

Abstract
Background
Inhibition of Glycogen synthase kinase-3 (GSK-3) protects the heart during ischemia/reperfusion (I/R). However, the underlying mechanisms of cardioprotection afforded by beta isoform-specific inhibition of GSK-3 remain to be elucidated.
Methods and Results
We studied the molecular mechanism mediating the effect of GSK-3β activation/inhibition upon myocardial injury during prolonged ischemia and I/R. Beta isoform-specific inhibition of GSK-3 by dominant negative GSK-3β in transgenic mice (Tg-DnGSK-3β) or in heterozygous GSK-3β knock-out mice (GSK-3β +/−) significantly increased, whereas activation of GSK-3β in constitutively active GSK-3β knock-in mice (βKI) significantly decreased, myocardial ischemic injury after prolonged ischemia. In contrast, inhibition of GSK-3β in Tg-DnGSK-3β or GSK-3β +/− significantly reduced, while activation of GSK-3β in βKI significantly enhanced, myocardial I/R injury. Inhibition of GSK-3β stimulated mTOR signaling and inhibited autophagy through a rapamycin-sensitive (mTOR-dependent) mechanism. Rapamycin enhanced autophagy and, at the same time, abolished the effects of GSK-3β inhibition on both prolonged ischemic injury and I/R injury. Importantly, the influence of rapamycin over the effects of GSK-3β inhibition on myocardial injury was reversed by inhibition of autophagy.
Conclusions
Our results suggest that beta isoform-specific inhibition of GSK-3 exacerbates ischemic injury but protects against I/R injury by modulating mTOR and autophagy.
Keywords: ischemia, reperfusion, apoptosis, autophagy, GSK-3, Beclin 1, rapamycin, mTOR
Despite the recent progress in clinical interventions to facilitate early myocardial reperfusion for patients who suffer from acute myocardial infarction (AMI), the death rate during the acute phase of AMI is about 10% and the incidence of heart failure reaches 25% during the chronic phase. Restoring the blood flow to the ischemic myocardium paradoxically causes myocardial damage (termed ischemia/reperfusion (I/R) injury), which potentially explains why many patients develop heart failure despite successful reperfusion. Although many interventions have been shown to protect the heart from I/R injury in experimental animals, virtually no intervention has shown clearly protective effects against I/R injury in the clinical setting, indicating that reevaluation of existing or development of new modalities to protect the heart from I/R injury is urgently needed.
GSK-3 is a serine/threonine kinase which is involved in many cellular functions in the heart, including gene expression, hypertrophy and apoptosis. GSK-3 is a particularly important target of drug therapy for I/R injury since phosphorylation/inhibition of mitochondrial GSK-3β during I/R is believed to be a final common mechanism mediating myocardial protection by many agents/interventions 1. However, many important issues require clarification before inhibition of GSK-3β can be considered to be an option in clinical therapy for I/R injury. First, the precise mechanism through which inhibition of GSK-3 affords cardiac protection remains to be elucidated. Phosphorylation of GSK-3β suppresses opening of the mitochondrial permeability transition pore (mPTP) by binding to adenine nucleotide translocase (ANT) and thereby reducing the affinity of ANT for cyclophilin D 2. GSK-3 inhibitors also reduce adenine nucleotide transport across the outer mitochondrial membrane by decreasing voltage-dependent anion channel phosphorylation 3, which preserves ATP by blocking its consumption by mitochondria and prevents mitochondrial Ca2+ overloading and oxidative stress 3. However, judging from the fact that GSK-3β phosphorylates a wide variety of cellular proteins, other mechanisms, including the tuberous sclerosis complex 2 (TSC2)-mammalian target of rapamycin (mTOR) pathway, may also mediate the function of GSK-3β. Second, previous investigations into the role of GSK-3 in regulating I/R injury heavily relied on chemical inhibitors, which would equally affect both GSK-3α and GSK-3β, the major isoforms of GSK-3 equally expressed in the heart. We have shown recently that these two isoforms have distinct subcellular localizations and functions in the heart 4. Thus, the isoform-specific functions of GSK-3 should be reevaluated with genetically altered mouse models, in which the function of each isoform is specifically modulated. Third, activity and the significance of signaling molecules/biological responses could be different in the ischemic phase and the reperfusion phase. For example, we have shown recently that although activation of autophagy during prolonged ischemia is beneficial, it could be detrimental during reperfusion 5. This issue is important because an effective treatment for I/R may not necessarily be ideal for prolonged ischemia. To our knowledge, the phase-specific roles of GSK-3β in regulating injury during prolonged ischemia and reperfusion have not been compared.
Thus, the purposes of this study were 1) to evaluate the activity of GSK-3β during prolonged ischemia and reperfusion, 2) to investigate the role of GSK-3β and downstream signaling mechanisms in regulating myocardial injury in response to prolonged ischemia and I/R, and 3) to evaluate the role of autophagy in mediating the effect of GSK-3β modulation upon myocardial injury during prolonged ischemia and I/R. The β-isoform specific function of GSK-3 was evaluated with genetically altered mouse models in which activity of GSK-3β is specifically modified.
Methods
An expanded Methods section is available online.
Animals
Generation of transgenic mice with cardiac-specific overexpression of dominant negative GSK-3β (Tg-DnGSK-3β) (FVB background) was previously reported 6. Heterozygous GSK-3β knock-out (GSK-3β +/−) mice (C57BL/6J background) were purchased from Jackson Laboratories. GSK-3βS9A knock-in (βKI) mice (C57BL/6J background) were provided by Dr. D.R. Alessi (University of Dundee, Dundee, UK) 4. Disruption of the TSC2 gene was achieved by a conventional gene targeting method. We used TSC2 heterozygous knock-out mice bred on a C57BL/6J background. Heterozygous green fluorescent protein/light chain 3 (GFP-LC3) transgenic mice (RIKEN BioResource Center, C57BL/6J background) were provided by Dr. N. Mizushima (Tokyo Medical Dental University, Tokyo, Japan) 7. Heterozygous beclin 1 knockout mice (C57BL/6J background) were provided by Dr. B. Levine (University of Texas Southwestern, Dallas, TX) 5. All experiments involving animals were approved by the New Jersey Medical School’s Institutional Animal Care and Use Committee.
I/R
Myocardial I/R was achieved by temporarily occluding the left anterior descending coronary artery (LAD) and then releasing the occlusion 5. The duration of ischemia was 45 min for mice on an FVB background and 20 min for mice on a C57BL/6J background. The reperfusion duration was 24 hours for mice used in determining MI and 2 hours for mice used in biochemical analyses, except for those used in a time course experiment. In some mice, rapamycin (1 mg/Kg, i.p.) was administered 10 min before ischemia or 10 min before reperfusion. For myocardial ischemia studies, the LAD was ligated and occluded for 2 hours, with the exception of a time course experiment.
Statistics
All values are expressed as mean±SEM. Statistical analyses were performed using ANOVA and Tukey-Kramer post-hoc test or the t test, with P<0.05 considered significant.
RESULTS
GSK-3β is activated during ischemia but inhibited during reperfusion
In order to examine the functional status of GSK-3β during ischemia and reperfusion, C57BL/6J mice were subjected to either prolonged ischemia (2h) or I/R (20 min/2h). Ischemia alone significantly reduced S9 phosphorylation of GSK-3β, but I/R increased S9 phosphorylation of GSK-3β in the mouse heart (Fig. 1A–B). Similar results were obtained when FVB mice were subjected to either prolonged ischemia (2h) or I/R (45 min/2h) (Fig. 1C–D). The progressive reduction in S9 phosphorylation of GSK-3β during ischemia started within 30 min of ischemia and persisted for the 2 hour duration (Fig. 1C and Online Fig. IA). The S473 phosphorylation of AKT, an upstream kinase of GSK-3β, was also decreased during ischemia, from 30 min onward (Fig. 1C). The increase in S9 phosphorylation of GSK-3β during reperfusion started within 30 min of reperfusion, persisted for 4 hours, and returned to baseline by 6 hours of reperfusion (Fig. 1D and Online Fig. IB). The S473 phosphorylation of AKT was increased within 30 min of reperfusion and remained increased for up to 6 hours of reperfusion (Fig. 1D). These results suggest that the activity of GSK-3β is differentially regulated by ischemia and I/R. Since the activity of GSK-3β is negatively regulated by S9 phosphorylation, GSK-3β is activated by prolonged ischemia, but is inhibited by I/R.
Figure 1.

Phosphorylation of GSK-3β after 2 hours of ischemia (A) and after 20 min of ischemia and 2 hours of reperfusion (B) in C57BL/6J mice. *P<0.01 vs. respective Sham. Data are mean ± SEM. Phosphorylation of GSK-3β and AKT at various time points during prolonged ischemia (C) and at various time points during reperfusion after 45 min ischemia (D) in FVB mice.
Inhibition of GSK-3β exacerbates, while activation of GSK-3β diminishes ischemic injury
In order to study the role of GSK-3β in prolonged ischemia, a 2-hour ischemia protocol was carried out. After 2 hours of ischemia without reperfusion, the infarct size/area at risk (AAR) in Tg-DnGSK-3β mice, which have diminished GSK-3β activity 6, was significantly increased (Fig. 2A). After ischemia, the infarct size/AAR in constitutively active GSK-3βS9A knock-in (βKI) mice was significantly decreased, but that in GSK-3β +/− mice was significantly increased (Fig. 2B–C). Since the use of TTC staining at an early point during ischemia without reperfusion could have underestimated the infarct size, we performed Hairpin-2 staining, a method of directly detecting necrosis 8. The percentage of Hairpin-2 positive nuclei after ischemia was significantly higher in Tg-DnGSK-3β than in non-transgenic (NTg) mice (Online Fig. IIA). The percentage of Hairpin-2 positive nuclei after ischemia was significantly lower in βKI mice, but was significantly higher in GSK-3β +/− mice, than in wild type (WT) mice (Online Fig. IIB–C). These results suggest that activation of GSK-3β during ischemia ameliorates ischemic myocardial injury, whereas interventions to inhibit GSK-3β during prolonged ischemia enhance ischemic injury.
Figure 2.
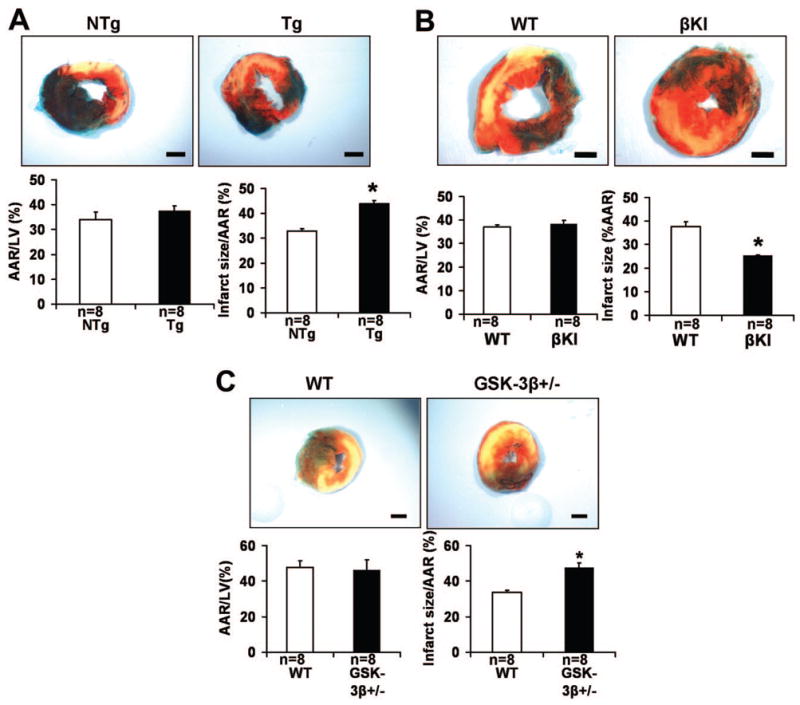
The role of GSK-3β in modulating myocardial injury caused by prolonged ischemia. In A–C, animals were subjected to 2 hours of ischemia. Representative images of LV slices with Alcian blue and TTC staining are shown. AAR/LV (left) and infarct size/AAR (right) are shown. Data are mean ± SEM. Scale bar = 1 mm. A. Tg-DnGSK-3β (Tg) and littermate NTg mice were used. *P<0.05 vs. NTg. B. βKI and WT were used. *P<0.05 vs. WT. C. GSK-3β +/− and WT mice were used. *P<0.05 vs. WT.
Inhibition of GSK-3β attenuates, while activation of GSK-3β enhances reperfusion injury
In order to study the role of GSK-3 in I/R injury, we used Tg-DnGSK-3β, GSK-3β +/−, and βKI mice. After I/R, the infarct size/AAR in Tg-DnGSK-3β was significantly reduced compared with that in NTg (Fig. 3A). On the other hand, infarct size/AAR after I/R was significantly greater in βKI than in WT mice (Fig. 3B). The number of TUNEL positive nuclei after I/R was significantly greater in βKI mice than in WT mice (Online Fig. III). After 20 minutes of ischemia and 24 hours of reperfusion, the infarct size/AAR was significantly smaller in GSK-3β +/− mice than in WT mice, although AAR/LV was not significantly different between them (Fig. 3C). To test whether the difference in ischemic duration between the ischemic protocol and I/R protocol makes inhibition of GSK-3β appear to have different effects, a protocol of 2 hour ischemia followed by 24 hour reperfusion was carried out in GSK-3β +/− mice. In this experimental condition, the infarct size/AAR was still significantly smaller in GSK-3β +/− mice than in WT mice (Fig. 3D). This result suggests that GSK-3β inhibition is protective during reperfusion even after prolonged ischemia. It should be noted that the extent of protection (% reduction in the MI size vs control mice) was significantly smaller after 2 hour ischemia/24 hour reperfusion than after 20 min ischemia/24 hour reperfusion (Fig. 3E). Thus, it is possible that the overall benefit of inhibiting GSK-3β during the entire period of I/R model may be attenuated if the period of ischemia is prolonged. In order to manipulate the activity of GSK-3β in a phase-specific manner, a protocol of 2 hour ischemia followed by 24 hour reperfusion was carried out in βKI mice with or without SB216763, a specific inhibitor of GSK-3, given at the time of reperfusion. Without SB216763, βKI mice exhibited a significantly greater infarct size than WT mice (Fig. 3F). SB216763 significantly decreased infarct size in both WT mice and βKI mice. Importantly, with SB216763, βKI mice had a significantly smaller infarct size than WT mice (Fig. 3F). These results further support the idea that activation of GSK-3β is detrimental during reperfusion but protective during ischemia.
Figure 3.

The role of GSK-3β in modulating myocardial injury caused by I/R. In A–D and F, animals were subjected to either 45 min (A), 20 min (B–C) or 2 hours (D, F) of ischemia, followed by 24 hours of reperfusion. In A–D, representative images of LV slices with Alcian blue and TTC staining are shown. The AAR/LV (left) and infarct size/AAR (right) are shown (A–D, F). Data are mean ± SEM. Scale bar = 1 mm. A. Tg-DnGSK-3β (Tg) and littermate NTg mice were used. *P<0.05 vs. NTg. B. βKI and WT mice were used. *P<0.05 vs. WT. C–D. GSK-3β +/− mice were used. *P<0.05 vs. WT. E. Reduction in infarct size in GSK-3β +/− mice after 20 min or 2 hours of ischemia followed by 24 hours of reperfusion, compared with the average infarct size of respective WT mice. *P<0.01 vs. 20 min/24 h I/R. F. GSK-3 inhibitor SB216763 (SB) was given to βKI mice at the time of reperfusion. *P<0.05 vs. respective WT. #P<0.05 vs. vehicle control.
Inhibition of GSK-3β increases, while activation of GSK-3β inhibits mTOR activity during both ischemia and reperfusion phases
Increasing lines of evidence suggest that mTOR regulates fundamental cellular functions relevant to hypoxia/ischemia 9, thereby serving as a critical regulator of cell survival/death under conditions of hypoxia/ischemia. In order to study the molecular mechanisms mediating the effects of GSK-3 on ischemic and I/R injury, we studied the activity of mTOR after prolonged ischemia and I/R. Both at baseline and after 2 hours of ischemia, the phosphorylation level of p70 S6 kinase (S6K), a downstream target phosphorylated by mTOR, in Tg-DnGSK-3β mice (Fig. 4A) was increased, but that in βKI mice was decreased (Fig. 4B), compared with that of WT mice, indicating that inhibition of GSK-3β increased, but activation of GSK-3β inhibited mTOR activity after prolonged ischemia. Importantly, ischemia decreased the phosphorylation level of S6K in NTg and WT mice, indicating that dephosphorylation of GSK-3β by ischemia might inhibit mTOR activity. I/R increased the phosphorylation level of S6K in NTg (Fig. 4C) and WT mice (Fig. 4D), indicating that I/R elevated mTOR activity. Both at baseline and after I/R, the phosphorylation level of S6K in Tg-DnGSK-3β mice was markedly increased compared with that of NTg mice (Fig. 4C). In contrast, both at baseline and after I/R, the phosphorylation level of S6K in βKI mice was significantly lower than that of WT mice (Fig. 4D). These results suggest that inhibition of GSK-3β increases mTOR activity, while activation of GSK-3β decreases mTOR activity during I/R.
Figure 4.
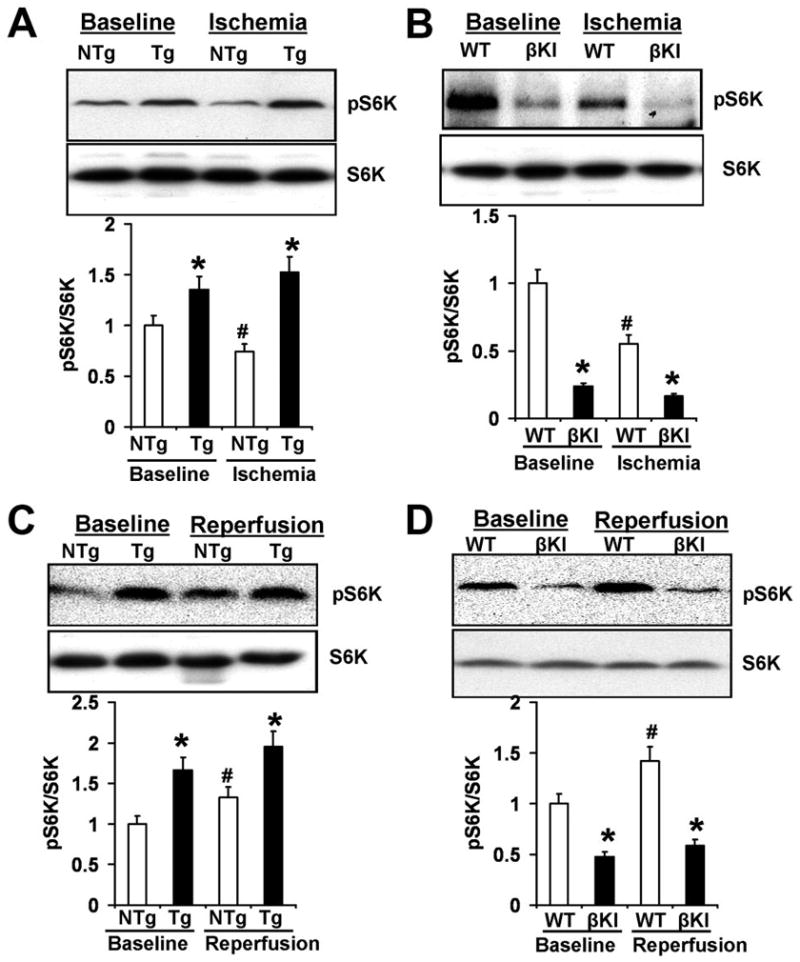
GSK-3β inhibits mTOR signaling. A. Phospho-p70 S6 kinase (pS6K) and total S6K in the ischemic myocardium of Tg-DnGSK-3β (Tg) and NTg mice at baseline and after 2 hours of ischemia. *P<0.05 vs. NTg. #P<0.05 vs. baseline. B. pS6K and total S6K in ischemic myocardium of βKI and WT mice at baseline and after 2 hours of ischemia. *P<0.05 vs. WT. #P<0.05 vs. baseline. C. pS6K and total S6K in Tg and NTg at baseline and after 45 min of ischemia and 2 hours of reperfusion. *P<0.05 vs. NTg. #P<0.05 vs. baseline. D. pS6K and total S6K in βKI and WT at baseline and after 20 min of ischemia and 2 hours of reperfusion. *P<0.05 vs. WT. #P<0.05 vs. baseline.
Down-regulation of TSC2 stimulates ischemic injury but protects against reperfusion injury
Increasing lines of evidence suggest that mTOR is controlled by ischemia through TSC2 as an essential mechanism of adaptation 10. Importantly, GSK-3β regulates mTOR through phosphorylation of TSC2 in cancer cell lines 11. In order to study whether GSK-3β regulates mTOR in cardiomyocytes, experiments were carried out in neonatal rat cardiomyocytes. Overexpression of GSK-3β reduced the phosphorylation of S6K in cardiomyocytes, and knocking down TSC2 abolished the inhibitory effect of GSK-3β overexpression on S6K phosphorylation (Fig. 5A). These results suggest that GSK-3β down-regulates mTOR through TSC2-dependent mechanisms in cardiomyocytes. In order to study the role of mTOR activation in mediating myocardial injury caused by prolonged ischemia and I/R, experiments were conducted in TSC2+/− mice. In TSC2+/− hearts, the phosphorylation level of S6K was increased (Fig. 5B) compared with that in WT mouse hearts, indicating that mTOR activity was stimulated by down-regulation of TSC2. After 2 hours of ischemia without reperfusion, the infarct size/AAR was significantly larger in TSC2+/− mice than in WT mice, although the AAR was not significantly different between them (Fig. 5C–E). The percentage of Hairpin-2 positive nuclei was significantly higher in TSC2+/− mice than in WT mice after 2 hours of ischemia (Online Fig. IV). In contrast, after 45 min of ischemia and 24 hours of reperfusion, the infarct size/AAR was significantly smaller in TSC2+/− mice than in WT mice, although the AAR was similar between them (Fig. 5F–H). These results indicate that activation of mTOR is protective during I/R, but detrimental during prolonged ischemia. In the absence of TSC2 knock-out, the protein level of TSC2 does not change during prolonged ischemia but is decreased during reperfusion in the mouse heart in vivo. Neither constitutively active GSK-3β nor heterozygous deletion of GSK-3β significantly affected baseline expression of TSC2 (Online Fig. V). Thus, down-regulation of endogenous TSC2 may also contribute to mTOR activation during I/R.
Figure 5.
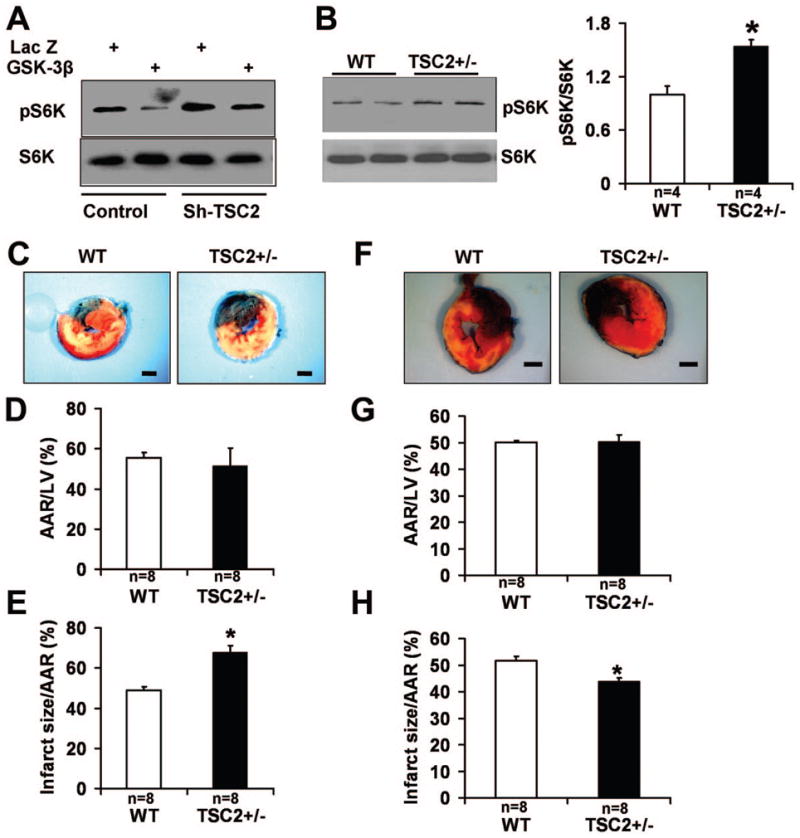
mTOR signaling and ischemic and ischemia/reperfusion injury. A. Phospho-p70 S6 kinase (pS6K) and total S6K in neonatal rat cardiomyocytes transduced with Lac Z or GSK-3β. GSK-3β decreased pS6K level. Knocking down TSC2 restored the pS6K level in GSK-3β-transduced myocytes. B. pS6K and total S6K in TSC2+/− and WT mice. *P<0.01 vs. WT. C–E. MI in TSC2+/− and WT after 2 hours of ischemia. Representative images of LV slices with Alcian blue and TTC staining (C), AAR/LV (D) and infarct size/AAR (E) are shown. Scale bar = 1 mm. *P<0.05 vs. WT. F–H. MI in TSC2+/− and WT mice after 45 min of ischemia and 24 hours of reperfusion. Representative images of LV slices with Alcian blue and TTC staining (F), AAR/LV (G) and infarct size/AAR (H) are shown. Scale bar = 1 mm. *P<0.05 vs. WT.
Inhibition of GSK-3β prevents autophagy through mTOR and is detrimental during ischemia
Autophagy is activated by nutrient starvation, and it is protective during the ischemic phase 5. In order to study the effect of GSK-3 inhibition on autophagy after prolonged ischemia, DnGSK-3β/GFP-LC3 bigenic mice were subjected to 2 hours of ischemia and the number of GFP-LC3 dots in the ischemic area was counted. The number of GFP-LC3 dots per microscopic field in DnGSK-3β/GFP-LC3 bigenic mice was significantly lower than in GFP-LC3 transgenic mice (Fig. 6A). Moreover, there was more p62, a protein degraded by autophagy, in the ischemic area of Tg-DnGSK-3β than in that of NTg mice (Fig. 6B). These results suggest that inhibition of GSK-3β by overexpression of dominant negative GSK-3β resulted in less autophagy in the heart after prolonged ischemia. Rapamycin abolished the inhibitory effects of dominant negative GSK-3β on the number of GFP-LC3 dots after prolonged ischemia (Fig. 6C), and reduced the infarct size/AAR and the percentage of Hairpin-2 positive nuclei in Tg-DnGSK-3β to the levels of those in NTg mice (Fig. 6D), indicating that GSK-3β inhibition prevents autophagy through an mTOR-dependent mechanism, and that GSK-3β inhibition may enhance ischemic injury via mTOR-dependent prevention of autophagy. Rapamycin also abolished the increase in infarct size caused by SB216763 (Fig. 6E). However, in beclin 1 heterozygous knock-out mice, which have a decreased level of autophagy, as well as a larger infarct/AAR and a higher percentage of Hairpin-2 positive nuclei than control WT mice, rapamycin failed to abolish the infarct-enhancing effect of SB216763 after ischemia (Fig. 6E). These results indicate that autophagy is one of the targets of mTOR inhibition mediating its protective effect during prolonged ischemia.
Figure 6.

GSK-3β inhibition and myocardial autophagy after 2 hours of ischemia. A. GFP-LC3 dots (arrow) in GFP-LC3 mice (GFP-LC3) and DnGSK-3β/GFP-LC3 mice (Bigenic, DnGSK/GFP-LC3). Scale bar = 100 μm. The bar graph shows the number of GFP-LC3 dots per microscopic field. *P<0.05 vs. GFP-LC3 Ischemia. B. Immunoblotting results of p62 in Tg-DnGSK-3β (Tg) and NTg mice. C. The number of GFP-LC3 dots per microscopic field in Tg-GFP-LC3 and DnGSK-3β/GFP-LC3 bigenic mice with or without rapamycin. *P<0.05 vs. GFP-LC3 Control. #P<0.05 vs. DnGSK-3β/GFP-LC3 Control. D. The AAR/LV (left), infarct size/AAR (middle), and percent Hairpin-2 positive nuclei (right) 2 hours after ischemia in Tg and NTg with or without rapamycin. *P<0.05 vs. NTg Control. #P<0.05 vs. Tg Control. E. AAR/LV (left), infarct size/AAR (middle), and percent Hairpin-2 positive nuclei (right) in WT mice and beclin1+/− mice after 2 hours of ischemia. SB, SB216763. Rap, rapamycin. *P<0.05 vs. Control. #P<0.05 vs. SB. $P<0.05 vs. SB+Rap.
Inhibition of GSK-3β is protective against reperfusion injury through mTOR-dependent down-regulation of autophagy
In order to study the involvement of mTOR in the effects of GSK-3β on reperfusion injury, rapamycin was used to inhibit mTOR. Rapamycin abolished the infarct-limiting effect observed in Tg-DnGSK-3β (Fig. 7A). Rapamycin also abolished the decrease in infarct size caused by SB216763 (Fig. 7B). These results suggest that stimulation of mTOR plays an important role in mediating the protective effects of GSK-3β inhibition during reperfusion. Importantly, the number of GFP-LC3 dots per microscopic field was significantly decreased after I/R in DnGSK-3β/GFP-LC3 bigenic mice compared with that in GFP-LC3 transgenic mice (Fig. 7C), indicating that reperfusion-induced autophagy was attenuated by GSK-3β inhibition. Rapamycin treatment restored the number of GFP-LC3 dots after I/R in DnGSK-3β/GFP-LC3 bigenic mice (Fig. 7D), indicating that inhibition of mTOR enhances autophagy in Tg-DnGSK-3β mice. However, in beclin 1 heterozygous knock-out mice, which have a decreased level of autophagy, rapamycin failed to abolish the infarct-limiting effect of SB216763 after I/R (Fig. 7B). These results suggest that GSK-3 inhibition protects against reperfusion injury through mTOR-dependent prevention of autophagy.
Figure 7.

GSK-3 inhibition, mTOR signaling, and autophagy after ischemia/reperfusion. A. AAR/LV (left) and infarct size/AAR (right) in Tg-DnGSK-3β mice (Tg) after 45 min of ischemia and 24 hours of reperfusion, with or without rapamycin. *P<0.05 vs. NTg. #P<0.05 vs. Tg Rapamycin. B. AAR/LV (left) and infarct size/AAR (right) in WT mice and beclin1+/− mice after I/R. SB, SB216763. Rap, rapamycin. *P<0.05 vs. Control. #P<0.05 vs. SB. $P<0.05 vs. SB+Rap. C. GFP-LC3 dots in Tg-GFP-LC3 and DnGSK-3β/GFP-LC3 bigenic mice (Bigenic, DnGSK/GFP-LC3) after 45 min of ischemia and 2 hours of reperfusion. Representative images are shown. Scale bar = 100 μm. The bar graph shows the number of GFP-LC3 dots per microscopic field. *P<0.05 vs. GFP-LC3. D. The number of GFP-LC3 dots per microscopic field in GFP-LC3 mice and DnGSK/GFP-LC3 bigenic mice, after 45 min of ischemia and 2 hours of reperfusion, with or without rapamycin. *P<0.05 vs. GFP-LC3 Control.
DISCUSSION
We here investigated the β isoform-specific function of GSK-3 in the heart subjected to either prolonged ischemia alone or a short period of ischemia followed by reperfusion, using genetically engineered mouse models of GSK-3β inhibition (Tg-DnGSK-3β and GSK-3β +/−) and activation (βKI). The main findings in this study are 1) that GSK-3β is dephosphorylated and activated during prolonged ischemia, whereas it is phosphorylated and inhibited during reperfusion, 2) that activation of GSK-3β during ischemia and inactivation of GSK-3β during reperfusion are both compensatory for the heart, and 3) that the compensatory role of GSK-3β activation and inactivation during ischemia and reperfusion, respectively, is mediated through regulation of mTOR and autophagy (Fig. 8).
Figure 8.
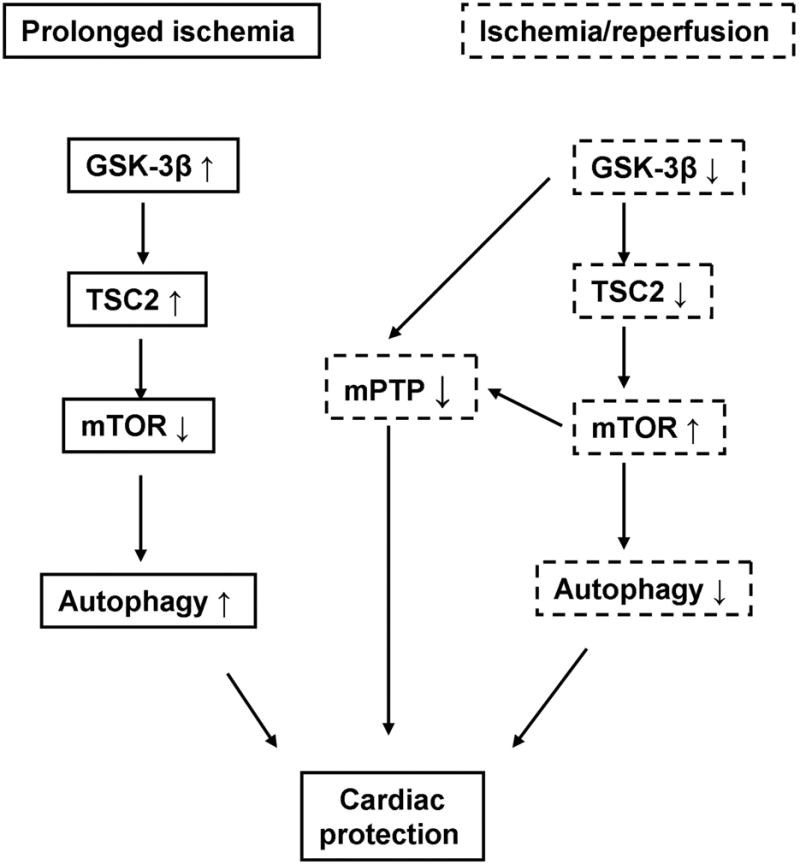
A scheme showing distinct roles of GSK-3β during ischemia and reperfusion. Solid-line boxes show the role of GSK-3β during ischemia. Dotted-line boxes show the role of GSK-3β during reperfusion.
The fact that phosphorylation of the S9 residue in endogenous GSK-3β is regulated by ischemia and reperfusion differentially but in a way which promotes cardioprotection during both phases is intriguing. The status of S9 phosphorylation in GSK-3β coincided with that of Akt phosphorylation, suggesting that Akt may control the activity of GSK-3β during ischemia and reperfusion. At present, however, phosphatases or kinases mediating either dephosphorylation during ischemia or phosphorylation during reperfusion remain to be identified 1. Interestingly, although activation of GSK-3β through dephosphorylation during prolonged ischemia is protective, inactivation of GSK-3β by phosphorylation during the reperfusion phase is also protective. To our knowledge, phase-dependent changes in modulation and function of GSK-3β between ischemia and reperfusion have not been demonstrated previously.
Hypoxia inhibits mTOR through multiple signaling mechanisms, most of which utilize TSC1/2-dependent mechanisms. For examples, brief exposures to modest hypoxia prevent insulin-mediated stimulation of mTORC1 through AMP-activated protein kinase (AMPK)-dependent activation of TSC1 – TSC2 12. Hypoxia induces expression of REDD1, which in turn re-activates TSC1 – TSC2 by releasing TSC2 from 14–3–3 10, 13, which could also lead to inactivation of mTOR. GSK-3β phosphorylates/activates TSC2, which in turn inhibits mTOR 11, where GSK-3β is required for mTOR regulation by energy starvation 11. Our results suggest that GSK-3β is activated during prolonged ischemia, which in turn inhibits mTOR in the heart. Suppression of GSK-3β activation significantly attenuated suppression of mTOR during ischemia, indicating a causative role of GSK-3β activation in mediating mTOR inhibition. Suppression of mTOR by GSK-3β was reversed when TSC2 was down-regulated, indicating that GSK-3β regulates mTOR through TSC2 in cardiomyocytes. Constitutive activation of GSK-3β inhibited activation of mTOR during reperfusion, suggesting that phosphorylation/suppression of GSK-3β plays an essential role in mediating mTOR activation during reperfusion as well. Taken together, our results suggest that GSK-3β is a critical upstream regulator of mTOR during both prolonged ischemia and reperfusion in the heart.
GSK-3β has many downstream targets, which can mediate both survival and death of cardiomyocytes. For example, suppression of GSK-3β stabilizes Mcl-1, a Bcl-2 family protein, thereby promoting survival 6. However, perhaps one of the most important mechanisms by which suppression of GSK-3β mediates cardioprotection during I/R is regulation of mPTP opening and other mitochondrial proteins 1–3. GSK-3β regulates components of the mPTP through direct phosphorylation, thereby inducing mitochondrial depolarization, release of cytochrome c, and eventual cell death 1. Interestingly, the enhancement of myocardial injury in response to prolonged ischemia by suppression of GSK-3β was reversed in the presence of rapamycin. Furthermore, the suppression of reperfusion injury by inhibition of GSK-3β was also completely reversed by rapamycin. These results suggest that, besides regulation of the mPTP, modulation of mTOR also plays a significant role in mediating the cardioprotective effects of GSK-3β activation and inactivation during prolonged ischemia and reperfusion, respectively.
Accumulating lines of evidence suggest that modulation of mTOR has a significant influence on I/R injury in the heart. However, the effect of mTOR modulation upon myocardial injury appears to be model-dependent. The infarct-reducing effects of insulin and ischemic preconditioning are abolished by rapamycin given before the onset of reperfusion in Langendorff-perfused rat hearts 14. In addition, rapamycin given prior to preconditioning abolished the second window of ischemic preconditioning in rabbits 15. On the other hand, rapamycin given to mice one hour before ischemia conferred infarct-limiting effects in Langendorff-perfused mouse hearts 16. In the present study, increased mTOR signaling in TSC2+/− mice induced a smaller MI after I/R, whereas it induced a greater MI after prolonged ischemia. Interestingly, although the detrimental effect of GSK-3β inhibition during prolonged ischemia was reversed by rapamycin, the cardioprotective effect of GSK-3β inhibition during I/R was also attenuated by rapamycin. Our results clearly suggest that the function of mTOR is distinct between prolonged ischemia and reperfusion.
What is the underlying mechanism mediating the effect of mTOR regulation upon myocardial injury during ischemia and reperfusion? mTOR is an essential regulator of protein synthesis and cardiac hypertrophy 17. Inhibiting protein synthesis during ischemia is beneficial because protein synthesis not only consumes energy, which is undesirable during ischemia, but also promotes ER stress 18. On the other hand, activation of mTOR during the reperfusion phase may upregulate cell survival signaling mechanisms 19 and mitochondrial biogenesis through peroxisome-proliferator-activated receptor gamma coactivator-1 alpha and Ying Yang 1, which may facilitate recovery from myocardial ischemia during reperfusion 20. Importantly, mTOR inhibits autophagy by negatively regulating the association between autophagy-related gene 1 (ATG1) and ATG13 21, which affects survival and death of cardiomyocytes during ischemia and reperfusion in a phase-dependent manner 5.
Autophagy is an intracellular degradation process through which long-lived cytosolic proteins and organelles are degraded by lysosomes and recycled 22. Hypoxia/reoxygenation and I/R are among the stresses known to prominently activate autophagy in the heart 5. Autophagy appears to be protective when it is activated during transient ischemia 5, ischemic preconditioning 23, and myocardial hibernation 24, possibly by regenerating ATP and removing protein aggregates and damaged organelles, including mitochondria. On the other hand, excessive activation of autophagy during reperfusion may lead to cell death 5, although autophagy also plays a salutary role during myocardial reperfusion in some experimental conditions 25. Our results suggest that GSK-3β inhibition and the resultant mTOR-dependent attenuation of autophagy are detrimental during prolonged ischemia, whereas they are protective during reperfusion. Furthermore, inhibition of mTOR signaling by rapamycin reversed the inhibitory effect of GSK-3β inhibition on autophagy and, at the same time, abolished the effect of GSK-3β inhibition on myocardial ischemic injury and reperfusion injury. These findings suggest that GSK-3β modulates autophagy through mTOR-dependent mechanisms, thereby mediating survival and death of cardiomyocytes in a phase-dependent manner. The fact that heterozygous beclin 1 knock-out, which inhibits autophagy, canceled the effect of rapamycin on GSK-3β inhibition during prolonged ischemia and I/R further indicates that GSK-3β inhibition indeed modulates myocardial injury due to prolonged ischemia and I/R through mTOR-dependent regulation of autophagy. Regulation of mTOR and that of mPTP opening may not be mutually exclusive as mechanisms mediating the effect of GSK-3β upon myocardial injury/protection. For example, mTOR-mediated regulation of mitochondrial genes 20 may indirectly affect mPTP opening, with mPTP opening being a critical mediator of mitochondrial autophagy 26.
In summary, GSK-3β plays an important role in modulating mTOR during both prolonged ischemia and reperfusion in the heart. Changes in autophagy contribute to the differential effects of GSK-3β upon myocardial injury during prolonged ischemia and I/R. Inhibition of GSK-3β by a small molecule inhibitor may be considered to reduce reperfusion injury due to its inhibitory effects upon mPTP opening. However, if ischemia is prolonged, GSK-3β inhibition may exacerbate myocardial injury before the heart is reperfused. In this case, an alternative strategy might be to limit the timing of GSK-3β inhibition to the time of reperfusion only.
Supplementary Material
Novelty and Significance.
What Is Known?
Inhibition of glycogen synthase kinase-3 (GSK-3) attenuates ischemia/reperfusion (I/R) injury by inhibiting mitochondrial permeability transition pore (mPTP) opening.
Autophagy is activated by both myocardial ischemia and reperfusion and affects survival and death of cardiac myocytes.
What New Information Does This Article Contribute?
Activation of the β-isoform of GSK-3, GSK-3β, is protective during ischemia whereas it is detrimental during reperfusion.
GSK-3β modulates autophagy through inhibition of mTOR, which significantly affects survival and death of cardiac myocytes during both prolonged ischemia and reperfusion.
Acknowledgments
The authors thank Daniela Zablocki for critical reading of the manuscript.
Funding Sources
This work was supported by U.S. Public Health Service Grants HL 59139, HL67724, HL69020, HL91469, and AG27211. This work was supported by Foundation of Leducq Transatlantic Network of Excellence. PZ is supported by an AHA grant 0930179N.
Non-standard Abbreviations
- GSK-3
glycogen synthase kinase-3
- Tg
transgenic
- NTg
non-transgenic
- Dn
dominant negative
- βKI
GSK-3βS9A knock-in
- mTOR
mammalian target of rapamycin
- MI
myocardial infarction
- I/R
ischemia/reperfusion
- mPTP
mitochondrial permeability transition pore
- TCS2
tuberous sclerosis complex 2
- GFP
green fluorescent protein
- LAD
left anterior descending coronary artery
- AAR
area at risk
- S6K
p70 S6 kinase
- WT
wild type
Footnotes
Disclosures
None
References
- 1.Juhaszova M, Zorov DB, Kim S-H, Pepe S, Fu Q, Fishbein KW, Ziman BD, Wang S, Ytrehus K, Antos CL, Olson EN, Sollott SJ. Glycogen synthase kinase-3{beta} mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J Clin Invest. 2004;113:1535–1549. doi: 10.1172/JCI19906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nishihara M, Miura T, Miki T, Tanno M, Yano T, Naitoh K, Ohori K, Hotta H, Terashima Y, Shimamoto K. Modulation of the mitochondrial permeability transition pore complex in GSK-3beta-mediated myocardial protection. J Mol Cell Cardiol. 2007;43:564–570. doi: 10.1016/j.yjmcc.2007.08.010. [DOI] [PubMed] [Google Scholar]
- 3.Das S, Wong R, Rajapakse N, Murphy E, Steenbergen C. Glycogen synthase kinase 3 inhibition slows mitochondrial adenine nucleotide transport and regulates voltage-dependent anion channel phosphorylation. Circ Res. 2008;103:983–991. doi: 10.1161/CIRCRESAHA.108.178970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Matsuda T, Zhai P, Maejima Y, Hong C, Gao S, Tian B, Goto K, Takagi H, Tamamori-Adachi M, Kitajima S, Sadoshima J. Distinct roles of GSK-3alpha and GSK-3beta phosphorylation in the heart under pressure overload. Proc Natl Acad Sci U S A. 2008;105:20900–20905. doi: 10.1073/pnas.0808315106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, Levine B, Sadoshima J. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res. 2007;100:914–922. doi: 10.1161/01.RES.0000261924.76669.36. [DOI] [PubMed] [Google Scholar]
- 6.Hirotani S, Zhai P, Tomita H, Galeotti J, Marquez JP, Gao S, Hong C, Yatani A, Avila J, Sadoshima J. Inhibition of glycogen synthase kinase 3beta during heart failure is protective. Circ Res. 2007;101:1164–1174. doi: 10.1161/CIRCRESAHA.107.160614. [DOI] [PubMed] [Google Scholar]
- 7.Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15:1101–1111. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chimenti C, Kajstura J, Torella D, Urbanek K, Heleniak H, Colussi C, Di Meglio F, Nadal-Ginard B, Frustaci A, Leri A, Maseri A, Anversa P. Senescence and death of primitive cells and myocytes lead to premature cardiac aging and heart failure. Circ Res. 2003;93:604–613. doi: 10.1161/01.RES.0000093985.76901.AF. [DOI] [PubMed] [Google Scholar]
- 9.Wouters BG, Koritzinsky M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer. 2008;8:851–864. doi: 10.1038/nrc2501. [DOI] [PubMed] [Google Scholar]
- 10.Brugarolas J, Lei K, Hurley RL, Manning BD, Reiling JH, Hafen E, Witters LA, Ellisen LW, Kaelin WG., Jr Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004;18:2893–2904. doi: 10.1101/gad.1256804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Inoki K, Ouyang H, Zhu T, Lindvall C, Wang Y, Zhang X, Yang Q, Bennett C, Harada Y, Stankunas K, Wang CY, He X, MacDougald OA, You M, Williams BO, Guan KL. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell. 2006;126:955–968. doi: 10.1016/j.cell.2006.06.055. [DOI] [PubMed] [Google Scholar]
- 12.Arsham AM, Howell JJ, Simon MC. A novel hypoxia-inducible factor-independent hypoxic response regulating mammalian target of rapamycin and its targets. J Biol Chem. 2003;278:29655–29660. doi: 10.1074/jbc.M212770200. [DOI] [PubMed] [Google Scholar]
- 13.DeYoung MP, Horak P, Sofer A, Sgroi D, Ellisen LW. Hypoxia regulates TSC1/2-mTOR signaling and tumor suppression through REDD1-mediated 14-3-3 shuttling. Genes Dev. 2008;22:239–251. doi: 10.1101/gad.1617608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fuglesteg BN, Tiron C, Jonassen AK, Mjos OD, Ytrehus K. Pretreatment with insulin before ischaemia reduces infarct size in Langendorff-perfused rat hearts. Acta Physiol (Oxf) 2009;195:273–282. doi: 10.1111/j.1748-1716.2008.01901.x. [DOI] [PubMed] [Google Scholar]
- 15.Kis A, Yellon DM, Baxter GF. Second window of protection following myocardial preconditioning: an essential role for PI3 kinase and p70S6 kinase. J Mol Cell Cardiol. 2003;35:1063–1071. doi: 10.1016/s0022-2828(03)00208-6. [DOI] [PubMed] [Google Scholar]
- 16.Khan S, Salloum F, Das A, Xi L, Vetrovec GW, Kukreja RC. Rapamycin confers preconditioning-like protection against ischemia-reperfusion injury in isolated mouse heart and cardiomyocytes. J Mol Cell Cardiol. 2006;41:256–264. doi: 10.1016/j.yjmcc.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 17.McMullen JR, Sherwood MC, Tarnavski O, Zhang L, Dorfman AL, Shioi T, Izumo S. Inhibition of mTOR signaling with rapamycin regresses established cardiac hypertrophy induced by pressure overload. Circulation. 2004;109:3050–3055. doi: 10.1161/01.CIR.0000130641.08705.45. [DOI] [PubMed] [Google Scholar]
- 18.Glembotski CC. Endoplasmic reticulum stress in the heart. Circ Res. 2007;101:975–984. doi: 10.1161/CIRCRESAHA.107.161273. [DOI] [PubMed] [Google Scholar]
- 19.Jonassen AK, Sack MN, Mjos OD, Yellon DM. Myocardial protection by insulin at reperfusion requires early administration and is mediated via Akt and p70s6 kinase cell-survival signaling. Circ Res. 2001;89:1191–1198. doi: 10.1161/hh2401.101385. [DOI] [PubMed] [Google Scholar]
- 20.Cunningham JT, Rodgers JT, Arlow DH, Vazquez F, Mootha VK, Puigserver P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature. 2007;450:736–740. doi: 10.1038/nature06322. [DOI] [PubMed] [Google Scholar]
- 21.Chan EY. mTORC1 phosphorylates the ULK1-mAtg13-FIP200 autophagy regulatory complex. Sci Signal. 2009;2:pe51. doi: 10.1126/scisignal.284pe51. [DOI] [PubMed] [Google Scholar]
- 22.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gurusamy N, Lekli I, Gorbunov NV, Gherghiceanu M, Popescu LM, Das DK. Cardioprotection by adaptation to ischaemia augments autophagy in association with BAG-1 protein. J Cell Mol Med. 2009;13:373–387. doi: 10.1111/j.1582-4934.2008.00495.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yan L, Vatner DE, Kim SJ, Ge H, Masurekar M, Massover WH, Yang G, Matsui Y, Sadoshima J, Vatner SF. Autophagy in chronically ischemic myocardium. Proc Natl Acad Sci U S A. 2005;102:13807–13812. doi: 10.1073/pnas.0506843102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hamacher-Brady A, Brady NR, Gottlieb RA. The interplay between pro-death and pro-survival signaling pathways in myocardial ischemia/reperfusion injury: apoptosis meets autophagy. Cardiovasc Drugs Ther. 2006;20:445–462. doi: 10.1007/s10557-006-0583-7. [DOI] [PubMed] [Google Scholar]
- 26.Gustafsson AB, Gottlieb RA. Autophagy in ischemic heart disease. Circ Res. 2009;104:150–158. doi: 10.1161/CIRCRESAHA.108.187427. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.