

Abstract
Mutations in genes that encode components of the sarcomere are well established as the cause of hypertrophic and dilated cardiomyopathies. Sarcomere genes, however, are increasingly being associated with other cardiomyopathies. One phenotype more recently recognized as a disease of the sarcomere is restrictive cardiomyopathy (RCM). We report on two patients with RCM associated with multiple mutations in sarcomere genes not previously associated with RCM. Patient 1 presented with NYHA Class III/IV heart failure at 22 years of age. She was diagnosed with RCM and advanced heart failure requiring heart transplantation. Sequencing of sarcomere genes revealed previously reported homozygous p.Glu143Lys mutations in MYL3, and a novel heterozygous p.Gly57Glu mutation in MYL2. The patient’s mother is a double heterozygote for these mutations, with no evidence of cardiomyopathy. Patient 2 presented at 35 years of age with volume overload while hospitalized for oophorectomy. She was diagnosed with RCM and is being evaluated for heart transplantation. Sarcomere gene sequencing identified homozygous p.Asn279His mutations in TPM1. The patient’s parents are consanguineous and confirmed heterozygotes. Her father was diagnosed with HCM at 42 years of age.
This is the first report of mutations in TPM1, MYL3 and MYL2 associated with primary, non-hypertrophied restrictive cardiomyopathy. The association of more sarcomere genes with RCM provides further evidence that mutations in the various sarcomere genes can cause different cardiomyopathy phenotypes. These cases also contribute to the growing body of evidence that multiple mutations have an additive effect on the severity of cardiomyopathies.
Keywords: Restrictive Cardiomyopathy, Hypertrophic Cardiomyopathy, Sarcomere, Genetic Testing, Genetic Counseling, Cardiovascular Genetics
INTRODUCTION
Sarcomere genes were first linked to primary myocardial disease in 1990 when a mutation in MYH7 (OMIM 160760) was implicated in hypertrophic cardiomyopathy (HCM) [Geisterfer-Lowrance et al., 1990]. Later that decade, another sarcomere gene, ACTC1 (OMIM 102540), was associated with dilated cardiomyopathy (DCM) [Olson et al., 1998]. Since then mutations in the various genes that encode the sarcomere have become a well established cause of HCM and DCM [Moller et al., 2009; Van Driest et al., 2005]. In recent years many of these genes, though not all, have also been associated with other primary cardiomyopathies, such as left ventricular non-compaction and restrictive cardiomyopathy (RCM), suggesting a common genetic etiology to these clinically diverse phenotypes [Perrot et al., 2007].
Cardiomyopathies are classified primarily based on morphological and physiological parameters [Maron et al., 2006]. RCM, in particular, is characterized by increased stiffness of the ventricular walls with impaired diastolic filling and preserved systolic function, without left ventricular hypertrophy but often with enlarged atria. While RCM is rare compared to other primary cardiomyopathies it is also more severe, usually leading to transplantation or death [Ammash et al., 2000; Maron et al., 2006; Mogensen and Arbustini, 2009].
RCM has diverse etiologies. A minority of patients has a secondary cardiomyopathy due to infiltrative disease, such as amyloidosis or hemochromatosis, or has a desmin cardiomyopathy, caused by mutations in DES (OMIM 125660). Most cases of RCM show no pathological evidence of infiltrative disease and have thus been considered primary cardiomyopathies of unknown etiology [Ammash et al., 2000]. Sarcomere genes were first linked to RCM in 2003 when mutations in TNNI3 were implicated in multiple cases of RCM [Mogensen et al., 2003]. Since then RCM has been associated with several (MYH7, OMIM 160760; MYPBC3, OMIM 600958; TNNT2, OMIM 191045; TNNI3, OMIM 191044; ACTC1, OMIM 102540), but not all, sarcomere genes and a significant proportion of “idiopathic” cases have been attributed to sarcomere mutations [Kaski et al., 2008; Kubo et al., 2007; Mogensen et al., 2003; Monserrat et al., 2007; Peddy et al., 2006; Ware et al., 2008].
We report on two patients with primary RCM associated with multiple mutations in sarcomere genes that have not previously been associated with this type of cardiomyopathy. The documentation of these reports further expand the association between the various sarcomere genes and the primary cardiomyopathies.
CLINICAL REPORTS
Patient 1
An El-Salvadoran 22-year-old female with a prior diagnosis of childhood asthma presented with worsening fatigue and dyspnea on exertion over the course of 12 months. She reported having frequent episodes of shortness of breath as a child. She also reported orthopnea, paroxysmal nocturnal dyspnea, and palpitations; her heart failure symptoms were classified as NYHA Class III–IV. She denied dizziness, syncope, and lower extremity edema but did have ascites suggestive of congestive hepatopathy. Her cardiovascular examination revealed a nondisplaced, nonsustained point of maximal impulse, with a mild parasternal lift without a pulmonary artery tap. She had a normal S1, but P2>A2. She had an S3, but no S4. She had a soft I/IV holosystolic murmur over the apex; no knock or rub. Her electrocardiogram demonstrated atrial flutter with an atrial rate of 250–300 and a ventricular rate of 85. The P-wave voltage approximated the QRS voltage in some leads. There was right-axis deviation with an incomplete RBBB. Chest x-ray was consistent with congestive heart failure.
A transthoracic echocardiogram revealed severe biatrial enlargement with preserved biventricular systolic function and no left ventricular hypertrophy or valvular disease (Table I). Doppler interrogation suggested advanced LV diastolic dysfunction. The pulmonary artery systolic pressure was estimated to be >55mm Hg, based on a right atrial pressure estimate of >15mm Hg. These findings were all consistent with restrictive physiology.
Table I.
Echocardiographic features of patients and family members
| LVEDV (35–75ml/m2) | RA Sizea (normal) | LA Volume (16–28ml/m2) | LVEF (55%–65%) | LVEDPW (0.6–1.0cm) | LVEDSW (0.7–1.0cm) | RVHb (none) | |
|---|---|---|---|---|---|---|---|
| Patient 1 | |||||||
| Proband | 25 | Severely enlarged | 55 | 66 | 0.9 | 0.8 | None |
| II-1 | |||||||
| I-2 | 43 | Normal | 32 | 73 | 0.8 | 0.9 | None |
| Patient 2 | |||||||
| Proband | 43 | Severely enlarged | 66 | 54 | 0.9 | 1.0 | Mild |
| II-3 | |||||||
| I-1 | 65 | Moderately enlarged | 72 | 55 | 1.1 | 2.3 | None |
| I-2 | 44 | Mildly enlarged | 63 | 68 | 1.0 | 1.0 | None |
LVEDV = Left ventricular end-diastolic volume, RA = right atrial, LA = left atrial, LVEF = left ventricular ejection fraction, LVEDPW = left ventricular end-diastolic posterior wall, LVEDSW = left ventricular end-diastolic septal wall, RVH = right ventricular hypertrophy (note normal range is noted in parentheses)
RA size is normal, mildly, moderately, or severely enlarged.
RVH is designated as none (normal), mild, moderate, or severe.
Her symptoms appeared to improve slightly after diuresis. A cardiac MRI mirrored the echocardiographic findings. In addition, areas of mild focal hypertrophy and juxtaposed wall thinning were noted in the septum. Following administration of gadolinium contrast, markedly delayed enhancement was noted in the interventricular septum, suggesting myocardial fibrosis. This was seen most prominently in the mid-myocardium but sparing the endocardium.
Left and right heart cardiac catheterization revealed elevated right-sided and left sided-filling pressures. There was a prominent y-descent, suggestion of a “dip-and-plateau,” and ventricular concordance, all classically described in restrictive cardiomyopathy. Right ventricular endomyocardial biopsy revealed marked myocyte hypertrophy and myofiber disarray with interstitial fibrosis, and no evidence of infiltrative or storage disease (Fig 1).
Figure 1.
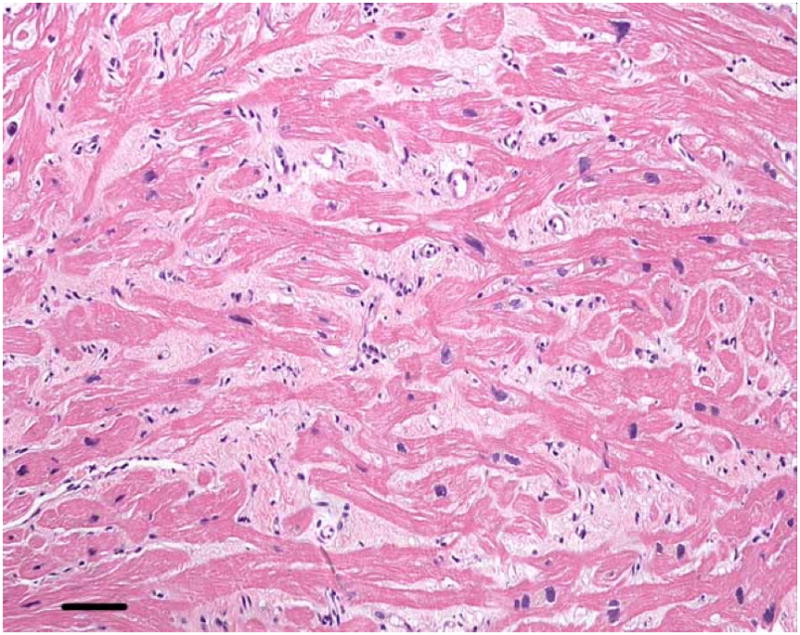
Right ventricular endomyocardial biopsy from Patient 1.
At high magnification the hypertrophied muscle fibers (dark pink) are separated by abundant interstitial fibrosis (light pink). Coursing in various directions, the myofibers in some areas are oriented orthogonally, characteristic of myofiber disarray. Hematoxylin and eosin, calibration bar = 45 microns. Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.
The patient denied a family history of cardiomyopathy but the lack of family history could not be confirmed because supporting records could not be obtained (Fig 2).
Figure 2.
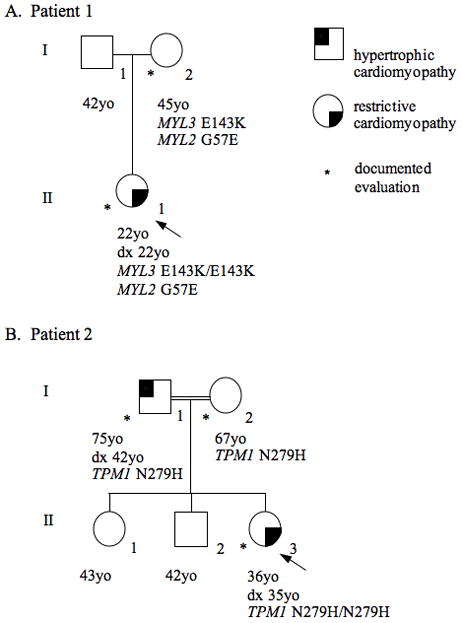
Pedigree from Patient 1 (A) and Patient 2 (B) demonstrating incomplete dominance, reduced penetrance, and variable expressivity.
Sequencing of the exons and intron-exon boundaries of the ACTC, MYBPC3, MYH7, MYL2, MYL3, TNNI3, TNNT2, and TPM1 genes was ordered from a commercial CLIA-certified laboratory (Correlagen Diagnostics, Inc.). This found previously reported homozygous mutations in the essential myosin light chain gene (MYL3, OMIM 160790), c.427G>A, p.Glu143Lys, and a novel heterozygous mutation in the regulatory myosin light chain (MYL2, OMIM 160781), c.170G>A, p.Gly57Glu (see supporting information Figure 1 which may be found in the online version of this article). The p.Glu143Lys mutation in MYL3 had been described previously in a Guatemalan family with HCM with restrictive physiology [Olson et al., 2002]. Olson et al reported this variant was absent in 150 controls (ancestry not reported), the clinical laboratory did not observe it in their 93 controls of mixed ancestry and we did not observe it in an additional 102 control individuals of Central American origin in our laboratory. The variant was entered into dbSNP(build 36) as rs104893750 based on the report by Olson et al. but has not been found independently by any other SNP discovery efforts. SNP rs104893750 is absent from HapMap data release 28.
The p.Gly57Glu mutation in MYL2 was also absent in the clinical laboratory’s 93 controls of mixed ancestry and our 102 control individuals of Central American origin. Codon 57 is part of a highly conserved EF-hand domain (Fig 3). A different SNP, rs2428140, was reported at the same site, which causes a p.Gly57Arg missense mutation. This SNP was discovered in a group of 5 individuals, from the following populations: European-American (two individuals), African-American, Hispanic-Mexican, and Asian-Chinese but was not seen in 60 Europeans (CEU), 45 East Asians and 60 Yoruban individuals used for HapMap data release 28 nor was it present in our 102 control individuals of Central American origin. There is also a mutation at neighboring codon 58, p.Arg58Gln, that significantly alters the calcium binding properties of the protein [Szczesna et al., 2001] and has been reported to co-segregate with HCM [Flavigny et al., 1998]. The MYL2 mutation found in Patient 1 changes the first nucleotide of exon 4 from G, which is the most frequent base at this position, to A, which is the next most frequent base [Cartegni et al., 2002]. The predicted effect of the variant on splicing at the 3’ splice site of intron 3 immediately preceding exon 4 was found to be minimal, based on analysis using splice prediction server Splicing Regulation Online Graphical Engine (http://sroogle.tau.ac.il/) [Schwartz et al., 2009].
Figure 3.

Alignment of MYL2 (A) and TPM1 (B) mutation-flanking regions showing evolutionary conservation of the mutated residues and neighboring amino acids across and within species.
Based on these data, the homozygous p.Glu143Lys mutations in MYL3 and the heterozygous p.Gly57Glu mutation in MYL2 were considered very likely to contribute to RCM in this patient.
The patient’s mother was the only family member available for evaluation (Fig 2). On genetic testing she was found to be a double heterozygote for the p.Glu143Lys mutation in MYL3 and the p.Gly57Glu mutation in MYL2. She had a normal transthoracic echocardiogram, electrocardiogram, and physical exam at 45 years of age (Table I).
The patient went on to develop recurrent syncope and had an automatic implantable cardiac defibrillator placed. She continued to suffer from severe heart failure and underwent orthotopic heart transplantation six months after she was initially diagnosed with RCM.
Patient 2
A 36-year-old female of Italian extraction was referred for chest pain, lightheadedness, shortness of breath, significant fatigue, dyspnea on exertion, three-pillow orthopnea and abdominal bloating. Eighteen months previously, she had been diagnosed with an unspecified form of cardiomyopathy after developing volume overload while hospitalized for a right oopherectomy and uterine fibroid removal. She denied syncope and lower extremity edema. Her symptoms were consistent with NYHA III–IV heart failure.
Transthoracic echocardiogram was consistent with restrictive cardiomyopathy; systolic function was preserved, and there was severe biatrial enlargement with no left ventricular hypertrophy or dilatation and only minimal mitral and tricuspid regurgitation (Table I). There was Doppler evidence of restrictive filling pattern.
Right and left heart catheterization on two occasions 16 months apart revealed consistently elevated left ventricular end diastolic pressure (24 and 32 mm Hg respectively) and elevated right ventricular end diastolic pressure of 18 mm Hg on the second catheterization. Cardiac index by Fick or thermodilution methods was markedly reduced at 1.4 L/min/m2 (Fick) and 1.66 L/min/m2 (thermodilution) during the first catheterization and 1.47 L/min/m2 and 1.75L/min/m2 at the second catheterization. The pattern was consistent with restrictive physiology although the dip-plateau (“square-root sign”) was notably absent.
An endomyocardial biopsy showed moderate myocyte hypertrophy and mild interstitial fibrosis. Myofiber disarray was not identified. There was no evidence of amyloid or iron.
Sequencing of the exons and intron-exon boundaries of MYH7, MYBPC3, TNNT2, TNNI3, TPM1, MYL3, MYL2, ACTC, GLA, LAMP2, and PRKAG2 was performed at a commercial CLIA-certified laboratory (Laboratory for Molecular Medicine). This testing revealed that she was homozygous for a novel missense mutation in α-tropomyosin (TPM1, OMIM 191010), c.835A>C p.Asn279His (see supporting information Figure 1 which may be found in the online version of this article). This mutation was absent in 600 Caucasian controls. Codon 279 is highly conserved across species (Fig 3). A variant at neighboring codon 281 has been reported in association with HCM: p.Met281Thr [Van Driest et al., 2003].
The patient’s parents are first cousins and confirmed heterozygotes for p.Asn279His (Fig 2). The patient’s 75-year-old father was diagnosed with HCM at 42 years of age. He reportedly has a history of heart failure but is currently asymptomatic. His most recent echocardiogram showed moderate asymmetric hypertrophy, mild pulmonary hypertension, mild left ventricular systolic dysfunction and moderate biatrial enlargement, indicating a chronic restrictive physiology (Table I). The patient’s 67-year-old mother had her first echocardiogram after the patient’s diagnosis. There was no evidence of asymmetric septal hypertrophy. Septal and posterior wall thicknesses were at the upper limits of normal and there was mild biatrial enlargement with normal systolic function and no significant evidence of restrictive physiology (Table I).
The patient has continued to have significant heart failure symptoms that have been difficult to control with medical therapy. Freedom from arrhythmias, however, has been notable. She is currently being considered for orthotopic heart transplantation but her evaluation has been complicated by psychiatric issues.
DISCUSSION
Mutations in several, but not all, of the sarcomere genes have previously been reported with RCM. To the best of our knowledge, these two cases are the first reports of mutations in TPM1, MYL3 and MYL2 with a purely restrictive phenotype. With the addition of these cases, mutations in all eight major sarcomere genes have now been associated with RCM.
The common genetic etiology underlying hypertrophic, dilated, restrictive and non-compaction cardiomyopathy has important implications for clinical care and research. Recognition that many cases of cardiomyopathy formerly thought to be idiopathic are in fact due to genetic mutations suggests that the relatives of individuals with any of the primary cardiomyopathies may be at risk to develop cardiomyopathy and should undergo cardiac screening for development of cardiomyopathy [Hershberger et al., 2009; Kaski et al., 2008]. While most cases of cardiomyopathy “run true” within a family it is important to keep in mind that the same mutation can cause a different cardiomyopathy, both in different families and within the same family [Menon et al., 2008; Moller et al., 2009], as was the case with patient 2, whose father carries a diagnosis of HCM. It is not yet known what modifying factors, genetic or environmental, determine which specific cardiomyopathy phenotype develops when someone has a sarcomere mutation.
Given the ever-broadening link between primary cardiomyopathies and the sarcomere, it may be advantageous for genetic testing for any one of hypertrophic, dilated, restrictive and non-compaction cardiomyopathy to include all of the sarcomere genes, as well as other non-sarcomere genes associated with that specific phenotype. This will increase the likelihood that genetic testing will find the causative mutation and thus permit predictive testing in at-risk relatives and ongoing cardiac screening targeted to relatives who carry the mutation [Hershberger et al., 2009]. Genetic testing can also help determine the precise cardiovascular diagnosis, particularly in the case of RCM. Historically, endomyocardial biopsies have been needed to differentiate between primary and secondary disease. In the future, this invasive test may no longer be needed if genetic testing can clarify the underlying cause of the disease by finding a mutation in a sarcomere gene or in a gene associated with infiltrative disease, such as TTR or DES.
Recognition of shared genetic etiology among primary cardiomyopathies may have implications for treatment and prevention of these conditions. For instance, recognition of the role of calcium handling in the development of HCM led to studies in rodent models demonstrating that the L-type Ca2+ channel inhibitor diltiazem can prevent development of hypertrophy in mutation carriers [Semsarian et al., 2002]. Clinical trials investigating the efficacy of diltiazem in the prevention of HCM in currently unaffected mutation carriers are ongoing (NCT00319982). Similar studies may be appropriate for the other sarcomere cardiomyopathies.
Cardiomyopathies have traditionally been classified based on cardiac function and morphology [Elliott et al., 2008; Maron et al., 2006]. Recognition that dilated, hypertrophic, restrictive and non-compaction cardiomyopathies can all be caused by sarcomere mutations raises the question of whether cardiomyopathies should instead be classified by genetic etiology [Thiene et al., 2008]. However, retaining classifications based on morphology and functions are important, as they facilitate prognostication and clinical management [Elliott et al., 2008]. For instance, HCM patients tend to have a reasonable prognosis if sudden cardiac death is prevented, whereas RCM patients often suffer a poor prognosis due to significant heart failure [Ammash et al., 2000; Cecchi et al., 1995; Elliott et al., 2000; Maron et al., 2000]. Perhaps the most effective way to classify cardiomyopathies to guide both clinical care and research endeavors is both by their molecular etiology and their clinical presentation, using genotype and phenotype.
About 5% of individuals with HCM have two sarcomere mutations [Kelly and Semsarian 2009; Van Driest et al., 2005]. Double heterozygotes and compound heterozygotes have been reported in RCM [Blok et al., 2005; Peddy et al., 2006]. To the best of our knowledge Patient 1 is the first reported case of three sarcomere mutations in RCM. Girolami et al recently reported four HCM cases with three mutations [Girolami et al., 2010]. In HCM cohorts, individuals with multiple mutations typically have a more severe disease with earlier age of onset, greater left ventricular hypertrophy and a higher incidence of sudden cardiac death [Girolami et al., 2010; Kelly and Semsarian 2009; Van Driest et al., 2005]. This pattern of incomplete dominance or ‘dosage effect’ is evident in the family history of Patient 2 (Fig 2). However, the case of Patient 1 demonstrates that even with two mutations, an at-risk relative can be free of disease well into adulthood. This suggests that the marked variability observed in cardiomyopathies due to one sarcomere mutation is also a factor when there are multiple mutations.
Performance of accurate genetic counseling depends on ascertaining all of the mutations that are segregating in a family. These cases underscore the importance of maximizing the likelihood of finding multiple mutations by starting genetic testing with the individual who is most severely affected and has the youngest onset of disease [Hershberger et al., 2009; Kelly and Semsarian, 2009]. The importance of finding multiple mutations argues for testing the index case with a multi-gene panel, opposed to using a step-wise approach to genetic testing that starts with the most common gene(s) [Hershberger et al., 2009; Kelly and Semsarian, 2009]. Testing with more comprehensive panels has become more feasible recently with the application of next-generation sequencing technologies to clinical genetic testing [Dellefave et al., 2010].
Limitations of this Clinical Report include the limited amount of segregation data, lack of functional data, and the limited number of ancestry-matched controls available for Patient 1. Associations between RCM and the identified mutations are based on conservation, prior reports of the same or related mutations, absence in ancestry-matched healthy controls, and biological plausibility given prior associations of other sarcomere genes and RCM.
These two patients provide further association between RCM and the sarcomere. With the addition of the patients we report here, all sarcomere genes have now been associated with RCM. These cases further our emerging understanding of the association between sarcomere mutations and a range of primary cardiomyopathies [Dellefave and McNally 2008; Perrot et al., 2007]. These patients also suggest that the phenotype of sarcomere mutations may differ within the same family, presenting either with hypertrophic or a restrictive disease. The reason for variable expressivity is unknown and may reflect the impact of mutant gene copy number, modifier genes, different environmental exposures or co-existing comorbidities, or some combination of these factors. While a few sarcomere genes have yet to be implicated in dilated cardiomyopathy and left ventricular noncompaction it seems reasonable to hypothesize that each of these genes could be associated with each primary cardiomyopathy.
Supplementary Material
Sequence traces for the mutations identified in the probands in MYL3 (A), MYL2 (B), and TPM1 (C).
Acknowledgments
We thank the patients and their families.
References
- Ammash NM, Seward JB, Bailey KR, Edwards WD, Tajik AJ. Clinical profile and outcome of idiopathic restrictive cardiomyopathy. Circulation. 2000;101:2490–2496. doi: 10.1161/01.cir.101.21.2490. [DOI] [PubMed] [Google Scholar]
- Blok R, van den Wijngaard A, Merckx D, Marcelis C, Rubio E, de Coo C, de Rie R, Jongbloed R, Smeets B. Two novel TNNI3 mutations in restrictive cardiomyopathy. Eur J Hum Genet. 2005;13:108. [Google Scholar]
- Cartegni L, Chew SL, Krainer AR. Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat Rev Genet. 2002;3:285–298. doi: 10.1038/nrg775. [DOI] [PubMed] [Google Scholar]
- Cecchi F, Olivotto I, Montereggi A, Santoro G, Dolara A, Maron BJ. Hypertrophic cardiomyopathy in Tuscany: clinical course and outcome in an unselected regional population. J Am Coll Cardiol. 1995;26:1529–1536. doi: 10.1016/0735-1097(95)00353-3. [DOI] [PubMed] [Google Scholar]
- Dellefave L, McNally EM. Sarcomere mutations in cardiomyopathy, noncompaction, and the developing heart. Circulation. 2008;117:2847–2849. doi: 10.1161/CIRCULATIONAHA.108.781518. [DOI] [PubMed] [Google Scholar]
- Dellefave L, McNally EM. The genetics of dilated cardiomyopathy. Curr Opin Cardiol. 2010;25:7. doi: 10.1097/HCO.0b013e328337ba52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, Dubourg O, Kuhl U, Maisch B, McKenna WJ, Monserrat L, Pankuweit S, Rapezzi C, Seferovic P, Tavazzi L, Keren A. Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2008;29:270–276. doi: 10.1093/eurheartj/ehm342. [DOI] [PubMed] [Google Scholar]
- Elliott PM, Poloniecki J, Dickie S, Sharma S, Monserrat L, Varnava A, Mahon NG, McKenna WJ. Sudden death in hypertrophic cardiomyopathy: identification of high risk patients. J Am Coll Cardiol. 2000;36:2212–2218. doi: 10.1016/s0735-1097(00)01003-2. [DOI] [PubMed] [Google Scholar]
- Flavigny J, Richard P, Isnard R, Carrier L, Charron P, Bonne G, Forissier JF, Desnos M, Dubourg O, Komajda M, Schwartz K, Hainque B. Identification of two novel mutations in the ventricular regulatory myosin light chain gene (MYL2) associated with familial and classical forms of hypertrophic cardiomyopathy. J Mol Med. 1998;76:208–214. doi: 10.1007/s001090050210. [DOI] [PubMed] [Google Scholar]
- Geisterfer-Lowrance AA, Kass S, Tanigawa G, Vosberg HP, McKenna W, Seidman CE, Seidman JG. A molecular basis for familial hypertrophic cardiomyopathy: a beta cardiac myosin heavy chain gene missense mutation. Cell. 1990;62:999–1006. doi: 10.1016/0092-8674(90)90274-i. [DOI] [PubMed] [Google Scholar]
- Girolami F, Ho CY, Semsarian C, Baldi M, Will ML, Baldini K, Torricelli F, Yeates L, Cecchi F, Ackerman MJ, Olivotto I. Clinical features and outcome of hypertrophic cardiomyopathy associated with triple sarcomere protein gene mutations. J Am Coll Cardiol. 2010;55:1444–1453. doi: 10.1016/j.jacc.2009.11.062. [DOI] [PubMed] [Google Scholar]
- Hershberger RE, Lindenfeld J, Mestroni L, Seidman CE, Taylor MR, Towbin JA. Genetic evaluation of cardiomyopathy--a Heart Failure Society of America practice guideline. J Card Fail. 2009;15:83–97. doi: 10.1016/j.cardfail.2009.01.006. [DOI] [PubMed] [Google Scholar]
- Kaski JP, Syrris P, Burch M, Tome-Esteban MT, Fenton M, Christiansen M, Andersen PS, Sebire N, Ashworth M, Deanfield JE, McKenna WJ, Elliott PM. Idiopathic restrictive cardiomyopathy in children is caused by mutations in cardiac sarcomere protein genes. Heart. 2008;94:1478–1484. doi: 10.1136/hrt.2007.134684. [DOI] [PubMed] [Google Scholar]
- Kelly M, Semsarian C. Multiple Mutations in Genetic Cardiovascular Disease. A Marker of Disease Severity? Circ Cardiovasc Genet. 2009:182–190. doi: 10.1161/CIRCGENETICS.108.836478. [DOI] [PubMed] [Google Scholar]
- Kubo T, Gimeno JR, Bahl A, Steffensen U, Steffensen M, Osman E, Thaman R, Mogensen J, Elliott PM, Doi Y, McKenna WJ. Prevalence, clinical significance, and genetic basis of hypertrophic cardiomyopathy with restrictive phenotype. J Am Coll Cardiol. 2007;49:2419–2426. doi: 10.1016/j.jacc.2007.02.061. [DOI] [PubMed] [Google Scholar]
- Maron BJ, Olivotto I, Spirito P, Casey SA, Bellone P, Gohman TE, Graham KJ, Burton DA, Cecchi F. Epidemiology of hypertrophic cardiomyopathy-related death: revisited in a large non-referral-based patient population. Circulation. 2000;102:858–864. doi: 10.1161/01.cir.102.8.858. [DOI] [PubMed] [Google Scholar]
- Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, Moss AJ, Seidman CE, Young JB. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 2006;113:1807–1816. doi: 10.1161/CIRCULATIONAHA.106.174287. [DOI] [PubMed] [Google Scholar]
- Menon SC, Michels VV, Pellikka PA, Ballew JD, Karst ML, Herron KJ, Nelson SM, Rodeheffer RJ, Olson TM. Cardiac troponin T mutation in familial cardiomyopathy with variable remodeling and restrictive physiology. Clin Genet. 2008;74:445–454. doi: 10.1111/j.1399-0004.2008.01062.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogensen J, Arbustini E. Restrictive cardiomyopathy. Curr Opin Cardiol. 2009;24:214–220. doi: 10.1097/hco.0b013e32832a1d2e. [DOI] [PubMed] [Google Scholar]
- Mogensen J, Kubo T, Duque M, Uribe W, Shaw A, Murphy R, Gimeno JR, Elliott P, McKenna WJ. Idiopathic restrictive cardiomyopathy is part of the clinical expression of cardiac troponin I mutations. J Clin Invest. 2003;111:209–216. doi: 10.1172/JCI16336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moller DV, Andersen PS, Hedley P, Ersboll MK, Bundgaard H, Moolman-Smook J, Christiansen M, Kober L. The role of sarcomere gene mutations in patients with idiopathic dilated cardiomyopathy. Eur J Hum Genet. 2009;17:1241–1249. doi: 10.1038/ejhg.2009.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monserrat L, Hermida-Prieto M, Fernandez X, Rodriguez I, Dumont C, Cazon L, Cuesta MG, Gonzalez-Juanatey C, Peteiro J, Alvarez N, Penas-Lado M, Castro-Beiras A. Mutation in the alpha-cardiac actin gene associated with apical hypertrophic cardiomyopathy, left ventricular non-compaction, and septal defects. Eur Heart J. 2007;28:1953–1961. doi: 10.1093/eurheartj/ehm239. [DOI] [PubMed] [Google Scholar]
- Olson TM, Karst ML, Whitby FG, Driscoll DJ. Myosin light chain mutation causes autosomal recessive cardiomyopathy with mid-cavitary hypertrophy and restrictive physiology. Circulation. 2002;105:2337–2340. doi: 10.1161/01.cir.0000018444.47798.94. [DOI] [PubMed] [Google Scholar]
- Olson TM, Michels VV, Thibodeau SN, Tai YS, Keating MT. Actin mutations in dilated cardiomyopathy, a heritable form of heart failure. Science. 1998;280:750–752. doi: 10.1126/science.280.5364.750. [DOI] [PubMed] [Google Scholar]
- Peddy SB, Vricella LA, Crosson JE, Oswald GL, Cohn RD, Cameron DE, Valle D, Loeys BL. Infantile restrictive cardiomyopathy resulting from a mutation in the cardiac troponin T gene. Pediatrics. 2006;117:1830–1833. doi: 10.1542/peds.2005-2301. [DOI] [PubMed] [Google Scholar]
- Perrot A, Dietz R, Osterziel KJ. Is there a common genetic basis for all familial cardiomyopathies? Eur J Heart Fail. 2007;9:4–6. doi: 10.1016/j.ejheart.2006.04.010. [DOI] [PubMed] [Google Scholar]
- Schwartz S, Hall E, Ast G. SROOGLE: webserver for integrative, user-friendly visualization of splicing signals. Nucleic Acids Res. 2009;37:W189–192. doi: 10.1093/nar/gkp320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semsarian C, Ahmad I, Giewat M, Georgakopoulos D, Schmitt JP, McConnell BK, Reiken S, Mende U, Marks AR, Kass DA, Seidman CE, Seidman JG. The L-type calcium channel inhibitor diltiazem prevents cardiomyopathy in a mouse model. J Clin Invest. 2002;109:1013–1020. doi: 10.1172/JCI14677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczesna D, Ghosh D, Li Q, Gomes AV, Guzman G, Arana C, Zhi G, Stull JT, Potter JD. Familial hypertrophic cardiomyopathy mutations in the regulatory light chains of myosin affect their structure, Ca2+ binding, and phosphorylation. J Biol Chem. 2001;276:7086–7092. doi: 10.1074/jbc.M009823200. [DOI] [PubMed] [Google Scholar]
- Thiene G, Corrado D, Basso C. Revisiting definition and classification of cardiomyopathies in the era of molecular medicine. Eur Heart J. 2008;29:144–146. doi: 10.1093/eurheartj/ehm585. [DOI] [PubMed] [Google Scholar]
- Van Driest SL, Ellsworth EG, Ommen SR, Tajik AJ, Gersh BJ, Ackerman MJ. Prevalence and spectrum of thin filament mutations in an outpatient referral population with hypertrophic cardiomyopathy. Circulation. 2003;108:445–451. doi: 10.1161/01.CIR.0000080896.52003.DF. [DOI] [PubMed] [Google Scholar]
- Van Driest SL, Ommen SR, Tajik AJ, Gersh BJ, Ackerman MJ. Sarcomeric genotyping in hypertrophic cardiomyopathy. Mayo Clin Proc. 2005;80:463–469. doi: 10.1016/S0025-6196(11)63196-0. [DOI] [PubMed] [Google Scholar]
- Ware SM, Quinn ME, Ballard ET, Miller E, Uzark K, Spicer RL. Pediatric restrictive cardiomyopathy associated with a mutation in beta-myosin heavy chain. Clin Genet. 2008;73:165–170. doi: 10.1111/j.1399-0004.2007.00939.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Sequence traces for the mutations identified in the probands in MYL3 (A), MYL2 (B), and TPM1 (C).