

Abstract
Early intervention using hypothermia treatment has been shown to reduce early inflammation, apoptosis and infarct size in animal models of cardiac ischemia/reperfusion. We have shown that 5'-adenosine monophosphate (5'-AMP) can induce a reversible deep hypothermia in mammals. We hypothesize that 5'-AMP-induced hypothermia (AIH) may reduce ischemic/reperfusion damage following myocardial infarct. C57BL/6J male mice were subjected to myocardial ischemia by ligating the left anterior descending coronary artery (LAD) followed by reperfusion. Compared to euthermic controls, mice given AIH treatment exhibited significant inhibition of neutrophil infiltration and a reduction in matrix metallopeptidase 9 (MMP-9) expressions in the infarcted myocardium. A decrease in terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)-positive nuclei in the left ventricle myocardium were also observed. The overall infarct size of the heart was significantly smaller in AIH treated mice. Myocardial ischemia in mice given 5'-AMP without hypothermia had similar ischemia/reperfusion injuries as the euthermic control. Thus, the AIH cardio-protective effects were primarily hypothermia based.
Keywords: Hypothermia, 5'-adenosine monophosphate, ischemia/reperfusion injury, myocardial infarct, inflammation
Introduction
Myocardial infarction is the leading cause of premature death in the United States and in many developed countries. Current therapies for acute myocardial infarction (AMI) involve early reperfusion by thrombolysis and percutaneous coronary intervention. An important and active area in cardiovascular research is therapeutic hypothermia after AMI. Mild hypothermia in animal myocardial infarction model studies showed beneficial results [1-4]. Myocardial infarct size has been shown to be smaller in animals treated by mild hypothermia [2, 3, 5, 6]. In 2005, the Advanced Life Support Task Force of the International Liaison Committee on Resuscitation recommended therapeutic hypothermia treatment for out-of-hospital cardiac arrest when the initial rhythm was ventricular fibrillation [7-9]. However, the authors of these studies found that this post-resuscitation treatment using hypothermia is not widely practiced [10].
A major factor limiting the use of hypothermia as a therapeutic tool is the technical difficulty of cooling a patient. We believe AIH can overcome this obstacle and will aid the application of hypothermia therapy.
In our previous studies, we observed that mice given a high dosage of 5'-adenosine monophosphate (5'-AMP) enter a hypometabolic state that allows for relative rapid lowering of body temperature [11-13]. We showed that mice given 5'-AMP can have their core body temperature (Tb) safely reduced to 16-17°C when kept at an ambient temperature (Ta) of 15°C [12, 13]. Our studies show that AIH is preceded by hypome-tabolism and is linked to decreased affinity for oxygen by the erythrocytes caused by excessive 5'-AMP influx. During this transient period of hypometabolism the Tb can be reduced rapidly without encountering a severe thermo-regulatory defense. We observed that the length of AIH is regulated in part by the decline in Tb, which is in turn mediated by Ta [12]. Mice remain in this deep hypothermia state for several hours if the ambient temperature is maintained at about 15°C. The aim of this study is to investigate whether such deep hypothermia induced by 5'-AMP reduces inflammation response, apoptosis and infarct size in a cardiac ischemia/reperfusion model. AMI was generated by temporary ligation of the left anterior descending coronary artery (LAD) followed by 24 h reperfusion. Here we show that AIH can have a cardio-protective effect in mice that have been subjected to myocardial ischemia. The heart tissue of AIH treated mice displayed a reduction in molecular and cellular indicators of inflammatory response, apoptosis and tissue repair. In addition the AIH treated mice also displayed smaller infarct size after AMI. Thus, our studies indicate that AIH has potential utility to limit cellular damage after ischemic assault.
Materials and methods
Animal model
Male mice (C57BL/6J) between 10-12 weeks, purchased from Jackson Laboratories, were used in these studies. Adenosine receptor knock-out mice were generously provided by Dr. Michael Blackburn [12]. Mice were housed in standard husbandry care in a 12h:12h dark-light cycle with ad labitum access to food and water. These studies were carried out under IACUC approved protocol AWC-07-011.
Induction of deep hypothermia and experimental groups
Adenosine 5'-monophosphate was obtained from Sigma (St Louis, MO, USA). A freshly prepared 5'-AMP solution in phosphate buffered saline (PBS) was injected intraperitoneally (IP) at a dosage of 0.5 mg/g body weight. The Tb was measured using a digital thermometer (Fisher Scientific, Pittsburgh, PA) with the probe placed about 1 cm into the anus. Electrocardiograms were recorded for all experimental animals, from the beginning till the end of all experiments.
Mice after AMI were randomized into 4 experimental groups (n=8): 1) normothermia: control group with Tb at 37°C. 2) normothermia + 5'-AMP: at the end of ischemia, 5'-AMP was administrated but the Tb of mice were kept at euthermia by maintaining Ta between 33-34°C for 3 h. 3) post-ischemia-hypothermia: 5'-AMP was administered at the end of ischemia and the mice were kept at 15°C Ta. 4) pre-reperfusion-hypothermia: 5'-AMP was injected 20 min before reperfusion and at the end of ischemia, the animals were maintained at 15°C Ta. After 6 h at 15°C, all AIH treated mice were returned to normal husbandry care. Mice in the AIH group returned to euthermia after 2-3 h in standard husbandry care environment.
Closed chest ischemia/reperfusion protocol
A closed-chest ischemia/reperfusion protocol was carried out as described previously [14]. Briefly, mice were anesthetized and ventilated with 2% isoflurane and 98% oxygen provided to the inflow of the ventilator (Vaporize Sales & Service Inc, Rockmart, GA; Harvard Apparatus, Harvard Model 683 Small Animal Ventilator, Holliston, MA). A left thoracotomy was created via the fourth intercostal space; the left lung was pulled back and the pericardium was then dissected to expose the heart. The two ends of the 8-0 suture were passed under the LAD and threaded through a 0.5-mm piece of PT-10 tubing. These ends were placed under the skin and the chest was closed. Antibiotics and Buprenor-phine HCl were given for 2 days post-surgery. After one week, mice were re-anesthetized and the skin in the chest was reopened. The two ends of the suture under the skin were taped to metal picks. The metal picks were carefully pulled apart until an obvious ST segment elevation was observed on the electrocardiogram. After 45 minutes of coronary occlusion, reperfusion was restored by releasing the ligation.
Tissue preparation and immunohistochemical staining
Mice were sacrificed at 24 h after the start of reperfusion. The hearts were dissected, and the left ventricles were sectioned transversely into 3 slices, fixed in 4% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA) and then embedded in paraffin. Paraffin sections (5 μm) were deparaffinized in histo-clear (National Diagnostics, Atlanta, GA) and then rehydrated. Sections were pre-incubated with 0.3% hydrogen peroxide in PBS to inhibit endogenous per-oxidase activity, washed twice in PBS, pre-incubated with blocking serum (5% normal serum) for 1 h, and then incubated at 4°C over-night with either rat anti-mouse neutrophil antibody (1:500 dilution; Serotec, Raleigh, NC) or goat anti-mouse MMP-9 antibody (1:100 dilution; R&D Systems, Inc. Minneapolis, MN). After washing 3 times with PBS, samples were incubated with biotinylated secondary antibodies for 1 h at room temperature, followed by addition of ABC reagent (Vector Laboratories, Burlin-game, CA). Then the sections were treated with SIGMA FAST 3, 3 - Diaminobenzidine (Sigma, St Louis, MO) and slides were mounted in Cytoseal XYL (Richard-Allan Scientific, Kalamazoo, MI). Control sections were incubated with PBS in place of the primary antibody. From each slice, 2 fields close to the infarct border were photographed at 100x magnification (Olympus microscopy BX60m, Olympus DP71). Quantitative evaluation of neutrophil or MMP-9 levels was performed using the Image-Pro plus software (Media Cybernetics, Silver Spring, MD, USA).
Apoptosis assay
The TUNEL assay was carried out according the manufacturer's instructions (Roche, Germany). Briefly, sections were de-waxed, rehydrated and then incubated with 20 μg/ml Proteinase K at 37°C for 30 min followed by 3 washes with PBS. Each slide was incubated with 50 μl TUNEL reaction mixture at 37°C for 1 h, followed by 3 washes with PBS. Slides were mounted with SlowFade Gold reagent with DAPI (Invitrogen Corp. Carlsbad, CA). From each slice, 2 fields close to the infarct border were photographed at 100x magnification (Olympus microscopy BX60m, Olympus DP71). Apoptotic cells were quantitatively evaluated using Image-Pro software (Media Cybernetics, Silver Spring, MD, USA).
Infarct size assessment
To measure the infarct size, 1% Evan's blue was infused into the aorta and washed with cardioplegic solution. The left ventricle was sliced transversally into five 1-mm-thick sections and incubated in a 1% solution of 2,3,5-triphenyltetrazolium chloride (TTC; Sigma-Aldrich, St Louis, MO) for 30 minutes at 37°C. After fixation in a 4% paraformaldehyde-phosphate-buffered saline solution, the slices were weighed, and each side was photographed. The infarct size and area at risk includes the whitish and pinkish areas of each tissue section, respectively. The ischemic risk area and the infarcted area which is unstained by TTC were quantified with the use of Image J (Scion Corp, Frederick, MD). Infarct size was expressed as a percentage of the ischemic risk area.
Q-PCR quantification of MMP9 expression from paraffin embedded tissues
mRNA was recovered from tissue sections of the same paraffin embedded tissue blocks used for MMP9 immunohistochemical staining, using the RecoverAII™ Total Nucleic Acid Isolation Kit for FFPE kit (Applied Biosystems/Ambion, Austin, TX, USA). First strand cDNA was synthesized using Superscript™ III Reverse Transcriptase from Invitrogen (Carlsbad, CA, USA) with 200 ng of total RNA for each sample in a 20 μl volume. Due to low levels of cDNA from recovered mRNA, an enrichment PCR of 33 cycles was carried out using 1 μl of each cDNA synthesis mix. Then each q-PCR reaction was set up with 2 μl of the enhancement PCR and the respective primer set using the Maxima SYBR Green qPCR reaction kit (Fermentas Life Sciences, Glen Burnie, MA, USA). A Stratagene MX3000p was used for running all qPCRs simultaneously, and MxPro software was used for data acquisition, tracking and preliminary analysis. A quantitative control reference, GAPDH, was included and processed in parallel. The relative amount of each transcript was determined using the ΔΔCt method [15].
Data analysis
All data were expressed as means ± SEM. Data were analyzed by GraphPad Prism software (GraphPad Software, San Diego CA) using oneway AVOVA for multiple group comparison tests.
Results
Reduction in heart rate during AIH treatment
Reduction of the work load of an injured heart would have a protective effect on its tissue due to lower metabolic demand [16]. Our studies revealed that AIH treatment resulted in a rapid decline in heart rate of wild type mice following administration of 5'-AMP via IP. The heart rates of anesthetized mice dropped from 550 beats/ min to about 200 beats/min within minutes after 5'-AMP administration (Figure 1A). However, mice deficient in the A1 receptor did not display this bradycardiac response to 5'-AMP. The heart rates of mice deficient in adenosine receptor subtypes A2a, A2b and A3 were similar to wild type mice in response to 5'-AMP injection. The Tb of both wild type and A1 receptor deficient mice declined in a similar fashion under the same Ta conditions (Figure 1B). These observations indicate that the rapid slowing of heart rate is not a prerequisite for mice to enter hypometabolism and hypothermia. Wild type mice in deep hypometabolism, with a Tb of 16°C, exhibit a heart rate of about 10 % that of euthermic state (Figure 1C, 1D). We conclude that this bradycardia response is mediated by the A1 receptor. We hypothesize that the bradycardia response and the accompanying hypothermia induced by AIH may be beneficial in alleviating metabolic stress of an injured heart. Therefore, a study was undertaken to examine such a possibility using a mouse model for AMI.
Figure 1.

Rapid bradycardia response to 5'-AMP is mediated by the adenosine A1 receptor. A) Heart rates of anesthesized mice with various adenosine receptor genetic deficiencies following administration of 5'-AMP. B) Corresponding core body temperature (Tb) of mice shown in (A) maintained at a Ta of 23°C. C) A representative heart rate of an anesthesized wild type mouse during the normothermic state. D) A representative heart rate of an AIH treated wild type mouse with a Tb of 16°C.
AIH decreases levels of neutrophil infiltration in the infarcted heart
Ischemia/reperfusion damage is typically associated with increased inflammation. The level of neutrophil infiltration in the infarct area is a good indicator of inflammatory response. Therefore, immuno-histochemical staining using mouse neutrophil specific antibodies was carried out with the four experimental groups described in Methods. The results demonstrated that there were different levels of neutrophil infiltration into heart tissue at 24 h after AMI among the four experimental groups (Figure 2A). Neutrophil infiltration in the AIH treated post-ischemia group was significantly reduced to about 65% of the normothermia control (Figure 2B). An even greater reduction of neutrophil infiltration was found in the pre-reperfusion AIH treated group, neutrophil infiltration of about 38% of the normothermia group was observed (Figure 2B). Differences between the normothermia group and the AIH treatment group were statistically significant. Group 2 mice that were given 5'-AMP but maintained at a Tb in the euthermia range showed a similar level of neutrophil infiltration to the control nor-mothermia group. This observation suggests that it is the reduction of Tb rather than 5'-AMP itself that controls the ischemia induced neutrophil response.
Figure 2.

AIH reduces neutro-phil infiltration in the infarcted area. (A) Representative immu-nocytochemical stain of neutro-phils in the infarct area at 24 h post-reperfusion in hearts from the respective experimental groups (magnification, x100). (B) Quantification of the number of neutrophil-positive staining cells for each experimental group. Data are presented as mean cell numbers per squared millimeter ± SEM; n = 8 for each group. #, P ≤ 0.01 compared with the normothermic group.
AIH reduces MMP-9 expression in the infarcted heart
Matrix metalloproteinase 9 (MMP-9) belongs to a family of collagenases that are released by cardiac cells following tissue injury. Therefore, the level of MMP-9 is a good indicator of ischemic tissue damage and remodeling [17, 18]. Similar to neutrophil infiltration, MMP-9 was highly up regulated in the control normothermic group and in group 2 mice, which were given 5'-AMP but Tb maintained at normothermia (Figure 3A). The level of MMP-9 in the post-ischemia AIH group was reduced to 54% (P<0.01) of the level in the control normothermia group (Figure 3B). A greater MMP-9 reduction was observed in the pre-reperfusion AIH group, which exhibited a level of MMP-9 18% (p<0.01) of that observed in the control normothermia group. To confirm that MMP-9 levels were elevated in group 1 and group 2, qPCR quantification of mRNA extracted from paraffin-embedded tissue samples was carried out. The qPCR studies demonstrate that the mean levels of MMP-9 mRNA was at least 2-3 fold higher in samples from group 1 and group 2 than those from group 3 and group 4 (Figure 3C). These findings provide further evidence that AIH treatment has a cardiac protective effect after myocardial infarction.
Figure 3.

AIH reduces MMP-9 expression in the infarcted area. (A) A representative immunocytochemical stain is shown for each experimental group. Samples were analyzed at 24 h post-reperfusion after 45 minutes LAD ischemia (magnification, x100). (B) Quantification of MMP-9 positive-stainingceiis in each of the experimental groups. Data are presented as mean cell numbers per squared millimeter ± SEM; n = 8 for each group. #, P < 0.01 compared with the normothermia group. Error bars indicate SEM, (n=8). (C). MMP-9 levels measured by qPCR analysis. Internal control was GAPDH transcript level. Error bars indicate SEM, (n=5).
AIH decreases apoptosis in the infarcted heart
Cell death by apoptosis is another indicator of ischemic damage since apoptosis is initiated shortly after AMI [19]. Hearts from mice in post-ischemia and pre-reperfusion groups displayed a significant reduction of TUNEL positive nuclei compared with the normothermia group 1 (Figure 4A). TUNEL positive nuclei in hearts of the post-ischemia group 3 animals were reduced to 75% of levels observed for the normothermia group 1. In pre-reperfusion group 4 hearts, the TUNEL positive nuclei were 32% of the normothermic group 1 levels (Figure 4B). Again, no difference in apoptotic levels was observed between hearts from group 2 and group 1 mice. Overall, these findings indicate that there was less apoptosis in hearts from mice that were treated with hypothermia than in the control normothermic group.
Figure 4.

AIH decreases apoptosis in the infarct area. (A) A representative immuno-cytochemical stain is shown for samples from each group at 24 h post-reperfusion after LAD ischemia (magnification, x100). The nuclei are stained by DAPI (blue) and the apoptotic cells are displayed using fluorescently modified nucleotide (pink). (B) Quantification of TUNEL positive-staining cells in each of the respective experimental groups. Data are presented as mean cell numbers per square millimeter ± SEM; n = 8 for each group. #, P < 0.01 compared with the normothermia group.
AIH reduces infarct size in the ischemia/reperfusion mouse heart
Another good indicator of ischemic damage is the infarct size of the heart after reperfusion. The average infarct size/area at risk ratio was determined at 24 hours post-reperfusion by TTC staining in all four experimental groups. The infarct size/area at risk of the 5'-AMP group 2 maintained at about normothermic Tb was 32.0%. This value is similar to a value of 33.8% observed for the normothermic control group 1. However, the average infarct size/area at risk dropped to 19.4% and 9.5% in the AIH post-ischemia group 3 and pre-reperfusion group 4, respectively (Figure 5). The difference in infarct size between the nor-mothermic and AIH treatment groups was highly significant (P<0.01). Therefore, our studies demonstrate that AIH treatment reduces the infarct size area in an AMI experimental model.
Figure 5.
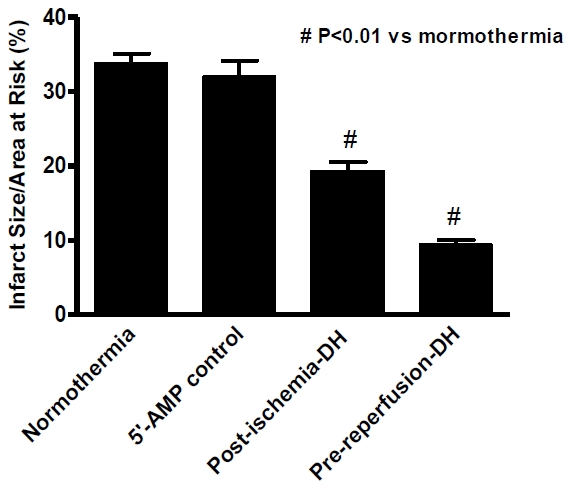
Average infarct size/area at risk 24 hours post- reperfusion after 45 min LAD ischemia. Data are presented as mean infarct size/area at risk ± SEM; n = 8 for each group. P < 0.01 compared with the normothermia group.
Discussion
Our previous studies have shown that mice given a high dosage of 5'-AMP enter a transient hypometabolic state [12, 13]. Under appropriate Ta, the Tb of mice in hypometabolic state can be safely reduced to about 15-16°C (Figure 6). The reduction in the animal's Tb can be regulated by controlling Ta. For example, mice given 5'-AMP but kept at a Ta of 33°C, such as group 2, can maintain their Tb at euthermic range (Figure 7). It has been shown that hypothermia can reduce ischemia and reperfusion damage [2, 3, 5, 6]. Therefore, we undertook this study to investigate whether AIH has a similar therapeutic value for AMI induced ischemia and reperfusion injuries. Our studies demonstrate that AIH treatment has a cardioprotective effect after myocardial infarct by the following criteria: 1) a significant inhibition of neutrophil infiltration into the infarcted myocardium; 2) a significant reduction of MMP-9 expression in the infarcted area; 3) a significant decrease in TUNEL positive nuclei in the left ventricle myocardium; and 4) a significant reduction of infarct size.
Figure 6.

The core body temperature measured by telemetry of mice given saline or 0.5g/gw of 5'-AMP and maintained at an ambient temperature of 15°C.
Figure 7.

The core body temperature measured by telemetry of mice given saline or 0.5g/gw of 5'-AMP and maintained at an ambient temperature of 33°C.
Following myocardial ischemia/reperfusion injury, an early inflammatory reaction is triggered. This is reflected by neutrophil infiltration into the damaged tissue to clear apoptotic cells and matrix debris, resulting in healing and replacement of damaged tissue by scar tissue. Studies have suggested that myocardial injury caused by coronary artery occlusion followed by reperfusion is neutrophil-dependent [20, 21]. One of the crucial mediators of cardiac damage during the reperfusion phase is the level of neutrophil infiltration [22]. One of the strategies to decrease myocardial injury is the use of anti-neutrophil treatments, including anti-neutrophil antibodies and anti-neutrophil drugs [23-28]. Consistent with previous observations, we observed a large increase in neutrophil infiltration in the infarcted area after 24 h of reperfusion. By contrast, the number of infiltrated neutro-phils was significantly decreased in hearts of mice from the AIH treatment groups. Neutrophil reduction was further enhanced in the pre-reperfusion group suggesting that the earlier the Tb can be reduced the better the outcome after ischemic damage. However, randomized clinical trials of anti-inflammatory agents in AMI patients have proven disappointing [29]. One reason for the poor outcome is that the inflammation cascade triggered by AMI involves many types of chemokines, and cytokines. Systemic hypothermia induced by AIH treatment might provide a better approach to inhibit inflammation by reducing a broad array of inflammatory responses. The observations from group 3 mice suggest that the inflammation response following reperfusion can be reduced by prolonged hypothermia following ischemia. How prolonged hypothermia reduces the inflammatory response to ischemia following reperfusion is not clear. Our recent studies suggest that over 99% of genes assayed by microarray analysis did not change their expression during deep hypothermia [13]. These observations would argue that changes in gene expression did not explain the reduced inflammatory response. It is has been shown that cardiac reperfusion damage is linked to a rapid built up of reactive oxygen species following ischemia damage [30]. Our recent studies have demonstrated that AIH is associated with decreased oxygen affinity by erythro-cytes due to increased levels of 2,3-bisphophoglycerate acting to enhances deoxy-hemoglobin levels [12]. We speculate that the decreased oxygen levels during deep hypothermia modulated the rapid built up of reactive oxygen species that could cause additional cellular damage following reperfusion. In addition, since AMP administration causes a global suppression of metabolism, we expect that mobility of neutrophils would be suppressed metaboli-cally as well as by hypothermia. Although AIH may suppress the acute phase of the inflammation response, a residual inflammation response is observed when hypometabolic animals were returned to normothemia.
Activation of MMPs plays an important role during the early stage of the healing process after myocardial ischemia/reperfusion injury. It has been shown that activated MMP-9 is co-localized with infiltrating neutrophils after myocardial ischemia/reperfusion [31, 32]. Ducharme et al found that the loss of the MMP-9 gene attenuated left ventricular dilation and collagen accumulation after myocardial infarction in mice [33]. The administration of MMP-inhibitors in animal models has been shown to attenuate left ventricle dilation and infarct expansion [34]. Our studies suggest that AIH treatment attenuates ischemia/reperfusion injury by reducing MMP-9 expression. Myocardial apop-tosis is typically initiated shortly after the onset of ischemia and is greatly enhanced during reperfusion [35, 36]. Reperfusion injury was attenuated by inhibition of apoptosis in dogs [37], mice and rats [38]. Hypothermia protects against endothelial cell apoptosis induced by ischemia/reperfusion by inhibiting both extrinsic and intrinsic signaling pathways in vitro [39]. Our studies show that cardiomyocyte apoptosis after ischemia/reperfusion in mice could be largely prevented by AIH treatment.
Reduction of the workload of an injured heart is thought to be beneficial after AMI [16]. Previous studies showed that extracellular 5'-ectonucleotidase/CD73 rapidly dephophory-lates 5'-AMP to adenosine, which induces bradycardia via the A1 adenosine receptor of the heart tissue [40]. Consistent with these findings, our studies demonstrate that the heart rate of adenosine A1 receptor deficient mice does not slow rapidly in response to 5'-AMP. However, adenosine A1 deficient mice can still attain hypothermia in a manner similar to wild type mice indicating that the bradycardia effect is independent of the hypometabolic response. In addition, AMI studies using CD73-/- mice demonstrated that AIH treatment resulted in similar outcome to that of wild type mice (data not shown). Thus, our studies suggest that the observed cardio-protective effects of AIH are not driven by dephosphorylation of 5'-AMP into adenosine but rather from the induction of hypothermia.
Mild hypothermia treatment has been shown to reduce myocardial infarct size in different animal models [2, 3, 5, 6]. Our studies demonstrate that infarct size in mice was significantly reduced by AIH treatment. Mice from the pre-reperfusion group gained greater protection against all measures of myocardial damage tested indicating that early intervention by AIH is highly beneficial. However, a recent study in rat using 5'-AMP to reduce Tb to about 33-34°C for 2.5 h did not show a beneficial outcome with a cerebral ischemia model [41]. One possible explanation is that 2.5 h of hypothermia may be too short a time to have significant impact on ischemic damage. In contrast our studies used at least 6 h. Another possible explanation is that mammals differ in their preference for the torpor state depending on their metabolic rate. Large mammals such as bears technically do not hibernate but instead undergo torpor at a Tb of about 32°C while small rodents can hibernate at Tb of less than 10°C [42]. Thus, we surmise that small rodents, such as mice and rats, with high metabolic rates need to be cooled much lower than 34°C to prevent ischemic damage.
Using an animal model of cardiac ischemia/reperfusion, we have shown that 5'-AMP induce a reversible deep hypothermia that reduces ischemia/reperfusion damage following myocardial infarct. The AIH cardio-protective effects were primarily a result of hypothermia.
Acknowledgments
This work was supported in part by NIH Pioneer grant to CCL. We thank Dr J. Lever for helpful suggestion and comments in the preparation of the manuscript. We thank Tomoko Miki, for her technical assistance for Figure 3C.
Conflict of Interest
The authors have neither financial nor personal gains with regard to the findings of this study.
References
- 1.Hale SL, Dave RH, Kloner RA. Regional hypothermia reduces myocardial necrosis even when instituted after the onset of ischemia. Basic Res Cardiol. 1997;92:351–357. doi: 10.1007/BF00788947. [DOI] [PubMed] [Google Scholar]
- 2.Duncker DJ, Klassen CL, Ishibashi Y, Herrlinger SH, Pavek TJ, Bache RJ. Effect of temperature on myocardial infarction in swine. Am J Physiol. 1996;270:H1189–1199. doi: 10.1152/ajpheart.1996.270.4.H1189. [DOI] [PubMed] [Google Scholar]
- 3.Dae MW, Gao DW, Sessler DI, Chair K, Stillson CA. Effect of endovascular cooling on myocardial temperature, infarct size, and cardiac output in human-sized pigs. Am J Physiol Heart Circ Physiol. 2002;282:H1584–1591. doi: 10.1152/ajpheart.00980.2001. [DOI] [PubMed] [Google Scholar]
- 4.Hale SL, Kloner RA. Myocardial temperature in acute myocardial infarction: protection with mild regional hypothermia. Am J Physiol. 1997;273:H220–227. doi: 10.1152/ajpheart.1997.273.1.H220. [DOI] [PubMed] [Google Scholar]
- 5.Miki T, Liu GS, Cohen MV, Downey JM. Mild hypothermia reduces infarct size in the beating rabbit heart: a practical intervention for acute myocardial infarction? Basic Res Cardiol. 1998;93:372–383. doi: 10.1007/s003950050105. [DOI] [PubMed] [Google Scholar]
- 6.Schwartz LM, Verbinski SG, Vander Heide RS, Reimer KA. Epicardial temperature is a major predictor of myocardial infarct size in dogs. J Mol Cell Cardiol. 1997;29:1577–1583. doi: 10.1006/jmcc.1997.0391. [DOI] [PubMed] [Google Scholar]
- 7.Safar PJ, Kochanek PM. Therapeutic hypothermia after cardiac arrest. N Engl J Med. 2002;346:612–613. doi: 10.1056/NEJM200202213460811. [DOI] [PubMed] [Google Scholar]
- 8.Nolan JP, Morley PT, Vanden Hoek TL, Hickey RW, Kloeck WG, Billi J, Bottiger BW, Morley PT, Nolan JP, Okada K, Reyes C, Shuster M, Steen PA, Weil MH, Wenzel V, Hickey RW, Carli P, Vanden Hoek TL, Atkins D. Therapeutic hypothermia after cardiac arrest: an advisory statement by the advanced life support task force of the International Liaison Committee on Resuscitation. Circulation. 2003;108:118–121. doi: 10.1161/01.CIR.0000079019.02601.90. [DOI] [PubMed] [Google Scholar]
- 9.Nolan JP, Hazinski MF, Steen PA, Becker LB. Controversial Topics from the 2005 International Consensus Conference on cardiopulmon-ary resuscitation and emergency cardiovascular care science with treatment recommendations. Resuscitation. 2005;67:175–179. doi: 10.1016/j.resuscitation.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 10.Merchant RM, Soar J, Skrifvars MB, Silfvast T, Edelson DP, Ahmad F, Huang KN, Khan M, Vanden Hoek TL, Becker LB, Abella BS. Therapeutic hypothermia utilization among physicians after resuscitation from cardiac arrest. Crit Care Med. 2006;34:1935–1940. doi: 10.1097/01.CCM.0000220494.90290.92. [DOI] [PubMed] [Google Scholar]
- 11.Zhang J, Kaasik K, Blackburn MR, Lee CC. Constant darkness is a circadian metabolic signal in mammals. Nature. 2006;439:340–343. doi: 10.1038/nature04368. [DOI] [PubMed] [Google Scholar]
- 12.Daniels IS, Zhang J, O'Brien WG, 3rd, Tao Z, Miki T, Zhao Z, Blackburn MR, Lee CC. A role of erythrocytes in adenosine monophos-phate initiation of hypometabolism in mammals. J Biol Chem. 2011;285:20716–20723. doi: 10.1074/jbc.M109.090845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao Z, Miki T, Van Oort-Jansen A, Matsumoto T, Loose DS, Lee CC. Hepatic gene expression profiling of 5'-AMP-induced hypometabolism in mice. Physiol Genomics. 2011;43:325–345. doi: 10.1152/physiolgenomics.00174.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nossuli TO, Lakshminarayanan V, Baumgarten G, Taffet GE, Ballantyne CM, Michael LH, Entman ML. A chronic mouse model of myocardial ischemia-reperfusion: essential in cytokine studies. Am J Physiol Heart Circ Physiol. 2000;278:H1049–1055. doi: 10.1152/ajpheart.2000.278.4.H1049. [DOI] [PubMed] [Google Scholar]
- 15.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 16.Neubauer S. The failing heart-an engine out of fuel. N Engl J Med. 2007;356:1140–1151. doi: 10.1056/NEJMra063052. [DOI] [PubMed] [Google Scholar]
- 17.Hlatky MA, Ashley E, Quertermous T, Boothroyd DB, Ridker P, Southwick A, Myers RM, Iribarren C, Fortmann SP, Go AS. Matrix metallopro-teinase circulating levels, genetic polymorphisms, and susceptibility to acute myocardial infarction among patients with coronary artery disease. Am Heart J. 2007;154:1043–1051. doi: 10.1016/j.ahj.2007.06.042. [DOI] [PubMed] [Google Scholar]
- 18.Romanic AM, Harrison SM, Bao W, Burns-Kurtis CL, Pickering S, Gu J, Grau E, Mao J, Sathe GM, Ohlstein EH, Yue TL. Myocardial protection from ischemia/reperfusion injury by targeted deletion of matrix metalloproteinase-9. Cardio-vasc Res. 2002;54:549–558. doi: 10.1016/s0008-6363(02)00254-7. [DOI] [PubMed] [Google Scholar]
- 19.Freude B, Masters TN, Robicsek F, Fokin A, Kostin S, Zimmermann R, Ullmann C, Lorenz-Meyer S, Schaper J. Apoptosis is initiated by myocardial ischemia and executed during reperfusion. J Mol Cell Cardiol. 2000;32:197–208. doi: 10.1006/jmcc.1999.1066. [DOI] [PubMed] [Google Scholar]
- 20.Jolly SR, Kane WJ, Hook BG, Abrams GD, Kunkel SL, Lucchesi BR. Reduction of myocardial infarct size by neutrophil depletion: effect of duration of occlusion. Am Heart J. 1986;112:682–690. doi: 10.1016/0002-8703(86)90461-8. [DOI] [PubMed] [Google Scholar]
- 21.Lift MR, Jeremy RW, Weisman HF, Winkelstein JA, Becker LC. Neutrophil depletion limited to reperfusion reduces myocardial infarct size after 90 minutes of ischemia. Evidence for neutrophil-mediated reperfusion injury. Circulation. 1989;80:1816–1827. doi: 10.1161/01.cir.80.6.1816. [DOI] [PubMed] [Google Scholar]
- 22.Vinten-Johansen J. Involvement of neutrophils in the pathogenesis of lethal myocardial reperfusion injury. Cardiovasc Res. 2004;61:481–497. doi: 10.1016/j.cardiores.2003.10.011. [DOI] [PubMed] [Google Scholar]
- 23.Simpson PJ, Todd RF, 3rd, Fantone JC, Mickelson JK, Griffin JD, Lucchesi BR. Reduction of experimental canine myocardial reperfusion injury by a monoclonal antibody (anti-Mol, anti-CDllb) that inhibits leukocyte adhesion. J Clin Invest. 1988;81:624–629. doi: 10.1172/JCI113364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gomoll AW, Lekich RF, Grove RI. Efficacy of a monoclonal antibody (MoAb 60.3) in reducing myocardial injury resulting from ischemia/reperfusion in the ferret. J Cardiovasc Pharmacol. 1991;17:873–878. doi: 10.1097/00005344-199106000-00003. [DOI] [PubMed] [Google Scholar]
- 25.Kohtani T, Abe Y, Sato M, Miyauchi K, Kawachi K. Protective effects of anti-neutrophil antibody against myocardial ischemia/reperfusion injury in rats. Eur Surg Res. 2002;34:313–320. doi: 10.1159/000063073. [DOI] [PubMed] [Google Scholar]
- 26.Mullane KM, Read N, Salmon JA, Moncada S. Role of leukocytes in acute myocardial infarction in anesthetized dogs: relationship to myocardial salvage by anti-inflammatory drugs. J Pharmacol Exp Ther. 1984;228:510–522. [PubMed] [Google Scholar]
- 27.Engler RL, Dahlgren MD, Morris DD, Peterson MA, Schmid-Schonbein GW. Role of leukocytes in response to acute myocardial ischemia and reflow in dogs. Am J Physiol. 1986;251:H314–323. doi: 10.1152/ajpheart.1986.251.2.H314. [DOI] [PubMed] [Google Scholar]
- 28.Romson JL, Hook BG, Kunkel SL, Abrams GD, Schork MA, Lucchesi BR. Reduction of the extent of ischemic myocardial injury by neutrophil depletion in the dog. Circulation. 1983;67:1016–1023. doi: 10.1161/01.cir.67.5.1016. [DOI] [PubMed] [Google Scholar]
- 29.Dirksen MT, Laarman GJ, Simoons ML, Duncker DJ. Reperfusion injury in humans: a review of clinical trials on reperfusion injury inhibitory strategies. Cardiovasc Res. 2007;74:343–355. doi: 10.1016/j.cardiores.2007.01.014. [DOI] [PubMed] [Google Scholar]
- 30.Dart RC, Sanders AB. Oxygen free radicals and myocardial reperfusion injury. Ann Emerg Med. 1988;17:53–58. doi: 10.1016/s0196-0644(88)80504-3. [DOI] [PubMed] [Google Scholar]
- 31.Lindsey M, Wedin K, Brown MD, Keller C, Evans AJ, Smolen J, Burns AR, Rossen RD, Michael L, Entman M. Matrix-dependent mechanism of neutrophil-mediated release and activation of matrix metalloproteinase 9 in myocardial ischemia/reperfusion. Circulation. 2001;103:2181–2187. doi: 10.1161/01.cir.103.17.2181. [DOI] [PubMed] [Google Scholar]
- 32.Tao ZY, Cavasin MA, Yang F, Liu YH, Yang XP. Temporal changes in matrix metalloproteinase expression and inflammatory response associated with cardiac rupture after myocardial infarction in mice. Life Sci. 2004;74:1561–1572. doi: 10.1016/j.lfs.2003.09.042. [DOI] [PubMed] [Google Scholar]
- 33.Ducharme A, Frantz S, Aikawa M, Rabkin E, Lindsey M, Rohde LE, Schoen FJ, Kelly RA, Werb Z, Libby P, Lee RT. Targeted deletion of matrix metalloproteinase-9 attenuates left ventricular enlargement and collagen accumulation after experimental myocardial infarction. J Clin Invest. 2000;106:55–62. doi: 10.1172/JCI8768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mukherjee R, Brinsa TA, Dowdy KB, Scott AA, Baskin JM, Deschamps AM, Lowry AS, Escobar GP, Lucas DG, Yarbrough WM, Zile MR, Spinale FG. Myocardial infarct expansion and matrix metalloproteinase inhibition. Circulation. 2003;107:618–625. doi: 10.1161/01.cir.0000046449.36178.00. [DOI] [PubMed] [Google Scholar]
- 35.Zhao ZQ, Nakamura M, Wang NP, Wilcox JN, Shearer S, Ronson RS, Guyton RA, Vinten-Johansen J. Reperfusion induces myocardial apoptotic cell death. Cardiovasc Res. 2000;45:651–660. doi: 10.1016/s0008-6363(99)00354-5. [DOI] [PubMed] [Google Scholar]
- 36.McCully JD, Wakiyama H, Hsieh YJ, Jones M, Levitsky S. Differential contribution of necrosis and apoptosis in myocardial ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2004;286:H1923–1935. doi: 10.1152/ajpheart.00935.2003. [DOI] [PubMed] [Google Scholar]
- 37.Zhao ZQ, Morris CD, Budde JM, Wang NP, Muraki S, Sun HY, Guyton RA. Inhibition of myocardial apoptosis reduces infarct size and improves regional contractile dysfunction during reperfusion. Cardiovasc Res. 2003;59:132–142. doi: 10.1016/s0008-6363(03)00344-4. [DOI] [PubMed] [Google Scholar]
- 38.Abbate A, Salloum FN, Vecile E, Das A, Hoke NN, Straino S, Biondi-Zoccai GG, Houser JE, Qureshi IZ, Ownby ED, Gustini E, Biasucci LM, Severino A, Capogrossi MC, Vetrovec GW, Crea F, Baldi A, Kukreja RC, Dobrina A. Anakinra, a recombinant human interleukin-1 receptor antagonist, inhibits apoptosis in experimental acute myocardial infarction. Circulation. 2008;117:2670–2683. [Google Scholar]
- 39.Yang D, Guo S, Zhang T, Li H. Hypothermia attenuates ischemia/reperfusion-induced en-dothelial cell apoptosis via alterations in apoptotic pathways and JNK signaling. FEBS Lett. 2009;583:2500–2506. doi: 10.1016/j.febslet.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 40.Castrop H, Huang Y, Hashimoto S, Mizel D, Hansen P, Theilig F, Bachmann S, Deng C, Briggs J, Schnermann J. Impairment of tu-buloglomerular feedback regulation of GFR in ecto-5'-nucleotidase/CD73-deficient mice. J Clin Invest. 2004;114:634–642. doi: 10.1172/JCI21851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang F, Wang S, Luo Y, Ji X, Nemoto EM, Chen J. When hypothermia meets hypotension and hyperglycemia: the diverse effects of adenosine 5'-monophosphate on cerebral ischemia in rats. J Cereb Blood Flow Metab. 2009;29:1022–1034. doi: 10.1038/jcbfm.2009.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Heldmaier G, Ortmann S, Elvert R. Natural hypometabolism during hibernation and daily torpor in mammals. Respir Physiol Neurobiol. 2004;141:317–329. doi: 10.1016/j.resp.2004.03.014. [DOI] [PubMed] [Google Scholar]