

Abstract
The single handedness of biological molecules has fascinated scientists and laymen alike since Pasteur's first painstaking separation of the enantiomorphic crystals of a tartrate salt over 150 years ago. More recently, a number of theoretical and experimental investigations have helped to delineate models for how one enantiomer might have come to dominate over the other from what presumably was a racemic prebiotic world. Mechanisms for enantioenrichment that include either chemical or physical processes, or a combination of both, are discussed in the context of experimental studies in autocatalysis and in the phase behaviour of chiral molecules.
Keywords: chirality, amino acids, RNA
1. Introduction
The property of chirality—non-superimposable forms that are mirror images of one another, as are left and right hands—is manifest in both molecular and macroscopic objects. As early as 1874, and a quarter century after Pasteur showed that salts of tartaric acid exist as mirror-image crystals, van't Hoff and Le Bel independently postulated the existence of chiral molecules [1]. Chiral molecules in living organisms in Nature exist almost exclusively as single enantiomers, a property that is critical for molecular recognition and replication processes and would thus seem to be a prerequisite for the origin of life. Yet left- and right-handed molecules of a compound will form in equal amounts (a racemic mixture) when we synthesize them in the laboratory in the absence of some type of directing template.
The fact of the single chirality of biological molecules—exclusively left-handed amino acids and right-handed sugars—presents us with two questions: First, what served as the original template for biasing production of one enantiomer over the other in the chemically austere, and presumably racemic, environment of the prebiotic world? And second, how was this bias sustained and propagated to give us the biological world of single chirality that surrounds us?
‘Symmetry breaking’ is the term used to describe the occurrence of an imbalance between enantiomeric molecules. This imbalance is traditionally measured in terms of the enantiomeric excess, or ee, where ee = (R – S)/(R + S) and R and S are concentrations of the right and left-hand molecules, respectively. Proposals for how an imbalance might have come about may be classified as either terrestrial or extraterrestrial, and then subdivided into either random or deterministic. Evidence of small enantiomeric excesses in amino acids found in chondritic meteor deposits [2] allows the hypothesis that the initial imbalance is not of our world. On the other hand, discussions of how an imbalance could have originated here on the Earth often debate the question of whether life was pre-ordained to be based on d-sugars and l-amino acids or whether this happened by chance, implying that a life form based on the opposite chirality might have been just as likely at the outset. Here, physics enters the picture: the discovery of parity violation and the elaboration of one of its consequences, that enantiomers have a very small energy difference between them, led many to consider the implications for biological homochirality [3]. Quantitative estimates of this energy difference have been made and revised in the intervening years, but it is clear that it is negligibly small; while experimental and theoretical work is ongoing, and the question is not yet settled, a relationship between biological homochirality and parity violation is not yet supported by experimental findings.
Proponents on the ‘chance’ side of this question point out that absolute asymmetric synthesis—defined as the production of enantiomerically enriched products in the absence of a chemical or physical chiral directing force—could occur stochastically [4]. A trivial example is that any collection of an odd number of enantiomeric molecules has, by definition, broken symmetry. Fluctuations in the physical and chemical environment could result in transient fluctuations in the relative numbers of left- and right-handed molecules. However, any small imbalance created in this way should average out as the racemic state unless some process intervenes to sustain and amplify it. Thus, whether or not the imbalance in enantiomers came about by chance, arising on the Earth or elsewhere, an amplification mechanism remains the key to sustaining enantioenrichment. A description of mechanisms for how this imbalance might be amplified is the main subject of this review.
Theoretical models for how a small initial imbalance in enantiomer concentrations might ultimately be turned into the subsequent production of a single enantiomer have been discussed for more than half a century [5,6], but only more recently have experimental studies begun to address this question directly. In the past two decades several distinct models with strikingly different features have emerged. These models draw upon both the chemical and the physical behaviour of chiral molecules, and they may be classified according to their relative emphasis on kinetics versus thermodynamics of the processes involved. ‘Far-from-equilibrium’ models involving autocatalytic chemical reactions or crystallization processes lie at one end of the spectrum. At the other end, a model based on equilibrium phase behaviour proposes a physical explanation. And in-between lies a model that invokes an interplay between thermodynamics and kinetics to explain how a combination of physical and chemical processes can drive a system of near-equal numbers of enantiomeric molecules to the left or to the right.
2. Autocatalysis
More than 60 years ago, Frank developed a mathematical model for an autocatalytic reaction mechanism for the evolution of homochirality. The model is based on a simple idea: a substance that acts as a catalyst in its own self-production and at the same time acts to suppress synthesis of its enantiomer enables the evolution of enantiopure molecules from a near-racemic mixture. The experimental challenge to discover a reaction with these features was posed in the last sentence of this purely theoretical paper: ‘A laboratory demonstration may not be impossible’ [6,7].
Frank's proposal serves to highlight the critical role played by an inhibition mechanism in autocatalytic models for the evolution of homochirality. Figure 1 illustrates this point using an example of a small group of l and d enantiomers that act as autocatalysts in an unlimited pool of substrate molecules. Each enantiomer is capable of reproducing itself in a reaction with a substrate molecule. In addition, there is ‘mutual antagonism’ between l and d such that when they react together, both become deactivated and lose their capacity to self-replicate. As shown in figure 1, the self-production of enantiomers will cause the ratio of l : d to grow as long as an initial imbalance was present at the beginning of the process. Together, autocatalysis and mutual antagonism propagate and amplify the imbalance in enantiomers. The only catch is that the smaller the initial imbalance, the greater the number of l and d molecules lost in the deactivation process before significant enantioenrichment can occur. If the substrate pool is large enough, however, the process can be sustained, and the selectivity of autocatalytic production of one enantiomer will eventually dominate.
Figure 1.

Schematic of the Frank model for the evolution of homochirality based on autocatalytic replication and mutual antagonism of enantiomers.
More than 40 years later, the first experimental proof of this concept was found when Soai et al. [8] reported the autocatalytic alkylation of pyrimidyl aldehydes with dialkylzincs (scheme 1), in which the reaction rate is accelerated by addition of catalytic amounts of its alcohol product. In addition, and most strikingly, this reaction was shown to yield the autocatalytic product in very high ee starting from a very low ee in the original catalyst.
Scheme 1.

The Soai autocatalytic reaction, where the product catalyses its own formation. The –CH3 group in the pyrimidyl aldehyde may be replaced with other groups such as alkynyl groups.
Since this initial discovery, Soai's group has gone on to present remarkable further observations of asymmetric amplification in the reaction that now bears his name. Enantiomeric excesses as high as 85 per cent were reported for a reaction initiated with an initiator produced at 0.1 per cent ee from exposure to circularly polarized light [9]. Asymmetric amplification has also been observed for the reaction initiated by inorganic chiral materials, such as quartz [10]. Most recently, Soai has shown that the reaction may be selectively triggered solely by the minute mirror-image difference provided by 12C/13C carbon isotope chirality of an initiator molecule [11], demonstrating that the reaction needs only an extremely small nudge to direct it consistently to the left or to the right.
Soai's observations continued to amaze and confound the community for several years before the first mechanistic rationalization of the reaction was reported by Blackmond et al. in 2001 [12]. A kinetic model was developed based on detailed experimental measurements of the autocatalytic rate profiles of the Soai reaction. The kinetic model rationalizes asymmetric amplification based on an extension of Kagan's model for nonlinear effects in catalytic reactions [13], that is, cases where the reaction product ee does not scale linearly with the catalyst ee. Such behaviour may ensue when the catalyst molecules aggregate to form higher order species, such as dimers. Blackmond and Brown's studies found that the Soai reaction R and S products form a stochastic distribution of homochiral and heterochiral dimers, with essentially no stereochemical bias between the dimers, and that the heterochiral dimer is inactive as a catalyst. Because each homochiral dimer catalyst produces more of itself in autocatalysis, and because mutual antagonism allows the minor enantiomer to be siphoned off as an inactive heterochiral dimer (serving the role of ‘mutual antagonism’ in the Frank model), catalyst concentration increases, and relative concentration of the two homochiral dimers changes, with reaction turnover. The kinetic model independently predicts both the temporal degree of asymmetric amplification, confirmed by compositional analysis, as well as the relative concentrations of the catalyst species, confirmed by nuclear magnetic resonance spectroscopy, as shown in figure 2 [14]. The ultimate product ee that may be achieved in such an autocatalytic reaction is limited only by the size of the substrate pool. This model demonstrates that effective amplification of ee in autocatalysis depends less on sophisticated stereoselection and more on higher relative activity for the homochiral dimers, repeated over many autocatalytic cycles [15].
Figure 2.

Product ee as a function of reaction progress in the Soai reaction of scheme 1 for reactions catalysed by 10 mol% of the reaction product with an initial ee of 6 and 22% [14]. Prediction of the Blackmond–Brown kinetic model (solid blue lines) and experimental values from chiral chromatographic analysis (filled magenta circles).
3. Physical models
Physical models for homochirality invoke the physical phase behaviour of chiral solids in dynamic interchange with their solution-phase molecules. The two main types of crystals formed by chiral molecules are (i) racemic compounds, where crystals contain a 1 : 1 ratio of d : l enantiomers; and (ii) conglomerates, where each crystal is composed of molecules of a single enantiomer, and the crystals themselves are mirror images (as were those Pasteur separated with his tweezers), and there is no direct molecular interaction between d and l molecules [16]. These types of compounds are illustrated schematically in figure 3. The type of crystal a chiral molecule forms in the solid phase is a fundamental property of that molecule at a given temperature and pressure. Racemic compounds are more prevalent than conglomerates by ca 10 : 1 on the planet Earth, including all but two of the 19 proteinogenic amino acids that are chiral (the 20th, glycine, is an achiral molecule). We discuss models for homochirality based on each of these types of crystal solids.
Figure 3.

Two types of crystalline solids formed by chiral compounds. Rectangles represent solid-phase enantiomeric molecules. (a) Conglomerates form separate crystals of each enantiomer; (b) racemic compounds form mixed crystals in a 1 : 1 ratio of the two enantiomers.
4. ‘Chiral amnesia’ model
A model describing a process for the evolution of single chirality in the solid phase is based on landmark work by Viedma, who first studied conglomerate crystals of NaClO3, an inorganic, achiral salt. NaClO3 happens to crystallize as mirror-image crystals that may be readily observed using circularly polarized light [17]. In a now classic experiment, Viedma stirred slurries containing equal amounts of left- and right-handed NaClO3 crystals in the presence of glass beads, which caused the solid particles to be finely ground. Under these conditions, he observed that over time the system evolved inexorably and randomly to a single enantiomorphic solid.
Viedma reasoned that the continual abrasion of the crystals enhanced both halves of the cycle of repetitive dissolution/crystallization that occurs in any dynamic solid-solution system. According to the Gibbs–Thomson rule, small crystals dissolve more readily than large crystals, and large crystals grow more readily than small ones. Attrition by glass beads produces a greater number of smaller crystals, whose increased dissolution, in turn, causes a slight supersaturation of NaClO3 in solution. Not sufficiently supersaturated to support primary nucleation of new crystals, the system strives to redress the balance between solid and solution by increasing the rate of re-accretion of solution-phase NaClO3 onto existing crystals (figure 4). A key point is that once a molecule of NaClO3 dissolves from a crystal, it no longer possesses chirality and it retains no memory of the chiral form it previously exhibited as part of a crystal. Solution-phase NaClO3 thus has no preference for re-accreting to a left- or right-handed crystal. If the system exhibits, by chance, a slight dominance of large crystals of one hand, the preference for solution molecules to add to larger crystals causes this enantiomorphic solid to increase relative to its smaller mirror-image form.
Figure 4.

‘Chiral amnesia’ process for the evolution of solid-phase homochirality for chiral molecules that form conglomerate solids. In this example, an initial imbalance towards larger l crystals helps to drive the dissolution/re-accretion process from d crystals to l crystals. The process is aided by solution-phase racemization, which converts the ‘excess’ dissolved d molecules to l molecules, equalizing the solution composition and enabling molecules that were formerly part of a d crystal to add as l molecules to l crystals.
Evolution of solid-phase homochirality in this case requires only an initial imbalance in the crystal size of left versus right-hand crystals. The achiral solution phase serves as the conduit through which the molecules from one hand of the crystal may readily become part of a crystal of the other hand, a feature that has been given the name ‘chiral amnesia’ (figure 5; [18,19]).
Figure 5.

Evolution of solid-phase homochirality for aspartic acid via solution-phase racemization. Energy input from grinding the crystals in the presence of enhanced attrition due to glass beads leads to a sigmoidal profile.
These results led many to consider a possible extension of this process from the achiral NaClO3 to intrinsically chiral molecules that form conglomerate solids. For these molecules, it was envisioned that solution-phase racemization could play the role of the solution phase conduit, transferring molecules from one crystal hand to the other by first interconverting them in solution. This process has been successfully demonstrated for intrinsically chiral molecules for an amino acid derivative [20] and for the proteinogenic amino acid aspartic acid [21] (scheme 2).
Scheme 2.

Transformation of aspartic acid crystals from one enantiomorphic solid to the other via solution-phase racemization. Mechanical or thermal energy input drives the dissolution/re-accretion process.
5. Crystal engineering model
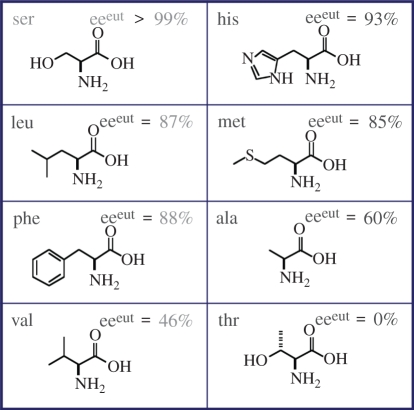
A model for the origin of biological homochirality for amino acids that form racemic compounds has been developed that provides solution-phase enantioenrichment based on the different solubility of homochiral and heterochiral crystals, as shown in figure 3b. When the numbers of enantiopure and d and l molecules are unequal, the minor enantiomer is present in the solution phase only to the extent that it can dissolve from the d : l crystal. The lower the solubility of the d : l crystals, the more strongly ‘trapped’ in the solid phase will be the minor enantiomer, and the higher the resulting solution phase partitioning of the major enantiomer. This will be manifested as a high solution phase ee, known as eeeut, and its value is a characteristic of a particular compound. Several of the proteinogenic amino acids form relatively insoluble d : l crystals, and therefore, exhibit high eutectic ee values, such as serine at greater than 99 per cent ee. This means that when a sample with nearly equal numbers of d and l serine molecules is partially dissolved, a virtually enantiopure solution results. Table 1 provides eutectic values for a number of amino acids [22].
Table 1.
Eutectic ee values for a number of proteinogenic amino acids, identified by their chemical structures and their three-letter names.
 |
Enantioenrichment in solution is thus dictated by thermodynamics for chiral compounds that happen to form relatively insoluble racemic compounds. This concept was first recognized by Morowitz [23] 40 years ago and was more recently elaborated by Klussmann et al. [22,24] and by Breslow & Levine [25]. Also Breslow & Cheng recently reported high eutectic ee values for several nucelosides of prebiotic importance [26]. A recent theoretical treatment based on a two-dimensional lattice model successfully predicts the ternary phase behaviour of amino acids based on the interactions that stabilize the racemic crystal, providing molecular-level insight into the observed enantiomer partitioning [27].
Further experimental work has expanded this model to include solution-phase enantioenrichment for chiral compounds with low intrinsic eeeut values by showing that in many cases eutectic ee composition may be ‘tuned’ through incorporation of a variety of small, achiral molecules into the solid-phase crystal structure via hydrogen-bonding [24,28]. If the incorporated molecule reduces the solubility of the racemic crystal relative to that of the enantiopure crystal, enhanced eutectic composition will result, as shown in figure 6. For example, d : l proline incorporates CHCl3 into its structure with a concomitant rise in eeeut from 50 to more than 99 per cent ee, and d : l valine and phenylalanine each form crystals incorporating fumaric acid; eeeut rose from 47 and 88 per cent, respectively, to more than 99 per cent in both cases. The structure of the d : l compound of proline with chloroform is shown in figure 7. Manipulation of the eutectic composition by additives may be thought of as an analogy to clathrate compounds, although here it is the amino acid enantiomers themselves that are trapped in the solvate–racemate structure, causing them to dissolve much less readily.
Figure 6.

Manipulation of eutectic ee value by formation of a solvate that reduces the solubility of the racemic compound.
Figure 7.

Crystal structure of ld proline incorporating one molecule of chloroform. (a) Five independent hydrogen bonds are shown; (b) long range structure with proline enantiomers in blue and magenta, chloroform in black [24].
The finding that the ee of an amino acid in solution may be significantly enhanced via solvate formation enables an approach to enantioenrichment for a wide range of chiral compounds. These studies suggest a general and facile route to homochirality that may have prebiotic relevance. Cycles of rain and evaporation create pools containing a small initial imbalance of amino acid enantiomers and appropriate hydrogen-bonding partner molecules that may form insoluble crystals. The resulting solution of enantioenriched molecules might then serve as efficient asymmetric catalysts or as building blocks themselves for construction of the complex molecules required for recognition, replication and ultimately for the chemical basis of life. [29].
6. Comparison of physical models
Comparison of the chiral amnesia and crystal engineering phase behaviour models for the origin of homochirality reveals that they are complementary in many ways: the former produces solid-phase homochirality while the latter provides enantioenrichment of the solution phase; the chiral amnesia model converts one enantiomer to the other, while the crystal engineering model simply partitions the existing molecules between phase. Chiral amnesia may be applied only to molecules that form conglomerates, which means that only about 10 per cent of known chiral compounds are candidates for enantioenrichment by this model. On the other hand, about 85 per cent of chiral compounds might be amenable to the selective partitioning provided by the crystal engineering model. Perhaps some combination of the two led to the initial enantioenrichment of biologically relevant molecules. Both models provide reasonable prebiotic scenarios, and further work to understand the mechanism of enantioenrichment in each case is underway.
The implications of the single chirality of biological molecules may be viewed in the general context of complexity, and the pathway to life may be seen as a saga of increasing chemical and physical complexity. The modern field of ‘systems chemistry’ [30] seeks to understand the chemical roots of biological organization by studying the emergence of system properties that may be different from those exhibited individually by the components in isolation. Whether or not we will ever know how the property of homochirality developed in the living systems represented on the Earth today, studies of how single chirality might have emerged will aid us in understanding the much larger question of how life might have, and might again, emerge as a complex system.
References
- 1.Heilbronner E., Dunitz J. D. 1993. Reflections on symmetry in chemistry … and elsewhere. Basel, Germany: Verlag Helvetica Chimica Acta [Google Scholar]
- 2.Pizzarello S. 2006. The chemistry of life's origin: a carbonaceous meteorite perspective. Acc. Chem. Res 39, 231–237 10.1021/ar050049f (doi:10.1021/ar050049f) [DOI] [PubMed] [Google Scholar]
- 3.Quack M. 2002. How important is parity violation for molecular and biomolecular chirality? Angew. Chem. Int. Ed. 41, 4618–4630 10.1002/anie.200290005 (doi:10.1002/anie.200290005) [DOI] [PubMed] [Google Scholar]
- 4.Mislow K. 2003. Absolute asymmetric synthesis: a commentary. Collect. Czech. Chem. Commun. 68, 849–864 [Google Scholar]
- 5.Calvin M. 1969. Molecular evolution. Oxford, UK: Oxford University Press [Google Scholar]
- 6.Frank F. C. 1953. On spontaneous asymmetric synthesis. Biochim. Biophys. Acta 11, 459–463 10.1016/0006-3002(53)90082-1 (doi:10.1016/0006-3002(53)90082-1) [DOI] [PubMed] [Google Scholar]
- 7.Wynberg H. 1989. Asymmetric autocatalysis: facts and fancy. J. Macromol. Sci. Chem A26, 1033–1041 10.1080/00222338908052033 (doi:10.1080/00222338908052033) [DOI] [Google Scholar]
- 8.Soai K., Shibata T., Morioka H., Choji K. 1995. Asymmetric autocatalysis and amplification of enantiomeric excess of a chiral molecule. Nature 378, 767–768 10.1038/378767a0 (doi:10.1038/378767a0) [DOI] [Google Scholar]
- 9.Shibata J., Yamamoto T., Matsumoto N., Yonekubo S., Osanai S., Soai K. 1998. Amplification of a slight enantiomeric imbalance in molecules based on asymmetric autocatalysis: the first correlation between high enantiomeric enrichment in a chiral molecule and circularly polarized light. J. Am. Chem. Soc. 120, 12 157–12 158 10.1021/ja980815w (doi:10.1021/ja980815w) [DOI] [Google Scholar]
- 10.Soai K., Osanai S., Kadowaki K., Yonekubo S., Shibata T., Sato I. 1999. d- and l-quartz-promoted highly enantioselective synthesis of a chiral compound. J. Am. Chem. Soc. 121, 11 235–11 236 10.1021/ja993128t (doi:10.1021/ja993128t) [DOI] [Google Scholar]
- 11.Kawasaki T., Matsumura Y., Tsutsumi T., Suzuki K., Ito M., Soai K. 2009. Asymmetric autocatalysis triggered by carbon isotope (13C/12C) chirality. Science 324, 492–495 10.1126/science.1170322 (doi:10.1126/science.1170322) [DOI] [PubMed] [Google Scholar]
- 12.Blackmond D. G., McMillan C. R., Ramdeehul S., Schorm A., Brown J. M. 2001. Origins of asymmetric amplification in autocatalytic alkylzinc additions. J. Am. Chem. Soc. 123, 10 103–10 104 10.1021/ja0165133 (doi:10.1021/ja0165133) [DOI] [PubMed] [Google Scholar]
- 13.Girard C., Kagan H. B. 1998. Nonlinear effects in asymmetric synthesis and stereoselective reactions: ten years of investigation. Angew. Chem. Int. Ed. 37, 2923–2959 [DOI] [PubMed] [Google Scholar]
- 14.Buono F. G., Blackmond D. G. 2003. Kinetic evidence for a tetrameric transition state in the asymmetric autocatalytic alkylation of pyrimidyl aldehydes. J. Am. Chem. Soc. 125, 8978. 10.1021/ja034705n (doi:10.1021/ja034705n) [DOI] [PubMed] [Google Scholar]
- 15.Blackmond D. G. 2004. Asymmetric autocatalysis and its implications for the origin of homochirality. Proc. Natl Acad. Sci. USA 101, 5732–5736 10.1073/pnas.0308363101 (doi:10.1073/pnas.0308363101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jacques J., Collet A., Wilen S. H. 1994. Enantiomers, racemates and resolutions, 2nd edn Melbourne, FL: Krieger Publishing Company [Google Scholar]
- 17.Viedma C. 2005. Chiral symmetry breaking during crystallization: complete chiral purity induced by nonlinear autocatalysis and recycling. Phys. Rev. Lett. 94, 065504. 10.1103/PhysRevLett.94.065504 (doi:10.1103/PhysRevLett.94.065504) [DOI] [PubMed] [Google Scholar]
- 18.Blackmond D. G. 2007. Chiral amnesia as a driving force for solid-phase homochirality. Chem. Eur. J. 13, 3290–3294 10.1002/chem.200601463 (doi:10.1002/chem.200601463) [DOI] [PubMed] [Google Scholar]
- 19.Viedma C. 2007. Chiral symmetry breaking and complete chiral purity by thermodynamic-kinetic feedback near equilibrium: implications for the origin of biochirality. Astrobiology 7, 312–319 10.1089/ast.2006.0099 (doi:10.1089/ast.2006.0099) [DOI] [PubMed] [Google Scholar]
- 20.Noorduin W. L., et al. 2008. Emergence of a single solid chiral state from a nearly racemic amino acid derivative. J. Am. Chem. Soc. 130, 1158–1159 10.1021/ja7106349 (doi:10.1021/ja7106349) [DOI] [PubMed] [Google Scholar]
- 21.Viedma C., Ortiz J. E., de Torres T., Izumi T., Blackmond D. G. 2008. Evolution of solid phase homochirality for a proteinogenic amino acid. J. Am. Chem. Soc. 130, 15 274–15 275 10.1021/ja8074506 (doi:10.1021/ja8074506) [DOI] [PubMed] [Google Scholar]
- 22.Klussmann M., Iwamura H., Mathew S. P., Wells D. H., Jr, Pandya U., Armstrong A., Blackmond D. G. 2006. Thermodynamic control of asymmetric amplification in amino acid catalysis. Nature 441, 621–623 10.1038/nature04780 (doi:10.1038/nature04780) [DOI] [PubMed] [Google Scholar]
- 23.Morowitz M. 1969. A mechanism for the amplification of fluctuations in racemic mixtures. J. Theor. Biol. 25, 491–494 10.1016/S0022-5193(69)80035-4 (doi:10.1016/S0022-5193(69)80035-4) [DOI] [PubMed] [Google Scholar]
- 24.Klussmann K., White A. J. P., Armstrong A., Blackmond D. G. 2006. Rationalization and prediction of solution enantiomeric excess in ternary phase systems. Angew. Chem. Int. Ed. 47, 7985–7989 10.1002/anie.200602520 (doi:10.1002/anie.200602520) [DOI] [PubMed] [Google Scholar]
- 25.Breslow R., Levine M. 2006. Amplification of enantiomeric concentrations under credible prebiotic conditions. Proc. Natl Acad. Sci. USA 103, 12 979–12 980 10.1073/pnas.0605863103 (doi:10.1073/pnas.0605863103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Breslow R., Cheng L. 2009. On the origin of terrestrial homochirality for nucleosides and amino acids. Proc. Natl Acad. Sci. USA 106, 9144–9146 10.1073/pnas.0904350106 (doi:10.1073/pnas.0904350106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lombardo T. G., Stillinger F. H., Debenedetti P. G. 2009. Thermodynamic mechanism for solution phase chiral amplification via a lattice model. Proc. Natl Acad. Sci. USA 106, 15 131–15 135 10.1073/pnas.0812867106 (doi:10.1073/pnas.0812867106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klussmann M., Izumi T., White A. J. P., Armstrong A., Blackmond D. G. 2007. Emergence of solution-phase homochirality via crystal engineering of amino acids. J. Am. Chem. Soc. 123, 7657–7660 10.1021/ja0708870 (doi:10.1021/ja0708870) [DOI] [PubMed] [Google Scholar]
- 29.Klussmann M., Blackmond D. G. 2007. Investigating the evolution of biomolecular homochirality. AICHE J 53, 2–8 10.1002/aic.11024 (doi:10.1002/aic.11024) [DOI] [Google Scholar]
- 30.von Kiedrowski G. 2005. Systems chemistry seeks to understand the chemical roots of biological organization based on the classical knowledge of chemistry—the language of molecules, their structures, reactions, and interactions—combined with aspects derived from the fields of theoretical biology and complex systems research. In European Science Foundation workshop at the European Center for Living Technology, Venice International University, Venice, Italy, 3–4 October, 2005. [Google Scholar]