

Abstract
The glmS ribozyme-riboswitch is the first known example of a naturally occurring catalytic RNA that employs a small molecule as a coenzyme. Binding of glucosamine-6-phosphate (GlcN6P) activates self-cleavage of the bacterial ribozyme, which is part of the mRNA encoding the metabolic enzyme GlcN6P-synthetase. Cleavage leads to negative feedback regulation. GlcN6P binds in the active site of the ribozyme, where its amine could function as a general acid and electrostatic catalyst. The ribozyme is pre-folded but inactive in the absence of GlcN6P, demonstrating it has evolved strict dependence on the exogenous small molecule. The ribozyme showcases the ability of RNA to co-opt non-covalently bound small molecules to expand its chemical repertoire. Analogue studies demonstrate that some molecules other than GlcN6P, such as l-serine (but not d-serine), can function as weak activators. This suggests how coenzyme use by RNA world ribozymes may have led to evolution of proteins. Primordial cofactor-dependent ribozymes may have evolved to bind their cofactors covalently. If amino acids were used as cofactors, this could have driven the evolution of RNA aminoacylation. The ability to make covalently bound peptide coenzymes may have further increased the fitness of such primordial ribozymes, providing a selective pressure for the invention of translation.
Keywords: catalytic RNA, enzymatic cofactor, RNA world
1. Introduction
The glmS ribozyme is a catalytic RNA derived from a ligand-dependent self-cleaving mRNA domain conserved among Gram-positive bacteria (reviewed in [1]). In these organisms, the 5′-untranslated region (5′-UTR) of the mRNA encoding the protein enzyme glucosamine-6-phosphate (GlcN6P) synthetase self-cleaves when it binds to GlcN6P [2]. Self-cleavage of the 5′-UTR leads to degradation of the mRNA, resulting in negative feedback regulation of the protein enzyme by its product metabolite [3]. GlcN6P synthetase is an essential enzyme that catalyzes the first committed step in the metabolic pathway that leads to synthesis of the bacterial cell wall. The activity is universally present, but is catalysed by two types of protein enzymes distributed in different taxa [4]. Eukaryotes and Gram-negative bacteria have a GlcN6P synthetase that is allosterically controlled by its reaction product. Gram-negative bacteria such as Escherichia coli also regulate expression of the enzyme at the mRNA using small antisense RNAs. The GlcN6P synthetase of Gram-positive bacteria, in contrast, is not an allosteric enzyme. Thus, the regulation of protein expression at the level of mRNA by the ribozyme-riboswitch domain is likely to be important [1]. Indeed, it has been shown that replacement of the ribozyme by a catalytically compromised mutant results in abrogation of sporulation by Bacillus subtilis [3].
In vitro, the self-cleaving domain of the glmS mRNA can be engineered to function as a multiple-turnover catalyst, that is, a ribozyme. Deletion analysis showed that the ribozyme is comprised of approximately 150 nucleotides (nt) and that a single nucleotide 5′ of the cleavage site suffices for maximum GlcN6P-induced activity. Furthermore, approximately 75 nt RNA constructs (with one nt 5′ of the cleavage site) are responsive to GlcN6P, but exhibit reduced catalytic activity [2]. Structure determination of the full-length glmS ribozyme [5] demonstrated that it is composed of a doubly pseudoknotted approximately 75 nt core domain that encompasses the cleavage site and the GlcN6P-binding site, and a peripheral element that appears to stabilize the core, fully explaining the biochemical results (figure 1).
Figure 1.
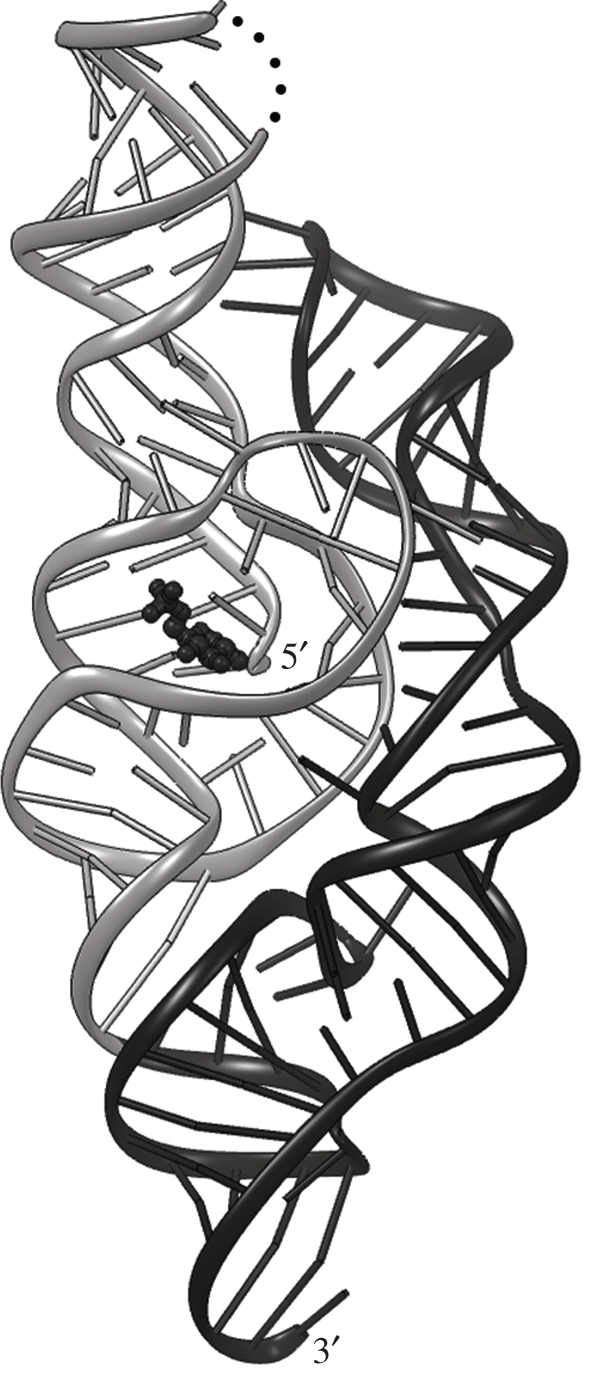
Overall structure of the glmS ribozyme. Cartoon representation of the 1.7 Å resolution structure of the ribozyme from T. tengcongensis [6]. The doubly pseudoknotted core domain is coloured grey; the peripheral stabilizing domain black. The cofactor GlcN6P is shown as a black ball-and-stick model. Cleavage takes place between the 5′-most two nucleotides. Black dots depict a break in the RNA construct used for crystallization.
2. Mechanism of glucosamine-6-phosphate activation of the ribozyme
GlcN6P could activate the glmS ribozyme by two mechanisms. The small molecule could function as an allosteric effector, its binding to an allosteric site leading to a conformational rearrangement that activates the ribozyme. Alternatively, the small molecule could function as a catalytic cofactor or coenzyme, binding to the active site of the ribozyme and providing chemical functionality that is indispensable for catalysis. Structural and biochemical evidence strongly support a coenzyme function for GlcN6P in glmS ribozyme activation. Crystal structures have been determined of glmS ribozymes from two different Gram-positive bacteria, each in multiple functional states (e.g. GlcN6P-free pre-cleavage, GlcN6P-bound pre-cleavage, GlcN6P-bound transition-state mimic, post-cleavage). The RNA structure is experimentally identical in all states [5–9]. In addition, it was shown that the glmS ribozyme crystallized in the absence of GlcN6P could be activated for cleavage in the crystalline state by briefly soaking the crystals in solutions containing GlcN6P. Structure determination using such soaked crystals revealed an RNA in a post-cleavage state that was structurally indistinguishable from structures obtained from crystals of ribozymes activated and cleaved in solution prior to crystallization [5]. Thus, global motions of the RNA (which would be incompatible with the crystal lattice) are not required for activation, catalysis or release of the 5′-cleavage product. Lack of GlcN6P-induced conformational changes of the glmS ribozyme is also supported by the results of hydroxy-radical footprinting [10] and fluorescence resonance energy transfer (FRET) experiments [11].
GlcN6P binds to the RNA with its amine group in van der Waals contact with the scissile phosphate; that is, the GlcN6P-binding site is the active site of the ribozyme, not an allosteric effector site [5]. The functional importance of the amine group of GlcN6P for glmS ribozyme activity is underscored by studies using the isosteric sugar, glucose-6-phosphate (Glc6P). This compound, which differs from GlcN6P only in having a hydroxyl rather than an amine group at position 2 of the pyranose ring, is not an activator of the ribozyme [12]. Cocrystal structures show that it binds precisely in the same location as GlcN6P. As expected from the foregoing, the conformation of the Glc6P-bound ribozyme is identical to those of the ribozyme in other functional states [6]. Biochemical studies show that Glc6P competes with GlcN6P for binding to the glmS ribozyme, functioning in effect as an antagonist [12]. Since the ribozyme is pre-folded but completely inactive in the absence of GlcN6P, the most parsimonious conclusion is that this compound functions as a catalytic cofactor of the RNA.
3. Plasticity of natural and artificial gene-regulatory RNAs
The glmS ribozyme is also a riboswitch, that is, an mRNA domain that controls gene expression in cis in response to the intracellular concentration of a small molecule metabolite or second messenger (reviewed in [13,14]). However, its mechanism of action and its structural rigidity make it an atypical riboswitch. Five distinct mechanisms of gene regulation have been documented for riboswitches. First, the majority of known bacterial riboswitches function by transcriptional attenuation. The ligand-binding (or ‘aptamer’) domain of the riboswitch and a ρ-independent terminator stem-loop compete for folding, with ligand binding altering the efficiency of formation of the latter. (Aptamers are RNAs evolved in vitro to bind to specific ligands [15].) Second, many bacterial riboswitches function at the level of translation initiation. In these, the ligand-bound conformation of the ligand-binding domain (the ‘aptamer’ domain) of the riboswitch competes with another that either occludes or exposes the Shine–Dalgarno element. Third, ligand binding by some riboswitches results in alternative splicing of introns. The thiamine pyrophosphate (TPP)-responsive thi-box riboswitch is the only riboswitch thus far described in eukaryotes. It is present in some introns in algae, fungi and plants, and it controls alternative splicing by exposing splice junctions or splicing enhancers depending on TPP concentration (reviewed in [16]). In bacteria, group I self-splicing introns have been described [17] that select between two alternative splice sites based on binding of cyclic diguanylate (c-di-GMP). Fourth, an S-adenosylmethionine (SAM)-responsive riboswitch has been described that initially functions through transcriptional attenuation, but in which the prematurely terminated transcript induced by ligand binding also functions in trans as an antisense RNA [18]. Finally, the glmS riboswitch is the first known example of a riboswitch that functions by ligand-induced self-cleavage. Whereas the first four mechanisms of riboswitch action require that the riboswitch (comprised of aptamer domain and the ‘expression platform’, the RNA sequences that interface with the transcription, translation or splicing machinery) adopt at least two mutually exclusive conformations, the glmS riboswitch is rigid and fully assembled prior to GlcN6P binding, and it is only its catalytic activity that switches (by approximately a million-fold).
4. Ligand selectivity of the glmS ribozyme
Riboswitches and artificial aptamers that recognize their ligands by partially or wholly enveloping them are known to undergo folding transitions upon ligand binding (e.g. [19–25]). Conversely, the rigid and pre-folded glmS ribozyme binds GlcN6P in an open, solvent-accessible pocket (figure 1) [5]. Analogue studies [12] underscore the importance of three of the functional groups of GlcN6P that interact with the glmS ribozyme: the anomeric hydroxyl, the amine and the phosphate. A variety of compounds that present vicinal amine and hydroxyl groups are weak activators of the glmS ribozyme. Such (presumably adventitious) activators include glucosamine (GlcN), l-serine, serinol, Tris and ethanolamine (figure 2). Importantly, the apparent pKa of the ribozyme reaction tracks the pKa of the amine group of the activating compound [12]. Thus, the pKa of the reaction rises from 7.9 to 8.7 when GlcN is replaced with serinol (the pKa values of their amine groups are 7.8 and 8.8, respectively). This confirms the importance of the amine group of the activator, suggested by the intimate contact between the amine of GlcN6P and the scissile phosphate [5,6,8]. However, analogues also point to the importance of the vicinal hydroxyl group, and to the precise stereochemical relationship between the amine and the hydroxyl groups. Thus, ammonia and methylamine are not activators, and d-serine, in which the stereochemical arrangement of the two functional groups is opposite that of GlcN6P (and l-serine) is not an activator [12]. The ability of GlcN or glucosameine-6-sulphate to serve as weak activators of the glmS ribozyme also indicates that the phosphate of GlcN6P is not chemically essential, even though it is important for increased binding affinity to the RNA. In addition to interacting with the RNA through outer-sphere coordinated metal ions, the phosphate of GlcN6P receives a hydrogen bond from the N1 imine of riboswitch residue G1 [5]. It was found that replacing G1 with purines with a non-protonated N1 position (such as A, 2-aminopurine, or dimethyladenine) resulted in riboswitches more strongly activated by GlcN than GlcN6P, presumably because of the clash between the phosphate of GlcN6P and the unprotonated N1 of these purines [6].
Figure 2.

Structures of cognate and adventitious activators [12] of the glmS ribozyme. Numbering system is indicated for glucosamine-6-phosphate (GlcN6P). Adventitious activators are drawn to emphasize their similarity to the key functional segment (thick lines) of GlcN6P.
5. Catalytic mechanism of the glmS ribozyme
The glmS ribozyme catalyzes the same overall chemical transformation as other well-characterized self-cleaving RNAs, such as the hepatitis delta virus (HDV), hairpin and hammerhead ribozymes (reviewed in [26]). RNA cleavage proceeds through a concerted (SN2) transesterification in which the 2′-OH of the nucleotide preceding the scissile phosphate (residue −1) attacks the phosphorus of the scissile phosphate. A transition state featuring a pentacovalent phosphorus is resolved by departure of the 5′-oxo group of residue +1, and formation of a 2′,3′-cyclic phosphate (figure 3). A variety of strategies are employed by RNA to catalyse this reaction. The HDV ribozyme employs a water molecule chelated by a magnesium ion to function as a specific base to deprotonate the nucleophilic 2′-OH, and the N3 amine of a cytosine residue (C75) to serve as a general acid, to protonate the leaving group [27,28]. In order that it functions effectively as a general acid, the pKa of the N3 of C75 is perturbed by over 2 pH units from its undisturbed value of approximately 4.2. Indeed, C75 is responsible for most of the catalytic rate enhancement achieved by the HDV ribozyme (figure 4) [29]. The hairpin ribozyme has two nucleobases, G8 and A38, positioned to serve as general base and general acid in the cleavage reaction [30,31]. This ribozyme, unlike the HDV or glmS ribozymes, also catalyzes the reverse ligation reaction. For catalysis of the ligation, the catalytic role of the nucleotides has to be reversed (because of the principle of microscopic reversibility). Raman spectroscopic studies of crystalline hairpin ribozymes show that the pKa of A38 is perturbed by nearly 2 pH units [32]. In addition, crystallographic studies show that G8 and A38 have the ability to preferentially bind to the transition state of the reaction, thus lowering the activation energy by hydrogen bonding [33]. The hammerhead ribozyme appears to use yet another strategy, in which N1 of G12 functions as a general base to deprotonate the nucleophilic 2′-OH of the cleavage reaction, and the 2′-OH of G8 functions as a general acid [34]. Like the hairpin ribozyme, the hammerhead ribozyme also catalyses ligation during which the role of the catalytic functional groups must be reversed. Importantly, it can be shown that near-neutral pKa values are not a strict condition for functional groups to serve as general acid–base catalysts [35,36].
Figure 3.

RNA cleavage through internal transesterification. The glmS ribozyme cleaves the phosphodiester between residues A − 1 and G + 1. :B and AH denote Brønsted–Lowry base and acid, respectively.
Figure 4.

Catalytic mechanisms of (a) the HDV and (b) glmS ribozymes. The HDV ribozyme employs a metal-ion bound hydroxide as a specific base and the protonated form of C75 as a general acid catalyst. The glmS ribozyme appears to employ the protonated form of GlcN6P as a general acid. The nucleobase of G40 might function as a general base catalyst.
The location of the amine group of GlcN6P in the active site of the glmS ribozyme is analogous to that of the N3 of C75 in the HDV ribozyme and A38 in the hairpin ribozyme [5,30,37]. This suggests that the small molecule would function as a general acid catalyst (figure 4). The simplest interpretation of the catalytic inactivity of the glmS ribozyme in the absence of GlcN6P is that the small molecule is responsible for all the catalytic power of the ribozyme. However, mutational analysis does not support this. All known glmS ribozyme isolates have a G residue at position 40 (Thermoanaerobacter tengcongensis numbering scheme). G40 is positioned with its N1 imine within hydrogen bonding distance of the nucleophilic 2′-OH of A − 1. Thus, it is possible that G40 functions as a general base in catalysis, or that it serves to orient the nucleophilic 2′-OH during the reaction. Mutation of G40 to A completely abrogates the activity of the ribozyme, underscoring its importance. Structure determination of the G40A mutant reveals that a ribozyme folds into a structure indistinguishable from that of the wild-type with, most surprisingly, GlcN6P bound in precisely the same position as in the active ribozyme [7]. This demonstrates that while GlcN6P is necessary for glmS ribozyme activity, it is not sufficient. It also indicates that whatever the precise role of G40 in catalysis might be, it becomes manifest only when GlcN6P binds the ribozyme. Thus, the active site of the glmS ribozyme is not simply a rigid, passive scaffold for GlcN6P binding. Rather, the RNA and the small molecule appear to tune each other's chemical properties to give rise to catalysis. Two molecular dynamics studies reached conflicting conclusions regarding the potential catalytic role of G40 [38,39]. The exact nature of this coupling between the glmS ribozyme active site and GlcN6P remains unresolved.
6. Expansion of RNA chemistry by exogenous small molecules
The glmS ribozyme is the first characterized example of a natural catalytic RNA that employs a small molecule cofactor. Such a role for small molecules has precedents in artificial RNAs. In vitro selection methodology has been employed by many groups to isolate RNAs capable of catalysing a variety of chemical transformations from pools of random sequences (reviewed in [40,41]). Several such experiments have isolated ribozymes that employ a non-covalently bound small molecule as a cofactor (rather than, or in addition to, a small molecule substrate). For instance, Meli et al. [42] mutagenized the hairpin ribozyme and isolated variants that are dependent on free adenine for catalytic activity. Biochemical analyses of these ribozymes suggest that the exogenous purine base participates in acid–base catalysis. Starting with a pool of random sequence RNA, Tsukiji et al. [43] selected alcohol dehydrogenase ribozymes that employ NAD+ as a redox cofactor. These ribozymes were capable of rate enhancements of 107-fold over the uncatalysed reaction. From a structural standpoint, the ability of RNA molecules to bind to adenine or NAD+ and employ them as cofactors is not surprising, given the chemical similarity of these molecules to nucleic acids. Indeed, White [44] proposed that the fact that many coenzymes (NAD, FAD, CoA, etc.) are nucleotides or contain heterocycles that could be derived from nucleotides (TPP, tetrahydrofolate, pyridoxal phosphate) could indicate that the metabolism of primordial organisms was catalysed by nucleic acids, and that these coenzymes are remnants of the active sites of such ancestral ribozymes.
Structural and biochemical studies of riboswitches and aptamers show that many of these RNAs undergo large conformational rearrangements concomitant with ligand binding. Likewise, the plasticity of RNA is a recurrent theme in RNA–protein interactions (e.g. [45,46]). Noller [47] has suggested that the fact that many peptide-binding RNAs display large ligand-driven folding transitions may hint at the primordial driving force for the invention of translation. Specifically, peptides could have first served as facilitators for the folding of primordial ribozymes, and translation evolved to make synthesis of such peptides more efficient and reproducible. The successful design of synthetic aptazymes, artificial catalytic RNAs that require binding of a small molecule (by an aptamer domain) in order to achieve catalytic activity, demonstrates that ligand-induced folding of RNA can be functionally coupled to the catalytic competence of ribozymes [48].
7. Amino acids as primordial ribozyme cofactors?
The mechanism of action of the glmS ribozyme, which does not fold concomitant with ligand binding and which can use cofactors that do not have obvious chemical similarity to nucleotides (amine sugars or even amino acids), suggests how small molecules may have expanded RNA chemistry in the RNA world, rather than functioning as folding chaperones. Activation of the glmS ribozyme l-serine is particularly suggestive in the context of speculation about cofactor use by primordial ribozymes for two reasons. First, it has often been posited that the greater chemical diversity of amino acids over nucleotides would have been a driving force for the transition from RNA catalysts to protein enzymes (reviewed in [49]). Serine provides the glmS ribozyme with two chemical groups (a primary amine and a primary hydroxyl) that are absent in RNA, indeed showing that amino acids have more chemical diversity than RNA. However, the greater chemical diversity of amino acids does not, per se, explain why the conventional set of 20 amino acids was chosen. It can be postulated that primordial metabolism was catalysed by numerous ribozymes that shared a set of amino acid cofactors, and that the specific set of contemporary amino acids is the result of a contingent choice of cofactors by early ribozymes that became fixed through the ‘principle of many users’ or ‘principle of continuity’ [50]. Once a particular set of amino acids became required for the function of many genetically distinct ribozymes, this set could not be altered. Any change would have necessitated the simultaneous coevolution of multiple amino acid-dependent ribozymes. This is analogous to the explanation given for the persistence of particular coenzymes that are essential for the function of the majority of protein enzymes [51]. Second, the glmS ribozyme is activated by l-serine but not d-serine. While this is a trivial stereochemical consequence of the chirality of RNA, it nonetheless suggests that the choice of l-amino acids in early evolution may have been the result of a contingent choice of cofactors by a group of primordial l-amino acid-dependent ribozymes. That choice would also have been fixed through the principle of many users (this does not provide an explanation for the choice of d-ribose to make RNA).
8. Covalent attachment of cofactors to RNA and the evolution of translation
Others have speculated that tRNA aminoacylation originated either as a means of conferring a replicative advantage to a primordial tRNA-like genomic tag [52] or as a form of post-transcriptional RNA modification that would confer new structural or functional capabilities to the nucleic acid [53]. If, as suggested above, primordial metabolism was catalysed by a set of ribozymes some of which evolved dependence on amino acids functioning as coenzymes, then RNA aminoacylation (and the subsequent evolution of translation) would have arisen as a consequence of the evolutionary pressure to improve the efficiency of such ribozymes by covalent attachment of their cofactors. In vitro selection experiments demonstrate that even fairly simple ribozymes can carry out aminoacylation with high specificity and regioselectivity (e.g. [54]).
In summary, the glmS ribozyme is a small-molecule-dependent catalytic RNA that is widespread among Gram-positive bacteria, where it regulates a key metabolic pathway. It employs the small molecule GlcN6P not to facilitate folding, but as part of the active site, as a cofactor or coenzyme. This shows that RNA can employ non-covalently bound small molecules to expand its chemical functionality. Moreover, in vitro mechanistic characterization of the glmS ribozyme demonstrates that it can employ l-serine as an adventitious coenzyme. This use of a free amino acid as a coenzyme by a ribozyme suggests how the use of amino acids in modern biology may have derived from the primordial use of these small molecules as coenzymes in the RNA world, and that the specific set of amino acids used throughout contemporary biology may reflect the ‘basis set’ of amino acid cofactors shared among the ribozymes of the RNA world in which translation evolved.
Acknowledgements
The author thanks past and current members of the Ferré-D'Amaré laboratory for their many contributions, and in particular D. Klein for his work on the glmS ribozyme. This research was supported by the intramural programme of the National Heart, Lung and Blood Institute, National Institutes of Health.
References
- 1.Ferré-D'Amaré A. R. 2010. The glmS ribozyme: use of a small molecule coenzyme by a gene-regulatory RNA. Q. Rev. Biophys. 43, 423–447 10.1017/S0033583510000144 (doi:10.1017/S0033583510000144) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Winkler W. C., Nahvi A., Roth A., Collins J. A., Breaker R. R. 2004. Control of gene expression by a natural metabolite-responsive ribozyme. Nature 428, 281–286 10.1038/nature02362 (doi:10.1038/nature02362) [DOI] [PubMed] [Google Scholar]
- 3.Collins J. A., Irnov I., Baker S., Winkler W. C. 2007. Mechanism of mRNA destabilization by the glmS ribozyme. Genes Dev. 21, 3356–3368 10.1101/gad.1605307 (doi:10.1101/gad.1605307) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Milewski S. 2002. Glucosamine-6-phosphate synthase–the multi-facets enzyme. Biochem. Biophys. Acta 1597, 173–192 10.1016/S0167-4838(02)00318-7 (doi:10.1016/S0167-4838(02)00318-7) [DOI] [PubMed] [Google Scholar]
- 5.Klein D. J., Ferré-D'Amaré A. R. 2006. Structural basis of glmS ribozyme activation by glucosamine-6-phosphate. Science 313, 1752–1756 10.1126/science.1129666 (doi:10.1126/science.1129666) [DOI] [PubMed] [Google Scholar]
- 6.Klein D. J., Wilkinson S. R., Been M. D., Ferré-D'Amaré A. R. 2007. Requirement of helix P2.2 and nucleotide G1 for positioning of the cleavage site and cofactor of the glmS ribozyme. J. Mol. Biol. 373, 178–189 10.1016/j.jmb.2007.07.062 (doi:10.1016/j.jmb.2007.07.062) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klein D. J., Been M. D., Ferré-D'Amaré A. R. 2007. Essential role of an active-site guanine in glmS ribozyme catalysis. J. Am. Chem. Soc. 129, 14 858–14 859 10.1021/ja0768441 (doi:10.1021/ja0768441) [DOI] [PubMed] [Google Scholar]
- 8.Cochrane J. C., Lipchock S. V., Strobel S. A. 2007. Structural investigation of the GlmS ribozyme bound to its catalytic cofactor. Chem. Biol. 14, 97–105 10.1016/j.chembiol.2006.12.005 (doi:10.1016/j.chembiol.2006.12.005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cochrane J. C., Lipchock S. V., Smith K. D., Strobel S. A. 2009. Structural and chemical basis for glucosamine 6-phosphate binding and activation of the glmS ribozyme. Biochemistry 48, 3239–3246 10.1021/bi802069p (doi:10.1021/bi802069p) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hampel K. J., Tinsley M. M. 2006. Evidence for preorganization of the glmS ribozyme ligand binding pocket. Biochemistry 45, 7861–7871 10.1021/bi060337z (doi:10.1021/bi060337z) [DOI] [PubMed] [Google Scholar]
- 11.Tinsley R. A., Furchak J. R., Walter N. G. 2007. Trans-acting glmS catalytic riboswitch: locked and loaded. RNA 13, 468–477 10.1261/rna.341807 (doi:10.1261/rna.341807) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McCarthy T. J., Plog M. A., Floy S. A., Jansen J. A., Soukup J. K., Soukup G. A. 2005. Ligand requirements for glmS ribozyme self-cleavage. Chem. Biol. 12, 1221–1226 10.1016/j.chembiol.2005.09.006 (doi:10.1016/j.chembiol.2005.09.006) [DOI] [PubMed] [Google Scholar]
- 13.Edwards T. E., Klein D. J., Ferré-D'Amaré A. R. 2007. Riboswitches: small-molecule recognition by gene regulatory RNAs. Curr. Opin. Struct. Biol. 17, 273–279 10.1016/j.sbi.2007.05.004 (doi:10.1016/j.sbi.2007.05.004) [DOI] [PubMed] [Google Scholar]
- 14.Zhang J., Lau M. W., Ferré-D'Amaré A. R. 2010. Ribozymes and riboswitches: modulation of RNA function by small molecules. Biochemistry 49, 9123–9131 10.1021/bi1012645 (doi:10.1021/bi1012645) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ellington A. D., Szostak J. W. 1990. In vitro selection of RNA molecules that bind specific ligands. Nature 346, 818–822 10.1038/346818a0 (doi:10.1038/346818a0) [DOI] [PubMed] [Google Scholar]
- 16.Wachter A. 2010. Riboswitch-mediated control of gene expression in eukaryotes. RNA Biol. 7, 67–76 10.4161/rna.7.1.10489 (doi:10.4161/rna.7.1.10489) [DOI] [PubMed] [Google Scholar]
- 17.Lee E. R., Baker J. L., Weinberg Z., Sudarsan N., Breaker R. R. 2010. An allosteric self-splicing ribozyme triggered by a bacterial second messenger. Science 329, 845–848 10.1126/science.1190713 (doi:10.1126/science.1190713) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Loh E., et al. 2009. A trans-acting riboswitch controls expression of the virulence regulator PrfA in Listeria monocytogenes. Cell 139, 770–779 10.1016/j.cell.2009.08.046 (doi:10.1016/j.cell.2009.08.046) [DOI] [PubMed] [Google Scholar]
- 19.Edwards T. E., Ferré-D'Amaré A. R. 2006. Crystal structures of the thi-box riboswitch bound to thiamine pyrophosphate analogs reveal adaptive RNA-small molecule recognition. Structure 14, 1459–1468 10.1016/j.str.2006.07.008 (doi:10.1016/j.str.2006.07.008) [DOI] [PubMed] [Google Scholar]
- 20.Lang K., Rieder R., Micura R. 2007. Ligand-induced folding of the thiM TPP riboswitch investigated by a structure-based fluorescence spectroscopic approach. Nucleic Acids Res. 35, 5370–5378 10.1093/nar/gkm580 (doi:10.1093/nar/gkm580) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klein D., Edwards T., Ferré-D'Amaré A. 2009. Cocrystal structure of a class I preQ1 riboswitch reveals a pseudoknot recognizing an essential hypermodified nucleobase. Nat. Struct. Mol. Biol. 16, 343–344 10.1038/nsmb.1563 (doi:10.1038/nsmb.1563) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rieder U., Kreutz C., Micura R. 2010. Folding of a transcriptionally acting preQ1 riboswitch. Proc. Natl Acad. Sci. USA 107, 10 804–10 809 10.1073/pnas.0914925107 (doi:10.1073/pnas.0914925107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kulshina N., Baird N. J., Ferré-D'Amaré A. R. 2009. Recognition of the bacterial second messenger cyclic diguanylate by its cognate riboswitch. Nat Struct. Mol. Biol. 16, 1212–1217 10.1038/nsmb.1701 (doi:10.1038/nsmb.1701) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baird N. J., Kulshina N., Ferré-D'Amaré A. R. 2010. Riboswitch function: flipping the switch or tuning the dimmer? RNA Biol. 7, 328–332 10.4161/rna.7.3.11932 (doi:10.4161/rna.7.3.11932) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiao H., Edwards T. E., Ferré-D'Amaré A. R. 2008. Structural basis for specific, high-affinity tetracycline binding by an in vitro evolved aptamer and artificial riboswitch. Chem. Biol. 15, 1125–1137 10.1016/j.chembiol.2008.09.004 (doi:10.1016/j.chembiol.2008.09.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ferré-D'Amaré A. R., Scott W. G. 2010. Small self-cleaving ribozymes. Cold Spring Harb. Perspect. Biol. 2, a003574. 10.1101/cshperspect.a003574 (doi:10.1101/cshperspect.a003574) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nakano S., Cerrone A. L., Bevilacqua P. C. 2003. Mechanistic characterization of the HDV genomic ribozyme: classifying the catalytic and structural metal ion sites within a multichannel reaction mechanism. Biochemistry 42, 2982–2994 10.1021/bi026815x (doi:10.1021/bi026815x) [DOI] [PubMed] [Google Scholar]
- 28.Das S. R., Piccirilli J. A. 2005. General acid catalysis by the hepatitis delta virus ribozyme. Nat. Chem. Biol. 1, 45–52 10.1038/nchembio703 (doi:10.1038/nchembio703) [DOI] [PubMed] [Google Scholar]
- 29.Bevilacqua P. C., Yajima R. 2006. Nucleobase catalysis in ribozyme mechanism. Curr. Opin. Chem. Biol. 10, 455–464 10.1016/j.cbpa.2006.08.014 (doi:10.1016/j.cbpa.2006.08.014) [DOI] [PubMed] [Google Scholar]
- 30.Rupert P. B., Ferré-D'Amaré A. R. 2001. Crystal structure of a hairpin ribozyme-inhibitor complex with implications for catalysis. Nature 410, 780–786 10.1038/35071009 (doi:10.1038/35071009) [DOI] [PubMed] [Google Scholar]
- 31.Rupert P. B., Massey A. P., Sigurdsson S. T., Ferré-D'Amaré A. R. 2002. Transition state stabilization by a catalytic RNA. Science 298, 1421–1424 10.1126/science.1076093 (doi:10.1126/science.1076093) [DOI] [PubMed] [Google Scholar]
- 32.Guo M., Spitale R. C., Volpini R., Krucinska J., Cristalli G., Carey P. R., Wedekind J. E. 2009. Direct Raman measurement of an elevated base pKa in the active site of a small ribozyme in a precatalytic conformation. J. Am. Chem. Soc. 131, 12 908–12 909 10.1021/ja9060883 (doi:10.1021/ja9060883) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ferré-D'Amaré A. R. 2004. The hairpin ribozyme. Biopolymers 73, 71–78 10.1002/bip.10516 (doi:10.1002/bip.10516) [DOI] [PubMed] [Google Scholar]
- 34.Martick M., Scott W. G. 2006. Tertiary contacts distant from the active site prime a ribozyme for catalysis. Cell 126, 309–320 10.1016/j.cell.2006.06.036 (doi:10.1016/j.cell.2006.06.036) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bevilacqua P. C. 2003. Mechanistic considerations for general acid-base catalysis by RNA: revisiting the mechanism of the hairpin ribozyme. Biochemistry 42, 2259–2265 10.1021/bi027273m (doi:10.1021/bi027273m) [DOI] [PubMed] [Google Scholar]
- 36.Wilson T. J., Li N.-S., Lu J., Frederiksen J. K., Piccirilli J. A., Lilley D. M. J. 2010. Nucleobase-mediated general acid–base catalysis in the Varkud satellite ribozyme. Proc. Natl Acad. Sci. USA 107, 11 751–11 756 10.1073/pnas.1004255107 (doi:10.1073/pnas.1004255107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ferré-D'Amaré A. R., Zhou K., Doudna J. A. 1998. Crystal structure of a hepatitis delta virus ribozyme. Nature 395, 567–574 10.1038/26912 (doi:10.1038/26912) [DOI] [PubMed] [Google Scholar]
- 38.Banás P., Walter N. G., Sponer J., Otyepka M. 2010. Protonation states of the key active site residues and structural dynamics of the glmS riboswitch as revealed by molecular dynamics. J. Phys. Chem. B 114, 8701–8712 10.1021/jp9109699 (doi:10.1021/jp9109699) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xin Y., Hamelberg D. 2010. Deciphering the role of glucosamine-6-phosphate in the riboswitch action of glmS ribozyme. RNA 16, 2455–2463 10.1261/rna.2334110 (doi:10.1261/rna.2334110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wilson D. S., Szostak J. W. 1999. In vitro selection of functional nucleic acids. Annu. Rev. Biochem. 68, 611–647 10.1146/annurev.biochem.68.1.611 (doi:10.1146/annurev.biochem.68.1.611) [DOI] [PubMed] [Google Scholar]
- 41.Chen X., Li N., Ellington A. D. 2007. Ribozyme catalysis of metabolism in the RNA world. Chem. Biodivers. 4, 633–655 10.1002/cbdv.200790055 (doi:10.1002/cbdv.200790055) [DOI] [PubMed] [Google Scholar]
- 42.Meli M., Vergne J., Maurel M.-C. 2003. In vitro selection of adenine-dependent hairpin ribozymes. J. Biol. Chem. 278, 9835–9842 10.1074/jbc.M213058200 (doi:10.1074/jbc.M213058200) [DOI] [PubMed] [Google Scholar]
- 43.Tsukiji S., Pattnaik S. B., Suga H. 2003. An alcohol dehydrogenase ribozyme. Nat. Struct. Biol. 10, 713–717 10.1038/nsb964 (doi:10.1038/nsb964) [DOI] [PubMed] [Google Scholar]
- 44.White H. B. 1976. Coenzymes as fossils of an earlier metabolic state. J. Mol. Evol. 7, 101–104 10.1007/BF01732468 (doi:10.1007/BF01732468) [DOI] [PubMed] [Google Scholar]
- 45.Ferré-D'Amaré A. R. 2003. RNA-modifying enzymes. Curr. Opin. Struct. Biol. 13, 49–55 10.1016/S0959-440X(02)00002-7 (doi:10.1016/S0959-440X(02)00002-7) [DOI] [PubMed] [Google Scholar]
- 46.Hoang C., Chen J., Vizthum C. A., Kandel J. M., Hamilton C. S., Mueller E. G., Ferré-D'Amaré A. R. 2006. Crystal structure of pseudouridine synthase RluA: indirect sequence readout through protein-induced RNA structure. Mol. Cell 24, 535–545 10.1016/j.molcel.2006.09.017 (doi:10.1016/j.molcel.2006.09.017) [DOI] [PubMed] [Google Scholar]
- 47.Noller H. F. 2010. Evolution of protein synthesis from an RNA world. Cold Spring Harb. Perspect. Biol. 2, a003681 (doi:10.1101/cshperspect.a003681) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Koizumi M., Soukup G. A., Kerr J. N., Breaker R. R. 1999. Allosteric selection of ribozymes that respond to the second messengers cGMP and cAMP. Nat. Struct. Biol. 6, 1062–1071 10.1038/14947 (doi:10.1038/14947) [DOI] [PubMed] [Google Scholar]
- 49.Benner S. A., Kim H.-J., Yang Z. 2010. Setting the stage: the history, chemistry, and geobiology behind RNA. Cold Spring Harb. Perspect. Biol. 2, a003541 (doi:10.1101/cshperspect.a003541) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Orgel L. E. 1968. Evolution of the genetic apparatus. J. Mol. Biol. 38, 381–393 10.1016/0022-2836(68)90393-8 (doi:10.1016/0022-2836(68)90393-8) [DOI] [PubMed] [Google Scholar]
- 51.White H. B. I. 1982. Evolution of coenzymes and the origin of pyridine nucleotides. In The pyridine nucleotide coenzymes (eds Everse J., Anderson B., You K.-S.), pp. 1–17 New York: Academic Press [Google Scholar]
- 52.Maizels N., Weiner A. M. 1993. The genomic tag hypothesis: modern viruses as molecular fossils of ancient strategies for genome replication. In The RNA world (eds Gesteland R. F., Atkins J. F.), pp. 577–602 Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press [Google Scholar]
- 53.Wong J. T. 1991. Origin of genetically encoded protein synthesis: a model based on selection for RNA peptidation. Orig. Life Evol. Biosph. 21, 165–176 10.1007/BF01809445 (doi:10.1007/BF01809445) [DOI] [PubMed] [Google Scholar]
- 54.Xiao H., Murakami H., Suga H., Ferré-D'Amaré A. R. 2008. Structural basis of specific tRNA aminoacylation by a small in vitro selected ribozyme. Nature 454, 358–361 10.1038/nature07033 (doi:10.1038/nature07033) [DOI] [PubMed] [Google Scholar]