

Abstract
We have determined 2.2 Å resolution crystal structure of Thermotoga maritima CheB methylesterase domain to provide insight into the interaction mode between CheB and chemoreceptors. T. maritima CheB methylesterase domain has identical topology of a modified doubly-wound α/β fold that was observed from the previously reported Salmonella typhimurium counterpart, but the analysis of the electrostatic potential surface near the catalytic triad indicated considerable charge distribution difference. As the CheB demethylation consensus sites of the chemoreceptors, the CheB substrate, are not uniquely conserved between T. maritima and S. typhimurium, such surfaces with differing electrostatic properties may reflect CheB regions that mediate protein-protein interaction. Via the computational docking of the two T. maritima and S. typhimurium CheB structures to the respective T. maritima and Escherichia coli chemoreceptors, we propose a CheB:chemoreceptor interaction mode.
Keywords: CheB, Methylesterase, Thermotoga maritima, Chemoreceptor, Methyl-accepting chemotaxis proteins, Protein-protein interaction
Introduction
Bacterial chemotaxis is a signal transduction cascade which allows bacteria to swim towards an attractant or away from a repellent chemical. Bacterial chemotaxis begins in response to the sensing of extracellular chemicals by the membrane-spanning chemoreceptors (also known as methyl-accepting chemotaxis proteins; MCPs), and bacteria use their flagella to either smooth-swim forward, or to tumble resulting in a randomly changing swimming direction. In bacteria such as Escherichia coli, the respective smooth-swimming and tumbling behavior is the result of alternation between a counter-clockwise (CCW) and a clockwise (CW) flagellar rotation [1–10]. In E. coli, a chemical ligand binding to the extracellular domain of the chemoreceptor is transmitted to the cytoplamsic CheA kinase, thus triggering kinase activity. CheA-bound ATP is auto-phosphorylated to a histidine of CheA, and is subsequently transferred to an aspartate of the two response-regulators, CheY and CheB. CheY and CheB bind competitively to CheA [11], and consequently determine two responses: “excitation” and “adaptation”. In the “excitation” process mediated by CheY, phospho-CheY detaches from CheA to interact with the switch protein FliM at the flagellar motor complex, which alters the rotational direction of the flagella.
However, after the CheY-mediated response, bacteria return to the pre-stimulus state via a slower “adaptation” process mediated by CheB. Adaptation can be viewed as a negative feedback mechanism that allows the bacteria to detect temporally increasing gradients of chemicals, by preventing saturation. In general, two chemoreceptor modifying enzymes - CheR and CheB - each catalyze the methylation and demethylation of glutamates in the chemoreceptors, and the level of reversible methylation during the “adaptation” behavior enables the chemoreceptor’s attenuated sensitivity to the ligand. Methyltransferase CheR constantly methylates the chemoreceptors via S-adenosyl-L-methionine, and methylesterase CheB functions in accordance with the activated CheA. CheB binds to CheA via an N-terminal CheY-like response-regulator domain which becomes phosphorylated by CheA. As is seen in CheY after CheA phosphorylation, phospho-CheB detaches from CheA, resulting in a concomitant opening of the C-terminal catalytic domain (CheBc) for catalytic methylesterase activity [12–15]. The activated CheBc subsequently hydrolyzes the methyl glutamates of the chemoreceptor, and results in the deactivation of the chemoreceptor, thus setting a new threshold compared to the previous chemical concentration.
Glutamates that undergo CheR-mediated methylation and CheB-mediated demethylation are located at the cytoplasmic regions of the membrane-spanning chemoreceptors. The CheR-mediated methylation consensus sites have been defined for chemoreceptors of Salmonella enterica Tar [16] and E. coli Tsr [17], and the protein-protein interaction surface harboring the glutamate methylation sites has been predicted via docking studies [18] using the structures of the E. coli Tsr chemoreceptor (PDB code: 1QU7) [19] and CheR (PDB code: 1BC5) [20]. A recent study has also defined the CheR-mediated methylation consensus sites of Thermotoga maritima chemoreceptors, and the results demonstrate that the sites are not uniquely conserved between distant bacterial species of S. enterica or E. coli [22].
Despite the known crystal structures of full-length Salmonella typhimurium CheB (PDB code: 1A2O) [15] and CheBc (PDB code: 1CHD) [21], only limited attempts have been made to model the CheBc and the chemoreceptor interaction. Provided that the interaction modes are conserved between distant bacterial species, an improved model may be generated by comparing the docking results from more than two sets of interacting protein pairs. For this approach, we have determined the crystal structure of T. maritima CheBc, and by using this structure together with the structures of S. typhimurium CheBc [15, 21], T. maritima chemoreceptor [23] and E. coli chemoreceptor [19], we propose a CheBc and chemoreceptor interaction model.
Materials and methods
Protein Preparation
The genes encoding T. maritima methylesterase domain of CheB (CheBc: residues 153–344) were PCR cloned into the vector pET28a (Novagen) using the T. maritima genomic DNA (ATCC). The protein was expressed with N-terminal His6-tag in E. coli strain BL21 (DE3) (Stratagene) using kanamycin selection (25 μg/mL). Using a conventional shaker, the transformed cells were grown at 37 °C in 2 L of Terrific Broth medium to OD600= ~0.6. The recombinant protein expression was induced with 0.5 mM isopropyl β-D-thiogalactopyranoside (IPTG), and the cells were further grown at 25 °C for 16 hrs. The cell pellets were harvested using centrifugation at 4500 g for 10 mins at 4°C, and were re-suspended in ice-cold lysis buffer (20 mM TRIS pH 7.5, 500 mM sodium chloride and 5 mM imidazole) prior to homogenization by sonication.
The cell lysate was centrifuged at 70000g for 30 mins at 4°C. The supernatants were loaded onto Nickel-NTA columns, washed with wash buffer (20 mM TRIS pH 7.5, 500 mM sodium chloride and 20 mM imidazole) and the recombinant proteins were eluted with elution buffer (20 mM TRIS pH 7.5, 500 mM sodium chloride and 200 mM imidazole). The His6-tag was removed by treating the eluted protein with human thrombin (Roche) for 16 hrs at 4°C. The protein was further purified using a Superdex 200 sizing column (GE Healthcare) equilibrated in gel-filtration buffer (50 mM TRIS pH 7.5 and 150 mM NaCl). The eluting fractions corresponding to CheBc were concentrated to ~50 mg/mL by centrifugation (Amicon Millipore). Protein purity determined on SDS-PAGE was >95% and the concentration was estimated by absorption at λ=280 nm by employing the calculated molar extinction coefficient of 7680 M−1 cm−1 (SWISS-PROT; http://www.expasy.ch/).
Crystallization and Data Collection
Conditions for growing T. maritima CheBc crystals were found in commercial screening solutions (Hampton Research). Crystallization screenings were performed at 25 °C by using hanging drop vapor diffusion method where the protein and the well solutions were mixed in 1:1 ratio of total 2 μL volumes. Diffraction quality crystals grew after two months against a reservoir of 0.2 M ammonium sulfate, 0.1 M cacodylate pH 6.5 and 30% (w/v) PEG 8K. Diffraction data were collected under a 100 K nitrogen stream at a NSLS beamline (X25) on a CCD detector (ADSC Quantum 315). The crystal belongs to the space group P212121 and contains one molecule per asymmetric unit. Data were processed by DENZO and SCALEPACK [24].
Structure determination and refinement
The position of CheBc was determined by AMoRe molecular replacement [25] with free catalytic domain of S. typhimurium CheB as a search model (PDB code: 1CHD) and the diffraction data of 4.0 – 10.0 Å resolution. The entire model of CheBc was automatically built with ARP/wARP and SIDEgui [26] in CCP4 suite [27]. The final model (one CheBc molecule in asymmetric unit, residues 156–344) was further refined in CNS (R-factor = 0.204, Rfree = 0.223 where Rfree is the R-factor for 10% of randomly selected reflections excluded from the refinement.) [28].
Computational modelling of CheB and chemoreceptor interaction
The structures of the S. typhimurium CheB methylesterase domain (PDB code: 1CHD) and the E. coli chemoreceptor Tsr (PDB code: 1QU7; edited only to include α-helices containing the methylation Glu/Gln of residues 294-320 and 464-493 in each subunit), both lacking all heteroatoms, were docked using the ZDOCK program [29]. This program carries out rigid-body docking at all possible orientations between the two proteins, CheBc and Tsr. Among them, 1,000 predicted complexes were selected based on the scoring function implemented in the program. The RDOCK program [30] was used for refinement and energy minimization of the complexes with a CHARMM force field [31]. The docked complexes were ranked based on the shape complementarities and electrostatic considerations implemented in the program, and only the top 40% were considered for the subsequent filtering processes. The top 40% complexes were filtered further on the basis of the constraints of the distance measured from three residues of the CheB catalytic triad (Ser164, His190 and Asp286) and each of the methylation Glu/Gln residues (Gln297, Glu304, Gln311). Cα-to-Cα distance constraints of at least one triad residue to Glu (or Gln) within 9 Å, and the sum of three triad residues to Glu (or Gln) within 32 Å were applied. This filtering step yielded two complexes for the methylation site Gln297, two complexes for site Glu304, and six complexes for site Gln311.
Similar steps were applied for the docking of T. maritima CheBc and the T. maritima chemoreceptor TM1143 (PDB code: 2CH7; edited to only to include α-helices containing the methylation Glu/Gln of residues 262-297 and 454-489 in each subunit). The two structures were docked using ZDOCK and minimized with RDOCK. The docked complexes were ranked and only the top 40% were considered for the subsequent filtering process by Cα-to-Cα distance. Distance constraints were based on the distance measured from three residues of the CheB catalytic triad (Ser166, His193 and Asp289) and each of the methylation Glu/Gln residues (Gln281 and Glu294). Cα-to-Cα distance constraints of at least one triad residue to Glu/Gln within 10 Å, and the sum of three triad residues to Glu-Gln within 30 Å were applied. This filtering step yielded only two complexes (no. 400 and no. 554) for the Gln281 methylation site. The 10 complexes resulting in the case of E. coli (or S. typhimurium) proteins and two complexes for the T. maritima proteins were assessed and compared using the PyMol molecular graphics display program [31]. All the structural figures were also rendered with PyMol [32].
Results and Discussion
Overall structure of T. maritima CheBc
The crystal structure of T. maritima CheB methylesterase domain was determined to a resolution of 2.2 Å using a S. typhimurium CheB structure (PDB code: 1CHD) as a molecular replacement model. The refined model harbours one α/β sandwich-fold CheBc molecule within the crystal asymmetric unit, which is composed of nine β-strands, six α-helices, and one 310-helix (Fig. 1). Among the 192 residues designed in the protein construct, 189 residues were built in the model. The three N-terminal CheB residues (153-KPA-155) were not included in the model, as their electron densities were not clearly discernable. 106 water molecules were also included. Data collection and refinement statistics are provided in Table 1 and Table 2.
Figure 1. Sequence alignment of CheBc from five organisms.

CheBc sequences from T. maritima, E. coli, S. typhimurium, Bacillus subtilis and Myxococcus xanthus were aligned on the basis of the amino acid sequences. Secondary structural elements are indicated as block rectangles (α-helix) and arrows (β-sheet) on top of the corresponding amino acid residues. Conserved residues are boxed around the amino acids. Catalytically critical residues (Ser-His-Asp triad) are indicated with an asterisk.
Table 1.
Data Collection and Phasing Statistics
| Wavelength(Å) | 1.100 |
| Resolution(Å) | 30–2.15 |
| Highest Shell | (2.23–2.15) |
| Completeness (%) | 99.9(99.9) |
| Rmerge1 | 0.106(0.382) |
| I/σ(I) | 35(8.4) |
Rmerge = ΣΣj |Ij − <I>|/ΣΣj Ij
Table 2.
Refinement Statistics
| Space Group | P212121 (a= 52.47 Å, b=65.08 Å, c=78.35 Å) |
| Resolution range (Å) | 30–2.2 |
| Unique Reflections (test set) | 13555(1382) |
| Wilson B (Å2) | 28.51 |
| R-factor1 (Rfree2) | 0.204 (0.223) |
| No. of Scatters (No. of residues) | 2928(189) |
| No. of Water molecules | 106 |
| RMSD bonds (Å) | 0.00624 |
| RMSD angles(°) | 1.405 |
| Average B-factor (main chain) (Å2) | 24.29 |
| Average B-factor (side chain) (Å2) | 26.40 |
| Average B-factor (water) (Å2) | 38.31 |
R-factor = Σ(|Fobs|−|Fcalc|)/Σ|Fobs|
R-factor for 10% of randomly selected reflections excluded from the refinement.
The fold of T. maritima CheBc is identical to that of S. typhimurium CheBc, which is a modified, doubly wound α/β fold containing the central seven parallel β-strands flanked by six α-helices and an anti-parallel β-hairpin (Fig. 2). In brief, the fold starts from β1 and the subsequently alternating α-helices and β-strands (α1-β2/α2-β3/β4) form one core of the central parallel β-sheet (β1/β2/β4/β3). The following two β-strands (β5-β6) form an anti-parallel β-hairpin distal to the central parallel β-sheet, which result in a modified α/β fold. Finally, the three following sets of alternating α-helix and the β-strand (α3-β7/α4-β8/α5-β9) complete the second half of the central seven-stranded β-sheet (β9/β8/β7/β1/β2/β4/β3). The catalytic Ser-His-Asp triad which generates the methylesterase activity lie at the surface created mainly by loops that connect the β-strands and the α-helices (β1-α1 loop, β2-α2 loop, β6-α3 loop, β7-α4 loop). Two other loops (β3-β4 and 310 helix-α5 loop) are also in the vicinity of the catalytic triad (Fig. 2).
Figure 2. Similarities in the overall fold do not account for the dissimilarities in the electrostatic potential surface of T. maritima and S. typhimurium CheBc.
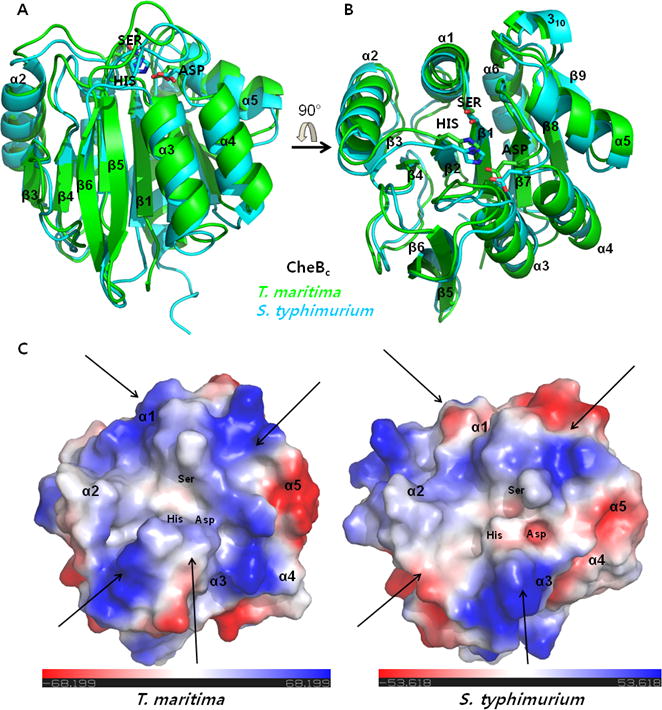
(A, B) Schematic ribbon diagrams of the superimposed structures of T. maritima and S. typhimurium CheBc are shown in two different orientations which are rotated 90° with respect to each other along the horizontal arrow. Three side chains of the catalytic triad are shown in sticks. (C) Differences in the electrostatic potential surface near the catalytic triad are highlighted using colors by charge. The surface was generated from the 90°-rotated orientation (B) which looks at the catalytic triad from the top. Dissimilar regions of charge distribution in T. maritima and S. typhimurium are mostly located around regions indicated with arrows. Since the consensus sequences adjacent to the methylation/demethylation glutamates of the chemoreceptors are not exactly conserved between the two species, such surface with electrostatic difference may be the sites of chemoreceptor interaction.
Surface electrostatic potential difference between T. maritima and S. typhimurium CheBc
As T. maritima and S. typhimurium CheBcs are conserved with 36% identity (54% similarity) in their protein sequences (Fig. 1), the superposition of T. maritima and S. typhimurium CheBcs (PDB code: 1CHD) is clearly indicative of an overlap of secondary structural elements (Cα RMSD=1.032 Å aligning 182 residues). Partial sequence conservation also occurs near the catalytic surface of the protein, which is created by the contiguous loops that connect the β-strands and the α-helices (Fig. 1, β1-α1 loop, β2-α2 loop, β6-α3 loop, β7-α4 loop). However, analysis of the electrostatic potential surface near the catalytic triad reflects considerable differences in the charge distribution between the two T. maritima and S. typhimurium CheBcs. The region of conspicuous differences is outlined along the arrows near the catalytic triad surface (Fig. 2C).
The CheR-mediated methylation consensus sites neighboring the modified glutamate have been defined for chemoreceptors of S. enterica Tar [17], E. coli Tsr [16], and several T. maritima chemoreceptors [22], and the results demonstrate that the sites are not uniquely conserved. As perfect complementarity of proteins, the fundamental driver of biological specificity, within biologically relevant complexes results during and requires co-evolution [33], such surfaces with differing electrostatic properties may reflect CheBc regions that mediate interactions with chemoreceptors.
Methylation-undergoing glutamates of chemoreceptors, which are also the substrates for methylesterase CheB, are located in the cytoplasmic regions of the membrane-spanning chemoreceptor. The methylation (or demethylation) consensus sequences (Glx-Glx-X-X-Ala-Ser/Thr, methylation residue is in bold) of S. enterica (Tar) and E. coli (Tsr) chemoreceptors [16, 17] have been shown to differ slightly from that of the T. maritima receptors (Ala/Ser-sm-X-Glx-Glu-X-sm-Ala/Ser, methylation residues are in bold; sm indicates small residues which are either Ala, Gly, Ser or Thr) [22]. Two CheBcs recognizing the different consensus sequence of the chemoreceptors can be mapped to the different surface complementarities which have co-evolved between the chemoreceptor substrate and the CheB enzyme. Electrostatic potential surfaces near the T. maritima and S. typhimurium CheBc catalytic triad with substantial differences suggested that two CheBc structures in conjunction with the chemoreceptor’s structural information may assist in generating a reliable CheB and chemoreceptor interaction model. A systematic docking study has been carried out using two sets of CheBc and chemoreceptor structures originating from T. maritima and S. typhimurium. Provided that the general interaction modes are conserved between the two distant bacterial species, an interaction model has been generated by comparing the overlapping docking results.
Prediction of chemoreceptor and CheB interaction mode
E. coli and S. typhimurium both belong to the Enterobacteriaceae family and are closely related in the bacterial phylogeny tree. As the CheBs from the two species also share 95% sequence identity, the structures of the cytoplasmic regions of the E. coli Tsr chemoreceptor (PDB code: 1QU7) and S. typhimurium CheBc (PDB code: 1CHD) were docked using ZDOCK and RDOCK. Docking was carried out solely using the truncated four-helix bundle of the chemoreceptor, which includes the methylation Glu/Gln (Gln297, Glu304, and Gln311). The docking results were sorted on the basis of the relevant complementarities and electrostatic considerations, and further filtered using distance constraints between the catalytic residues and methylation glutamates. The final result yielded 10 CheBc and chemoreceptor complexes in three Tsr methylation Glu/Gln residues (two complexes for site Gln297, two complexes for site Glu304, and six complexes for site Gln311). However, when the 10 complexes were superposed relative to CheBc, the positions of the chemoreceptor relative to CheBc - i.e. the directionality of the chemoreceptor four-helix bundle - in the 10 complexes were heterogeneous, which indicates that generating a single interaction mode is not possible (results not shown).
Similar docking steps were applied for the case of T. maritima CheBc and the TM1143 chemoreceptor (PDB code: 2CH7). A recently reported cytoplasmic structure of the T. maritima chemoreceptor TM0014 (PDB code: 3G67) [34] was not included in the docking set due to the absence of the methylation consensus sequences. Similar to the previous case, docking was carried out solely using the truncated four-helix bundle of the chemoreceptor which includes the methylation Glu/Gln (Gln294 and Gln281). After sorting and filtering processes using distance constraints, the final docking result with the T. maritima proteins yielded two CheBc and chemoreceptor complexes (no. 400 and no. 554) around only the Gln281 methylation residue. The positions of the chemoreceptor relative to CheBc in the two resultant complexes are practically identical, with only a slight ~1° shift in the four-helix bundle around Gln281 (results not shown). When the two docking complexes from T. marimita proteins were overlaid with the 10 docking complexes from E. coli proteins, only one E. coli docking pair (no. 640 from site Gln311) matched exactly with an identical helical arrangement of the chemoreceptor substrate relative to CheBc (Fig. 3). Surprisingly, when the α-helix of the chemoreceptor containing the demethylation site was observed outside the context of the four-helix bundle, it sits within a groove created at the adjunction of the four CheBc α-helices (α1, α2, α3, and α4, Fig. 4). The interaction surface is created by the contacting loops that connect the β-strands to the α-helices (β1-α1 loop, β2-α2 loop, β6-α3 loop, β7-α4 loop), and is the region where majority of the electrostatic potential surface differed between the two T. maritima and S. typhimurium CheBcs (Fig. 2C & Fig. 4). It is worth noting that this CheBc groove has also been previously predicted to be the chemoreceptor interface, which was concluded from the manual inspection of a 10 Å - diameter channel that accommodates an α-helix [21].
Figure 3. Proposed chemoreceptor docking site for T. maritima and S. typhimurium CheBc.
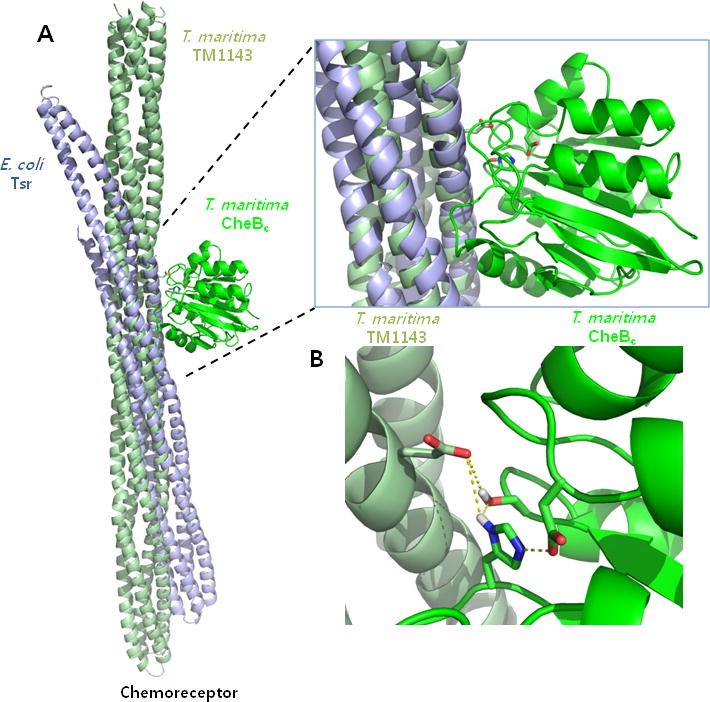
(A) Docking models were generated from the PDB coordinates of the chemoreceptors and the CheBcs from T. maritima and S. typhimurium using ZDOCK and RDOCK. Overall docking modes are shown with the chemoreceptor positions relative to the superimposed CheBc (Only T. maritima CheBc is shown for clarity). (B) The methylation residue for the T. maritima chemoreceptor (Gln281) is shown with sticks in relation to the catalytic triad. Distances indicate the measured bonding distance between atoms in the model.
Figure 4. View of the proposed docking site with chemoreceptor α-helix shown over the T. maritima CheBc electrostatic potential surface.
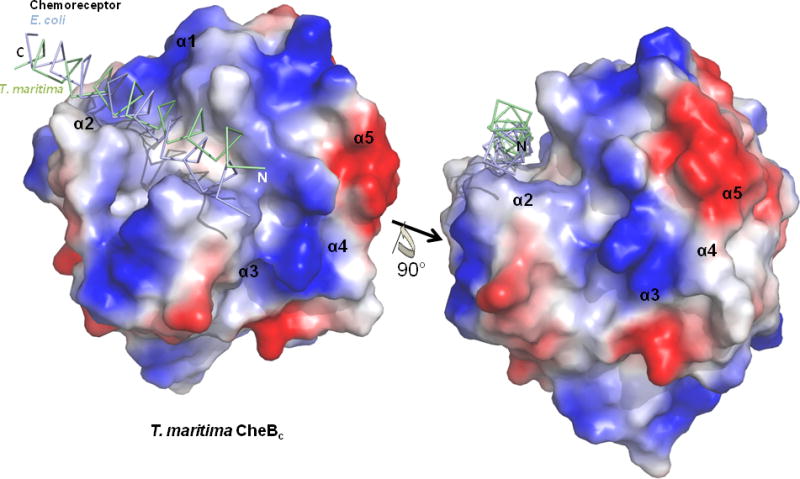
View of the truncated α-helical substrate of the T. maritima and E. coli chemoreceptor overlaid on top of the T. maritima CheBc electrostatic potential surface are shown in two different orientations, which are rotated 90° with respect to each other along the indicated axis (Only T. maritima CheBc is shown for clarity). The α-helical substrate of the chemoreceptor sits within a groove created at the active site.
We have succeeded in generating a probable model between the two interacting proteins using the systematic docking approach that defines the helical orientation of the chemoreceptor relative to CheBc. However, since T. maritima CheB demethylates on at least two different methyl glutamates of the chemoreceptor, more than one interaction mode – an interface covering methylation residues Gln294 as well as Gln281 – should be predicted from the docking methods. As no docking results satisfy the constraints on Gln294, we are unable to suggest multiple modes of CheBc interaction between the different demethylation glutamates.
In summary, we have determined the crystal structure of T. maritima CheBc and used it to generate a chemoreceptor interaction model by carrying out docking studies. We have clearly demonstrated that the limited efficiencies of the docking processes can be enhanced in cases in which two or more structure sets of interacting proteins are known from different species. Although we had aimed to crystallize T. maritima CheBc in complex with the TM1143 chemoreceptor, to date this has not proven successful. A more complete picture of methylesterase CheBc and the chemoreceptor substrate interaction thus awaits a successful determination of the complex structure.
We report the structure of T. maritima CheB methylesterase domain.
We have compared the structure with the S. typhimurium CheB.
Analysis indicates differing electrostatic surfaces near the catalytic region.
We carry out computational docking using the two respective chemoreceptor structures.
We thereby propose a CheB:chemoreceptor interaction mode.
Acknowledgments
This work was supported by the Human Resources Development of the Korea Institute of Energy Technology Evaluation and Planning (KETEP) grant funded by the Ministry of Knowledge Economy, Republic of Korea (No. 20104010100610) to K.W.C., and NIH grant GM066775 to B.R.C. This work was supported by the Research carried out (in part) at the National Synchrotron Light Source, Brookhaven National Laboratory, which is supported by the U.S. Department of Energy, Division of Materials Sciences and Division of Chemical Sciences, under Contract No. DE-AC02-98CH10886. We thank J. Y. Park for graphical assistance during figure preparation.
Footnotes
Accession Code: Worldwide Protein Data Bank coordinate for T. maritima CheBc has been deposited with an accession code 3SFT.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Borkovich KA, Simon MI. The dynamics of protein phosphorylation in bacterial chemotaxis. Cell. 1990;63:1339–1348. doi: 10.1016/0092-8674(90)90429-i. [DOI] [PubMed] [Google Scholar]
- 2.Parkinson JS, Kofoid EC. Communication modules in bacterial signaling proteins. Annu Rev Genet. 1992;26:71–112. doi: 10.1146/annurev.ge.26.120192.000443. [DOI] [PubMed] [Google Scholar]
- 3.Appleby JL, Parkinson JS, Bourret RB. Signal transduction via the multistep phosphorelay: not necessarily a road less travelled. Cell. 1996;86:845–848. doi: 10.1016/s0092-8674(00)80158-0. [DOI] [PubMed] [Google Scholar]
- 4.Goudreau PN, Stock AM. Signal transduction in bacteria: molecular mechanisms of stimulus-response coupling. Curr Opin Microbiol. 1998;1:160–169. doi: 10.1016/s1369-5274(98)80006-4. [DOI] [PubMed] [Google Scholar]
- 5.Berg HC. The rotary motor of bacterial flagella. Annu Rev Biochem. 2003;72:19–54. doi: 10.1146/annurev.biochem.72.121801.161737. [DOI] [PubMed] [Google Scholar]
- 6.Kojima S, Blair DF. The bacterial flagellar motor: structure and function of a complex molecular machine. Int Rev Cytol. 2004;233:93–134. doi: 10.1016/S0074-7696(04)33003-2. [DOI] [PubMed] [Google Scholar]
- 7.Wadhams GH, Armitage JP. Making sense of it all: bacterial chemotaxis. Nat Rev Mol Cell Biol. 2004;5:1024–1037. doi: 10.1038/nrm1524. [DOI] [PubMed] [Google Scholar]
- 8.Sourjik V. Receptor clustering and signal processing in E. coli chemotaxis. Trends Microbiol. 2004;12:569–576. doi: 10.1016/j.tim.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 9.Parkinson JS, Ames P, Studdert CA. Collaborative signaling by bacterial chemoreceptors. Curr Opin Microbiol. 2005;8:116–121. doi: 10.1016/j.mib.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 10.Inouye M, Dutta R. Histidine Kinases in Signal Transduction. Academic Press; San Diego: 2002. [Google Scholar]
- 11.Li J, Swanson RV, Simon MI, Weis RM. The response regulators CheB and CheY exhibit competitive bind to the kinase CheA. Biochemistry. 1995;34:14626–14636. doi: 10.1021/bi00045a003. [DOI] [PubMed] [Google Scholar]
- 12.Lupas A, Stock J. Phosphorylation of an N-terminal regulatory domain activates the CheB methylesterase in bacterial chemotaxis. J Biol Chem. 1989;264:17337–17342. [PubMed] [Google Scholar]
- 13.Stewart RC. Activating and inhibitory mutations in the regulatory domain of CheB, the methylesterase in bacterial chemotaxis. J Biol Chem. 1993;268:1921–1930. [PubMed] [Google Scholar]
- 14.Anand GS, Goudreau PN, Stock AM. Activation of methylesterase CheB: Evidence of a dual role for the regulatory domain. Biochemistry. 1998;37:14038–14047. doi: 10.1021/bi980865d. [DOI] [PubMed] [Google Scholar]
- 15.Djordjevic S, Goudreau PN, Xu Q, Stock AM, West AH. Structural basis for methylesterase CheB regulation by a phosphorylation-activated domain. Proc Natl Acad Sci USA. 1998;95:1381–1386. doi: 10.1073/pnas.95.4.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Terwilliger TC, Koshland DE., Jr Sites of methyl esterification and deamination on the aspartate receptor involved in chemotaxis. J Biol Chem. 1984;259:7719–7725. [PubMed] [Google Scholar]
- 17.Kehry MR, Bond MW, Hunkapiller MW, Dahlquist FW. Enzymatic deamidation of methyl-accepting chemotaxis proteins in Escherichia coli catalyzed by the cheB gene product. Proc Natl Acad Sci U S A. 1983;80:3599–3603. doi: 10.1073/pnas.80.12.3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perez E, West AH, Stock AM, Djordjevic S. Discrimination between different methylation states of chemotaxis receptor Tar by receptor methyltransferase CheR. Biochemistry. 2004;43:953–961. doi: 10.1021/bi035455q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim KK, Yokota H, Kim SH. Four-helical-bundle structure of the cytoplasmic domain of a serine chemotaxis receptor. Nature. 1999;400:787–792. doi: 10.1038/23512. [DOI] [PubMed] [Google Scholar]
- 20.Djordjevic S, Stock AM. Chemotaxis receptor recognition by protein methyltransferase CheR. Nat Struct Biol. 1998;5:446–450. doi: 10.1038/nsb0698-446. [DOI] [PubMed] [Google Scholar]
- 21.West AH, Martinez-Hackert E, Stock AM. Crystal structure of the catalytic domain of the chemotaxis receptor methylesterase, CheB. J Mol Biol. 1995;250:276–290. doi: 10.1006/jmbi.1995.0376. [DOI] [PubMed] [Google Scholar]
- 22.Perez E, Zheng H, Stock AM. Identification of methylation sites in Thermotoga maritima chemotaxis receptors. J Bacteriol. 2006;188:4093–4100. doi: 10.1128/JB.00181-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Park S-Y, Borbat PR, Gonzalez-Bonet G, Bhatnagar J, Pollard AM, Freed JH, Bilwes AM, Crane BR. Reconstruction of the chemotaxis receptor-kinase assembly. Nat Struct Mol Biol. 2006;13:400–407. doi: 10.1038/nsmb1085. [DOI] [PubMed] [Google Scholar]
- 24.Otwinowski Z, Minor W. Processing of X-ray diffraction data in oscillation mode. Methods Enzymol. 1997;276:307–325. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 25.Navaza J. AMoRe: an automated package for molecular replacement. Acta Cryst. 1994;A50:157–163. [Google Scholar]
- 26.Perrakis A, Morris RM, Lamzin VS. Automated protein model building combined with iterative structure refinement. Nat Struct Biol. 1999;6:458–463. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- 27.Collaborative Computational Project Number 4, The CCP4 suite: programs for protein crystallography. Acta Cryst. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 28.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 29.Chen R, Li L, Weng Z. ZDOCK: an initial-stage protein-docking algorithm. Proteins. 2003;52:80–87. doi: 10.1002/prot.10389. [DOI] [PubMed] [Google Scholar]
- 30.Li L, Chen R, Weng Z. RDOCK: refinement of rigid-body protein docking predictions. Proteins. 2003;53:693–707. doi: 10.1002/prot.10460. [DOI] [PubMed] [Google Scholar]
- 31.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J Comput Chem. 1983;4:187–217. [Google Scholar]
- 32.DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific; San Carlos, CA, USA: 2002. http://www.pymol.org. [Google Scholar]
- 33.Park SY, Jin W, Woo JR, Shoelson SE. Crystal structures of Human TBC1D1 and TBC1D4 (AS160) RabGAP domains reveal critical elements for GLUT4 translocation. J Biol Chem. 2011;286:18130–18138. doi: 10.1074/jbc.M110.217323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pollard AM, Bilwes AM, Crane BR. The structure of a soluble chemoreceptor suggests a mechanism for propagating conformational signals. Biochemistry. 2009;48:1936–1944. doi: 10.1021/bi801727m. [DOI] [PMC free article] [PubMed] [Google Scholar]