

Abstract
Positron emission tomography (PET) is a nuclear imaging technique, which allows in vivo estimations of important physiological parameters such as, glucose metabolism and neuroreceptor binding enabling greater understanding of the pathophysiology of Parkinson's disease (PD). The review will evaluate the role of PET in assessing both the dopaminergic (DA) and non-DA systems in relation to the pathophysiology of PD, differential diagnosis, progression of disease and pre-clinical disease. Medication side effects, genetic forms of PD, the non-motor symptoms of PD and alternative restorative approaches will also be discussed in relation to how PET imaging can enhance our understanding of these aspects of the disease. PET neuroimaging has to date, provided an excellent tool to assess the underlying mechanisms of the disease as well as evaluating the complications and management of PD and has the potential to be of great clinical value if the current limitations of costing and availability are resolved.
Keywords: Positron Emission Tomography, PET, Parkinson's disease
Introduction
Parkinson's disease (PD) is a neurodegenerative disorder characterised by the motor features of tremor, rigidity and bradykinesia. These features are associated with the loss of dopaminergic (DA) neurons in the substantia nigra pars compacta and the subsequent deficiency in striatal DA, which is required for the effective control of movements. However, there is evidence of a more diffuse pathology in PD [1] with other, non-DA neutotransmitter systems possibly playing a role [2-6]. Furthermore, PD is also associated with many non-motor features including behavioural and psychiatric problems such as dementia [7] sleep disturbances and fatigue [8], depression [9], addiction and compulsion [10] and psychosis [11].
PD is the second most common neurodegenerative disorder after Alzheimer's disease (AD) [12, 13] occurring at a median age of 62.4 years although up to 10% of cases begins by the age of 40 [14]. PD is uncommon before the age of 40. The prevalence of PD increases with age from 1% to up to 3% after 80 years of age [15].
The pathological hallmark of PD is characterised by the DA neuronal loss together with the presence of ubiquinated protein deposits in the cytoplasm of neurons, called Lewy bodies [16, 17].
Brain imaging techniques provide a useful tool of in vivo investigation of the pathogenesis of PD and pathophysiology of PD processes. Positron Emission Tomography (PET) imaging is a nuclear medicine technique enabling the estimation of important physiological parameters, such as, glucose metabolism and neuroreceptor binding. In PET, radioisotopes bound to specific tracers are administered to an individual via an intravenous (IV) injection whereby the estimation of the distribution of the radiotracer over time in the brain can be calculated.
PET is a relatively expensive technique and to date is not widely available. However, it does offer high sensitivity with admirable spatial and temporal resolution compared to other imaging techniques.
Dopaminergic imaging
Presynaptic DA system imaging studies
Presynaptic DA terminal functionality can be investigated using PET by measuring aromatic amino acid decarboxylase (AADC) activity, dopa-mine transporter (DAT) activity and vescular monoamine transporter (VMAT2) density.
18F-DOPA PET is a radiotracer which can be used to assess AADC in the DA terminals [18]. As AADC activity permits the conversion of L-DOPA to DA, 18F-DOPA PET can be used as a measure of DA terminal functionality. This tracer has been utilised to assess the correlation between 18F-DOPA binding and motor disability. It has been demonstrated that striatal 18F-DOPA binding and motor disability (as measured by the Unified Parkinson's Disease Rating Scale [UPDRS]) are inversely correlated [19, 20], i.e. loss of DA terminals correlates with an increase in motor disability. There has also been an attempt with 18F-DOPA PET to delineate the pathogenesis of the core features of PD. This study reported 18F-DOPA uptake correlated with increased bradykinesia and rigidity scores but not with tremor scores, indicating that the pathogenesis for tremor may not be solely implicated by the DA system [21].
Furthermore, 18F-DOPA PET has been used to determine the stages of DA degeneration in PD. It has been demonstrated that the decline of DA function starts in the dorsalcaudal putamen contralateral to the clinically affected side [22] and that the rate of degeneration in the caudate nucleus is slower than that of the putamen [23] in early PD (Figure 1).
Figure 1.

Transverse 18F-DOPA PET images of a healthy control (left) and a patient with idiopathic PD (right). In PD, there is asymmetric loss of uptake of the tracer, and a more pronounced loss in the caudal putamen than in the rostral putamen and the caudate nucleus.
A recent 18F-DOPA PET study of ten early PD patients over 3 years reported reductions in 18F-DOPA binding in the putamen (8.1%), locus coeruleus (7.8%), globus pallidus interna (7.7%), caudate nucleus (6.3%) and hypothalamus (6.1%) [24].
Presynaptic synthesis and release of DA can be investigated by assessing the uptake and storage procedure of DA into the storage granules, which can be imaged using the radiotracer 11C-dihydrotetrabenazine (DTBZ). This tracer labels the VMAT2 located in the presynaptic vesicles and as such can be used to evaluate the presynaptic status of the nigrostriatal system in PD [25]. It has been shown in advanced cases of PD that that striatal DTBZ binding decreases following L-3, 4-dihydroxphenylalanine (L-DOPA) administration, likely reflecting an increase in vesicular DA levels [26]. Striatal uptake of DTBZ binding has also been shown to correlate with motor disability as measured by the UPDRS [27].
Labelling DATs, which are located on the presynaptic DA nerve terminals and facilitate the reuptake of released DA into the synaptic cleft, allows another method of assessing the presynaptic DA system. Various radiotracers have been developed in order to measure DAT in vivo, including, 11C-nomifensine, 11C-RT132, 11C-CFT, 18F-CFT [28-33]. These radiotracers are also capable of providing a measure of presynaptic DA terminal function [30, 34].
Postsynaptic DA system imaging studies
Postsynaptic DA receptors have a lower affinity to agonists than for antagonists and as such, most PET studies utilise antagonist ligands [35]. A study of early PD patients used the radiotracers 11C-SCH23390 to assess striatal D1 receptors and 11C-raclopride (RAC) to assess striatal D2 receptors [36]. It was demonstrated that although the patient groups clinically presented with unilateral symptoms, there was a symmetric binding of 11C-SCH23390 across both hemispheres compared to an asymmetric binding of RAC (contralateral to the clinically affected side), suggesting that there is an abnormal binding of D2 and not D1 receptors in early PD. Similarly, a PET study using both 11C-SCH23390 and 18F-DOPA demonstrated no difference in D1 receptor density between PD patients and healthy controls [37].
A two-scan RAC PET study has reported, in advanced PD cases that, improvement in bradykinesia and rigidity scores following oral DA medication administration were significantly correlated with reductions in RAC binding suggesting an effect of increased DA on the striatal D2 receptors [38].
RAC PET has also been employed in de novo PD patients and demonstrated a 10-20% increase in D2 receptor availability in the putamen contralateral to the clinically affected side whereas the caudate nucleus appears to remain relatively intact [32, 39-40].
Further RAC PET studies in previously untreated PD patients, has demonstrated that 3-4 months post-initiation of L-DOPA or lisuride treatment resulted in no change in D2 receptor density [41]. However, 3-5 years post-treatment, RAC binding was demonstrated to be significantly reduced in the putamen and caudate nucleus compared to the baseline assessment [42]. This is indicative of a process of down-regulation of the striatal D2 receptor binding in PD related to long-term treatment. It has been suggested that D2 receptor changes observed in the putamen may be a consequence of the reduction in pre-synaptic DA nerve terminals as an association between RAC PET and 18F-DOPA binding has been found [43]. However, serial RAC PET studies have indicated that as the disease progresses, and thus patients are exposed to DA medications, D2 binding in the putamen stabilises, whereas the caudate nucleus displays a reduction in D2 binding by approximately 20% [40, 44-45].
Extrastriatal areas have also been investigated with RAC PET, with one study demonstrating a significant decrease in RAC binding in the hypothalamus of PD patients compared to a group of healthy controls [46] (Figure 2). This finding may be suggestive of a hypothalamic involvement in non-motor features commonly observed in PD, such as sleep, endocrine and autonomic disturbances.
Figure 2.
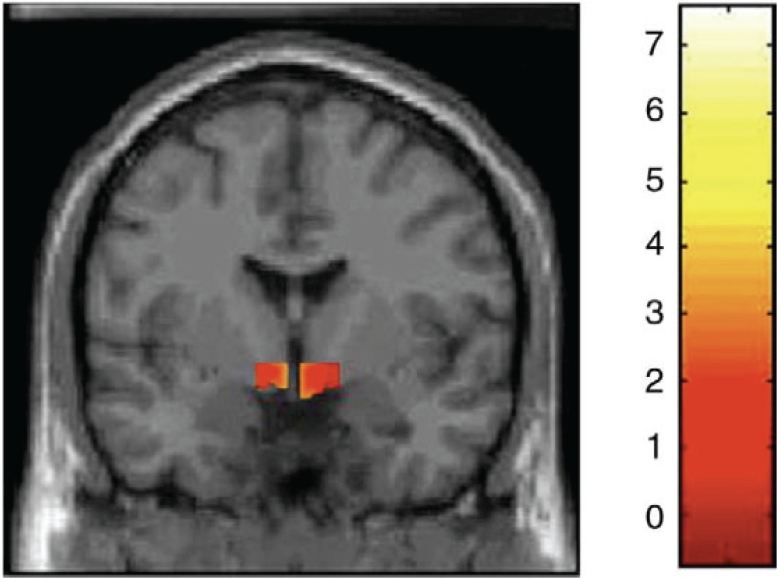
Coronal section of statistical parametric map. Yellow-red areas represent voxel clusters with significant decreases in 11C-raclopride binding within the hypothalamic region mask in PD patients compared with the group of healthy controls.
RAC PET can also be used to assess DA release. This can be achieved by administering a pharmacological challenge, which inhibits DAT function, such as, methamphetamine. A study which employed this method in a group of healthy controls and a group of advanced PD patients reported that RAC binding was reduced in both groups but more pronounced in the PD group for the striatal regions (caudate nucleus: 8% v 17% and putamen: 7% v 25%) [47]. Moreover, RAC binding percentage reductions correlated with motor disability as measured by the UPDRS.
The endogenous release of DA can be assessed during the performance of motor tasks while in the scanner. A RAC PET study was able to assess DA release via a visuomotor task during which the subject may or may not gain a financial reward [48]. This study demonstrated with a group of healthy controls and a group of advanced PD patients that striatal DA release was apparent only in the healthy control group. However, in both groups, significant increases in the prefrontal cortical DA levels were detected, indicating although striatal DA release during a motor task is impaired in early PD patients, it appears to be somewhat preserved in the prefrontal cortex.
Non-dopaminergic imaging
Imaging the serotonergic system
The suggestion that the serotonergic system may be implicated in PD has arisen from both post-mortem and biochemical studies [2, 3]. Various radiotracers have been developed in order to assess the integrity of the serotonergic system in vivo including, 11C-WAY100635, which specifically binds to the 5-HT1A receptors. These receptors are expressed both presynaptically and postsynaptically on 5-HT cell bodies in the midbrain raphe nuclei and on cortical pyramidal neurons and glia respectively. One study utilising this radiotracer has reported that 5-HT1A binding in the midbrain of PD patients was reduced by 29% compared to healthy controls [49]. Furthermore, the decreased 5-HT1A raphe binding was correlated with UPDRS tremor scores, but not rigidity or bradykinesia scores, suggesting that the serotonergic system may have a role in the development of tremor.
11C-DASB and 11C-McN5652 are two radiotracers, which bind specifically to the serotonin transporter (SERT, 5-HTT) in the presynaptic terminals, thus providing a good measure of the integrity of serotonergic innervation. However to date, most studies utilising these radiotracers have reported that striatal serotonergic denervation is relatively moderate compared to striatal DA denervation [5, 33, 50]. It should be noted though that these studies are limited by the small sample size, which may implicate any correlations between regional binding measurements and motor disability scores. Furthermore, a study using single photon emission computed tomography (SPECT) has reported that striatal serotonergic innervation in PD is within normal range [51] suggesting that the complete picture of how the serotonergic system is implicated in PD is still unclear.
More recently, one 11C-DASB PET study with a larger sample size of 30 PD patients, has reported significant reductions in regional 11C-DASB binding in striatal, brainstem and cortical regions [6]. Furthermore, the authors demonstrated that 11C-DASB uptake was affected in the caudate nucleus, hypothalamus, thalamus and anterior cingulate cortex (ACC) early in the disease followed by the putamen, insular cortex, posterior cingulate cortex (PCC) and pre-frontal cortex (PFC) once PD is established, with advanced cases displaying further reductions in the ventral striatum, raphe nuclei and amygdala. These results are strongly suggestive of a progressive, non-linear serotonergic dysfunction in PD. Moreover, the regional 11C-DASB binding did not correlate with UPDRS scores, Hoehn and Yahr (H&Y) staging, disease duration or DA medication implying that serotonergic dysfunction does not influence motor disability.
Interestingly, findings from animal studies have suggested that the serotonin neurons possess the ability to convert exogenous L-DOPA to DA and subsequently store and release DA in an activity-dependent manner [52, 53].
Imaging the cholinergic system
11C-PMP and 11C-MP4A are radiotracers which measure acetylcholinesterase (AChE) levels as they are analogues for acetylcholine and as such serve as a selective substrate for AChE hydrolysis thus providing a measure of the cholinergic system integrity [54]. These radiotracers can be used in conjunction with 11C-NMPB (a marker of postsynaptic muscarinic receptor availability) [55] to assess cholinergic function in dementia in PD.
Imaging the opioid system
11C-diprenorphine is a radiotracer, which allows the measurement of μ, κ and δ opioid sites and has been demonstrated to show sensitivity to endogenous opioids [56], which are found in high densities in the caudate nucleus and putamen. Opioid neuropeptides are abundant in the basal ganglia [57] and it is known that the opioid system is involved in the pathophysiology of PD [58].
Imaging the noradrenergic system
There are currently no PET radiotracers to assess specifically the noradrenergic neurons in vivo. An attempt to use reboxetine derivatives as a marker of noradrenergic transporter (NAT) binding failed due to fact that they were lipophilic and related to nonspecific signal [59, 60].
18F-DOPA PET can be used to assess serotonergic and noradrenergic system function and integrity as both contain AADC. Yet, considering DA terminals also contain a high degree of AADC, the use of 18F-DOPA PET for non-DA system evaluation should be restricted to regions where a high innervation of serotonin and noradrenaline is established, i.e. the raphe nucleus and locus coeruleus respectively.
Assessment of microglial activation in PD
Microglia are resident immune cells within the central nervous system (CNS) constituting approximately 20% of the total glial population within the brain. Microglia act as the brains' first line defence mechanism and as such remain in a quiescent state until required to be activated following trauma, ischemia, tumour, inflammation, and neurodegeneration. Peripheral benzo-diazepine receptors (PBR) develop on the surface of the mitochondria of activated microglia, therefore the use of the radiotracer, 11C-PK11195 (a selective marker of peripheral BDZ sites) allows the in vivo measurement of microglia activation and can be used to investigate the contribution of neuroinflammatory glial response in the degenerative process in PD.
A combined 11C-PK11195 and 11C-CFT (A DAT marker serving as a tool to measure the density of DA terminals) study in early de novo PD patients has reported that levels of midbrain 11C-PK11195 binding contralateral to the clinically affected side was significantly higher in the PD group compared to the 10 age-matched healthy controls [61]. Furthermore, midbrain 11C-PK1195 binding was significantly correlated inversely with 11C-CFT binding in the putamen and significantly positively correlated with motor disability as measured by the UPDRS. These findings demonstrate that parallel changes in microglia activation correspond with DA terminal loss in the nigrostriatal pathway in early PD, suggesting a role of microglia activation in the progression of PD. A follow up study reported that as the disease progresses, microglia activation as measured by 11C-PK1195 extends to meso-basal ganglia-tha la mo-cortical loop and extrastriatal regions, especially in the occipital cortex [62]. This is suggestive of microglia activation not dissipating as the disease progresses but actually extending beyond the nigrostriatal pathway.
Another study which used both 11C-PK11195 and 18F-DOPA PET reported that 11C-PK1195 binding was significantly increased in the pons, basal ganglia, frontal and temporal cortical regions [63]. It was also reported in the eight PD patients which were examined longitudinally that levels of microglia activation remained stable over two years and did not correlate with clinical severity of disease or with 18F-DOPA uptake. These results suggest that there is diffuse microglia activation corresponding with the pathological PD process and that microglia may be contributing to the continued degeneration via cytokine release.
Imaging differential diagnosis of PD from other movement disorders
Clinical features of PD may be shared with other disorders, thus creating some issues surrounding the correct diagnosis. For example, multiple system atrophy (MSA) and progressive supranu-clear palsy (PSP) are often confused with PD at the early stages. Although, essential tremor, drug-induced parkinsonism, corticobasal gangli-onic degeneration, dementia with Lewy bodies (DLB) and Alzheimer's disease (AD) also share common features with PD. Furthermore, some these disorders will initially produces a somewhat mild to moderate response to DA therapy.
PET imaging can be useful in differentiating PD from other disorders. 18F-DOPA PET has been used to as a utility in the diagnosis of PD, although it is not clear how useful this method is in the differential diagnosis of other parkinsonism disorders [64]. 18F-DOPA PET has been used to differentiate between post-neuroleptics parkinsonism and idiopathic PD as the former will display intact DA terminals [65]. Furthermore, 18F-DOPA PET has been applied to disorders other than PD to demonstrate that in PSP, MSA and corticobasal ganglionic degeneration, the average 18F-DOPA uptake in the caudate nucleus is reduced compared to idiopathic PD patients although putaminal 18F-DOPA binding is similarly decreased across all disorders [66, 67].
18F-FDG PET has been shown to be useful in differentiating PD from MSA (Kwon et al., 2007, 2008). In MSA and PSP, striatal metabolism is low compared to PD patients, where it is either within normal range or increased [42, 66, 68, 69].
RAC PET has also been used in the differential diagnosis of PD from PSP and MSA. It has been reported that PSP and MSA patients show a putaminal D2 receptor availability reduction compared to PD patients [70-72].
Imaging preclinical and genetic forms of PD
Preclinical PD
In order to assess preclinical PD, the subject under investigation must be known to be a carrier of a known causative gene associated with parkinsonism, have a relative with parkinsonism, be an elderly individual with idiopathic hyposomia or REM sleep behaviour disorders (RBD). The latter two may be harbingers of PD.
The radiotracers DTBZ, 11C-nomifensine, 11C-RT132, 11C-CFT, 18F-CFT and 18F-DOPA PET can be used to assess preclinical PD [25, 73, 74]. These studies revealed that DAT binding was the earliest indicator of DA dysfunction in these individuals suggesting that DAT imaging may provide a useful and sensitive tool in detecting subclinical deficits associated with abnormalities in the nigrostriatal pathway. DTBZ PET has also demonstrated the ability to detect nigrostriatal deficits in preclinical PD [75].
An 18F-DOPA PET study has demonstrated putaminal DA terminal loss in 25% of adult asymptomatic individuals who had family members with PD [76]. Moreover, at a five-year follow up, one third of these individuals went on to develop clinical parkinsonism.
Asymptomatic co-twins of idiopathic PD patients have been studies with 18F-DOPA PET [77]. This study reported significant putaminal DA terminal loss in both dizygotic co-twins (18%) and monozygotic co-twins (55%). At the four year follow up, advanced putaminal DA terminal loss was observed in all asymptomatic monozygotic co-twins, of which two had developed clinical parkinsonism, whereas, the asymptomatic dizygotic co-twins did not display any advancement in DA denervation.
Carriers of a single parkin mutation have also been assessed with 18F-DOPA PET where a reported decrease in 18F-DOPA binding in the putamen, caudate nucleus, and ventral and dorsal midbrain were apparent in the PD group compared to the healthy controls [78]. However, this study did not complete a follow-up so data relating to the progression to clinical parkinsonism is unknown.
Although using PET for the preclinical diagnosis of PD is expensive and not widely available, continued longitudinal follow-up of the asymptomatic at-risk subjects is essential in studying the conversion from preclinical DA dysfunction to clinical disease. As such, an improvement in estimating the duration of the preclinical period can be made as well as increasing understanding of the progression of PD.
Genetic forms of PD
PET imaging provides a useful tool is delineating differences between familial and idiopathic PD as it can be difficult to distinguish clinically. The main causative genes implicated in individuals with a family history of PD include autosomal recessive mutations in Parkin (PARK2), PINK1 (PARK6), DJ1 (PARK7) and APT13A2 (PARK9). Autosomal dominant mutations also exist in mutations in alpha-synuclein (PARK1/4), ubiquitin carboxyhydrolase L1-UCH-L1 (PARK5) and leucine rich repeat kinase 2 -LRRK2 (PARK8). All of these mutations can lead to young onset PD.
Both 18F-DOPA and RAC PET have been used to image genetic forms of PD in comparison to idiopathic PD. These studies have shown that 18F-DOPA uptake in PARK1/PARK8 compared to idiopathic PD cases is reduced [79, 80]. Furthermore, it has been shown that PARK2, PARK6 and PARK7 PD patients display rather more symmetrical reduction of 18F-DOPA [81-83] and 18F-CIT [84] uptake compared to idiopathic PD cases. One of these studies provides further information by demonstrating that reductions in striatal 18F-DOPA uptake is slower in parkin PD cases compared to idiopathic PD patients [81].
A combined 18F-DOPA, 11C-PE2I and RAC PET study in a group of young-onset PD patients, with and without parkin mutations, has demonstrated that uptake of all three radiotracers were similar between parkin and non-parkin PD patients [85]. This is suggestive of PET DA markers being indistinguishable between carriers of parkin mutations and other young-onset PD patients with long disease duration.
More recently, an 18F-DOPA PET study has investigated both symptomatic and asymptomatic parkin PD patients over five years [86]. It was demonstrated that the symptomatic parkin PD patients showed an annual 18F-DOPA binding reduction in the putamen of 0.5% and in the caudate nucleus of 2%, compared to asymptomatic parkin PD patients who showed an annual reduction of 0.56% and 0.62% respectively. Another 18F-DOPA PET study from the same group detected reduced 18F-DOPA binding in parkin PD patients compared to healthy controls in the caudate nucleus, putamen, ventral striatum, locus coeruleus, midbrain raphe and pallidum [87]. Furthermore, it was shown that the hypothalamus was targeted in idiopathic PD patients compared to the midbrain raphe nuclei in parkin PD patients.
RAC PET has also been applied to genetic forms of PD to assess D2 receptor availability. One study of de novo PD patients has demonstrated increased putaminal RAC binding compared to healthy controls, which displayed similar uptake values to idiopathic PD patients [88]. Furthermore, this study demonstrated that parkin PD patients may be more responsive to DA medication than idiopathic PD cases as by following disease progression (and thus further exposure to DA medication), it was detected in the parkin PD patients that D2 receptor availability significantly decreased in the putamen and caudate nucleus whereas idiopathic PD patients normalized in the same regions.
Monitoring the progression of PD with PET
Monitoring the clinical progression of PD can be assessed using 18F-DOPA PET. However, monitoring progression is complicated by the administration of symptomatic medication, thus disguising the PD symptoms. Furthermore, current scales to assess motor disability are relatively biased towards detecting bradykinesia rather than the other common features of PD. Ideally; studies would aim to assess patients following a period of medication wash-out. However, this is a difficult process due to patient intolerance, and the likelihood that a substantial amount of withdrawal time is required to achieve a successful and complete wash-out of any medication.
Nonetheless, clinical progression has been investigated with 18F-DOPA PET. It has been reported that 18F-DOPA uptake in the putamen is correlated with disease progression [20, 21]. Furthermore, that this correlation is particularly related to bradykinesia and rigidity (and not tremor) severity [66]. It is also suggested that DA terminal loss in the caudate nucleus occurs at a slower rate than in the putamen [89].
The ability to monitor PD progression effectively ultimately leads to the ability to track modification of PD progression with DA medications. Some attempts have been made to track the neuroprotective effect of DA agonists, the agents possessing the possible ability to modify disease progression. The REAL PET trial administered either ropinirole or L-DOPA to a cohort of de novo PD patients. This trial demonstrated that patients in the ropinirole group demonstrated a slower reduction of putaminal 18F-DOPA uptake over two years compared to patients in the L-DOPA group. On the other hand, symptomatic improvement was superior in the L -DOPA group.
However, a different trial utilising 18F-DOPA PET failed to detect any effect of riluzole, a glutamate inhibitor, in PD progression [90].
Imaging motor complications in PD
Currently regular administration of the direct metabolic precursor for DA, L-DOPA, L-3, 4-dihydroxyphenylalanine (L-DOPA) remains the most effective treatment of PD symptomatology. However, long term use often leads to motor fluctuations and the appearance of motor complications such as involuntary movements, so-called L-DOPA-induced dyskinesia (LIDs). The exact underlying mechanisms precipitating these complications are not clear, although it is thought that both pre- and postsynaptic DA systems play a role with further evidence emerging that non-DA systems are also implicated.
To assess the role of presynaptic DA system in motor complications in PD, 18F-DOPA PET has been utilised in comparing two groups of PD patients; one with a fluctuating motor response to L-DOPA and another with a stable response to L-DOPA. This study reported a 28% decrease in presynaptic terminal function in the putamen of PD patients with a fluctuating response to L-DOPA compared to the stable responders [91]. These results were suggested to be due to i) an inability to store and release DA for use in the nigrostriatal pathway causing motor complications and ii) an altered ‘buffering’ capacity of the DA terminals as a response of differences in nigrostriatal damage between groups. A combined 11C-methylphenidate (MP) and DTBZ PET study has shown that putaminal MP/DHBZ is decreased in PD patients with motor fluctuations compared to stable responder PD patients [92]. These data support the hypothesis that presynaptic alterations play a role in the appearance of motor complications in PD due to a continued DAT downregulation, which ultimately leads to an increase in extracellular DA levels.
Postsynaptic DA mechanisms have also been investigated utilising 11C-SCH23390 and RAC PET as measures of the D1 and D2 receptor subtype availability respectively comparing a group of stable responder PD patients and fluctuating responder PD patients [40, 93]. However, the findings from these studies reported that mean D1 receptor availability was within the normal range in the caudate nucleus and putamen and mean D2 receptor availability in the putamen at baseline for both groups. Mean D2 receptor availability was reduced by approximately 15% within the caudate nucleus for both groups suggesting that this reduction may not be a precipitating factor of motor complications but an observation of disease progression. RAC PET used in conjunction with an L-DOPA challenge can monitor striatal DA release alterations. An early study reported a decrease of 23% in putaminal RAC binding following a single L-DOPA dose in PD patients with motor complications compared to stable responders [94]. Furthermore, UPDRS scores during the 'off medication state were inversely correlated with the reduction of putaminal RAC binding suggestive of an increasing inability to regulation DA release effectively as the disease progresses in patients with a fluctuating response to L-DOPA. A further RAC PET study reported that synaptic DA levels were three times higher in patients with motor fluctuations compared to stable responders following a single dose of L-DOPA [91]. Furthermore, UPDRS scores were corresponded with increases in synaptic DA levels. More specifically, it was reported that bradykinesia and rigidity scores were correlated with putaminal DA release, whereas tremor scores were not.
Non-DA systems have also received some attention regarding the pathophysiology of motor fluctuations and dyskinesia development. 11C-diprenorphine, a marker of μ, κ and δ opioid sites, has demonstrated that binding was reduced in both striatal (caudate nucleus and putamen) and extra-striatal (thalamus and anterior cingulate) in PD patients experiencing LID compared to stable responders [76]. Neurokinin -1 (NK1) receptor availability has been assessed in vivo with 18F-L829165 and reported that thalamic NK1 receptor availability in patients with LID was reduced [95]. A preliminary 11C-SCH442416 PET study (marker for A2A receptors) in an attempt to assess the adenosi-nergic systems has revealed that a significant increase of striatal A2A binding in patients with LID compared to patients without LID and healthy controls which had a similar degree of binding [96]. The authors also reported that thalamic A2A binding was similar across all three groups and suggest that the use of A2A receptor agonists in the clinical management of LID is justified. Only one study to date has examined the glutamatergic system in vivo with 11C-CNS5161 PET, which binds to the MK801 site. This two-scan (one ‘on’ medication and one ‘off’ medication) study had 18 PD patients divided into one group with LID and one group of stable responders. It was reported that binding was reduced in the caudate nucleus, putamen and motor cortex of the stable responders suggesting that the PD LID group may have relatively enhanced glutamate receptor activity in these areas [97]. Finally, the role of serotonin terminals potentially mishandling exogenous L-DOPA and subsequently releasing DA as a false neuro-transmitter in LID has recently been assessed in vivo in patient groups for the first time using RAC PET and suprathreshold L-DOPA and buspirone (5-HT1A agonist) challenges. The authors demonstrated in 16 PD patients with LID that buspirone administration which precedes L-DOPA administration, led to a reduction in putaminal RAC binding compared to the L-DOPA challenge and to the stable responders [98]. This is in contrast to the stable responders who appeared to be unaffected following the buspirone challenge. Clinically, the PD LID group demonstrated attenuation of LID following administration of buspirone. These findings indicate that serotonin terminals are likely to play a key role in LID, thus justifying the use of 5-HT agonists in the clinic in order to dampen the excessive DA release and as such attenuate LID.
Contribution of PET in restorative therapeutic strategies
The aim of restorative approaches in PD is to restore DA function in the affected areas. Potential strategies to achieve this include transplantation of striatal grafts of human or fetal mesencephalic cells, stem cells, gene therapy and nerve growth factors.
18F-DOPA PET can be used monitor the outcome of striatal graft transplantations in humans. This technique has shown that 18F-DOPA uptake increases in the striatum following the transplantation [77, 99] (Figure 3). Furthermore, it has been demonstrated with 18F-DOPA PET that transplantation of midbrain fetal cells into the putamen of PD patients' results in graft survival of up to ten years and the ability to release DA following a methamphetamine challenge is normalised [77]. Moreover, activation of the dor-salateral prefrontal cortex (DLFPC) and supplementary motor area (SMA) is restored [100].
Figure 3.

Transverse 18F-DOPA PET images of a PD patient who received striatal transplant with fetal ventral mesencephalic tissue, before the transplantation (left), 8 months after transplantation (middle) and 21 months after transplantation (right).
Two double-blind trials have been conducted to assess the efficacy of human fetal transplants [101, 102]. However, although the 18F-DOPA binding levels appeared to be successfully restored there did not appear to be any clinical improvement amongst the transplanted PD patients. One of the most troubling side effects of these trails was the emergence of graft-induced dyskinesias (GID). It has been suggested that the transplanted grafts were over-producing DA, thus causing the GID [103]. However, there were suggestions that other factors played a role [104, 105].
Glial cell line-derived neurotrophic factor (GDNF) infusion has been infused into the putamen of PD patients. It is established that GDNF protect DA neurons in rodents and non-human primates and 18F-DOPA PET has been used to assess it efficacy in humans [106]. It was shown that 18F-DOPA uptake increased in line with UPDRS scores over 12 months, suggesting that the use of GDNF may be a viable restorative approach in PD.
Finally, the suggestion that GID pathogenesis is related to the serotonergic system has been recently assessed for the first time utilising 11C-DASB PET. Two patients with good recovery of motor symptoms but who developed GID following neural transplantation were studied and demonstrated excessive serotonergic innervation in the grafted striatum [107]. The patients demonstrated an increase of 11C-DASB binding of 172% and 285% each compared to the mean binding values of the non-transplanted PD patients and healthy controls. The degree of serotonergic hyperinnervation in the two transplanted patients was consistent with the size of graft originally transplanted and severity of GID, i.e. the patient who received 42% more tissue than the other patient demonstrated more serotonergic hyperinnervation as well as more severe GIDs. It was also discovered that the ratio of serotonergic to DA innervation (as measured by 11C-DASB and 18F-DOPA binding) was increased up to 230% compared to healthy controls. Furthermore, the authors attempted to address the claim that serotonin terminals possess the ability to convert L-DOPA to DA, thus acting as a false neurotransmitter by administering the 5-HT1A agonist, buspirone, to the two transplanted patients. Indeed, GIDs were successfully attenuated following the administration of buspirone, indicating that the motor complications arose from serotonergic hyperinnervation and dysregulated release of DA in the grafted striatum.
Contribution of PET for the assessment of non-motor symptoms
Sleep and fatigue
Sleep disorders in PD manifest in a variety of forms including, disruptions in nocturnal sleep, insomnia and excessive daytime sleepiness (EDS) that often occurs due to the two former sleep disruptions mentioned.
18F-DOPA PET has been used to study PD patients with sleep problems [108]. This study reported a significant inverse correlation between 18F-DOPA uptake in the mesopontine and rapid eye movement (REM) sleep as measured by polysomnography. This finding indicates that monominergic activity in the mesopontine may be related to the inability to maintain nocturnal REM sleep in PD patients.
Disabling fatigue is estimated to occur in approximately one third of PD patients [109]. To date only one PET study has been conducted which specifically investigated fatigue in PD [8]. This combined 18F-DOPA and 11C-DASB PET study sought to investigate both the DA and serotonergic systems in a group of PD patients with and without fatigue. Results from a region of interest analysis approach indicate that the PD group with fatigue showed significantly reduced 11C-DASB binding compared to the PD group without fatigue in the putamen, caudate nucleus, ventral striatum and thalamus. However, 18F-DOPA uptake was similar in all regions in both groups. A voxel based analysis revealed further reductions of 11C-DASB binding in the cingulate and amygdale and further 18F-DOPA binding reductions in the caudate and insula in the PD group with fatigue. The authors suggest that these results are indicative of a possible association between fatigue in PD with reduced serotonergic function in the basal ganglia and limbic structures as well as a possible insular DA dysfunction.
Depression
Depression in PD is reported to occur in approximately 45% of patients, however the pathophysiology is unclear. It is also not clear if depression in PD may be part of the disease course for certain patients or in fact a reaction of the patient due to the diagnosis itself. Because Lewy body pathology is known to effect the serotonergic, noradrenergic and DA systems, it is possible that dysfunction in any of these systems may be responsible for the occurrence of depression in PD.
11C-RTI 32 PET is a marker of both DAT and noradrenergic transporter binding. A study utilizing this radiotracer showed that PD patients without depression demonstrated reduced putaminal 11C-RTI 32 uptake, but PD patients with depression demonstrated additional reductions in the noradrenergic locus coeruleus, thalamus, and the limbic system (amygdala, ventral striatum, and anterior cingulate) [4]. Severity of anxiety inversely correlates with 11C-RTI 32 binding in these regions.These results suggest that depression in PD may be associated with noradrenergic and limbic DA dennervation in addition to striatal DA dennenrvation.
123β-CIT is a radiotracer, which binds with nanomolar affinity to DA, noradrenaline, and serotonin transporters. One study using 123β-CIT PET in PD patients with depression found no binding differences between patients with and without depression or between binding and Hamilton Depression Rating Scale scores (HDRS) [51].
Altered serotonergic neurotransmission in PD patients with depression has been investigated using 11C-WAY100635 PET [49]. This study reported that despite a decrease in 5-HT1A binding in the raphe nucleus or PD patients compared to controls, there was no difference between depressed and non-depressed PD patients.
One 11C-DASB PET study has reported an increase in 5-HTT binding in the OFC region of depression patients with early PD [110].
More recently, another study utilising 11C-DASB PET has reported a relationship between PD patients exhibiting depressive symptoms and 5-HTT binding in limbic regions and the raphe nucleus [111]. This study used a cohort of 34 anti-depressant-naïve PD patients and 10 matched healthy controls. Depressive symptoms were systematically assessed using the Beck Depression Inventory-II (BDI-II), HRDS and a structured clinical interview for DSM - IV Axis I Disorders (SCID-I). It was demonstrated that PD patients with depressive symptoms, had significantly increased 11C-DASB binding in the amygdale, hypothalamus, caudal raphe nuclei and posterior cingulate cortex. Furthermore, all other brain regions demonstrated similarly decreased 11C-DASB binding in both the high and low depressive symptom PD groups compared to healthy controls. The authors suggest that raised 5-HTT availability in limbic areas is implicated in the pathophysiology of depression in PD and justifies the use of agents acting on 5-HTT in the treatment of PD depression.
Dementia
The presence of dementia in PD is estimated to be approximately 40% [112]. PET imaging has been used to investigate both DA and cholinergic dysfunction in PD patients with dementia.
18F-DOPA PET has been used to compare mesofrontal DA projections in PD patients and PD patients with dementia (PDD). All patients were matched for age, disease duration and disease severity and both groups showed a similar level of 18F-DOPA binding in the putamen [113]. However, the PDD group showed additional reductions in the right caudate and bilaterally in the ventral striatum and the anterior cingulate, suggesting a potential role of mesolimbic and mesocortical dysfunction in demented PD patients.
18F-FDG PET studies have indicated that there is a pattern of reduced glucose metabolism in frontal, temporal and parietal areas when comparing PDD and AD patients [7, 114, 115]. Furthermore, comparing PDD patients and patients with dementia Lewy bodies (DLB), both groups appear to display hypometabolism in parietal, temporal, occipital, frontal areas and in the anterior cingulate when they are compared to healthy controls. Although when PDD and DLB groups are directly compared, hypometabolism in the anterior cingulate is more pronounced in the DLB group [116].
Cholinergic dysfunction has been investigated using the radiotracers; 11C-PMP and 11C-MP4A, which can be used to assess acetylcholi-nesterase (AChE) activity. An 11C-MP4A PET study has reported a reduction in cortical binding in PD patients of 11% which increases to 30% in PDD patients (particularly in parietal areas) [117].
An 11C-MP4A PET study has reported that 11C-MP4A binding correlated with levels of striatal 18F-DOPA uptake in a group of PD patients with and without dementia [54]. This is suggestive of a parallel reduction in DA and cholinergic function in PD. Interestingly; cortical AChE deficiency correlated with cognitive testing scores but did not correlate with motor symptoms. This is suggestive of a dysfunctional cholinergic system contributing to PDD.
11C-PIB PET has been utilised in determining the prevalence of raised amyloid load in DLB and PDD. This study reported that 11/13 DLB and 2/13 PDD patients has significantly raised amyloid plaque levels, therefore, it may not be a factor in the development of PDD [118].
Psychosis
Visual hallucinations are the most common form of psychosis observed in PD, which tend to occur later in the disease stage alongside some form of cognitive impairment. PET studies investigating psychosis in PD is limited.
18F-FDG PET has been used to investigate visual hallucinations in PD [12]. This study reported that the frontal areas, and in particular, the superior frontal gyrus demonstrated increased cerebral glucose metabolic rate in PD patients with visual hallucinations compared to PD patient without any form of visual hallucinations. More recently, the radiotracer, 18F-setoperone, a selective marker for the serotonin 2A receptor, has been implemented in a pilot study of seven PD patients with visual hallucinations and seven age and sex matched PD controls without any history of visual hallucinations [119]. This study reported that increased 18F-setoperone binding mainly clustered in the ventral visual pathway, including the bilateral inferooccipital gyrus, right fusiform gyrus, and inferotemporal cortex. Binding increases were also detected bilaterally in the insula and dorsolateral prefrontal cortex.
Impulse control disorders and dopamine dysregulation syndrome
DA medications, including L-DOPA are the most effective method of PD symptom management. However, following long-term use of DA medications, it has now become apparent that addictive and compulsive behaviours are associated with chronic treatment [120]. The most commonly seen maladaptive behaviours in clinic include punding and the so-called impulse control disorders (ICDs); hypersexuality (HS), pathological gambling (PG), compulsive shopping (CS), binge eating (BE). Also seen, although to a lesser extent is the phenomenon, dopamine dysregulation syndrome (DDS) which is a consequence of the patient becoming addicted to their DA medication with an increased degree of craving [121], thus seeking to self-administer without the clinicians' knowledge. These behaviours are difficult to manage as often they are not apparent to the patient's family or to the clinician until they have become quite advanced, often due to the patient themselves concealing the acquired behaviours. Once the issue becomes apparent they require immediate attention.
DDS has been investigated using RAC PET [122]. It has been demonstrated that following an L-DOPA challenge, DDS PD patients have increased DA release in the ventral striatal areas compared to a group of PD patients without DDS. Furthermore, RAC binding in the DDS group correlated with an increase in ‘wanting’ and not ‘liking’ the drug indicative of an addictive behaviour. It is suggested that this finding is consistent with the theory of incentive sensitisation of compulsive drug use. This theory suggests that compulsive drug use occurs due to an increased attribution of incentive salience for rewards (via drug use) due to mesolimbic neuro-circuitry adaptations [123].
Recently, ICDs have begun receiving attention. Another RAC PET study with a PD cohort consisting of two groups, one with PD patients with either DDS or an ICD (n= 11) and another PD group without any DDS or ICDs (n= 7) has been recently published [10]. This study conducted three RAC PET scans; one while patients were in the ‘off’ medication state and were exposed to neutral cues, another one while patients were in the ‘on’ medication state and exposed to the neutral cues and a third which consisted of the patients being in an ‘on’ medication state and exposed to reward-related cues. During the ‘on’ scans, the patients were administered an oral dose of L-DOPA. It was reported that patients with DDS or ICD demonstrated a greater decrease of RAC binding in the ventral striatum compared to the PD patients without any evidence of DDS or ICD during the scan with reward cues. This group difference was not observed during the L-DOPA challenge alone scan. The authors suggest that their findings are consistent with the incentive sensitisation theory.
PG in PD has been investigated with RAC PET using a gambling task [124]. It was reported that patients with PG showed greater decreases in RAC binding in the ventral striatum during gambling (13.9%) compared to control patients (8.1%), suggesting greater DA release.
An H215O PET study has also been conducted in PD patients with PG [125]. It was reported that a reduction in cerebral blood flow in lateral orbitofrontal cortex, rostral cingulate, amygdala and pallidum was detected following administration of DA medication.
Cardiac sympathetic denervation
It has been reported that most patients with idiopathic PD show a significant loss of sympathetic innervation of the heart [126]. Myocardial dennervation in early PD patients has been detected even when cardiovascular reflexes are still intact. Cardiac sympathetic denervation may contribute to symptoms of autonomic failure such as orthostatic hypotension.
The only PET study to investigate orthostatic hypotension in PD has utilised 11C-MHED PET [127] which is able to visualise sympathetic neurons. This study reported 11C-MHED uptake was reduced in PD patients compared to healthy controls. Furthermore, PD patients with orthostatic hypotension displayed further reduction in 11C-MHED uptake.
Olfactory function
Olfactory dysfunction is a frequent non-motor symptom of PD, which has been attributed to early pathological deposition of Lewy bodies, and Lewy neurites in primary olfactory centers and involves deficits in odor detection, discrimination and identification [128]. Hyposmia may be related to neuronal degeneration with deposition of alpha-synuclein in primary olfactory areas as a very early component of the pathology of PD [129, 130].
11C-CFT PET has been utilised in the investigation of DA denervation in the nigrostriatal pathway and olfactory deficits in PD [131]. It was reported that reduced 11C-CFT in the nigrostriatal pathway was more pronounced in a group of PD patients with olfactory dysfunction compared to a group of healthy controls. Furthermore, uptake values correlated with poorer smell identification scores.
More recently, a study using the same method has reported that selective hyposmia in PD is mostly correlated with hippocampal rather than amygdala, ventral or dorsal striatal DA innervation as measured by 11C-CFT binding. These findings are suggestive of a hippocampal mesolimbic DA involvement in selective hyposmia in PD.
Conclusions
PET imaging has provided an excellent tool to investigate the many facets of PD in vivo. The technique has increased understanding surrounding the differential diagnosis of PD, the progression of the disease, complications arising from DA medications, as well as the non-motor symptoms of the disease. Furthermore, PET imaging will allow greater advancement of alternative restorative approaches to therapy in PD as it can be used to evaluate the efficacy of a therapy and monitor improvements (or declines) longitudinally.
However, there are many gaps in our knowledge still remaining, which could be closed by using PET imaging techniques. Future PET studies should focus on understanding the role of non-DA neurotransmitter systems in not only the pathogenesis of the PD motor disease and arising medication related complications, but also the non-motor features, such as addiction and compulsion as they greatly impact on the quality of life of PD patients.
Of course, to achieve all of this, new radio-tracers need to be developed for biological specificity and to enable us to image chemical pathways, which are currently out of reach, such as noradrenergic function.
References
- 1.Braak H, Ghebremedhin E, Rüb U, Bratzke H, Del Tredici K. Stages in the development of Parkinson's disease-related pathology. Cell Tissue Res. 2004;318:121–34. doi: 10.1007/s00441-004-0956-9. [DOI] [PubMed] [Google Scholar]
- 2.Kish SJ. Biochemistry of Parkinson's disease: is a brain serotonergic deficiency a characteristic of idiopathic Parkinson's disease? Adv Neurol. 2003;91:39–49. [PubMed] [Google Scholar]
- 3.Kish SJ, Tong J, Hornykiewicz O, Rajput A, Chang LJ, Guttman M, Furukawa Y. Preferential loss of serotonin markers in caudate versus putamen in Parkinson's disease. Brain. 2008;131:120–131. doi: 10.1093/brain/awm239. [DOI] [PubMed] [Google Scholar]
- 4.Remy P, Doder M, Lees AJ, Turjanski N, Brooks DJ. Depression in Parkinson's disease: loss of dopamine and noradrenaline innervation in the limbic system. Brain. 2005;128:1314–1322. doi: 10.1093/brain/awh445. [DOI] [PubMed] [Google Scholar]
- 5.Politis M, Wu K, Loane C, Kiferle L, Molloy S, Brooks DJ, Piccini P. Staging of serotonergic dysfunction in Parkinson's disease: An in vivo 11C-DASB PET study. Neurobiol Dis. 2010;40:216–21. doi: 10.1016/j.nbd.2010.05.028. [DOI] [PubMed] [Google Scholar]
- 6.Albin RL, Koeppe RA, Bohnen NI, Wernette K, Kilbourn MA, Frey KA. Spared caudal brain-stem SERT binding in early Parkinson's disease. J Cereb Blood Flow Metab. 2008;8:441–4. doi: 10.1038/sj.jcbfm.9600599. [DOI] [PubMed] [Google Scholar]
- 7.Peppard RF, Martin WR, Carr GD, Grochowski E, Schulzer M, Guttman M, McGeer PL, Phillips AG, Tsui JK, Calne DB. Cerebral glucose metabolism in Parkinson's disease with and without dementia. Arch Neurol. 1992;49:1262–8. doi: 10.1001/archneur.1992.00530360060019. [DOI] [PubMed] [Google Scholar]
- 8.Pavese N, Metta V, Bose SK, Chaudhuri KR, Brooks DJ. Fatigue in Parkinson's disease is linked to striatal and limbic serotonergic dysfunction. Brain. 2010;133:3434–43. doi: 10.1093/brain/awq268. [DOI] [PubMed] [Google Scholar]
- 9.Burn DJ. Depression in Parkinson's disease. Eur J Neurol. 2002;9(Suppl 3):44–54. doi: 10.1046/j.1468-1331.9.s3.6.x. [DOI] [PubMed] [Google Scholar]
- 10.O'Sullivan SS, Wu K, Politis M, Lawrence AD, Evans AH, Bose SK, Djamshidian A, Lees AJ, Piccini P. Cue-induced striatal dopamine release in Parkinson's disease-associated impulsive-compulsive behaviours. Brain. 2011;134:969–78. doi: 10.1093/brain/awr003. [DOI] [PubMed] [Google Scholar]
- 11.Nagano-Saito A, Washimi Y, Arahata Y, Iwai K, Kawatsu S, Ito K, Nakamura A, Abe Y, Yamada T, Kato T, Kachi T. Visual hallucination in Parkinson's disease with FDG PET. Mov Disord. 2004;19:801–6. doi: 10.1002/mds.20129. [DOI] [PubMed] [Google Scholar]
- 12.Wimo A, Jonsson L, Winblad B. An estimate of the worldwide prevalence and direct costs of dementia in 2003. Dement Geriatr Cogn Disord. 2006;21:175–81. doi: 10.1159/000090733. [DOI] [PubMed] [Google Scholar]
- 13.Thomas B, Beal MF. Parkinson's disease. Hum Mol Genet. 2007;16:183–194. doi: 10.1093/hmg/ddm159. [DOI] [PubMed] [Google Scholar]
- 14.Bower JH, Maraganore DM, McDonnell SK, Rocca WA. Incidence and distribution of park-insonism in Olmsted County, Minnesota, 1976-1990. Neurology. 1999;2:1214–20. doi: 10.1212/wnl.52.6.1214. [DOI] [PubMed] [Google Scholar]
- 15.Tanner CM, Goldman SM. Epidemiology of Parkinson's disease. Neurol Clin. 1996;14:317–335. doi: 10.1016/S0733-8619(05)70259-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kuzuhara S, Mori H, Izumiyama N, Yoshimura M, Ihara Y. Lewy bodies are ubiquitinated: a light and electron microscopic immunocytochemical study. Acta Neuropathol (Berl) 1988;75:345–353. doi: 10.1007/BF00687787. [DOI] [PubMed] [Google Scholar]
- 17.Pollanen MS, Dickson DW, Bergeron C. Pathology and biology of the Lewy body. J Neuropathol Exp Neurol. 1993;52:183–191. doi: 10.1097/00005072-199305000-00001. [DOI] [PubMed] [Google Scholar]
- 18.Garnett ES, Firnau G, Nahmias C. Dopamine visualized in the basal ganglia of living man. Nature. 1983;305:137–8. doi: 10.1038/305137a0. [DOI] [PubMed] [Google Scholar]
- 19.Vingerhoets FJ, Schulzer M, Calne DB, Snow BJ. Which clinical sign of Parkinson's disease best reflects the nigrostriatal lesion? Ann Neurol. 1997;41:58–64. doi: 10.1002/ana.410410111. [DOI] [PubMed] [Google Scholar]
- 20.Broussolle E, Dentresangle C, Landais P, Garcia-Larrea L, Pollak P, Croisile B, Hibert O, Bonnefoi F, Galy G, Froment JC, Comar D. The relation of putamen and caudate nucleus 18FDopa uptake to motor and cognitive performances in Parkinson's disease. J Neurol Sci. 1999;166:141–51. doi: 10.1016/s0022-510x(99)00127-6. [DOI] [PubMed] [Google Scholar]
- 21.Otsuka M, Ichiya Y, Kuwabara Y, Hosokawa S, Sasaki M, Yoshida T, Fukumura T, Masuda K, Kato M. Differences in the reduced 18F-Dopa uptakes of the caudate and the putamen in Parkinson's disease: correlations with the three main symptoms. J Neurol Sci. 1996;136:169–73. doi: 10.1016/0022-510x(95)00316-t. [DOI] [PubMed] [Google Scholar]
- 22.Fearnley JM, Lees AJ. Ageing and Parkinson's disease: substantia nigra regional selectivity. Brain. 1991;114:2283–301. doi: 10.1093/brain/114.5.2283. [DOI] [PubMed] [Google Scholar]
- 23.Brück A, Aalto S, Rauhala E, Bergman J, Marttila R, Rinne JO. A follow-up study on 6-[18F]fluoro-L-dopa uptake in early Parkinson's disease shows nonlinear progression in the putamen. Mov Disord. 2009;24:1009–15. doi: 10.1002/mds.22484. [DOI] [PubMed] [Google Scholar]
- 24.Pavese N, Rivero-Bosch M, Lewis SJ, Whone AL, Brooks DJ. Progression of monoaminergic dysfunction in Parkinson's disease: A longitudinal (18) F-dopa PET studies. Neuroimage. 2011;6:1463–8. doi: 10.1016/j.neuroimage.2011.03.012. [DOI] [PubMed] [Google Scholar]
- 25.Stoessl AJ. Positron emission tomography in premotor Parkinson's disease. Parkinsonism Relat Disord. 2007;13(suppl 3):S421–S424. doi: 10.1016/S1353-8020(08)70041-5. [DOI] [PubMed] [Google Scholar]
- 26.de la Fuente-Fernández R, Sossi V, McCormick S, Schulzer M, Ruth TJ, Stoessl AJ. Visualizing vesicular dopamine dynamics in Parkinson's disease. Synapse. 2009;63:713–6. doi: 10.1002/syn.20653. [DOI] [PubMed] [Google Scholar]
- 27.Lee CS, Samii A, Sossi V, Ruth TJ, Schulzer M, Holden JE, Wudel J, Pal PK, de la Fuente-Fernandez R, Calne DB, Stoessl AJ. In vivo positron emission tomographic evidence for compensatory changes in presynaptic dopaminergic nerve terminals in Parkinson's disease. Ann Neurol. 2000;47:493–503. [PubMed] [Google Scholar]
- 28.Salmon E, Brooks DJ, Leenders KL, Turton DR, Hume SP, Cremer JE, Jones T, Frackowiak RS. A two-compartment description and kinetic procedure for measuring regional cerebral [11C]nomifensine uptake using positron emission tomography. J Cereb Blood Flow Metab. 1990;10:307–16. doi: 10.1038/jcbfm.1990.59. [DOI] [PubMed] [Google Scholar]
- 29.Frost JJ, Rosier AJ, Reich SG, Smith JS, Ehlers MD, Snyder SH, Ravert HT, Dannals RF. Positron emission tomographic imaging of the dopamine transporter with 11C-WIN 35,428 reveals marked declines in mild Parkinson's disease. Ann Neurol. 1993;34:423–31. doi: 10.1002/ana.410340331. [DOI] [PubMed] [Google Scholar]
- 30.Marié RM, Barré L, Rioux P, Allain P, Lechevalier B, Baron JC. PET imaging of neocortical monoaminergic terminals in Parkinson's disease. J Neural Transm Park Dis Dement Sect. 1995;9:55–71. doi: 10.1007/BF02252963. [DOI] [PubMed] [Google Scholar]
- 31.Tedroff J, Aquilonius SM, Laihinen A, Rinne U, Hartvig P, Anderson J, Lundqvist H, Haaparanta M, Solin O, Antoni G, Gee AD, Ullin J, Långström B. Striatal kinetics of [11C]-(+)-nomifensine and 6-[18F]fluoro-L-dopa in Parkinson's disease measured with positron emission tomography. Acta Neurol Scand. 1990;81:24–30. doi: 10.1111/j.1600-0404.1990.tb00926.x. [DOI] [PubMed] [Google Scholar]
- 32.Leenders KL, Salmon EP, Tyrrell P, Perani D, Brooks DJ, Sager H, Jones T, Marsden CD, Frackowiak RS. The nigrostriatal dopaminergic system assessed in vivo by positron emission tomography in healthy volunteer subjects and patients with Parkinson's disease. Arch Neurol. 1990;47:1290–8. doi: 10.1001/archneur.1990.00530120034007. [DOI] [PubMed] [Google Scholar]
- 33.Guttman M, Burkholder J, Kish SJ, Hussey D, Wilson A, DaSilva J, Houle S. 11C]RTI-32 PET studies of the dopamine transporter in early dopa-naive Parkinson's disease: implications for the symptomatic threshold. Neurology. 1997;48:1578–83. doi: 10.1212/wnl.48.6.1578. [DOI] [PubMed] [Google Scholar]
- 34.Aquilonius SM. What has PET told us about Parkinson's disease? Acta Neurol Scand Suppl. 1991;136:37–9. doi: 10.1111/j.1600-0404.1991.tb05018.x. [DOI] [PubMed] [Google Scholar]
- 35.Schreckenberger M, Hägele S, Siessmeier T, Buchholz HG, Armbrust-Henrich H, Rösch F, Gründer G, Bartenstein P, Vogt T. The dopamine D2 receptor ligand 18F-desmethoxyfallypride: an appropriate fluorinated PET tracer for the differential diagnosis of parkinsonism. Eur J Nucl Med Mol Imaging. 2004;31:1128–35. doi: 10.1007/s00259-004-1465-5. [DOI] [PubMed] [Google Scholar]
- 36.Rinne JO, Laihinen A, Ruottinen H, Ruotsalainen U, Någren K, Lehikoinen P, Oikonen V, Rinne UK. Increased density of dopamine D2 receptors in the putamen, but not in the caudate nucleus in early Parkinson's disease: a PET study with [11C]raclopride. J Neurol Sci. 1995;132:156–61. doi: 10.1016/0022-510x(95)00137-q. [DOI] [PubMed] [Google Scholar]
- 37.Cropley VL, Fujita M, Bara-Jimenez W, Brown AK, Zhang XY, Sangare J, Herscovitch P, Pike VW, Hallett M, Nathan PJ, Innis RB. Pre- and post-synaptic dopamine imaging and its relation with frontostriatal cognitive function in Parkinson disease: PET studies with [11C]NNC 112 and [18F]FDOPA. Psychiatry Res. 2008;163:171–82. doi: 10.1016/j.pscychresns.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 38.Pavese N, Evans AH, Tai YF, Hotton G, Brooks DJ, Lees AJ, Piccini P. Clinical correlates of levodopa-induced dopamine release in Parkinson disease: a PET study. Neurology. 2006;67:1612–7. doi: 10.1212/01.wnl.0000242888.30755.5d. [DOI] [PubMed] [Google Scholar]
- 39.Rinne JO, Laihinen A, Rinne UK, Någren K, Bergman J, Ruotsalainen U. PET study on striatal dopamine D2 receptor changes during the progression of early Parkinson's disease. Mov Disord. 1993;8:134–8. doi: 10.1002/mds.870080203. [DOI] [PubMed] [Google Scholar]
- 40.Turjanski N, Lees AJ, Brooks DJ. In vivo studies on striatal dopamine D1 and D2 site binding in L-dopa-treated Parkinson's disease patients with and without dyskinesias. Neurology. 1997;49:717–23. doi: 10.1212/wnl.49.3.717. [DOI] [PubMed] [Google Scholar]
- 41.Antonini A, Schwarz J, Oertel WH, Beer HF, Madeja UD, Leenders KL. [11C]raclopride and positron emission tomography in previously untreated patients with Parkinson's disease: Influence of L-dopa and lisuride therapy on striatal dopamine D2-receptors. Neurology. 1994;44:1325–9. doi: 10.1212/wnl.44.7.1325. [DOI] [PubMed] [Google Scholar]
- 42.Antonini A, Schwarz J, Oertel WH, Pogarell O, Leenders KL. Long-term changes of striatal dopamine D2 receptors in patients with Parkinson's disease: a study with positron emission tomography and [11C]raclopride. Mov Disord. 1997;12:33–8. doi: 10.1002/mds.870120107. [DOI] [PubMed] [Google Scholar]
- 43.Antonini A, Vontobel P, Psylla M, Günther I, Maguire PR, Missimer J, Leenders KL. Complementary positron emission tomographic studies of the striatal dopaminergic system in Parkinson's disease. Arch Neurol. 1995;52:1183–90. doi: 10.1001/archneur.1995.00540360061017. [DOI] [PubMed] [Google Scholar]
- 44.Dentresangle C, Veyre L, Le Bars D, Pierre C, Lavenne F, Pollak P, Guerin J, Froment JC, Brousolle E. Striatal D2 dopamine receptor status in Parkinson's disease: an [18F]dopa and [11C]raclopride PET study. Mov Disord. 1999;14:1025–30. doi: 10.1002/1531-8257(199911)14:6<1025::aid-mds1020>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 45.Brooks DJ, Ibanez V, Sawle GV, Playford ED, Quinn N, Mathias CJ, Lees AJ, Marsden CD, Bannister R, Frackowiak RS. Striatal D2 receptor status in patients with Parkinson's disease, striatonigral degeneration, and progressive supranuclear palsy, measured with 11Craclopride and positron emission tomography. Ann Neurol. 1992;31:184–92. doi: 10.1002/ana.410310209. [DOI] [PubMed] [Google Scholar]
- 46.Politis M, Piccini P, Pavese N, Koh SB, Brooks DJ. Evidence of dopamine dysfunction in the hypothalamus of patients with Parkinson's disease: an in vivo 11C-raclopride PET study. Exp Neurol. 2008;214:112–6. doi: 10.1016/j.expneurol.2008.07.021. [DOI] [PubMed] [Google Scholar]
- 47.Piccini P, Pavese N, Brooks DJ. Endogenous dopamine release after pharmacological challenges in Parkinson's disease. Ann Neurol. 2003;53:647–53. doi: 10.1002/ana.10526. [DOI] [PubMed] [Google Scholar]
- 48.Sawamoto N, Piccini P, Hotton G, Pavese N, Thielemans K, Brooks DJ. Cognitive deficits and striato-frontal dopamine release in Parkinson's disease. Brain. 2008;131:1294–302. doi: 10.1093/brain/awn054. [DOI] [PubMed] [Google Scholar]
- 49.Doder M, Rabiner EA, Turjanski N, Lees AJ, Brooks DJ. Brain serotonin HT1A receptors in Parkinson's disease with and without depression measured by positron emission tomography and 11C-WAY100635. Mov Disord. 2000;15(Suppl 3):213. [Google Scholar]
- 50.Kerenyi L, Ricaurte GA, Schretlen DJ, McCann U, Varga J, Mathews WB, Ravert HT, Dannals RF, Hilton J, Wong DF, Szabo Z. Positron emission tomography of striatal serotonin transporters in Parkinson disease. Arch Neurol. 2003;60:1223–9. doi: 10.1001/archneur.60.9.1223. [DOI] [PubMed] [Google Scholar]
- 51.Kim SE, Choi JY, Choe YS, Choi Y, Lee WY. Serotonin transporters in the midbrain of Parkinson's disease patients: a study with 123Ibeta-CIT SPECT. J Nucl Med. 2003;44:870–876. [PubMed] [Google Scholar]
- 52.Tanaka H, Kannari K, Maeda T, Tomiyama M, Suda T, Matsunaga M. Role of serotonergic neurons in L-DOPA-derived extracellular dopamine in the striatum of 6-OHDA-lesioned rats. Neuroreport. 1999;10:631–4. doi: 10.1097/00001756-199902250-00034. [DOI] [PubMed] [Google Scholar]
- 53.Maeda T, Nagata K, Yoshida Y, Kannari K. Serotonergic hyperinnervationinto the dopaminergic denervated striatum compensates for dopamine conversion from exogenously administered L-DOPA. Brain Res. 2005;1046:230–3. doi: 10.1016/j.brainres.2005.04.019. [DOI] [PubMed] [Google Scholar]
- 54.Bohnen NI, Albin RL, Koeppe RA, Kannari K. Positron emission tomography of monoaminergic vesicular binding in aging and Parkinson disease. J Cereb Blood Flow Metab. 2006;26:1198–1212. doi: 10.1038/sj.jcbfm.9600276. [DOI] [PubMed] [Google Scholar]
- 55.Asahina M, Suhara T, Shinotoh H, Inoue O, Suzuki K, Hattori T. Brain muscarinic receptors in progressive supranuclear palsy and Parkinson's disease: a positron emission tomographic study. J Neurol Neurosurg Psychiatry. 1998;65:155–63. doi: 10.1136/jnnp.65.2.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Koepp MJ, Duncan JS. PET: opiate neuroreceptor mapping. Adv Neurol. 2000;83:145–56. [PubMed] [Google Scholar]
- 57.Haber SN, Watson SJ. The comparative distribution of enkephalin, dynorphin and substance P in the human globus pallidus and basal forebrain. Neuroscience. 1985;14:1011–24. doi: 10.1016/0306-4522(85)90272-6. [DOI] [PubMed] [Google Scholar]
- 58.Fernandez A, de Ceballos ML, Jenner P, Marsden CD. Neurotensin, substance P, delta and mu opioid receptors are decreased in basal ganglia of Parkinson's disease patients. Neuroscience. 1994;61:73–9. doi: 10.1016/0306-4522(94)90061-2. [DOI] [PubMed] [Google Scholar]
- 59.Ding YS, Lin KS, Garza V, Carter P, Alexoff D, Logan J, Shea C, Xu Y, King P. Evaluation of a new norepinephrine transporter PET ligand in baboons, both in brain and peripheral organs. Synapse. 2003;50(4):345–52. doi: 10.1002/syn.10281. Dec 15. [DOI] [PubMed] [Google Scholar]
- 60.Ding YS, Fowler J. New-generation radiotracers for nAChR and NET. Nucl Med Biol. 2005;32:707–18. doi: 10.1016/j.nucmedbio.2005.04.017. [DOI] [PubMed] [Google Scholar]
- 61.Ouchi Y, Yoshikawa E, Sekine Y, Futatsubashi M, Kanno T, Ogusu T, Torizuka T. Microglial activation and dopamine terminal loss in early Parkinson's disease. Ann Neurol. 2005;57:168–75. doi: 10.1002/ana.20338. [DOI] [PubMed] [Google Scholar]
- 62.Ouchi Y, Yagi S, Yokokura M, Sakamoto M. Neuroinflammation in the living brain of Park inson's disease. Parkinsonism Relat Disord. 2009;15(Suppl 3):S200–4. doi: 10.1016/S1353-8020(09)70814-4. [DOI] [PubMed] [Google Scholar]
- 63.Gerhard A, Pavese N, Hotton G, Turkheimer F, Es M, Hammers A, Eggert K, Oertel W, Banati RB, Brooks DJ. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson's disease. Neurobiol Dis. 2006;21:404–12. doi: 10.1016/j.nbd.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 64.Puñal-Riobóo J, Serena-Puig A, Varela-Lema L, Alvarez-Páez AM, Ruano-Ravina A. Clinical utility of (18)F-DOPA-PET in movement disorders. A systematic review] Rev Esp Med Nucl. 2009;28:106–13. [PubMed] [Google Scholar]
- 65.Burn DJ, Brooks DJ. Nigral dysfunction in druginduced parkinsonism: an 18F-dopa PET study. Neurology. 1993;43:552–6. doi: 10.1212/wnl.43.3_part_1.552. [DOI] [PubMed] [Google Scholar]
- 66.Otsuka M, Ichiya Y, Hosokawa S, Kuwabara Y, Tahara T, Fukumura T, Kato M, Masuda K, Goto I. Striatal blood flow, glucose metabolism and 18F-dopa uptake: difference in Parkinson's disease and atypical parkinsonism. J Neurol Neurosurg Psychiatry. 1991;54:898–904. doi: 10.1136/jnnp.54.10.898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sawle GV, Leenders KL, Brooks DJ, Harwood G, Lees AJ, Frackowiak RS, Marsden CD. Dopa-responsive dystonia: [18F]dopa positron emission tomography. Ann Neurol. 1991;30:4–30. doi: 10.1002/ana.410300106. [DOI] [PubMed] [Google Scholar]
- 68.Ghaemi M, Hilker R, Rudolf J, Sobesky J, Heiss WD. Differentiating multiple system atrophy from Parkinson's disease: contribution of striatal and midbrain MRI volumetry and multitracer PET imaging. J Neurol Neurosurg Psychiatry. 2002;73:517–23. doi: 10.1136/jnnp.73.5.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Eidelberg D, Takikawa S, Moeller JR, Dhawan V, Redington K, Chaly T, Robeson W, Dahl JR, Margouleff D, Fazzini E, Przedborski S, Fahn S. Striatal hypometabolism distinguishes striatonigral degeneration from Parkinson's disease. Ann Neurol. 1993;33:518–27. doi: 10.1002/ana.410330517. [DOI] [PubMed] [Google Scholar]
- 70.Pierot L, Desnos C, Blin J, Raisman R, Scherman D, Javoy-Agid F, Ruberg M, Agid Y. D1 and D2-type dopamine receptors in patients with Parkinson's disease and progressive supranuclear palsy. J Neurol Sci. 1988;86:291–306. doi: 10.1016/0022-510x(88)90106-2. [DOI] [PubMed] [Google Scholar]
- 71.Landwehrmeyer B, Palacios JM. Alterations of neurotransmitter receptors and neurotransmitter transporters in progressive supranuclear palsy. J Neural Transm Suppl. 1994;42:229–46. doi: 10.1007/978-3-7091-6641-3_18. [DOI] [PubMed] [Google Scholar]
- 72.Joyce JN, Ryoo HL, Beach TB, Caviness JN, Stacy M, Gurevich EV, Reiser M, Adler CH. Loss of response to levodopa in Parkinson's disease and co-occurrence with dementia: role of D3 and not D2 receptors. Brain Res. 2002;955:38–52. doi: 10.1016/s0006-8993(02)03396-6. [DOI] [PubMed] [Google Scholar]
- 73.Hilker R, Schweitzer K, Coburger S, Ghaemi M, Weisenbach S, Jacobs AH, Rudolf J, Herholz K, Heiss WD. Nonlinear progression of Parkinson disease as determined by serial positron emission tomographic imaging of striatal fluorodopa F 18 activity. Arch Neurol. 2005;62:378–382. doi: 10.1001/archneur.62.3.378. [DOI] [PubMed] [Google Scholar]
- 74.Panzacchi A, Moresco RM, Garibotto V, Antonini A, Gobbo C, Isaias IU, Goldwurm S, Bonaldi L, Carpinelli A, Pezzoli G, Fazio F, Perani D. A voxel-based PET study of dopamine transporters in Parkinson's disease: relevance of age at onset. Neurobiol Dis. 2008;31:102–109. doi: 10.1016/j.nbd.2008.03.012. [DOI] [PubMed] [Google Scholar]
- 75.Bohnen NI, Kaufer DI, Hendrickson R, Ivanco LS, Lopresti BJ, Constantine GM, Mathis ChA, Davis JG, Moore RY, Dekosky ST. Cognitive correlates of cortical cholinergic denervation in Parkinson's disease and parkinsonian dementia. J Neurol. 2006;253:242–247. doi: 10.1007/s00415-005-0971-0. [DOI] [PubMed] [Google Scholar]
- 76.Piccini P, Morrish PK, Turjanski N, Sawle GV, Burn DJ, Weeks RA, Mark MH, Maraganore DM, Lees AJ, Brooks DJ. Dopaminergic function in familial Parkinson's disease: a clinical and 18F-dopa positron emission tomography study. Ann Neurol. 1997;41:222–9. doi: 10.1002/ana.410410213. [DOI] [PubMed] [Google Scholar]
- 77.Piccini P, Burn DJ, Ceravolo R, Maraganore D, Brooks DJ. The role of inheritance in sporadic Parkinson's disease: evidence from a longitudinal study of dopaminergic function in twins. Ann Neurol. 1999;45:577–82. doi: 10.1002/1531-8249(199905)45:5<577::aid-ana5>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 78.Khan NL, Jain S, Lynch JM, Pavese N, Abou-Sleiman P, Holton JL, Healy DG, Gilks WP, Sweeney MG, Ganguly M, Gibbons V, Gandhi S, Vaughan J, Eunson LH, Katzenschlager R, Gayton J, Lennox G, Revesz T, Nicholl D, Bhatia KP, Quinn N, Brooks D, Lees AJ, Davis MB, Piccini P, Singleton AB, Wood NW. Mutations in the gene LRRK2 encoding dardarin (PARK8) cause familial Parkinson's disease: clinical, pathological, olfactory and functional imaging and genetic data. Brain. 2005;128:2786–96. doi: 10.1093/brain/awh667. [DOI] [PubMed] [Google Scholar]
- 79.Samii A, Markopoulou K, Wszolek ZK, Sossi V, Dobko T, Mak E, Calne DB, Stoessl AJ. PET studies of parkinsonism associated with mutation in the alpha-synuclein gene. Neurology. 1999;53:2097–102. doi: 10.1212/wnl.53.9.2097. [DOI] [PubMed] [Google Scholar]
- 80.Adams JR, van Netten H, Schulzer M, Mak E, Mckenzie J, Strongosky A, Sossi V, Ruth TJ, Lee CS, Farrer M, Gasser T, Uitti RJ, Calne DB, Wszolek ZK, Stoessl AJ. PET in LRRK2 mutations: comparison to sporadic Parkinson's disease and evidence for presymptomatic compensation. Brain. 2005;128:2777–85. doi: 10.1093/brain/awh607. [DOI] [PubMed] [Google Scholar]
- 81.Khan NL, Brooks DJ, Pavese N, Sweeney MG, Wood NW, Lees AJ, Piccini P. Progression of nigrostriatal dysfunction in a parkin kindred: an [18F]dopa PET and clinical study. Brain. 2002;125:2248–56. doi: 10.1093/brain/awf237. [DOI] [PubMed] [Google Scholar]
- 82.Scherfler C, Khan NL, Pavese N, Eunson L, Graham E, Lees AJ, Quinn NP, Wood NW, Brooks DJ, Piccini PP. Striatal and cortical preand postsynaptic dopaminergic dysfunction in sporadic parkin-linked parkinsonism. Brain. 2004;127:1332–42. doi: 10.1093/brain/awh150. [DOI] [PubMed] [Google Scholar]
- 83.Hernandez DG, Paisán-Ruíz C, McInerney-Leo A, Jain S, Meyer-Lindenberg A, Evans EW, Berman KF, Johnson J, Auburger G, Schäffer AA, Lopez GJ, Nussbaum RL, Singleton AB. Clinical and positron emission tomography of Parkinson's disease caused by LRRK2. Ann Neurol. 2005;57:453–456. doi: 10.1002/ana.20401. [DOI] [PubMed] [Google Scholar]
- 84.Hering R, Strauss KM, Tao X, Bauer A, Woitalla D, Mietz EM, Petrovic S, Bauer P, Schaible W, Müller T, Schöls L, Klein C, Berg D, Meyer PT, Schulz JB, Wollnik B, Tong L, Krüger R, Riess O. Novel homozygous p.E64D mutation in DJ1 in early onset Parkinson disease (PARK7) Hum Mutat. 2004;24:321–329. doi: 10.1002/humu.20089. [DOI] [PubMed] [Google Scholar]
- 85.Ribeiro MJ, Thobois S, Lohmann E, du Montcel ST, Lesage S, Pelissolo A, Dubois B, Mallet L, Pollak P, Agid Y, Broussolle E, Brice A, Remy P, French Parkinson's Disease Genetics Study Group A multitracer dopaminergic PET study of young-onset parkinsonian patients with and without parkin gene mutations. J Nucl Med. 2009;50:1244–50. doi: 10.2967/jnumed.109.063529. [DOI] [PubMed] [Google Scholar]
- 86.Pavese N, Khan NL, Scherfler C, Cohen L, Brooks DJ, Wood NW, Bhatia KP, Quinn NP, Lees AJ, Piccini P. Nigrostriatal dysfunction in homozygous and heterozygous parkin gene carriers: an 18F-dopa PET progression study. Mov Disord. 2009:2260–6. doi: 10.1002/mds.22817. [DOI] [PubMed] [Google Scholar]
- 87.Pavese N, Moore RY, Scherfler C, Khan NL, Hotton G, Quinn NP, Bhatia KP, Wood NW, Brooks DJ, Lees AJ, Piccini P. In vivo assessment of brain monoamine systems in parkin gene carriers: a PET study. Exp Neurol. 2010;222:120–4. doi: 10.1016/j.expneurol.2009.12.021. [DOI] [PubMed] [Google Scholar]
- 88.Scherfler C, Khan NL, Pavese N, Lees AJ, Quinn NP, Brooks DJ, Piccini PP. Upregulation of dopamine D2 receptors in dopaminergic drug-naive patients with Parkin gene mutations. Mov Disord. 2006;21:783–8. doi: 10.1002/mds.20811. [DOI] [PubMed] [Google Scholar]
- 89.Morrish PK, Rakshi JS, Bailey DL, Sawle GV, Brooks DJ. Measuring the rate of progression and estimating the preclinical period of Parkinson's disease with [18F]dopa PET. J Neurol Neurosurg Psychiatry. 1998;64:314–9. doi: 10.1136/jnnp.64.3.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rascol O, Brooks DJ, Korczyn AD, De Deyn PP, Clarke CE, Lang AE. A five-year study of the incidence of dyskinesia in patients with early Parkinson's disease who were treated with ropinirole or levodopa. 056 Study Group. N Engl J Med. 2000;342:1484–91. doi: 10.1056/NEJM200005183422004. [DOI] [PubMed] [Google Scholar]
- 91.de la Fuente-Fernández R, Pal PK, Vingerhoets FJ, Kishore A, Schulzer M, Mak EK, Ruth TJ, Snow BJ, Calne DB, Stoessl AJ. Evidence for impaired presynaptic dopamine function in parkinsonian patients with motor fluctuations. J Neural Transm. 2000;107:49–57. doi: 10.1007/s007020050004. [DOI] [PubMed] [Google Scholar]
- 92.Troiano AR, de la Fuente-Fernandez R, Sossi V, Schulzer M, Mak E, Ruth TJ, Stoessl AJ. PET demonstrates reduced dopamine transporter expression in PD with dyskinesias. Neurology. 2009;72:1211–6. doi: 10.1212/01.wnl.0000338631.73211.56. [DOI] [PubMed] [Google Scholar]
- 93.Kishore A, De la Fuente-Fernandez R, Snow BJ, Schulzer M, Mak E, Huser J, Stoessl AJ, Calne DB. Levodopa-induced dyskinesias in idiopathic Parkinsonism (IP): a simultaneous PET study of dopamine D1 and D2 receptors. Neurology. 1997;48:A327. [Google Scholar]
- 94.Torstenson R, Hartvig P, Långström B, Westerberg G, Tedroff J. Differential effects of levodopa on dopaminergic function in early and advanced Parkinson's disease. Ann Neurol. 1997;41:334–40. doi: 10.1002/ana.410410308. [DOI] [PubMed] [Google Scholar]
- 95.Whone AL, Rabiner EA, Arahata Y, Luthra SK, Hargreaves R, Brooks DJ. Reduced substance P binding in Parkinson's disease complicated by dyskinesias: an F-18-L829165 PET study. Neurology. 2002;58:A488–49. [Google Scholar]
- 96.Ramlackhansingh AF, Bose SK, Ahmed I, Turkheimer FE, Pavese N, Brooks DJ, Adenosine A. (2A) Receptor Availability in Parkinson's Disease Patients with and without Levodopa Induced Dyskinesias Studied with [C-11]SCH442416 PET. 62nd Annual Meeting of the American-Academy-of-Neurology. 2010;74:A588–A588. [Google Scholar]
- 97.Ahmed I, Bose SK, Pavese N, Ramlackhansingh A, Turkheimer F, Hotton G, Hammers A, Brooks DJ. Glutamate NMDA receptor dysregulation in Parkinson's disease with dyskinesias. Brain. 2011;134:979–86. doi: 10.1093/brain/awr028. [DOI] [PubMed] [Google Scholar]
- 98.Politis M, Wu K, Loane C, Kiferle L, Molloy S, Bain P, Brooks DJ, Piccini P. Serotonergic involvement in L-DOPA-induced dyskinesia [abstract] Mov Disord. 2010;5:S658. [Google Scholar]
- 99.Wenning GK, Odin P, Morrish P, Rehncrona S, Widner H, Brundin P, Rothwell JC, Brown R, Gustavii B, Hagell P, Jahanshahi M, Sawle G, Björklund A, Brooks DJ, Marsden CD, Quinn NP, Lindvall O. Short- and long-term survival and function of unilateral intrastriatal dopaminergic grafts in Parkinson's disease. Ann Neurol. 1997;42:95–107. doi: 10.1002/ana.410420115. [DOI] [PubMed] [Google Scholar]
- 100.Piccini P, Lindvall O, Björklund A, Brundin P, Hagell P, Ceravolo R, Oertel W, Quinn N, Samuel M, Rehncrona S, Widner H, Brooks DJ. Delayed recovery of movement-related cortical function in Parkinson's disease after striatal dopaminergic grafts. Ann Neurol. 2000;48:689–95. [PubMed] [Google Scholar]
- 101.Freed CR, Greene PE, Breeze RE, Tsai WY, DuMouchel W, Kao R, Dillon S, Winfield H, Culver S, Trojanowski JQ, Eidelberg D, Fahn S. Transplantation of embryonic dopamine neurons for severe Parkinson's disease. N Engl J Med. 2001;344:710–719. doi: 10.1056/NEJM200103083441002. [DOI] [PubMed] [Google Scholar]
- 102.Olanow CW, Goetz CG, Kordower JH, Stoessl AJ, Sossi V, Brin MF, Shannon KM, Nauert GM, Perl DP, Godbold J, Freeman TB. A doubleblind controlled trial of bilateral fetal nigral transplantation in Parkinson's disease. Ann Neurol. 2003;54:403–14. doi: 10.1002/ana.10720. [DOI] [PubMed] [Google Scholar]
- 103.Ma Y, Feigin A, Dhawan V, Fukuda M, Shi Q, Greene P, Breeze R, Fahn S, Freed C, Eidelberg D. Dyskinesia after fetal cell transplantation for Parkinsonism: a PET study. Ann Neurol. 2002;52:628–34. doi: 10.1002/ana.10359. [DOI] [PubMed] [Google Scholar]
- 104.Hagell P, Piccini P, Björklund A, Brundin P, Rehncrona S, Widner H, Crabb L, Pavese N, Oertel WH, Quinn N, Brooks DJ, Lindvall O. Dyskinesias following neural transplantation in Parkinson's disease. Nat Neurosci. 2002;5:627–8. doi: 10.1038/nn863. [DOI] [PubMed] [Google Scholar]
- 105.Piccini P, Pavese N, Hagell P, Reimer J, Björklund A, Oertel WH, Quinn NP, Brooks DJ, Lindvall O. Factors affecting the clinical outcome after neural transplantation in Parkinson's disease. Brain. 2005;128:2977–86. doi: 10.1093/brain/awh649. [DOI] [PubMed] [Google Scholar]
- 106.Lang AE, Langston JW, Stoessl AJ, Brodsky M, Brooks DJ, Dhawan V, Elias WJ, Lozano AM, Moro E, Nutt JG, Stacy M, Turner D, Wooten GF. GDNF in treatment of Parkinson's disease: response to editorial. Lancet Neurol. 2006;5:200–2. doi: 10.1016/S1474-4422(06)70359-3. [DOI] [PubMed] [Google Scholar]
- 107.Politis M, Wu K, Loane C, Quinn NP, Brooks DJ, Rehncrona S, Bjorklund A, Lindvall O, Piccini P. Serotonergic Neurons Mediate Dyskinesia Side Effects in Parkinson's Patients with Neural Transplants. Sci Transl Med. 2010;2:38–46. doi: 10.1126/scitranslmed.3000976. [DOI] [PubMed] [Google Scholar]
- 108.Hilker R, Razai N, Ghaemi M, Weisenbach S, Rudolf J, Szelies B, Heiss WD. [18F]fluorodopa uptake in the upper brainstem measured with positron emission tomography correlates with decreased REM sleep duration in early Parkinson's disease. Clin Neurol Neurosurg. 2003;105:262–9. doi: 10.1016/s0303-8467(03)00058-1. [DOI] [PubMed] [Google Scholar]
- 109.Herlofson K, Larsen JP. Measuring fatigue in patients with Parkinson's disease - the Fatigue Severity Scale. Eur J Neurol. 2002;9:595–600. doi: 10.1046/j.1468-1331.2002.00444.x. [DOI] [PubMed] [Google Scholar]
- 110.Boileau I, Warsh JJ, Guttman M, Saint-Cyr JA, McCluskey T, Rusjan P, Houle S, Wilson AA, Meyer JH, Kish SJ. Elevated serotonin transporter binding in depressed patients with Parkinson's disease: a preliminary PET study with [11C]DASB. Mov Disord. 2008;23:1776–80. doi: 10.1002/mds.22212. [DOI] [PubMed] [Google Scholar]
- 111.Politis M, Wu K, Loane C, Turkheimer FE, Molloy S, Brooks DJ, Piccini P. Depressive symptoms in PD correlate with higher 5-HTT binding in raphe and limbic structures. Neurology. 2010;75:1920–7. doi: 10.1212/WNL.0b013e3181feb2ab. [DOI] [PubMed] [Google Scholar]
- 112.Emre M. Dementia associated with Parkinson's disease. Lancet Neurol. 2003;2:229–237. doi: 10.1016/s1474-4422(03)00351-x. [DOI] [PubMed] [Google Scholar]
- 113.Ito K, Nagano-Saito A, Kato T, Arahata Y, Nakamura A, Kawasumi Y, Hatano K, Abe Y, Yamada T, Kachi T, Brooks DJ. Striatal and extrastriatal dysfunction in Parkinson's disease with dementia: a 6-[18F]fluoro-L-dopa PET study. Brain. 2002;125:1358–65. doi: 10.1093/brain/awf134. [DOI] [PubMed] [Google Scholar]
- 114.Goto I, Taniwaki T, Hosokawa S, Otsuka M, Ichiya Y, Ichimiya A. Positron emission tomographic (PET) studies in dementia. J Neurol Sci. 1993;114:1–6. doi: 10.1016/0022-510x(93)90040-6. [DOI] [PubMed] [Google Scholar]
- 115.Vander Borght T, Minoshima S, Giordani B, Foster NL, Frey KA, Berent S, Albin RL, Koeppe RA, Kuhl DE. Cerebral metabolic differences in Parkinson's and Alzheimer's diseases matched for dementia severity. J Nucl Med. 1997;38:797–802. [PubMed] [Google Scholar]
- 116.Yong SW, Yoon JK, An YS, Lee PH. A comparison of cerebral glucose metabolism in Parkinson's disease, Parkinson's disease dementia and dementia with Lewy bodies. Eur J Neurol. 2007;14:1357–62. doi: 10.1111/j.1468-1331.2007.01977.x. [DOI] [PubMed] [Google Scholar]
- 117.Hilker R, Thomas AV, Klein JC, Weisenbach S, Kalbe E, Burghaus L, Jacobs AH, Herholz K, Heiss WD. Dementia in Parkinson disease: functional imaging of cholinergic and dopaminergic pathways. Neurology. 2005;65:1716–1722. doi: 10.1212/01.wnl.0000191154.78131.f6. [DOI] [PubMed] [Google Scholar]
- 118.Edison P, Rowe CC, Rinne JO, Ng S, Ahmed I, Kemppainen N, Villemagne VL, O'Keefe G, Någren K, Chaudhury KR, Masters CL, Brooks DJ. Amyloid load in Parkinson's disease dementia and Lewy body dementia measured with [11C]PIB positron emission tomography. J Neurol Neurosurg Psychiatry. 2008;79:1331–1338. doi: 10.1136/jnnp.2007.127878. [DOI] [PubMed] [Google Scholar]
- 119.Ballanger B, Strafella AP, van Eimeren T, Zurowski M, Rusjan PM, Houle S, Fox SH. Serotonin 2A receptors and visual hallucinations in Parkinson disease. Arch Neurol. 2010;67:416–21. doi: 10.1001/archneurol.2010.35. [DOI] [PubMed] [Google Scholar]
- 120.Weintraub D, Koester J, Potenza MN, Siderowf AD, Stacy M, Voon V, Whetteckey J, Wunderlich GR, Lang AE. Impulse control disorders in Parkinson disease: a cross-sectional study of 3090 patients. Arch Neurol. 2010;67:589–95. doi: 10.1001/archneurol.2010.65. [DOI] [PubMed] [Google Scholar]
- 121.O'Sullivan SS, Evans AH, Lees AJ. Dopamine dysregulation syndrome: an overview of its epidemiology, mechanisms and management. CNS Drugs. 2009;23:157–70. doi: 10.2165/00023210-200923020-00005. [DOI] [PubMed] [Google Scholar]
- 122.Evans AH, Pavese N, Lawrence AD, Tai YF, Appel S, Doder M, Brooks DJ, Lees AJ, Piccini P. Compulsive drug use linked to sensitized ventral striatal dopamine transmission. Ann Neurol. 2006;59:852–8. doi: 10.1002/ana.20822. [DOI] [PubMed] [Google Scholar]
- 123.Berridge KC, Robinson TE, Aldridge JW. Dissecting components of reward: ‘liking’, ‘wanting’, and learning. Curr Opin Pharmacol. 2009;9:65–73. doi: 10.1016/j.coph.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Steeves TD, Miyasaki J, Zurowski M, Lang AE, Pellecchia G, Van Eimeren T, Rusjan P, Houle S, Strafella AP. Increased striatal dopamine release in Parkinsonian patients with pathological gambling: a [11C]raclopride PET study. Brain. 2009;132:1376–85. doi: 10.1093/brain/awp054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Van Eimeren T, Pellecchia G, Cilia R, Ballanger B, Steeves TD, Houle S, Miyasaki JM, Zurowski M, Lang AE, Strafella AP. Drug-induced deactivation of inhibitory networks predicts pathological gambling in PD. Neurology. 2010;75:1711–6. doi: 10.1212/WNL.0b013e3181fc27fa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Goldstein DS, Holmes C, Li ST, Bruce S, Metman LV, Cannon RO., III Cardiac sympathetic denervation in Parkinson disease. Ann Intern Med. 2000;133:338–347. doi: 10.7326/0003-4819-133-5-200009050-00009. [DOI] [PubMed] [Google Scholar]
- 127.Berding G, Schrader CH, Peschel T, van den Hoff J, Kolbe H, Meyer GJ, Dengler R, Knapp WH. [N-methyl 11C]meta-Hydroxyephedrine positron emission tomography in Parkinson's disease and multiple system atrophy. Eur J Nucl Med Mol Imaging. 2003;30:127–31. doi: 10.1007/s00259-002-1019-7. [DOI] [PubMed] [Google Scholar]
- 128.Kranick SM, Duda JE. Olfactory dysfunction in Parkinson's disease. Neurosignals. 2008;16:35–40. doi: 10.1159/000109757. [DOI] [PubMed] [Google Scholar]
- 129.Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol. Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 130.Hawkes CH, Del Tredici K, Braak H. Parkinson's disease: a dual-hit hypothesis, Neuropathol. Appl. Neurobiol. 2007;33:599–614. doi: 10.1111/j.1365-2990.2007.00874.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bohnen NI, Gedela S, Kuwabara H, Constantine GM, Mathis CA, Studenski SA, Moore RY. Selective hyposmia and nigrostriatal dopaminergic denervation in Parkinson's disease. J Neurol. 2007;254:84–90. doi: 10.1007/s00415-006-0284-y. [DOI] [PubMed] [Google Scholar]