It may be seen that during both time periods, larger percentages of new plant compounds were reported in this journal when compared with compounds derived from terrestrial microorganisms and fungi, and all types of marine and aquatic organisms. There are many reasons for this, and paramount among these is a strong interest in investigating the chemistry and biological properties of the constituents of medicinal plants, in countries representative of every continent except Antarctica.e.g.,27–32 In addition, among other plant-focused studies, investigators have scrutinized the constituents of botanical dietary supplements and food plants,e.g.,33–36 followed up on plant ethnomedicinal leads,e.g.37,38 and elucidated the constituents of toxic plants.e.g.,39,40
Figure 1.

Comparison of the types of organisms of origin of new compounds published in the Journal of Natural Products in the years 2001 and 2010. (Totals of 1,142 and 1,369 natural products from all types of organisms were published in 2001 and 2010, respectively. All types of organisms with a marine or aquatic habitat are grouped together. An example of an unspecified organism is propolis).
In the next two major sections of this review, work in the senior author’s laboratory directed towards the search for two distinct groups of new bioactive agents from plants will be featured, namely, highly sweet compounds and taste modifiers, and potential cancer chemotherapeutic agents.
Naturally Occurring Sucrose Substitutes and Taste Modifying Agents
Commercially Used Plant-derived Sweet Substances
Owing to health concerns caused by consuming calorie-rich food sweetened by sugars, there is a great demand low-caloric and non-cariogenic sucrose substitutes in the human diet.41 Such compounds are typically some fifty to several hundred times sweeter than sucrose on a weight basis.41 In the United States, there are five synthetic “high potency” or “intense” sweeteners currently approved, namely, acesulfame K, aspartame, neotame, saccharin, and sucralose.42,43 Approximately 100 discrete natural products are also highly sweet, and are based on about 20 different structural types, with most of these being either isoprenoids, flavonoids, and proteins.42 The structures of several of these sweet-tasting plant constituents were determined by the group of the late Prof. Osamu Tanaka, of Hiroshima University in Japan, most notably rebaudioside A (see below).42 All of the highly sweet compounds to date from Nature have been found in green (vascular) plants, with there having been no reports of such compounds from either terrestrial microbes or marine organisms.42,44 Several highly sweet compounds from plants are approved as sucrose substitutes in various countries around the world, as summarized in Table 2. The sweet-tasting natural products concerned will be discussed briefly in turn in the following paragraphs. The so-called “bulk” or “reduced calorie” sweeteners of plant origin used as food additives, inclusive of several monosaccharides, disaccharides, and polyols, will not be described below, since they have been dealt with by others recently.45,46
Table 2.
Commercially Available Plant-Derived Intense Sweetenersa
| compound name | examples of countries in which approved |
|---|---|
| glycyrrhizin (1)b | Japan |
| monoglucuronide of glycyrrhetinic acid (MGGR)c (2) | Japan |
| stevioside (3)d | Argentina, Brazil, Japan, Paraguay, People’s Republic of China, South Korea |
| rebaudioside A (4) | France,e Japan, United Statesf |
| mogroside V (5)g | Japan |
| phyllodulcin (6) | Japan |
| neohesperidin dihydrochalcone (7)h | European Union, Switzerland, Turkey |
| thaumatinh | Australia, European Union, Japan |
The purity levels for the compounds included in this table differ from country to country.
The salts, ammoniated glycyrrhizin and monoammonium glycyrrhizinate, have GRAS status as flavoring agents in the United States.
Semi-synthetic compound.
Refined Stevia rebaudiana extracts with stevioside as the major ent-kaurene glycoside component, are used as a dietary supplement in the United States.
Approved in September 2009 in anticipation of general EU approval (source: www.sweeteners.org).
Steviol glycoside mixtures with rebaudioside A as the major component received GRAS status in the United States in December 2008.
GRAS status accorded for a Siraitia grosvenorii extract of a standardarized mogroside V content accorded in January 2010 in the United States (source: www.biovitorria.com).
Has GRAS status as a flavoring agent in the United States.
Glycyrrhizin (1), also known as glycyrrhizic acid, is an oleanane-type triterpene diglucuronic acid glycoside constituent of the roots and stolons of licorice (Glycyrrhiza glabra L. and other Glycyrrhiza species) (Fabaceae), and based on the aglycone, glycyrrhetinic acid. Glycyrrhizin is approximately 50–100 times sweeter than sucrose on a weight basis, depending on concentration.47 A Glycyrrhiza sp. decoction containing glycyrrhizin (1) became the first natural product intense sweetener used in Japan in the early part of the 20th century, when it was also found to suppress the salty taste of pickled foods and dried seafoods.48 Efforts have been made to modify the structural units of compound 1 in order to enhance its sweetness potency, and the monoglucuronide of glycyrrhetinic acid (MGGR; 2) was found to be over 900 times sweeter than sucrose. MGGR may be produced from glycyrrhizin (1) by hydrolysis using a microbial enzyme, and has use in Japan to sweeten certain dairy products and soft drinks.47 In the United States, ammoniated glycyrrhizin, monoammonium glycyrrhizinate, and licorice extract are not approved sweetening agents, but are used as flavoring agents, and are included on the Generally Regarded as Safe (GRAS) list.49 Licorice root (“Glycyrrhizae Radix”) is used very widely as an ingredient in Oriental systems of traditional medicine, since in addition to its flavoring properties, it has reported harmonizing and detoxifying effects.50 However, the Ministry of Health, Labor and Welfare in Japan has imposed a daily upper limit on glycyrrhizin consumption of 200 mg, due to its well-known glucocorticomimetic effects, which may lead to pseudoaldosteronism.47
The intense sweetness of leaves of the Paraguayan herb, Stevia rebaudiana (Bertoni) Bertoni (Asteraceae), was reported initially in the western literature at the turn of the 20th century.51 Altogether, nine sweet-tasting ent-kaurane glycosides have been isolated and structurally characterized from S. rebaudiana leaves, and are all based on the aglycone, steviol (ent-13-hydroxykaur-16-en-19-oic acid).42 The two most abundant of these diterpene glycosides in the plant are stevioside (3) and rebaudioside A (4).42 Extracts of S. rebaudiana began to be used in Japan in the early 1970s, due in part to perceived difficulties in obtaining Glycyrrhiza species from other Asian countries, and in an attempt to overcome the latent sweet-tasting effect of glycyrrhizin (1).48 Three types of refined S. rebaudiana leaf preparations have use in the sweetening and flavoring of a wide range of foods and beverages in Japan. These are “stevia extract” (consisting of about 90% steviol glycosides and 50–55% stevioside), “rebaudioside A-enriched stevia extract” (containing a higher proportion of rebaudioside A than the 20–25% in “stevia extract”), and “sugar-transformed stevia extract” (a trans-α-1,4-glucosylated product of “stevia extract” using an enzymatic procedure). It is considered that the last two-mentioned of these products exhibit improved taste characteristics when compared to “stevia extract”.48 Following the successful utilization of S. rebaudiana refined leaf extracts containing stevioside (3) and rebaudioside A (4) in Japan, several other countries also began to permit the use of sweet-tasting products from this plant in foodstuffs and/or beverages, including Brazil and Korea (Table 2).52 A major step forward in the more widespread use of sweet products from S. rebaudiana was establishment of temporary specifications established for the acceptable daily intake (ADI) of steviol glycosides in humans by the Joint FAO/WHO Expert Committee on Food Additives (JECFA) in Europe in 2004, which were updated recently.53 In 2008, steviol glycoside sweetener mixtures with rebaudioside A (4) as the major constituent were accorded GRAS status by the U.S. Food and Drug Administration, leading to the introduction of many products on the market, including table-top sweetener brands.53 This positive decision was underpinned by a series of new safety and metabolism studies on rebaudioside A (4).53,54 Progress in the recent development of marketed rebaudioside A-containing food and beverage products has been very rapid, especially when it is considered that from 1991–1995 there was a ban by the FDA on the import of S. rebaudiana leaves to the United States.52
There are several other highly sweet plant-derived natural sweeteners, with some having a more restricted commercial use than either glycyrrhizin (1) and the steviol glycosides, stevioside (3) and rebaudioside A (4). These substances comprise the triterpenoid glycoside, mogroside V (5), the phenols, phyllodulcin (6) and neohesperidin dihydrochalcone (7), and the protein, thaumatin.42,47 Mogroside V (5) is the major cucurbitane glycoside constituent of the dried and roasted fruits of the Chinese plant Siraitia grosvenorii (Swingle) C. Jeffrey ex A.M. Lu & Zhi Y. Zhang (Cucurbitaceae), and has been rated as 250–425 times the sweetness potency of sucrose, with a relatively pleasant taste, and having improved solubility in water when compared with many other plant glycosides. Its plant of origin is used in traditional Chinese medicine, and is known as “lo han guo”, while refined extracts containing compound 5 are used in Japan to sweeten certain foods and beverages.42,47 Safety studies on extracts of S. grosvenorii and mogroside V have not been numerous, but are supportive of a wider use for sweetening purposes.42,47 Phyllodulcin (6) is a dihydroisocoumarin aglycone produced from its natural glycosidic form by crushing or fermenting the leaves of Hydrangea macrophylla Seringe var. thunbergii (Siebold) Makino (Saxifragaceae). This species is used to make a sweet-tasting ceremonial tea in Japan, with its sweet constituent, compound 6, having been evaluated as about 400 times the sweetness potency of sucrose. However, phyllodulcin is very water insoluble, and exhibits a delay in the onset of sweetness after being tasted, and has a licorice-like aftertaste. There have been several attempts to improve on the sweetness parameters of phyllodulcin (6) by synthetic modification, but none of these compounds has present commercial use.42,47 Neohesperidin dihydrochalcone (7) is a semisynthetic dihydrochalcone glycoside prepared from a flavonoid constituent of Citrus auranticum L. (Rutaceae) (Seville orange).42 This compound was approved as a sweetener in the European Union countries in 1994, and also has GRAS status in the U.S. as a flavoring ingredient. Compound 7 is approximately 400–600 times sweeter than sucrose at concentrations used in foodstuffs, but it suffers from a delay in sweetness onset after first being tasted and also has an aftertaste.55 Finally, thaumatin is sweet protein isolated from the fruits of the west African plant, Thaumatococcus danielli Benth. (Maranaceae). Of five different thaumatin analogs known, the major compounds, thaumatins I and II, have 207 amino acid residues each.56 The sweetness potency of thaumatin protein has been rated as high as 1,800–3,000 times in comparison to sucrose. Thaumatin is a approved as a sweetener in several countries, and has GRAS status in the U.S. as a flavor adjunct, and is used in a number of applications, including chewing gum.47
Natural Product Sweetener Discovery
For several years while at the University of Illinois at Chicago (UIC), a major focus of the laboratory research of the senior author was on the discovery of new sweetening agents from higher plants, with this work performed in collaboration with plant taxonomist Dr. Djaja Djendoel Soejarto and other faculty colleagues. More recently, laboratory work has begun on naturally occurring taste-modifying substances at The Ohio State University. Over the years, several reviews and book chapters have been published by our group on plant-derived sweetenerse.g.,42,47,53,57–62 and sweetness inhibitors.e.g.,42,47,58,63
It is only relatively recently that the human taste receptor has been elucidated as a G-protein coupled receptor, composed of a heterodimer of two proteins, TAS1R2 and TAS1R3 (formerly T1R2 and T1R3).64 Indeed, it is only in the last few years that high-throughput screening procedures using receptor-binding techniques have become available for new sweetener discovery.65 Therefore, in our work performed prior to this on natural sweetener discovery, it was necessary to use volunteer human subjects to taste crude extracts, chromatographic fractions, and pure compounds for the presence or absence of sweetness. Prior to tasting any fractions or pure compounds, their crude extract of origin was evaluated for acute toxicity in mice and for mutagenicity in a forward mutation assay.57,59,66,67 This required prior approval from both the Institutional Review Board (responsible for human subjects) and the Animal Care Committee at UIC. Pure compounds were evaluated for sweetness potency by a small human taste panel of three members, by dilution in water until perceived to taste equally sweet to a 2% w/v aqueous sucrose solution standard.e.g.,66 However, prior to being evaluated in this manner, lack of activity in both mouse acute toxicity and mutagenicity tests were established, requiring the consumption of some 50–100 mg of compound for this purpose.42,57,59 Therefore, it was not always possible to evaluate a sweet compound for potency relative to sucrose if only a few milligrams were isolated, especially with limited water solubility evident.
One of the approaches our group used towards locating candidate sweet-tasting plants was by making inquiries in medicinal plant marketplaces, and then following up on this by collecting the same species in the field.e.g.,67 This work was carried out before the ratification of the United Nations Convention of Biological Diversity in Rio de Janeiro in 1992, a topic described in detail in a recent book chapter by Dr. Geoffrey Cordell.68 However, as a consequence of the “Rio Convention”, in order to carry out this same approach today, relevant benefit-sharing agreements would have to be worked out in advance of performing field work, and “prior informed consent” would need to be approved by an Institutional Review Board, so that the relevant indigenous traditional knowledge would not be at risk of being exploited unfairly.59 Given that many sweet-tasting plants turn out to contain high concentration levels of either sugars and polyols69 or phenylpropanoids like trans-anethole,70 the chance of actually discovering a potently sweet substance representing a new structural type as a result of following up an ethnobotanical lead is quite low.
In the paragraphs following, a number of compounds discovered in our laboratory with sweet-tasting or taste-modifying properties are described briefly.
Hernandulcin and 4β-Hydroxyhernandulcin
In the early 1980’s, Cesar Compadre came to graduate school at the University of Illinois at Chicago with a sample of the aerial parts of the sweet-tasting plant, Lippia dulcis Trev. (Verbenaceae), purchased from a marketplace near Mexico City. He succeeded in isolating and structurally characterizing a bisabolane sesquiterpenoid (8) as a minor constituent of this plant, which was rated as being about 1,000 times more potently sweet than sucrose by a human taste panel, on a weight basis. However, the compound also exhibits some bitterness and has a rather unpleasant aftertaste. (+)-Hernandulcin proved to be the first highly sweet-tasting sesquiterpenoid reported in the literature, and was assigned its trivial name in honor of Dr. Francisco Hernández, a Spanish physician from the 16th century who first documented L. dulcis as a sweet-tasting plant under its Aztec name in a monograph entitled “Natural History of New Spain”.71–73 This sweet compound was synthesized in racemic form in our laboratory by directed-aldol condensation, from two commercially available ketones.71,73 It was found that only the 6S,1′S- configuration of hernandulcin exhibits a sweet taste, when all four diastereomers were synthesized by Mori and Kato.74 In a follow-up study performed by former postdoctoral Norito Kaneda on a Panamanian sample of the leaves and flowers of L. dulcis provided by Dr. Mahabir Gupta, a related sweet sesquiterpenoid, (+)-4β-hydroxyhernandulcin (9), was isolated.75 Although established as being sweet-tasting, the sweetness potency of compound 9 was not determined due to the small quantity of this substance isolated. It was found that (+)-hernandulcin (8) was present in this Panamanian sample in a much higher yield (0.154% w/w) than in the original Mexican sample investigated (0.004% w/w), with the latter sample accumulating high levels of camphor.71,75 In keeping with this observation, it has been confirmed by others that there is more than one chemotype of L. dulcis.76,77 In 2006, Ono and colleagues isolated six new and two known bisabolane sesquiterpenoids closely related in structure to hernandulcin from a freshly collected sample of the aerial parts of L. dulcis cultivated in Fukuoka, Japan, with three of these compounds containing a hydroperoxy group. The occurrence of hernandulcin (8) itself and the presence or absence of sweetness for the eight sesquiterpenoids isolated were not reported.78
Over 25 years have passed since the initial discovery by our group of the structure and highly sweet properties of (+)-hernandulin (8). The compound has been synthesized on several occasions, both as a racemate and as the naturally occurring (+)-isomer, and hernandulcin has also been produced by both hairy root and shoot cultures of L. dulcis, as summarized recently.42 As a result of producing several derivatives of hernandulcin (8) modified structurally at various functionalities, our group established that three key functionalities are necessary in mediating the sweet-tasting effect of this compound, namely, the C-6 carbonyl, the C-1′ hydroxy group, and the C-4′, C-5′ double bond.79 The intermolecular distances of these three functionalities conform with the Shallenberger-Acree-Kier model for sweet compounds.80 Unfortunately, it does not look as though this compound will be developed further for commercial use as a sweetener, because of its inherent thermal instability, a lack of significant water solubility, and unacceptable hedonic taste attributes. However, there has been some interest in developing hernandulcin as a sweetener for use in dentifrices, wherein its unpleasant taste profile could be masked by other flavor components.81 In view of its structural simplicity and high sweetness potency, hernandulcin (8) remains of interest as a standard molecule in studies on how sweet substances bind to the human taste receptor.e.g.,82
Abrusosides A–E
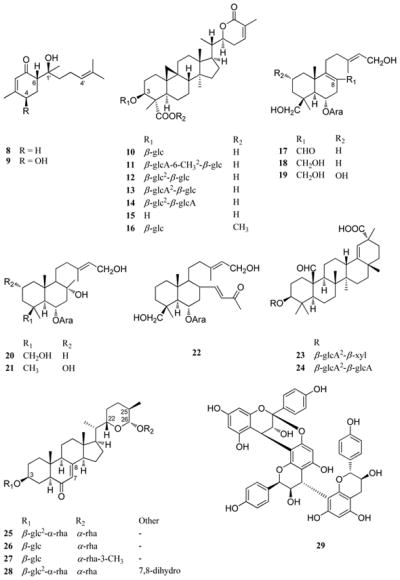
Abrusoside A (10) is the prototype member of a group of sweet-tasting cycloartane-type triterpenoid glycoside constituents of the leaves of Abrus precatorius L. (Fabaceae) (rosary pea; precatory bean), one of 12 species in the genus Abrus.83,84 While native to India, A. precatorius has become a commonly introduced species in various tropical and subtropical regions of the world. The roots of this plant are sweet-tasting, and have been used as a substitute for licorice.84 It was suggested our group work on A. precatorius leaves by the late Dr. Julia F. Morton, Director, Morton Collectanea, University of Miami, Miami, Florida, and the initial isolation work was performed by then graduate student Young-Hee Choi. Prior to our work, glycyrrhizin (1) had been regarded as the sweet-tasting principle of this species.e.g.,85,86 In our hands, when examining A. precatorius leaves collected in southern Florida, no glycyrrhizin was detected, with instead four sweet principles, abrusosides A–D (10–13) being isolated in the initial work.83,84 In a follow-up phytochemical study, another compound of this type, abrusoside E (14), was purified by former postdoctoral Ed Kennelly from a recollection of the leaves in Florida.87 Compounds 10–13 were identified in a second species in the genus, Abrus fruticulosus Wall., collected in Thailand.88
Abrusosides A–E (10–14) are based on a common aglycone with a new carbon skeleton, abrusogenin (15), for which the structure was established by single-crystal X-ray crystallography of the methyl ester.83 These five glycosides vary from one another structurally only in terms of their respective saccharide substituent attached to C-3 on the aglycone. After initial safety testing, the ammonium salts of compounds 10–13 were rated as between 30–100 times sweeter than 2% sucrose solution.84 In abrusoside E (14), there is a transposition of the two sugar units when compared to abrusoside D (13). However, compound 14 was found to have very poor solubility properties in several solvents, requiring its NMR data to be measured in the form of a monomethyl ester derivative.57,87 Abrusoside A methyl ester (16) was partially synthesized from abrusogenin (15) at the University of Illinois at Chicago, by former graduate student Nam-Cheol Kim, in work conducted in collaboration with Dr. Darrick Kim. Compound 16 was produced through methylation of abrusogenin (15), coupling with 1-chloro-2,3,4,6-tetra-O-acetylglucopyranose in the presence of AgOTf and TMU in CH2Cl2, and by deacetylation performed with methanolic K2CO3.89 The abrusosides have several positive aspects as sweet compounds, in that they are generally thermostable and can be rendered water soluble using ammonium hydroxide, and they exhibit a pleasant sweet taste. However, these compounds produce a long-lasting sweet sensation when tasted, and occur in the leaves only as minor constituents, representing less than a 1% w/w combined yield.57 Moreover, the plant of origin of the abrusosides, A. precatorius, is a vine, and has very light leaves that must be carefully separated from the seeds of this plant, which contain a well-known type II ribosome-inactivating protein toxin, abrin, occurring as four discrete isoforms.90,91
Miscellaneous Sweet-tasting and Taste-modifying Plant Constituents
A number of additional highly sweet-tasting substances of plant origin have been isolated and characterized in our laboratory, representative of the terpenoid glycoside, steroidal saponin, and phenolic types, and several examples will be mentioned briefly in turn. The labdane-type diterpene glycoside, gaudichaudioside A (17), was isolated by then graduate student Fekadu Fullas from the aerial parts of the Paraguayan plant, Baccharis gaudichaudiana DC. (Asteraceae), and found to exhibit a sweetness intensity 55 times that of 2% w/w sucrose solution, with a pleasant taste profile having limited bitterness.67 Also obtained from the same plant sample were five additional labdane arabinosides, gaudichaudiosides B–F (18–22), which proved to be sweet-bitter, neutral tasting, wholly bitter, sweet-bitter, and wholly bitter, respectively.67,92 On comparing the structures of compounds 17–22, while all six compounds have an allylic primary hydroxy group, the sweet-tasting substance gaudichaudioside A (17) is the only one with a primary aldehyde group at C-8, with the remaining compounds being variously substituted at this key functionality.67,92
A further sweet oleanane-type triterpenoid glycoside, periandrin V (23), was purified from a sample of Periandra dulcis roots collected in Brazil, provided by the late Prof. Yohei Hashimoto, of Kobe Women’s College of Pharmacy (now Kobe Pharmaceutical University) in Japan. This compound was characterized structurally by former graduate student Rutt Suttisri, and found to contain a terminal D-xylopyranosyl sugar unit in place of the terminal D-glucuronopyranosyl moiety of the parent compound in this series, periandin I (24).93 Periandin V (23) was rated as possessing a sweetness potency of about 200 times that of 2% w/v sucrose, and about twice that of compound 24.57,94
One of the most potently sweet compounds found in our laboratory work is polypodoside A (25), isolated from the rhizomes of Polypodium glycyrrhiza D.C. Eaton (Polypodiaceae) (licorice fern), a plant long suspected of containing glycyrrhizin as the sweet principle.66 From a plant sample collected in Oregon, former graduate student Jinwoong Kim isolated three related bidesmosidic steroidal saponins, polypodosides A-C (25–27), with saccharide substitution at C-3 and C-26.66,95 The structure of compound 25 was determined as the 7,8-dehydro derivative of osladin, a sweet constituent of the fern, Polypodium vulgare L.66 However, the structure of osladin (28) was reassigned by the late Professor Mugio Nishizawa (Tokushima Bunri University, Tokushima, Japan) as a result of a synthetic and X-ray crystallographic investigation.96 Working with Prof. Nishizawa, the configurational assignments of compounds 25 and 28 were shown to be the same (22R,25S,26R), from a chemical interconversion experiment.97 In this work, an intermediate obtained during the synthesis of osladin (28) was converted to an enone of the same structure produced upon the hydrolysis and silylation of compound 25.57,97 Polypodoside A (25) was rated as 500–600 times sweeter than sucrose, with polypodoside B (26) having a monosaccharide sugar unit at C-3, being marginally sweet. Polypodoside C (27), the 3″-O-methyl derivative of 26, was devoid of any sweet taste.57,66,97 Despite the promising sweetness intensity of polypodoside A (25), it is unlikely this will be used commercially as a sweetener, since extraction from a rather fragile epiphytic fern, and separation from a bitter-tasting constituent, (+)-afzelechin 7-O-β-D-apioside, would be needed.98
From the mature rhizomes of another fern, Selliguia feei Bory (Polypodiaceae), collected in Java, Indonesia, selligueain A (29) was isolated by former postdoctoral Nam-Im Baek as a new sweet-tasting ring-A double linked proanthocyanidin of the propelargonidin (afzelechin/epiafzelechin) sub-group.99 This was obtained as a major constituent of the rhizomes in 0.69% w/w yield by chromatography over silica gel, and the structure was characterized by the application of spectroscopic methods after peracetylation, thiolysis, and desulfurization steps.99 Selligueian A (29) was rated as exhibiting about 35 times the sweetness potency of a 2% w/v sucrose solution.99 Four other trimeric proanthocyanidins have been found to be sweettasting previously, two each from the fronds of the fern Arachniodes sporadosora (Kunze) Naikaike (Drypopteridaceae) and the root bark of Cinnamomum sieboldii Meisn. (Lauraceae).42 However, the sweetness potencies of these compounds relative to sucrose have not been determined, and compound 29 differs from them structurally in possessing an afzelechin C (terminal) unit.99 Further isolation work on S. feei led to the purification and structural characterization of selligueain B, a further new ring-A doubly linked proanthocyanidin that was not sweet.100 Selligueian A (29) was later isolated from Polypodium triseriale Sw. (Polypodiaceae) by Bohlin and co-workers, and shown to be inhibitory towards elastase, a proteolytic enzyme.101 Proanthocyanidins are widely distributed in common foods, and several health benefits have been attributed to this compound class.102 Therefore, a sweet representative of this series like selligueain A (29) might be of future use as a “functional food” ingredient.
In more recent work carried out at The Ohio State University, a search for bitterness-blocking agents103 has been performed on the leaves of Eriodictyon californicum Decne. (Hydrophyllaceae).104,105 This is a native U.S. plant known as “Herba Santa”, and its leaves exhibit an initial bitter taste, before turning sweetish when chewed, and hence this species may be regarded as a promising lead to search for constituents with taste-modifying constituents.104,105 In previous related work on the taste-modifying flavonoid constituents of this plant carried out in Germany, the sodium salt of the known flavanone, homoeriodictyol (30) was found to be able to block the bitter taste effected by caffeine.106 Hesperetin (31), a further flavanone from E. californicum, has also been documented as a sweetness-enhancing agent.107
In the work performed in our laboratory on E. californicum, nine flavanones and one flavone were isolated from the leaves, all of previously known structure.104,105 The plant specimen used was collected originally in California by World Botanical Associates (Laurel, MD) for work on its potential chemopreventive constituents performed by the group of Dr. John M. Cassady.108 The compounds were screened against HEK-293T cells expressing a chimeric G-protein α-subunit, transfected with the human bitterness receptor, hTAS2R31 (formerly hTAS2R44).109 Of the compounds evaluated, the C-7 methoxylated flavanones, sakuranetin (32) and 6-methoxysakuranetin (33), and the flavone, jaceosidin (34), were found to be selective inhibitors to the response to saccharin, with IC50 values of 3.6, 2.5, and 11.3 μM, respectively.105 In addition, 4′-isobutyrylhomoeriodictyol (35) was determined to be a potent agonist against a human bitterness receptor, with an IC50 value of ca. 2 μM (Fletcher, J. N., Kinghorn, A. D., Slack, J. P., Zia, J., unpublished results). Therefore, the change in taste profile of E. californicum leaves when chewed can be explained, at least in part, due to its flavonoid constituents. The practical utility of the purified flavonoid taste-modifying agents in E. californicum seems to be limited by the water insolubility of these substances.
Outlook for Future Research on Naturally Occurring Sweeteners and Taste-Modifying Agents
It may be confidently predicted that there will be a continued public demand for low-caloric foods and beverages in both the near- and long-term future, so there will be a need for the inclusion in these products of new sweeteners, sweetness enhancers, and bitterness blockers of natural origin. Criteria for the development of a new sweetener, however, are quite daunting, and include considerations of hedonic qualities, potency, stability, and water solubility, as well as economic aspects.110 So far as prospects for discovering new sweeteners are concerned, it is probable that most of the highly sweet plants in the world are already known, since their sensory properties would have become known to taxonomists at the time of their classification. It is still possible that plants with highly sweet constituents may be found in remote locations that have not become widely known, however. One method of finding new sweet compounds is to investigate further already known sweet-tasting plants obtained in large quantities. For example, very recently, several minor ent-kaurenoid diterpenoids have been isolated and characterized from Stevia rebaudiana leaves obtained from Malaysia, although it was not stated if the compounds obtained are sweet-tasting or not.111 Now that receptor-binding methods are available for high-throughput in vitro screening, it can be predicted that many more natural compounds will be detected as being sweet-tasting, although their sweetness will still need to be confirmed by sensory methods with human taste panels. Analogous to work that has been performed on flavonoid “co-effectors” of St. John’s Wort (Hypericum perforatum L., Clusiaceae), which modify the water solubility of the antidepressant principles,35 it is possible that other constituents of the plants of origin of sweet-tasting and other taste-modifying substances may enhance the overall effects experienced.
Potential Cancer Chemotherapeutic Agents from Selected Tropical Plants
Introductory Remarks
Cancer remains one of the major causes of death among the populations of developed and developing countries alike. For example, in the United States in 2010, it was determined that for persons under 85 years of age, cancer now accounts for more deaths than heart disease, with over half a million fatalities occurring annually.112 For some 50 years, substances from terrestrial micro-organisms and higher plants have afforded some relief from this scourge, and are representative of some of the most important anticancer agents available. Thus, from microbial sources, important cancer chemotherapeutic agents include the anthracyclines, bleomycin, dactinomycin, and mitomycin C, while four major classes of plant-derived compounds are used therapeutically in the U.S. (camptothecin derivatives; Catharanthus indole alkaloids; epidophyllotoxins; taxanes).113 The first approved marine-derived antitumor agent, trabectidin, is now in clinical use in Europe,21,114 and more recently eribulin mesylate, a synthetic analogue of the sponge constituent, halochondrin B, has been approved by the U.S. FDA for metastatic breast cancer treatment.115 Several authors have pointed to the future promise of additional new natural product oncology agents.e.g.,1,4,113,116,117
When potentially new antitumor agents of plant origin are concerned specifically, a number of compounds representative of a variety of skeletal types are currently in clinical trial. The continued influence of the seminal discoveries of camptothecin (36)118 and taxol (37)119 (now known as paclitaxel) by the group of the late Dr. Monroe Wall and of Dr. Mansukh Wani, of Research Triangle Institute in North Carolina, remains in evidence, as a result of the several camptothecin and taxane analogues now in advanced clinicial trials.120 Other plant-derived compounds under development are flavopiridol, indirubin, phenoxidiol, and analogues of combretastatin, homoharringtonine, and vinblastine.4,120,121 In spite of this, there is a perception among some members of the natural products community that plants have not been so productive as other types of organisms in yielding structurally diverse compounds, and therefore that additional work on plants as sources of new anticancer agents may not be so beneficial. Newman and Cragg have stated the following: “On the basis of recent experience using the National Cancer Institute (NCI) 60-cell line screen, the potential for the discovery of a new chemotype from plants, comparable to the taxanes or camptothecins, appears to be relatively low. In spite of work covering vascular plants from extensive collections in the tropics in the last 18+ years, no novel chemotypes have, as yet, been reported.”122 However, there is more than one way of interpreting these two sentences, even when it is considered how exceptional the discoveries of both camptothecin and taxol (paclitaxel) and their derivatives have been to biomedical research and clinical practice. For example, it could be that the use of even a very informative in vitro screen is not a substitute for the use of relevant in vivo testing early in the drug discovery process of a plant anticancer lead. It should be noted that several important lead antitumor agents from plants, many of which are still of interest today, were discovered in the 1970s and 1980s, including homoharringtonine (38),123 maytansine (39),124 bruceantin (40),125 pancratistatin (41),126 and combretastatin A-4 (42).127 For each of these five compounds, there was the early involvement of in vivo testing during the discovery process, and this is also true for camptothecin (36)118 and taxol (paclitaxel) (37).119 Today, there are many groups all over the world working on the elucidation of additional examples of plant-derived anticancer agents, all aiming at the a discovery of a compound of eventual clinical oncological prominence.
In the following paragraphs, recent progress made in our laboratory in the discovery of new potential anticancer agents from tropical plants will be described. It has been our practice for phytochemists and biologists to work closely together on this project, in order to obtain as much information as soon as possible on promising lead compound potency, selectivity, in vivo efficacy, and mechanism of action. A simple standardized solvent extraction scheme modified by the late Dr. Monroe Wall has been used.25 Another feature of our work has been the use of the in vivo hollow fiber assay as a secondary bioassay, to maximize the chances of obtaining compounds that have shown earlier in vitro activity in a cell- or mechanism-based bioassay, with subsequent in vivo activity in a xenograft model.128,129 We have been very fortunate over the years to have obtained significant funding from the U.S. National Cancer Institute, both through the “National Collaborative Drug Discovery Groups” (NCDDG) and program project (P01) mechanisms, involving in the last five years groups from several universities (The Ohio State University; University of Illinois at Chicago; University of North Carolina-Greensboro; North Carolina Central University), a private, non-profit research institute (Research Triangle Institute, North Carolina), a large pharmaceutical corporation (Bristol-Myers Squibb, Pharmaceutical Research Institute, Princeton, New Jersey), and a small biotechnology company (Mycosynthetix, Inc., Hillsborough, North Carolina). Several reviews have documented earlier the organization of this project team, the procedures used, and compounds of promise as antitumor agents obtained.130–132 As a result of an earlier organizational grouping (University of Illinois at Chicago; Research Triangle Institute; Glaxo Wellcome Medicines Research Centre, U.K.) of this same NCDDG project, the triterpenoid, betulinic acid, was isolated as a constituent of the plant, Ziziphus mauritiana Lam. (Rhamnaceae), collected in Zimbabwe. This apoptotic compound inhibited selectively the growth of human melanoma cancer cells, and in athymic mice bearing human melanoma cells it exhibited in vivo activity.133 Betulinic acid is now in phase I/II clinical trials at the College of Medicine, University of Illinois at Chicago, under the direction of Dr. Tapas Das Gupta, for the potential topical therapy for dysplastic melanocytic nevi.121
Aromatic Tropane Alkaloid Esters from Erythroxylum Species
The genus Erythroxylum (family Erythroxylaceae) is comprised of about 200 species that occur principally in tropical regions of South America and Africa.134 While cocaine appears to be a rare constituent within this genus, several species contain other tropane alkaloids.134 Phytochemical work by former postdoctorals Gloria Silva and Baoliang Cui in the laboratory of the senior author when at the University of Illinois at Chicago (UIC) led to the initial isolation of nine tropane aromatic ester alkaloids from the roots of Erythroxylum pervillei Baillon, including the six new compounds, pervilleines A–F (43–48), of interest as potential multidrug resistance (MDR) inhibitors.135 Thus far, there have been no MDR inhibitors approved as adjuncts to cancer chemotherapy, although some compounds have reached clinical trials for this purpose.136 The plant material was selected for research and collected in southern Madagascar by our collaborator, Prof. Philippe Rasoanaivo, of IMRA, Antananarivo, Madagascar. Recently, the stereostructure proposed for (+)-pervilleine C (45) was confirmed by enantiomeric total synthesis.137 When evaluated against a small panel of human tumor cells, pervilleines A–F (43–48) were found to be more or less selectively inhibitory for the KB-V1+ (vinblastine-resistant oral epidermoid carcinoma) cell line in the presence of vinblastine, while being inactive for the parent KB and other cell lines.135 In work conducted at UIC by then graduate student Qiuwen Mi, under the supervision of Dr. John M. Pezzuto, the parent compound, pervilleine A (43) was found to restore the sensitivity of CEM/VLB100 (multidrug-resistant human leukemic lymphoblast CEM cells (IC50 0.2 μM) and to be an effective inhibitor of P-glycoprotein.138 Further, this same alkaloid was demonstrated to be active as a MDR inhibitor when co-administered with vinblastine to KB- V1+ and KB-8–5 cells in hollow fibers implanted into NCr nu/nu mice.138 Pervilleines B (44), C (45), and F (48) were also found to reverse multidrug resistance when coadministered with vinblastine, in an analogous manner to pervilleine A (43).139,140
Phytochemical work was carried out by former postdoctoral Daniel Chavez on the stems of Erythroxylum rotundifolium Lunan, collected in the Dominican Republic. Five additional cytotoxic tropane aromatic esters (49–53) were isolated in this work, with the new compound 49 containing a Z-3,4,5-trimethoxycinnamyl ester unit at C-3.141 One of the tropane ester alkaloids (53) isolated from E. rotund0ifolium proved to be identical to pervilleine A (43), except for the aromatic ester groups at C-3 and C-6 being transposed, which led to a loss of specificity for the KB-V1+ cell line and weak broad cytotoxicity.135,141 Work-up of a large-scale recollection of the stem bark of E. pervillei (see below) at The Ohio State University by former Research Scientist Young-Won Chin led to the isolation and characterization of pervilleines G (54) and H (55), in addition to cis-pervilleines B (56) and F (57).142 In the latter two compounds, a Z-3,4,5-trimethoxycinnamyl ester unit occurred at C-6. Compounds 54–57 were not evaluated for their MDR modulatory activities.142
The most potent and selective MDR-inhibitory pervilleine derivatives of the tropane alkaloid type found in our work (43–48) all contain a E-3,4,5-trimethoxycinnamoyl unit attached to C-6, and are consistent structurally with other modulators of the MDR response in possessing a nitrogen atom flanked by two planar aromatic rings.143 Prior to our phytochemical work, tropane alkaloids had not been associated with cancer chemotherapy. Teodori and associates have developed a new class of highly potent synthetic MDR inhibitors in part using pervilleine A (43) as a model compound.144 In a pharmacological study performed by Dr. Popat N. Patil at The Ohio State University, the hydrochloride salt of pervilleine A (43) was found to show weakly competitive inhibition of the cholinergic response in the guinea pig ileum and also did not affect carbachol-induced contractions of the rat anoncoccygeus smooth muscle.146 The free base form of pervilleine H (55) was inactive in both these evaluations. The absence of any significant cholinergic or adrenergic effects by alkaloid 43 is supportive of its further development as a MDR inhibitor.145
Pervilleines A–C, and F (43–45,48) were found to be all of approximately equal abundance in the roots of E. pervillei, their plant of origin (yields in the range 0.0035–0.0043% w/w).135 As a result of their promising MDR inhibitory activities, which were comparable in potency to that of a positive control, verapamil, these compounds were selected for further development through the former RAID (“Rapid Access to Intervention Development”) program of the U.S. National Cancer Institute.146 Dr. Gordon Cragg served as the Program Director of this peer-reviewed award. The primary purpose of this project was to select one of the pervilleines (A–C or F) for preclinical development as a MDR inhibitor. Therefore, with the collaboration of Prof. Rasoanaivo, as well as staff members of the Missouri Botanical Garden, St. Louis, and its Madagascar office, 50 kg of both the stem bark and roots of E. pervillei were recollected from southern Madagascar in 2003, with the aim of purifying gram quantities of compounds 43–45, and 48. As a result of further testing carried out by Dr. Melinda Hollingshead at NCI-Frederick, it was determined that pervilleine A (43) is the best tropane alkaloid ester from E. pervillei as a potential MDR inhibitor.144 Scale-up isolation of the hydrochloride salt of pervilleine A (43) was performed, with the compound then sent to NCI-Frederick for additional testing. Thus, following a further successful hollow fiber test, pervilleine A HCl salt was evaluated in a HCT-15 colon tumor xenograft model in combination with vincristine. However, while there was a slowing of tumor growth in a mixture of the HCl salt of 43 with vincristine, compared with the same level of the latter compound alone, this was found not to be statistically significant at the doses used.
Rocaglate Derivatives from Aglaia Species
The genus Aglaia Lour. is the largest in the family Meliaceae, with over 100 timber species occurring in southeast Asia, tropical regions of east Asia, Papua New Guinea, northern Australia, and certain islands of Polynesia and Micronesia.147 The taxonomy of this genus has presented several problems in terms of species delimitation.147 In a review of antileukemic natural product extracts of plant and marine origin as evaluated biologically at the U.S. National Cancer Institute, Aglaia was designated as a “hot” genus and thus worthy of further phytochemical investigation.148 Similarly, in work carried out by former graduate student Marcy Balunas, in which cytotoxicity screening data were correlated over a 15-year period for extracts of plants that were largely of tropical origin, several Aglaia species were found to be exhibit promising ED50 values.149 Members of a series of aromatic compounds, known most commonly as “rocaglate” or “flavagline” derivatives, have been found as bioactive principles from members of the genus Aglaia.150,151 Somewhat unusually for this now quite large group of plant secondary metabolites, the structure of the first rocaglate derivative, rocaglamide (58), was elucidated as recently as 1982. In this initial paper, King et al. characterized the structure and relative configuration of (−)-rocaglamide by single-crystal X-ray diffraction, and demonstrated the activity of this compound in a P338 (murine lymphocytic leukemia) model.152 Using an enantiomeric synthetic procedure, Trost et al. determined the absolute configuration of (−)-rocaglamide (58) as (1R,2R,3S,3aR,8bS) in 1990.153 Spectroscopic data for this cyclopenta[b]benzofuran derivative were first published by Isman and Towers in 1993, who also showed rocaglamide to exhibit potent insecticidal activity against two variegated cutworms.154 Since this initial work, more than 100 rocaglate derivatives have been isolated from some 35 Aglaia species.150,151 These compounds have been classified into three basic types, namely, the cyclopenta[b]benzopyrans, the cyclopenta[bc]benzopyrans, and the benzo[b]oxepines, with most of the known biologically active representatives of the “flavaglines” being members of the first of these three groups.151
In our initial laboratory work at the University of Illinois at Chicago on a species in the genus Aglaia, the stems and fruits of A. elliptica Blume, collected in Thailand, were purified by former postdoctoral Baoliang Cui to afford the known compound, methyl rocaglate (59) and four new flavaglines (60–63). All five of these compounds showed strong growth inhibitory effects against a panel of 12 human cancer cell lines, with compound 60 (4′-demethoxy-3′,4′-methyldeoxy-methyl rocaglate) being the most potent.155 In a follow up biological study performed by then graduate student Sang Kook Lee in the laboratory of Dr. John Pezzuto, compound 60 was found to inhibit the growth of BC1 (breast) human cancer cells implanted in athymic mice (administered at 10 mg/kg body weight, three times a week, ip).156 It was also shown that in Lu2 lung cancer cells compounds 59–63 are cytostatic inhibitors of protein biosynthesis.156 Methyl rocaglate (59) was found also as a biologically active constituent of the leaves and twigs of Aglaia rubiginosa (Hiern) Pannell collected in Indonesia, together with two other known rocaglate derivatives and the new cytotoxic compound 1-O-acetylrocagloal (64).157 Rocaglaol (65), the parent compound of 64, was isolated as the only rocaglate constituent of the bark of Aglaia crassinervia Kurz ex Hiern collected in Indonesia.158 In a follow-up biological study on rocaglaol (65), carried out by then postdoctoral Qiuwen Mi under the supervision of Dr. Steven M. Swanson, this compound was found to be a potent cytotoxic agent that causes G2/M cell cycle arrest through the mitochondrial pathway.159
Activity-guided isolation work on the bark of Aglaia edulis from Indonesia was carried out by former graduate student Soyoung Kim, leading to the isolation of two new cytotoxic cyclopenta[b]benzofuran derivatives, compounds 66 and 67.160 It was found that compound 67 was considerably more cytotoxic than 66 for three cancer cell lines in a small panel, but both compounds were equally cytotoxic when tested against the non-tumorigenic HUVEC line. However, when evaluated in the P-388 lymphocytic leukemia in vivo model (4.5 mg/kg/injection, ip), compound 67 was found to be inactive.160 Several additional new compounds were isolated from A. edulis bark, representative of the cyclopenta[bc]benzopyran, benzo[b]oxepine, and amide classes, but none of these proved to be appreciably cytotoxic.160,161 Some compounds of the cyclopenta[bc]benzopyran type have been found to exhibit potent biological activity, however. As an example, former postdoctoral Angela Salim isolated and characterized ponapensin (68) as a new member of this class from the stems of Aglaia ponapensis Kaneh., collected in the Eastern Caroline Islands. Compound 68 exhibited potent NF-κB inhibitory activity in an enzyme-based Elisa assay, as evaluated in the laboratory of Dr. Esperanza Carcache de Blanco at The Ohio State University (IC50 0.06 μM).162 Other compounds of the rocaglate class have previously been found to exhibit inhibition of NF-κB.163
The fruits and twigs of Aglaia foveolata Pannell [originally misidentified as Aglaia silvestris (M. Roemer) Merrill] were found to contain a new compound, (−)-silvestrol (69), a compound with an interesting structural modification of the cyclopenta[b]benzofuran structural motif, in possessing also a C-6 attached dioxanyl ring.164 Silvestrol was found to have a similar nanomolar cytotoxic potency to camptothecin and paclitaxel for a small panel of cancer cell lines at the University of Illinois at Chicago (UIC). It proved to be substantially more cytotoxic than its analogue, methyl rocaglate (59), which has the same cyclopenta[b]benzofuran unit but with a C-6 methoxy group.164 Also isolated from the stems of A. foveolata was (−)-episilvestrol [(−)-5‴-episilvestrol, 70)], which also proved to be a very potent cytotoxic agent.164 The initial isolation of silvestrol was performed by former postdoctoral Bang Yeon Hwang, while then Research Scientist Baoning Su completed the initial phytochemical investigation of A. foveolata and performed scale-up isolation work. The structure and absolute configuration of silvestrol were determined as a result of the X-ray crystallographic analysis of the 5‴,6‴-di-p-bromobenzoate of 69, performed at UIC by Drs. Bernard Santarsiero and Andrew Mesecar.164 Silvestrol (69) was evaluated in vivo in the P-388 murine leukemia model by Drs. William Rose and Robert Wild at Bristol-Myers Squibb, Pharmaceutical Research Institute, Princeton, New Jersey. When administered ip daily at a maximal tolerated dose of 2.5 mg/kg/injection for five days, silvestrol was active and exhibited a maximum increase in lifespan equivalent to a T/C of 150%. In the P-388 iv leukemia model, the compound was also active when injected iv twice daily (T/C 129%; total of 2 mg/kg/day).164 In addition, silvestrol (69) showed activity when evaluated in the in vivo hollow fiber assay at UIC, using KB (human nasopharygeal), LNCaP (human prostate), and Col2 (human colon cancer) cells, at the dose range 0.625–5 mg/kg body weight), with better activity in each case at the ip site of administration, when compared with the sc site.164 In preliminary mechanistic work carried out at UIC under the supervision of Dr. Steven Swanson, in LNCaP cells, silvestrol (69) produced a p53-independent blockade of the G2/M checkpoint,165 and also induced apoptosis through the mitochondrial/apoptosome pathway, without the activation of caspases-3 and 7.166 Silvestrol has now been subjected to total synthesis by the groups of Porco167 and Rizzacasa168,169 with the stereostructure our team proposed being confirmed as a result. It should be pointed out that in a U.S. patent that predated our work on A. foveolata, two cyclopenta[b]benzofuran constituents were documented from Aglaia leptantha Miq., and shown subsequently to have the same NMR parameters as silvestrol (69) and episilvestrol (70), although only their planar structures were proposed.170 In a murine xenograft study, “compound A” from A. leptantha was found to inhibit the growth of PC-3 human prostate cancer cells.170
In view of an inability to obtain intellectual property protection on its potential anticancer properties, interest on silvestrol (69) declined significantly for some time. However, after the senior author moved to The Ohio State University in 2004, work began in the laboratory of Drs. Michael Grever and David Lucas on the effects of this compound on human leukemic cell lines from cancer patients. It was found that silvestrol exhibited a LC50 value of 7 nM in chronic lymphocytic leukemia (CLL) cells, with the compound being more potent against B-cells than T-cells, and showed depletion of the anti-apoptotic protein Mcl-1.171 Furthermore, when given at a dose of 1.5 mg/kg/body weight every other day on a three-week dosing schedule, in an aggressive in vivo mode of acute lymphoblastic leukemia, silvestrol treatment showed a significant improvement in survival.171 A pharmacokinetic study has been performed on silvestrol (69) formulated in hydroxypropyl-β-cyclodextrin at The Ohio State University Medical Center, in collaboration with the National Cancer Institute, using C57BL/6 mice. It was found that the systemic availability this compound was 100% when administered intraperitoneally, but there was only 1.7% bioavailability when given orally. Silvestrol proved to be more stable in mouse and human plasma than in rat plasma.172 While it possible that silvestrol has multiple cellular targets, independent work by Pelletier and colleagues has shown silvestrol to be an inhibitor of translation.173,174 A more detailed review of the biological activity of silvestrol has been included in a review by Lucas et al.175
More recent phytochemical work on A. foveolata has been carried out at The Ohio State University, in which postdoctoral Angela Salim showed that silvestrol (69) occurs in the leaves of this plant, albeit at a lower yield (0.002% w/w) than in the stem bark (0.02% w/w).176 This is a potentially important result, since it might be possible to grow the source plant of silvestrol (69) and periodically crop the leaves for the extraction of this compound as a renewable resource. A number of other flavaglines were isolated from A. foveolata leaves, but the only one of these that proved to be cytotoxic for a panel of cancer cells was foveoglin B (71), a cyclopenta[b]benzopyran derivative with a benzoyl-1,4-butanediamide moiety.176 When a largescale recollection of A. foveolata obtained in 2007 from Kalimantan, Indonesia, was processed phytochemically, two additional minor constituents of silvestrol (69) were isolated, namely, 5‴-episilvestrol (72) and 2‴, 5‴-diepisilvestrol (73).177 On evaluation of these new compounds for their cytotoxicity against the HT-29 cell line, they were much less active than the parent compounds 69 and 70. This indicates that the chiral carbon C-2‴ in the 1,4-dioxanyloxy moiety of silvestrol (69) is important for the mediation of optimum cytotoxicity in this compound class.177 Figure 2 shows a diagram summarizing the effects of structural modification of the molecule of silvestrol (69) on the resultant cytotoxicity.165,170,177,178
Figure 2.

Preliminary structure-activity relationship diagram for silvestrol (69)
Owing to its promise in treating B-cell malignancies such as CLL and ALL, silvestrol (69) is now undergoing preclinical development under the auspices of the U.S. NCI, as part of the NExT program.179 Dr. David Newman is the Project Director, and, in view of the existing patent assigned to the government of the State of Sarawak,171 the plant source is Aglaia stellatopilosa Pannell (previously identified as Aglaia leptantha Miq.) from Malaysia,180,181 and this will be supplied for evaluation through the Sarawak Biodiversity Centre, Kuching.
Miscellaneous Potential Anticancer Agents from Plants
In this section, a number of other new compounds that have provided structural interest and/or shown potential as cancer chemotherapeutic agents will be described briefly. Former graduate student Yuny Seo isolated vatdiospyroidol (74), a complex resveratrol tetramer, from the stems of Vatica diospyroides Sym. (Dipterocarpaceae), collected in Thailand.182 The structure elucidation process for this structurally complex polyhydroxylated compound was protracted, and involved 2D NMR data interpretation, derivatization, and molecular modeling steps. At the time of its isolation, compound 74 was one of two resveratrol tetramers found to exhibit cytotoxic activity against a panel of human tumor cells [best activity, oral epidermoid carcinoma (KB) cells, EC50, 1.0 μg/mL].182 In the same year as our report (1999), another resveratrol tetramer, hopeaphenol, was shown to be cytotoxic for KB and two other types of cancer cells.183 The resveratrol tetramer, vaticanol C, obtained as a constituent of Vatica rassak Blume demonstrated a cytotoxic effect for the HL60 leukemia and SW480 colon cell lines, and induced apoptosis in the latter cell line by affecting mitochondrial transport.184 The antineoplastic potential of the resveratrol oligomer compound class has been supported by work on a plant extract (Vateria indica L; Dipterocarpaceae), containing hopeapheol and vaticanol C, which inhibited tumor growth in an allografted sarcoma S-180 model in mice.185 Vaticaphenol A, a second new resveratrol oligomer was isolated and characterized in our work on V. diosyroides.182 Although this compound was not cytotoxic for any of the cell lines in which it was evaluated, in more recent work this same compound as a constituent of Vatica oblongifolia var. oblongifolia Hook. exhibited inhibitory activity against methicillin-resistant Staphyloccococcus aureus (MIC value 25 μg/mL).186
As a result of input from several isolation chemists, work has been performed on the stems of Amomum aceuleatum Roxb. (Zingiberaceae), collected in Indonesia, leading to the isolation of several compounds based on the unusual 1,7-dioxadispiro[5,1,5.2]pentadecane skeleton, including the parent compounds aculeatins A (75) and B (76).187,188 Also isolated were two new compounds, amomols A (77) and B (78), having a 1-oxospiro[4,5]decane skeleton, but for which it was not possible to establish the C-2 configuration, due to the small quantities of each compound isolated.188 These two compounds and some analogues were recently obtained by total synthesis.189 We have previously reviewed the work on the initial isolation and the determination of structure of aculeatin A (75).133 When seven natural products from A. aculeatum and nine semi-synthetic derivatives of compounds 75 and 76 were evaluated against a small panel of three human cancer cell lines in our laboratory, it was found that most of the substances were quite cytotoxic, with the most active compound determined to be aculeatin A (75). This compound was evaluated in the in vivo hollow fiber assay at the University of Illinois at Chicago, and was found to inhibit the growth of MCF-7 breast cancer cells by 10–60% when administered ip in the dose range 6.25–50 mg/kg body weight.188 However, aculeatin A (75) was not active for two further cell lines in the hollow fiber assay, or against two in vivo models (P388 murine lymphocytic leukemia and a human A2780 ovarian carcinoma xenograft) as evaluated at Bristol-Myers Squibb.188 It would seem worth following up the positive result of 75 in the hollow fiber test system with a breast cancer xenograft model. The aculeatins have proven to be attractive targets for synthetic chemists,e.g.,190–192 Some of the aculeatin and amomol derivatives synthesized have shown promising in vitro antiplasmodial inhibitory activities.189,190
Investigation of the stem bark of Garcinia lateriflora Blume (Clusiaceae) by postdoctoral Yulin Ren, a species collected in Indonesia, led to the isolation and structural characterization of several biflavonoids with proteasome-inhibitory activity, with the known compound, morelloflavone (79) being the most active (IC50 1.3 μM). In addition, several “caged” xanthones proved to be cytotoxic for HT-29 human colon cancer cells, with another known compound, (−)-morellic acid (80), found to be the most potent of these (ED50 0.36 μM).193 In work on compound 80, conducted in collaboration with the group of Dr. Daneel Ferreira, of the University of Mississippi, the absolute configuration for a “caged” xanthone at carbons C-5, C-7, C-8, C-8, C-10a, and C-27 was determined for the first time using electronic circular dichroism (ECD) spectroscopy.193 However, (−)-morellic acid (80) was not active when evaluated in the in vivo hollow fiber assay at the University of Illinois at Chicago (ip; highest dose tested, 20 mg/kg body weight).193 (−)-Gambogic acid (81) is a structurally related compound, and by comparison of its CD spectrum with that of compound 80, its absolute configuration could be established.194 Compound 81 is undergoing clinical trial in the People’s Republic of China as an anticancer agent.195
Outlook for Future Work on Plant-derived Anticancer Agents
When the entire past track record of the discovery of anticancer agents derived from plants is considered over the last 50 years, it can be stated confidently that outstanding progress has been made, with such compounds having affected in a positive manner the lives of millions of cancer patients all over the world. Plant-derived anticancer agents have frequently shown novel mechanisms of action, and have proven to be tantalizing targets for synthetic chemists. Therefore, efforts to purify and elucidate the structures of new promising lead compounds from plants should be enhanced in the future. However, such work needs to be tightly focused, with activity-guided fractionation occurring preferentially on leads that have been prioritized for potential novelty from a taxonomic point of view, and when having shown selectivity and potency in relevant bioassays at the crude extract stage. Purified natural product lead compounds with potential anticancer activity need to be optimized using modern medicinal chemistry approaches. Work of this type is necessarily multidisciplinary, but a great deal can be accomplished successfully by highly committed smaller teams of investigators.e.g.,119,120,124–128 Cragg, Grosthaus, and Newman have summarized work showing that certain anticancer agents of plant origin may be biosynthesized also by associated endophytic fungi, which in the future may offer a means of controlled drug production by fermentation.118 However, as evidenced by a recent study on the production of camptothecin (36) by the endophytic fungus, Fusarium solani, the instability of the biosynthetic genes of this organism resulted in decreased amounts of the desired alkaloid on repeated subculturing.196 Therefore, present-day methods of manipulation of endophytic fungi will need to be enhanced in order for commercial fermentation of important plant-derived compounds to occur, but the potential for this is clearly evident. Another approach that offers the possibility of increasing the potential the chemical diversity based on a given anticancer lead compound is directed biosynthesis conducted by feeding with unnatural substrates.e.g.,197
Conclusions
Even today, a majority of the world’s population relies on plants as a primary source of traditional medicine.15 As a consequence, investigating the scientific basis for the use of these medicinal plants is expected to be of prime interest to natural product researchers for the foreseeable future. As judged by the biomedical literature over the last 15 years, there has been an explosion of interest in the analysis, biological testing, chemical composition, clinical evaluation, and utilization of botanical dietary supplements. New specific avenues of inquiry for these agents are still being elucidated, such as active principle water solubility enhancement35 and compound synergy research.198 In the United States, the opportunity of developing additional examples of the new class of “botanical drugs” will be challenging but could be very rewarding.18 In terms of searching for new single chemical entities from plants, still only a relatively small percentage of higher plants have ever been investigated for any type of biological activity. Unfortunately, it has been predicted that as a result of potential climate change, significant losses in the global vegetative species will occur by 2050.199 Therefore, the search for such new biologically active compounds should be accelerated rather than be deemphasized in the next few years. In this review, plant-derived secondary metabolites have been described for just two (sweet-tasting/taste-modifying and potential anticancer) general types of biological activity. However, in vitro and in vivo bioassays have been developed for numerous other types of health-related effects. In an era when hundreds if not thousands of samples can be evaluated a single day through high-throughput screening, and the data obtained catalogued thought advances in informatics, the opportunities for new drug discovery from the small-molecule constituents of temperate and tropical plants seem highly propitious.







Acknowledgments
The senior author was fortunate enough to work as a postdoctoral at the University of Illinois at Chicago under Dr. Norman R. Farnsworth (1976–1977), and then as a faculty colleague the same institution (1977–2004). Dr. Farnsworth is thanked for his continuous inspiration, leadership, and conviviality. Many other faculty colleagues, graduate students, and postdoctorals who have contributed to the work described in this review, whose names are included in the bibliography, are they are very gratefully acknowledged. We are grateful to Dr. David J. Newman, NCI-Frederick, for providing updated research data on pervilleine A. The research described herein was generously supported over the years by NIH grant and contract awards DE-02425, DE-07560, DE-08937, CA-52956, and CA-125066, and through the NCI RAID program, and by grant support by General Foods Corporation, White Plains, New York, and Givaudan Flavors Corporation, Cincinnati, Ohio.
Footnotes
Adapted from a Norman R. Farnsworth Research Achievement Award address, 51st Annual Meeting of the American Society of Pharmacognosy, St. Petersburg, Florida, July 10-14, 2010.
References and Notes
- 1.Newman DJ, Cragg GM. J Nat Prod. 2007;70:461–477. doi: 10.1021/np068054v. [DOI] [PubMed] [Google Scholar]
- 2.Li JW-H, Vederas JC. Science. 2009;325:161–165. doi: 10.1126/science.1168243. [DOI] [PubMed] [Google Scholar]
- 3.Chin Y-W, Balunas MJ, Chai H-B, Kinghorn AD. AAPS J. 2006;8:E239–E253. doi: 10.1007/BF02854894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Butler MS. Nat Prod Rep. 2008;25:475–516. doi: 10.1039/b514294f. [DOI] [PubMed] [Google Scholar]
- 5.Williams DH, Stone MJ, Hauck PR, Rahman SK. J Nat Prod. 1989;52:1189–1208. doi: 10.1021/np50066a001. [DOI] [PubMed] [Google Scholar]
- 6.Henkel T, Brunne RM, Müller H, Riechel F. Angew Chem Int Ed. 1999;38:643–647. doi: 10.1002/(SICI)1521-3773(19990301)38:5<643::AID-ANIE643>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 7.Feher M, Schmid JM. J Chem Inf Comp Sci. 2003;43:218–227. doi: 10.1021/ci0200467. [DOI] [PubMed] [Google Scholar]
- 8.Koehn FE, Carter GT. Nature Rev Drug Discov. 2005;4:206–220. doi: 10.1038/nrd1657. [DOI] [PubMed] [Google Scholar]
- 9.Lam KS. Trends Microbiol. 2007;15:279–289. doi: 10.1016/j.tim.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 10.Sneader W. Drug Discovery A History. John Wiley & Sons; Chichester, West Sussex, U. K: [Google Scholar]
- 11.Tan G, Gyllenhaal C, Soejarto DD. Curr Drug Targets. 2006;7:265–277. doi: 10.2174/138945006776054942. [DOI] [PubMed] [Google Scholar]
- 12.McChesney JD, Venkataraman SK, Henri JT. Phytochemistry. 2007;68:2015–2022. doi: 10.1016/j.phytochem.2007.04.032. [DOI] [PubMed] [Google Scholar]
- 13.Balunas MJ, Kinghorn AD. Life Sci. 2005;78:431–441. doi: 10.1016/j.lfs.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 14.Heinrich M, Gibbons S. J Pharm Pharmacol. 2001;53:425–432. doi: 10.1211/0022357011775712. [DOI] [PubMed] [Google Scholar]
- 15.Fabricant DS, Farnsworth NR. Envir Health Pers. 2001;69(Suppl 1):69–75. doi: 10.1289/ehp.01109s169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jones WP, Chin Y-W, Kinghorn AD. Curr Drug Targets. 2006;7:247–264. doi: 10.2174/138945006776054915. [DOI] [PubMed] [Google Scholar]
- 17.Samuelsson G, Bohlin L. Drugs of Natural Origin – A Treatise of Pharmacognosy. 6. Swedish Pharmaceutical Press; Stockholm: 2009. [Google Scholar]
- 18.Chen ST, Dou J, Temple R, Agarwal R, Wu K-M, Walker S. Nature Biotechnol. 2008;26:1077–1083. doi: 10.1038/nbt1008-1077. [DOI] [PubMed] [Google Scholar]
- 19.Demain AL, Sanchez S. J Antibiot. 2009;62:5–16. doi: 10.1038/ja.2008.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Buss AD, Butler MS, editors. Natural Product Chemistry for Drug Discovery, RSC Biomolecular Sciences Publication No. 18. Royal Society of Chemistry; London: 2010. [Google Scholar]
- 21.Sashidhara KV, White KN, Crews P. J Nat Prod. 2009;72:588–603. doi: 10.1021/np800817y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Molinski TF, Dalisay DS, Lievens SL, Saludes JP. Nat Rev Drug Disc. 2009;8:69–85. doi: 10.1038/nrd2487. [DOI] [PubMed] [Google Scholar]
- 23.Lee K-H. J Nat Prod. 2010;73:500–516. doi: 10.1021/np900821e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dixon RA, Strack D. Phytochemistry. 2003;62:815–816. doi: 10.1016/s0031-9422(02)00712-4. [DOI] [PubMed] [Google Scholar]
- 25.Wall ME, Wani MC, Brown DM, Fullas F, Oswald JB, Josephson FF, Thornton NM, Pezzuto JM, Beecher CWW, Farnsworth NR, Cordell GA, Kinghorn AD. Phytomedicine. 1996;3:281–285. doi: 10.1016/S0944-7113(96)80067-5. [DOI] [PubMed] [Google Scholar]
- 26.Appendino G, Pollastro F. In: Natural Product Chemistry for Drug Discovery; RSC Biomolecular Sciences Publication No. 18. Buss AD, Butler MS, editors. Royal Society of Chemistry; London: 2010. pp. 140–173. [Google Scholar]
- 27.Nono ECN, Mkounga P, Kuete V, Marat K, Hultin PG, Nkengfack AE. J Nat Prod. 2010;73:213–216. doi: 10.1021/np9007393. [DOI] [PubMed] [Google Scholar]
- 28.Tsai SF, Lee S-S. J Nat Prod. 2010;73:1632–1635. doi: 10.1021/np100247r. [DOI] [PubMed] [Google Scholar]
- 29.Wohlmuth H, Deseo MA, Brushett DJ, Thompson DR, MacFarlane G, Stevenson LM, Leach DN. J Nat Prod. 2010;73:743–746. doi: 10.1021/np900688r. [DOI] [PubMed] [Google Scholar]
- 30.Saroglou V, Karioti A, Rancic A, Dimas K, Koukoulitsa C, Zervou M, Skaltsa H. J Nat Prod. 2010;73:242–246. doi: 10.1021/np9004129. [DOI] [PubMed] [Google Scholar]
- 31.Inui T, Wang Y, Nikolic D, Smith DC, Franzblau SG, Pauli GF. J Nat Prod. 2010;73:563–567. doi: 10.1021/np900674d. [DOI] [PubMed] [Google Scholar]
- 32.Serrano MAR, Pivatto M, Francisco W, Danuello A, Regasini LO, Lopes EMC, Lopes MN, Young MCM, Bolzani VS. J Nat Prod. 2010;73:482–484. doi: 10.1021/np900644x. [DOI] [PubMed] [Google Scholar]
- 33.Wang P-C, Ran X-H, Chen R, Luo H-R, Liu Y-Q, Zhou Y-Q, Zhao Y-Z. J Nat Prod. 2010;73:1563–1567. doi: 10.1021/np100452a. [DOI] [PubMed] [Google Scholar]
- 34.Cicek SS, Kohm S, Taferner B, Hering S, Stuppner H. J Nat Prod. 2010;73:2024–2028. doi: 10.1021/np100479w. [DOI] [PubMed] [Google Scholar]
- 35.Nahrstedt A, Butterweck V. J Nat Prod. 2010;73:1015–1021. doi: 10.1021/np1000329. [DOI] [PubMed] [Google Scholar]
- 36.Li Y, Shibahara A, Matsuo Y, Tanaka T, Kuono I. J Nat Prod. 2010;73:33–39. doi: 10.1021/np900618v. [DOI] [PubMed] [Google Scholar]
- 37.Yang MC, Kwon HC, Kim Y-J, Lee KR, Yang HO. J Nat Prod. 2010;73:801–805. doi: 10.1021/np900628j. [DOI] [PubMed] [Google Scholar]
- 38.Guerro-Analco JA, Martineau L, Saleem A, Madiraju P, Muhammad A, Durst T, Haddard P, Arnason JT. J Nat Prod. 2010;73:1519–1523. doi: 10.1021/np1003005. [DOI] [PubMed] [Google Scholar]
- 39.Chen W, Wang R, Shi Y-P. J Nat Prod. 2010;73:1398–1403. doi: 10.1021/np100339u. [DOI] [PubMed] [Google Scholar]
- 40.Hayes PY, Chow S, Somerville MJ, Fletcher MT, de Vos JJ. J Nat Prod. 2010;73:1907–1913. doi: 10.1021/np1005746. [DOI] [PubMed] [Google Scholar]
- 41.Grenby TH, editor. Developments in Sweeteners – 3. Elsevier Applied Science; London and New York: 1991. [Google Scholar]
- 42.Kinghorn AD, Chin Y-W, Pan L, Jia Z. Comprehensive Natural Products Chemistry II: Chemistry and Biology. In: Mander L, Liu H-W, Verpoorte R, editors. Development & Modification of Bioactivity. Vol. 3. Elsevier; Oxford, U. K: 2010. pp. 269–315. [Google Scholar]
- 43.Brown RJ, de Banate MA, Rother KI. Int J Ped Obes. 2010;5:305–312. doi: 10.3109/17477160903497027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Behrens M, Meyerhof W, Hellfritsch C, Hofmann T. Angew Chem Int Ed. 2011;50:2220–2242. doi: 10.1002/anie.201002094. [DOI] [PubMed] [Google Scholar]
- 45.von Rymon Lipinski G-W. In: Optimising Sweet Taste in Foods. Spillane WJ, editor. Woodhead Publishing Ltd./Taylor and Francis; Boca Raton, FL: 2006. pp. 252–280. [Google Scholar]
- 46.Embuscado ME. In: Optimising Sweet Taste in Foods. Spillane WJ, editor. Woodhead Publishing Ltd./Taylor and Francis; Boca Raton, FL: 2006. pp. 153–174. [Google Scholar]
- 47.Kinghorn AD, Compadre CM. In: Alternative Sweeteners, Fourth Edition. O’Brien Nabors L, editor. Taylor and Francis; Boca Raton, FL: 2011. pp. 223–246. [Google Scholar]
- 48.Mizutani K, Tanaka O. In: Stevia: the Genus Stevia; Medicinal and Aromatic Plants – Industrial Profiles. Kinghorn AD, editor. Vol. 19. Taylor & Francis; London: 2002. pp. 178–195. [Google Scholar]
- 49.Anonymous. Fed Reg. 1985;50(99):21043–21045. [Google Scholar]
- 50.Kitagawa I. Pure Appl Chem. 2002;74:1189–1198. [Google Scholar]
- 51.Soejarto DD. In: Stevia: the Genus Stevia; Medicinal and Aromatic Plants – Industrial Profiles. Kinghorn AD, editor. Vol. 19. Taylor & Francis; London: 2002. pp. 40–67. [Google Scholar]
- 52.Kinghorn AD. In: Stevia: the Genus Stevia; Medicinal and Aromatic Plants – Industrial Profiles. Kinghorn AD, editor. Vol. 19. Taylor & Francis; London: 2002. pp. 1–17. [Google Scholar]
- 53.Karacostas MC, Prakash I, Kinghorn AD, Wu CD, Soejarto DD. In: Alternative Sweeteners. 4. O’Brien Nabors L, editor. Taylor and Francis; Boca Raton, FL: 2011. pp. 159–180. [Google Scholar]
- 54.Karacostas MC, Curry LL, Boileau AC, Brusik DJ. Food Chem Toxicol. 2008;46(Suppl 7S):S1–S10. doi: 10.1016/j.fct.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 55.Kemp SE. In: Optimising Sweet Taste in Foods. Spillane WJ, editor. Woodhead Publishing Ltd./Taylor and Francis; Boca Raton, FL: 2006. pp. 175–251. [Google Scholar]
- 56.Crammer B. In: Selected Topics in the Chemistry of Natural Products. Ikan R, editor. World Scientific; Singapore: 2008. pp. 189–208. [Google Scholar]
- 57.Kinghorn AD, Kaneda N, Baek N-I, Kennelly EJ, Kinghorn AD. Med Res Rev. 1998;18:347–360. doi: 10.1002/(sici)1098-1128(199809)18:5<347::aid-med5>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 58.Kinghorn AD, Kim N-C, Kim DHSL. In: Naturally Occurring Glycosides: Chemistry, Distribution, and Biological Properties. Ikan R, editor. John Wiley & Sons; Chichester, U. K: 1999. pp. 399–429. [Google Scholar]
- 59.Kinghorn AD, Soejarto DD. Pure Appl Chem. 2002;74:1169–1179. [Google Scholar]
- 60.Kim N-C, Kinghorn AD. In: Studies in Natural Products Chemistry, vol. 27. Bioactive Natural Products (Part H) Atta-ur-Rahman, editor. Elsevier Scientific Publishers; Amsterdam: 2002. pp. 3–57. [Google Scholar]
- 61.Kim N-C, Kinghorn AD. Arch Pharm Res. 2002;25:725–746. doi: 10.1007/BF02976987. [DOI] [PubMed] [Google Scholar]
- 62.Kinghorn AD, Kim N-C. In: Optimising Sweet Taste in Foods. Spillane W, editor. Woodhead Publishing Ltd./CRC Press; Cambridge, U. K: 2006. pp. 292–306. [Google Scholar]
- 63.Suttisri R, Lee I-S, Kinghorn AD. J Ethnopharmacol. 1995;15:9–26. doi: 10.1016/0378-8741(95)01248-c. [DOI] [PubMed] [Google Scholar]
- 64.Li X, Staszewski L, Xu H, Durick K, Zoller M, Adler E. Proc Natl Acad Sci USA. 2002;99:4692–4696. doi: 10.1073/pnas.072090199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li X, Servant G. In: Sweetness and Sweeteners (ACS Symposium Series No. 979) Weerasinghe DK, DuBois GE, editors. Oxford University Press; New York: 2008. pp. 368–385. [Google Scholar]
- 66.Kim J, Pezzuto JM, Soejarto DD, Lang FA, Kinghorn AD. J Nat Prod. 1988;51:1166–1172. doi: 10.1021/np50060a019. [DOI] [PubMed] [Google Scholar]
- 67.Fullas F, Hussain RA, Bordas E, Pezzuto JM, Soejarto DD, Kinghorn AD. Tetrahedron. 1991;47:8515–8522. [Google Scholar]
- 68.Cordell GA. In: Natural Product Chemistry for Drug Discovery; RSC Biomolecular Sciences Publication No. 18. Buss AD, Butler MS, editors. Royal Society of Chemistry; London: 2010. pp. 81–139. [Google Scholar]
- 69.Hussain RA, Lin Y-M, Poveda LJ, Bordas E, Chung BS, Pezzuto JM, Soejarto DD, Kinghorn AD. J Ethnopharmacol. 1990;28:103–115. doi: 10.1016/0378-8741(90)90067-4. [DOI] [PubMed] [Google Scholar]
- 70.Hussain RA, Poveda LJ, Pezzuto JM, Soejarto DD, Kinghorn AD. Econ Bot. 1990;44:174–182. [Google Scholar]
- 71.Compadre CM, Pezzuto JM, Kinghorn AD, Kamath SK. Science. 1985;227:417–419. doi: 10.1126/science.3880922. [DOI] [PubMed] [Google Scholar]
- 72.Compadre CM, Robbins EF, Kinghorn AD. J Ethnopharmacol. 1986;15:89–106. doi: 10.1016/0378-8741(86)90105-4. [DOI] [PubMed] [Google Scholar]
- 73.Compadre CM, Hussain RA, Lopez de Compadre RL, Pezzuto JM, Kinghorn AD. J Agric Food Chem. 1987;35:273–279. [Google Scholar]
- 74.Mori K, Sato M. Tetrahedron. 1986;42:5895–5900. [Google Scholar]
- 75.Kaneda N, Lee I-S, Gupta MP, Soejarto DD, Kinghorn AD. J Nat Prod. 1992;55:1136–1141. doi: 10.1021/np50086a016. [DOI] [PubMed] [Google Scholar]
- 76.Souto-Bachiller FA, De Jesus-Echevevarría M, Cadenas-González OE, Acuña-Rodriguez MF, Meléndez PA, Romero-Ramsey L. Phytochemistry. 1997;44:1077–1086. [Google Scholar]
- 77.Nayal R, Abajy MY, Melzig MF. Int J Essent Oil Ther. 2009;3:91–100. [Google Scholar]
- 78.Ono M, Tsuru T, Abe H, Eto M, Okawa M, Abe F, Kinjo J, Ikeda T, Nohara T. J Nat Prod. 2006;69:1417–1420. doi: 10.1021/np068010m. [DOI] [PubMed] [Google Scholar]
- 79.Compadre CM, Hussain RA, Lopez de Compadre RL, Pezzuto JM, Kinghorn AD. Experientia. 1988;44:447–449. doi: 10.1007/BF01940543. [DOI] [PubMed] [Google Scholar]
- 80.Basoli A, Merlini L, Morini G. Pure Appl Chem. 2002;74:1181–1187. [Google Scholar]
- 81.Gomi T, Shimida T. 04074114 A. Jpn Kokai Tokkyo Koho JP. 1992
- 82.Morini G, Bassoli A, Temissi PA. J Med Chem. 2005;48:5520–5529. doi: 10.1021/jm0503345. [DOI] [PubMed] [Google Scholar]
- 83.Choi Y-H, Kinghorn AD, Shi X, Zhang H, Teo B-K. J Chem Soc, Chem Commun. 1989:887–888. [Google Scholar]
- 84.Choi Y-H, Hussain RA, Pezzuto JM, Kinghorn AD, Morton JF. J Nat Prod. 1989;52:1118–1127. doi: 10.1021/np50065a032. [DOI] [PubMed] [Google Scholar]
- 85.Guignet ME. C R Hebd Séances Acad Sci. 1885;100:151–153. [Google Scholar]
- 86.Akinloye BA, Adalumo LA. Nigerian J Pharm. 1981;12:405. [Google Scholar]
- 87.Kennelly EJ, Cai L, Kim N-C, Kinghorn AD. Phytochemistry. 1996;41:1381–1383. [Google Scholar]
- 88.Fullas F, Choi Y-H, Kinghorn AD, Bunyapraphatsara N. Planta Med. 1990;56:332–333. doi: 10.1055/s-2006-960974. [DOI] [PubMed] [Google Scholar]
- 89.Kim N-C, Kinghorn AD, Kim DHSL. Org Lett. 1999;1:223–224. doi: 10.1021/ol9905598. [DOI] [PubMed] [Google Scholar]
- 90.Tahirov TH, Lu T-H, Liaw Y-C, Chen Y-L, Lin J-Y. J Mol Biol. 1995;250:354–367. doi: 10.1006/jmbi.1995.0382. [DOI] [PubMed] [Google Scholar]
- 91.Olnes S. Toxicon. 2004;44:361–370. doi: 10.1016/j.toxicon.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 92.Fullas F, Soejarto DD, Kinghorn AD. Phytochemistry. 1992;31:2543–2545. [Google Scholar]
- 93.Suttisri R, Chung M-S, Kinghorn AD, Sticher O, Hashimoto Y. Phytochemistry. 1993;34:405–408. doi: 10.1016/0031-9422(93)80018-n. [DOI] [PubMed] [Google Scholar]
- 94.Suttisri R. PhD dissertation. University of Illinois at Chicago; Chicago, IL: 1993. Phytochemical Studies on the Constituents of Periandra dulcis and Baccharis genistelloides. [Google Scholar]
- 95.Kim J, Kinghorn AD. Phytochemistry. 1989;28:1225–1228. [Google Scholar]
- 96.Yamada H, Nishizawa M, Katayama C. Tetrahedron Lett. 1992;33:4009–4010. [Google Scholar]
- 97.Nishizawa M, Yamada H, Yamaguchi Y, Hatakayama S, Lee I-S, Kennelly EJ, Kim J, Kinghorn AD. Chem Lett. 1994:1555–1558 . ibid. 1994, 1979. [Google Scholar]
- 98.Kim J, Kinghorn AD. Tetrahedron Lett. 1987;28:3655–3658. [Google Scholar]
- 99.Baek N-I, Chung M-S, Shamon L, Kardono LBS, Tsauri S, Padmawinata K, Pezzuto JM, Soejarto DD, Kinghorn AD. J Nat Prod. 1993;56:1532–1538. doi: 10.1021/np50099a011. [DOI] [PubMed] [Google Scholar]
- 100.Baek N-I, Kennelly EJ, Kardono LBS, Tsauri S, Padmawinata K, Soejarto DD, Kinghorn AD. Phytochemistry. 1994;36:513–518. [Google Scholar]
- 101.Vasänge M, Liu B, Welch CJ, Rolfsen W, Bohlin L. Planta Med. 1997;63:511–517. doi: 10.1055/s-2006-957753. [DOI] [PubMed] [Google Scholar]
- 102.Rasmussen SE, Fredericksen H, Krogholm KS, Poulsen L. Mol Nutr Food Res. 2005;49:159–174. doi: 10.1002/mnfr.200400082. [DOI] [PubMed] [Google Scholar]
- 103.Ley JP. Chem Percept. 2008;1:58–77. [Google Scholar]
- 104.Fletcher JN, Kinghorn AD, Slack J, Odley A, Jia Z. Presentation at the 50th Annual Meeting of the American Society of Pharmacognosy; Honolulu, HI. July 31-August 4, 2009; p. Abstract P-249. [Google Scholar]
- 105.Fletcher JN, Kinghorn AD, Slack J, Odley A, Jia Z. Presentation at the 51st Annual Meeting of the American Society of Pharmacognosy; St. Petersburg, FL. July 10–14, 2010; p. Abstract P-12. [Google Scholar]
- 106.Ley JP, Krammer G, Reinders G, Gatfield IL, Bertram H-J. J Agric Food Chem. 2005;53:6061–6066. doi: 10.1021/jf0505170. [DOI] [PubMed] [Google Scholar]
- 107.Ley J, Kindel G, Paetz S, Reiss T, Haug M, Schmidtmann R, Krammer G. 2007014879. PCT Int Appl WO. 2007:A1. 97.
- 108.Liu Y-L, Ho DK, Cassady JM, Cook VM, Baird WM. J Nat Prod. 1992;55:357–363. doi: 10.1021/np50081a012. [DOI] [PubMed] [Google Scholar]
- 109.Slack JP, Brockhoff A, Batram C, Menzel S, Sonnabend C, Born S, Galindo MMM, Kohl S, Thalmann S, Ostopovici-Halap L, Simons CT, Ungureanu I, Duineveld K, Bologa CG, Behrens M, Furrer S, Oprea TI, Meyerhof W. Curr Biol. 2010;20:1104–1120. doi: 10.1016/j.cub.2010.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hough CAM, Parker KJ, Vlitos AJ, editors. Developments in Sweeteners – 1. Applied Science Publishers; London: 1979. [Google Scholar]
- 111.Chaturvedula VSP, Clos JF, Rhea J, Milanowski S, Mocek U, DuBois GE, Prakash I. Phytochem Lett. 2011 in press. [Google Scholar]
- 112.Jemal A, Siegel R, Xu J, Ward E. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 113.Cragg GM, Kingston DGI, Newman DJ. Anticancer Agents from Natural Sources. CRC Press/Taylor & Francis; Boca Raton, FL: 2005. [Google Scholar]
- 114.D’Incalci M, Galmarini GM. Mol Cancer Ther. 2010;9:2157–2163. doi: 10.1158/1535-7163.MCT-10-0263. [DOI] [PubMed] [Google Scholar]
- 115.Hucyk TK, Gradishar W, Manuguid F, Kirkpatrick P. Nat Rev Drug Discov. 2011;10:173–174. doi: 10.1038/nrd3389. [DOI] [PubMed] [Google Scholar]
- 116.Bailly C. Biochem Pharmacol. 2009;77:1447–1457. doi: 10.1016/j.bcp.2008.12.013. [DOI] [PubMed] [Google Scholar]
- 117.Cragg GM, Grosthaus PG, Newman DJ. Chem Rev. 2009;109:3012–3043. doi: 10.1021/cr900019j. [DOI] [PubMed] [Google Scholar]
- 118.Wall ME, Wani MC, Cook CE, Palmer KH, McPhail AT, Sim GA. J Am Chem Soc. 1966;88:3888–3890. [Google Scholar]
- 119.Wani MC, Taylor HL, Wall ME, Coggon P, McPhail AT. J Am Chem Soc. 1971;93:2325–2327. doi: 10.1021/ja00738a045. [DOI] [PubMed] [Google Scholar]
- 120.Butler MS. In: Natural Product Chemistry for Drug Discovery; RSC Biomolecular Sciences Publication No. 18. Buss AD, Butler MS, editors. Royal Society of Chemistry; London: 2010. pp. 321–354. [Google Scholar]
- 121.Pan L, Chai H, Kinghorn AD. Phytochem Lett. 2010;3:1–8. doi: 10.1016/j.phytol.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Newman DJ, Cragg GM. In: Anticancer Agents from Natural Sources. Cragg GM, Kingston DGI, Newman DJ, editors. CRC Press/Taylor & Francis; Boca Raton, FL: 2005. pp. 553–571. [Google Scholar]
- 123.Powell RG, Weisleder D, Smith CR, Jr, Rohwedder WK. Tetrahedron Lett. 1970:815–818. doi: 10.1016/s0040-4039(01)97839-6. [DOI] [PubMed] [Google Scholar]
- 124.Kupchan SM, Komoda Y, Court WA, Thomas GJ, Smith RM, Karim A, Gilmore CJ, Haltiwanger RC, Bryan RF. J Am Chem Soc. 1972;94:1354–1356. doi: 10.1021/ja00759a054. [DOI] [PubMed] [Google Scholar]
- 125.Kupchan SM, Britton RW, Sigel CW. J Org Chem. 1973;38:178–179. doi: 10.1021/jo00941a049. [DOI] [PubMed] [Google Scholar]
- 126.Pettit GR, Gaddamidi V, Herald DL, Singh SB, Cragg GM, Schmidt JM, Boettner FE, Williams M, Sagawa Y. J Nat Prod. 1986;49:995–1002. doi: 10.1021/np50048a005. [DOI] [PubMed] [Google Scholar]
- 127.Pettit GR, Singh SB, Hamel E, Lin CM, Alberts DS, Garcia-Kendall D. Experientia. 1989;45:209–211. doi: 10.1007/BF01954881. [DOI] [PubMed] [Google Scholar]
- 128.Mi Q, Lantvit DD, Reyes-Lim E, Chai H, Zhao W, Lee I-S, Peraza-Sanchez S, Ngassapa O, Kardono LBS, Riswan S, Hollingshead MG, Mayo JG, Farnsworth NR, Kinghorn AD, Pezzuto JM. J Nat Prod. 2002;65:842–850. doi: 10.1021/np010322w. [DOI] [PubMed] [Google Scholar]
- 129.Mi Q, Pezzuto JM, Farnsworth NR, Wani MC, Kinghorn AD, Swanson SM. J Nat Prod. 2009;72:573–580. doi: 10.1021/np800767a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kinghorn AD, Farnsworth NR, Soejarto DD, Cordell GA, Pezzuto JM, Udeani GO, Wani MC, Wall ME, Navarro HA, Kramer RA, Menendez AT, Fairchild CR, Lane KE, Forenza S, Vyas DM, Lam KS, Shu Y-Z. Pure Appl Chem. 1999;71:1611–1618. [Google Scholar]
- 131.Kinghorn AD, Farnsworth NR, Soejarto DD, Cordell GA, Swanson SM, Pezzuto JM, Wani MC, Wall ME, Oberlies NC, Kroll DJ, Kramer AD, Rose WC, Vite GD, Fairchild CR, Peterson RW, Wild R. Pharm Biol. 2003;41(Suppl):53–67. [Google Scholar]
- 132.Kinghorn AD, Carcache-Blanco EJ, Chai H-B, Orjala J, Farnsworth NR, Soejarto DD, Oberlies NH, Wani MC, Kroll DJ, Pearce CJ, Swanson SM, Kramer RA, Rose WC, Emanuel S, Vite GD, Jarjoura J, Cope FO. Pure Appl Chem. 2009;81:1051–1063. doi: 10.1351/PAC-CON-08-10-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Pisha E, Chai H, Lee I-S, Chagwedera TE, Farnsworth NR, Cordell GA, Beecher CWW, Fong HHS, Kinghorn AD, Brown DM, Wani MC, Wall ME, Heijken TJ, Das Gupta TK, Pezzuto JM. Nature Med. 1995;1:1046–1051. doi: 10.1038/nm1095-1046. [DOI] [PubMed] [Google Scholar]
- 134.Griffin WJ, Lin GD. Phytochemistry. 2000;53:623–637. doi: 10.1016/s0031-9422(99)00475-6. [DOI] [PubMed] [Google Scholar]
- 135.Silva GL, Cui B, Chávez D, You M, Chai H-B, Rasoanaivo P, Lynn SM, O’Neill MJ, Lewis JA, Besterman JL, Monks A, Farnsworth NR, Cordell GA, Pezzuto JM, Kinghorn AD. J Nat Prod. 2001;64:1514–1520. doi: 10.1021/np010295+. [DOI] [PubMed] [Google Scholar]
- 136.Avendaño C, Menéndez JC. Curr Med Chem. 2002;9:159–193. doi: 10.2174/0929867023371175. [DOI] [PubMed] [Google Scholar]
- 137.Kulkarni K, Zhao AY, Purcell AW, Perlmutter P. Synlett. 2008:2209–2212. [Google Scholar]
- 138.Mi Q, Cui B, Silva GL, Lantvit D, Lim E, Chai H, You M, Hollingshead MG, Mayo JG, Kinghorn AD, Pezzuto JM. Cancer Res. 2001;61:4030–4037. [PubMed] [Google Scholar]
- 139.Mi Q, Cui B, Silva GL, Lantvit D, Lim E, Chai H, Hollingshead MG, Mayo JG, Kinghorn AD, Pezzuto JM. Cancer Lett. 2002;184:13–20. doi: 10.1016/s0304-3835(02)00202-1. [DOI] [PubMed] [Google Scholar]
- 140.Mi Q, Cui B, Lantvit D, Reyes-Lim E, Chai H, Pezzuto JM, Kinghorn AD, Swanson SM. Anticancer Res. 2003;23:3607–3616. [PubMed] [Google Scholar]
- 141.Chávez D, Cui B, Chai H-B, García R, Mejía M, Farnsworth NR, Cordell GA, Pezzuto JM, Kinghorn AD. J Nat Prod. 2002;65:606–610. doi: 10.1021/np0104774. [DOI] [PubMed] [Google Scholar]
- 142.Chin Y-W, Jones WP, Waybright TJ, McCloud TG, Rasoanaivo P, Cragg GM, Cassady JM, Kinghorn AD. J Nat Prod. 2006;69:414–417. doi: 10.1021/np050366v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Mi Q, Cui B, Chávez D, Chai H, Cordell GA, Hedayat S, Zhu H, Kinghorn AD, Pezzuto JM. Anticancer Res. 2002;22:1385–1397. [PubMed] [Google Scholar]
- 144.Teodori E, Dei S, Garnier-Suillerot A, Gualtieri F, Manetti D, Martelli C, Romanelli MN, Scapecchi S, Sudwan P, Salerno M. J Med Chem. 2005;48:7426–7436. doi: 10.1021/jm050542x. [DOI] [PubMed] [Google Scholar]
- 145.Chin Y-W, Kinghorn AD, Patil PN. Phytother Res. 2007;21:1002–1005. doi: 10.1002/ptr.2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Cragg GM, Boyd MR, Khanna R, Kneller R, Mays TD, Mazan KD, Newman DJ, Sausville EA. Pure Appl Chem. 1999;71:1619–1633. [Google Scholar]
- 147.Pannell CM. Kew Bulletin Additional Series XVI. HMSO; London: 1992. A Taxonomic Monograph of the Genus Aglaia Lour. (Meliaceae) [Google Scholar]
- 148.Cragg GM, Newman DJ, Yang SS. J Nat Prod. 2006;69:488–498. doi: 10.1021/np0581216. [DOI] [PubMed] [Google Scholar]
- 149.Balunas MJ, Jones WP, Chin Y-W, Mi Q, Farnsworth NR, Soejarto DD, Cordell GA, Swanson SM, Pezzuto JM, Chai H-B, Kinghorn AD. Chem Biodiver. 2006;3:897–915. doi: 10.1002/cbdv.200690092. [DOI] [PubMed] [Google Scholar]
- 150.Proksch P, Edrada RA, Ebel R, Bohnenstengel FI, Nugroho BW. Curr Org Chem. 2001;5:923–938. [Google Scholar]
- 151.Kim S, Salim A, Swanson SM, Kinghorn AD. Anti-cancer Agents Med Chem. 2006;6:319–345. doi: 10.2174/187152006777698123. [DOI] [PubMed] [Google Scholar]
- 152.King ML, Chiang C-C, Ling H-C, Fujita H, Ochiai M, McPhail AT. J Chem Soc Chem Commun. 1982:1150–1151. [Google Scholar]
- 153.Trost BM, Greenspan PD, Yang BY, Saulnier MG. J Am Chem Soc. 1990;112:9022–9024. [Google Scholar]
- 154.Janprasert J, Satasook C, Sukumalanand P, Champagne DE, Isman MB, Towers GHN. Phytochemistry. 1993;32:67–69. [Google Scholar]
- 155.Cui B, Chai H, Santisuk T, Reutrakul V, Farnsworth NR, Cordell GA, Pezzuto JM, Kinghorn AD. Tetrahedron. 1997;52:17625–17632. [Google Scholar]
- 156.Lee SK, Cui B, Mehta RR, Kinghorn AD, Pezzuto JM. Chem-Biol Interact. 1998;115:215–228. doi: 10.1016/s0009-2797(98)00073-8. [DOI] [PubMed] [Google Scholar]
- 157.Rivero-Cruz JF, Chai H-B, Kardono LBS, Setyowati FM, Afriastini JJ, Riswan S, Farnsworth NR, Cordell GA, Pezzuto JM, Swanson SM, Kinghorn AD. J Nat Prod. 2004;67:343–347. doi: 10.1021/np0304417. [DOI] [PubMed] [Google Scholar]
- 158.Su B-N, Chai H, Mi Q, Riswan S, Kardono LBS, Afriastini JJ, Santarsiero BD, Mesecar AD, Farnsworth NR, Cordell GA, Swanson SM, Kinghorn AD. Bioorg Med Chem. 2006;14:960–972. doi: 10.1016/j.bmc.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 159.Mi Q, Su B-N, Chai H, Cordell GA, Farnsworth NR, Kinghorn AD, Swanson SM. Anticancer Res. 2006;26:947–952. [PubMed] [Google Scholar]
- 160.Kim S, Chin Y-W, Su B-N, Riswan S, Kardono LBS, Afriastini JJ, Chai H, Farnsworth NR, Cordell GA, Swanson SM, Kinghorn AD. J Nat Prod. 2006;69:1769–1775. doi: 10.1021/np060428x. [DOI] [PMC free article] [PubMed] [Google Scholar]; 2007;70:714. [Google Scholar]
- 161.Kim S, Su B-N, Riswan S, Kardono LBS, Afriastini JJ, Gallucci JC, Chai H, Farnsworth NR, Cordell GA, Swanson SM, Kinghorn AD. Tetrahedron Lett. 2005;46:9021–9024. [Google Scholar]
- 162.Salim AA, Pawlus AD, Chai H-B, Farnsworth NR, Kinghorn AD, Carcache-Blanco EJ. Bioorg Med Chem Lett. 2007;17:109–112. doi: 10.1016/j.bmcl.2006.09.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Baumann B, Bohnenstengel F, Siegmund D, Wajant H, Weber C, Herr I, Debatin KM, Proksch P, Wirth T. J Biol Chem. 2002;277:4479–44800. doi: 10.1074/jbc.M208003200. [DOI] [PubMed] [Google Scholar]
- 164.Hwang BY, Su B-N, Chai H, Mi Q, Kardono LBS, Afriastini JJ, Riswan S, Santarsiero BD, Mesecar AD, Fairchild CR, Wild R, Vite GD, Rose WC, Farnsworth NR, Cordell GA, Pezzuto JM, Swanson SM, Kinghorn AD. J Org Chem. 2004;69:3350–3358. doi: 10.1021/jo040120f. [DOI] [PubMed] [Google Scholar]; 69:6156. [Google Scholar]
- 165.Mi Q, Kim S, Hwang BY, Su B-N, Chai H, Arbieva ZH, Kinghorn AD, Swanson SM. Anticancer Res. 2006;26:3349–3356. [PubMed] [Google Scholar]
- 166.Kim S, Hwang B-Y, Su B-N, Chai H, Mi Q, Kinghorn AD, Wild R, Swanson SM. Anticancer Res. 2007;27:2175–2184. [PMC free article] [PubMed] [Google Scholar]
- 167.Gerard B, Cencic R, Pelletier J, Porco JA., Jr Angew Chem Int Ed. 2007;46:7831–7834. doi: 10.1002/anie.200702707. [DOI] [PubMed] [Google Scholar]
- 168.El Sous M, Khoo ML, Holloway G, Owen D, Scammels PJ, Rizzacasa MA. Angew Chem Int Ed. 2007;46:7835–7838. doi: 10.1002/anie.200702700. [DOI] [PubMed] [Google Scholar]
- 169.Adams TE, El Sous M, Hawkins BC, Hirner S, Holloway G, Khoo ML, Owen DJ, Savage GP, Scammels PJ, Rizzacasa MA. J Am Chem Soc. 2009;131:1607–1616. doi: 10.1021/ja808402e. [DOI] [PubMed] [Google Scholar]
- 170.Meurer-Grimes BM, Yu J, Vairo GL. 6710075. U S Pat US. 2004:B2.
- 171.Lucas DM, Edwards RB, Lozanski G, West DA, Shin JD, Vargo MA, Davis ME, Rozewski DM, Johnson AJ, Su B-N, Goettl VM, Lin TS, Lehman A, Zhang X, Jarjoura D, Newman DJ, Byrd JC, Kinghorn AD, Grever MR. Blood. 2009;113:4656–4666. doi: 10.1182/blood-2008-09-175430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Vijaya Saradhi UVR, Gupta SV, Chiu M, Wang J, Ling Y, Liu Z, Newman DJ, Covey JM, Kinghorn AD, Marcucci G, Lucas DM, Grever MR, Phelps MA, Chan KK. AAPS J. 2011 doi: 10.1208/s12248-011-9273-x. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Bordeleau M-E, Robert F, Gerard B, Lindqvist L, Chen SMH, Wendel H-G, Brem B, Greger H, Lowe SW, Porco JA, Jr, Pelletier J. J Clin Invest. 2008;118:2651–2660. doi: 10.1172/JCI34753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Cencic R, Carrier M, Galicia-Vázquez G, Bordeleau M-E, Sukarieh R, Bourdeau A, Brem B, Teodoro JG, Greger H, Tremblay ML, Porco JA, Jr, Pelletier J. PLoS One. 2009;4:e5223. doi: 10.1371/journal.pone.0005223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Lucas DM, Still PC, Bueno Pérez L, Grever MR, Kinghorn AD. Curr Drug Targets. 2010;11:812–822. doi: 10.2174/138945010791320809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Salim AA, Chai H-B, Rachman I, Riswan S, Kardono LBS, Farnsworth NR, Carcache de Blanco EJ, Kinghorn AD. Tetrahedron. 2007;63:7926–7934. doi: 10.1016/j.tet.2007.05.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Pan L, Kardono LBS, Riswan S, Chai H, Carcache de Blanco EJ, Pannell CM, Soejarto DD, McCloud TG, Newman DJ, Kinghorn AD. J Nat Prod. 2010;73:1873–1878. doi: 10.1021/np100503q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Salim AA, Fletcher JN, Pan L, Chai H-B, Riswan S, Kardono LBS, Ruskandi A, Farnsworth NR, Li P-K, Kinghorn AD. Presentation at the 48th Annual Meeting of the American Society of Pharmacognosy; Portland, ME. July 14–18, 2007; p. Abstract P-150M. [Google Scholar]
- 179.Doroshow JT. NCI Cancer Bull. 2009;20(6):4. [Google Scholar]
- 180.Pannell CM. Kew Bull. 2004;59:87–94. [Google Scholar]
- 181.Chia YT. In: Biodiversity and National Development: Achievements, Opportunities and Challenges. Sen YH, editor. Akademi Siens Malaysia; Kuala Lumpur: 2009. pp. 90–95. [Google Scholar]
- 182.Seo E-K, Chai H, Constant HL, Santisuk T, Reutrakul V, Beecher CWW, Farnsworth NR, Cordell GA, Pezzuto JM, Kinghorn AD. J Org Chem. 1999;64:6976–6983. [Google Scholar]
- 183.Ohyama M, Tanaka T, Ito T, Iinuma M, Bastow KF, Lee K-H. Bioorg Med Chem Lett. 1999;9:3057–3060. doi: 10.1016/s0960-894x(99)00520-x. [DOI] [PubMed] [Google Scholar]
- 184.Ito T, Akao Y, Yi H, Ohguchi K, Matsumoto K, Tanaka T, Iinuma M, Nozawa Y. Carcinogenesis. 2003;24:1489–1497. doi: 10.1093/carcin/bgg105. [DOI] [PubMed] [Google Scholar]
- 185.Mishima S, Matsumoto K, Futamura Y, Araki Y, Ito T, Tanaka T, Iinuma M, Nozawa Y, Akao Y. J Exp Ther Oncol. 2003;3:283–288. doi: 10.1111/j.1533-869x.2003.01102.x. [DOI] [PubMed] [Google Scholar]
- 186.Zgoda-Pols JR, Freyer AJ, Killmer LB, Porter JR. J Nat Prod. 2002;65:1554–1559. doi: 10.1021/np020198w. [DOI] [PubMed] [Google Scholar]
- 187.Salim AA, Su B-N, Chai H-B, Riswan S, Kardono LBS, Ruskandi A, Farnsworth NR, Swanson SM, Kinghorn AD. Tetrahedron Lett. 2007;48:1849–1853. doi: 10.1016/j.tetlet.2007.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Chin Y-W, Salim AA, Su B-N, Mi Q, Chai H-B, Riswan S, Kardono LBS, Ruskandi A, Farnsworth NR, Swanson SM, Kinghorn AD. J Nat Prod. 2008;71:390–395. doi: 10.1021/np070584j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Traoré M, Maynadier M, Souard F, Choisnard L, Vial H, Wong Y-S. J Org Chem. 2011;76:1409–1411. doi: 10.1021/jo102414s. [DOI] [PubMed] [Google Scholar]
- 190.Peuchmaur M, Saidani N, Botte D, Marechal E, Vial H, Wong Y-S. J Med Chem. 2008;51:4870–4873. doi: 10.1021/jm8007322. [DOI] [PubMed] [Google Scholar]
- 191.Malanthong V, Rychnovsky S. Org Lett. 2009;11:4220–4223. doi: 10.1021/ol901623h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Yadav JS, Gopala Rao Y, Chandrakanth D, Ravindar K, Subba Reddy BV. Helv Chim Acta. 2010;93:2426–2432. [Google Scholar]
- 193.Ren Y, Lantvit DD, Carcache de Blanco EJ, Kardono LBS, Riswan S, Chai H, Cottrell CE, Farnsworth NR, Swanson SM, Ding Y, Li X-C, Marais JPJ, Ferreira D, Kinghorn AD. Tetrahedron. 2010;66:5311–5320. doi: 10.1016/j.tet.2010.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Ren Y, Yuan C-H, Chai H-B, Ding Y-Q, Li X-C, Ferreira D, Kinghorn AD. J Nat Prod. 2011;74:460–463. doi: 10.1021/np100422z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Zhou ZT, Wang JW. Chin J New Drugs. 2007;16:79–83. [Google Scholar]
- 196.Kusari S, Zühlke S, Spiteller M. J Nat Prod. 2011;74:764–775. doi: 10.1021/np1008398. [DOI] [PubMed] [Google Scholar]
- 197.McCoy E, O’Connor SE. J Am Chem Soc. 2006;128:14276–14277. doi: 10.1021/ja066787w. [DOI] [PubMed] [Google Scholar]
- 198.Wagner H. Fitoterapia. 2011;82:34–37. doi: 10.1016/j.fitote.2010.11.016. [DOI] [PubMed] [Google Scholar]
- 199.Thomas CD, Cameron A, Green RE, Bakkenes M, Beaumont LJ, Collingham YC, Erasmus BFN, Ferreira de Siqueira M, Grainger A, Hannah L, Hughes L, Huntley B, van Jaarsveld AS, Midgley GF, Miles L, Ortega-Huerta MA, Peterson AT, Phillips OL, Williams SE. Nature. 2004;427:145–148. doi: 10.1038/nature02121. [DOI] [PubMed] [Google Scholar]