

Abstract
A variety of macromolecules and small molecules—(oligo)nucleotides, proteins, lipids and metabolites—are collectively considered essential to early life. However, previous schemes for the origin of life—e.g. the ‘RNA world’ hypothesis—have tended to assume the initial emergence of life based on one such molecular class followed by the sequential addition of the others, rather than the emergence of life based on a mixture of all the classes of molecules. This view is in part due to the perceived implausibility of multi-component reaction chemistry producing such a mixture. The concept of systems chemistry challenges such preconceptions by suggesting the possibility of molecular synergism in complex mixtures. If a systems chemistry method to make mixtures of all the classes of molecules considered essential for early life were to be discovered, the significant conceptual difficulties associated with pure RNA, protein, lipid or metabolism ‘worlds’ would be alleviated. Knowledge of the geochemical conditions conducive to the chemical origins of life is crucial, but cannot be inferred from a planetary sciences approach alone. Instead, insights from the organic reactivity of analytically accessible chemical subsystems can inform the search for the relevant geochemical conditions. If the common set of conditions under which these subsystems work productively, and compatibly, matches plausible geochemistry, an origins of life scenario can be inferred. Using chemical clues from multiple subsystems in this way is akin to triangulation, and constitutes a novel approach to discover the circumstances surrounding the transition from chemistry to biology. Here, we exemplify this strategy by finding common conditions under which chemical subsystems generate nucleotides and lipids in a compatible and potentially synergistic way. The conditions hint at a post-meteoritic impact origin of life scenario.
Keywords: prebiotic chemistry, systems chemistry, chemoselectivity, phosphorylation
1. Introduction
Although life is ineluctably associated with the chemistry of complex mixtures, the majority of organic chemistry thus far practised in the laboratory has been of the two component A + B → C variety. This is for a number of reasons: early organic chemists had limited purification and analytical tools, and so found such reactions—especially if they proceeded in high yields—easiest to manage; single products of such reactions can themselves be mixed with other single components in a subsequent step of a linear multi-step synthesis; and, commercially, single products can conveniently be sold as such. For these and other reasons, much of modern chemistry has focused ‘above the arrow’ i.e. not on facilitating multi-reactant experiments, but on the reagents of transformation. There are a few multi-component reactions known—e.g. the Ugi reaction [1]—but these again tend to be directed towards single products, and the concept of a multi-component reaction giving many products has been an anathema to most organic chemists until recently.
When it comes to origins of life chemistry, studies tend to be of two sorts: conventional A + B → C reactions directed towards prebiotically important molecules; or multi-component reactions such as Miller–Urey type syntheses that lead to hugely complex product mixtures [2]. In reality, neither sort of reaction scheme is likely to be conducive to the emergence of life. The former involves implausibly simple mixtures of starting materials and gives only one of many required product molecules. The latter, though often involving more plausible reactant mixtures, lacks intrinsic chemical control, and results in mixtures so complex that useful products are obtained in minimal yield interspersed with numerous by-products.
What is needed is Goldilocks chemistry: chemistry that works on plausible reactant mixtures to give multiple required products in good yield without creating too many deleterious by-products. This is because diminutive yields of myriad products leads to system degeneration, as subsequent productive multi-molecular reactions are kinetically disfavoured through dilution raised to the power of the molecularity. The early work perhaps closest to being Goldilocks chemistry is a study of the Strecker multi-component reaction and associated chemistry in a prebiotic context by authors who can also be credited with the first use of the systems chemistry concept in this area [3]. Finding similar chemistry leading to a broader spectrum of biologically relevant structures—including nucleotides and lipids—has been the goal of our laboratory in recent years [4,5].
Our approach has been to study chemical subsystems, and ultimately to assemble these subsystems into a prebiotically plausible overall system with the goal of observing emergent behaviour. In prebiotic chemistry, as in traditional biochemistry, catalysis has proved to be key to directing reaction kinetics; even low-level catalysis by components of mixtures can have a profound effect on reaction outcomes [4]. If products of subsystems turn out to be catalysts of reactions in other subsystems, a complex temporally controlled series of reactions can ensue in the complete system. Reactions need not occur in cycles for such control, though cycles can provide a mechanism for molecular (auto-)amplification and control [6]. The chemical behaviour of the components of such a system may appear to be choreographed, but it is crucial to recognize that the behaviour is implicit in the structures of the input reagents under the defined conditions through the laws of chemistry.
The behaviour of the system can also be influenced by variation in reaction conditions over time. This is an important point in the prebiotic chemistry field because too often it has been assumed that only fixed conditions prevailing over a long time should be considered for prebiotic synthesis. Temperature, humidity and light flux must have varied on the early Earth, and so it is perfectly reasonable to invoke variations in conditions over the course of a prebiotic systems chemistry experiment. One is not seeking to recreate exactly what happened on early Earth, but to show what is plausible. Given the huge range of possible conditions, and sequences of conditions, we caution against the tendency to rigidly invoke one set or sequence of conditions, and then to limit the chemistry one explores to only that which is possible under the chosen conditions.
What seems a much better approach is to explore all sorts of different geochemically plausible (sequences of) conditions in an attempt to elucidate scenarios in which chemistry giving all the products needed for life takes place. If one set, range, or sequence of conditions is thereby uncovered that gives all the products, then it is akin to triangulation in navigation wherein multiple pointers provide better evidence than a single pointer to define the relevant chemical space.
2. Prebiotic ribonucleotide synthesis in a subsystem
We recently demonstrated a prebiotically plausible systems chemistry route to pyrimidine ribonucleotides in which phosphate, a dormant reagent in the early steps, acts as a general acid–general base catalyst, pH buffer and chemical buffer (scheme 1) [4]. The sequence of reaction conditions—mild heating in water, followed by evaporation, dry state heating, rehydration and UV irradiation—under which this synthesis proceeds serves as a starting point in our triangulation strategy to find conditions for an overall systems chemistry synthesis of multiple prebiotic target molecules. Accordingly, we are investigating other subsystems to synthesize lipids, peptides and metabolites. Should we find that the conditions under which these other subsystems are synthetically productive match any of the conditions of ribonucleotide synthesis, it will strengthen the case for the occurrence of the common conditions on the early Earth, and further support the prebiotic plausibility of the syntheses. Thus far we have investigated the synthesis of alkyl phosphates as lipids, and report our results herein. To appreciate the results of these latter experiments in the context of our prebiotic ribonucleotide synthesis, it is first necessary to briefly review that synthesis.
Scheme 1.

‘Systems chemistry’ prebiotic synthesis of the canonical pyrimidine ribonucleotides. An indication of the dissociative mechanism for the phosphorylation of anhydronucleosides 8 and 9 is shown in the box. Pi, inorganic phosphate.
Cyanamide 1 reacts with glycolaldehyde 2 in the presence of phosphate to give 2-aminooxazole 3. If the stoichiometry of 1 : 2 is greater than 1 : 1, excess 1 is then hydrated to urea 4 in a slower phosphate-catalysed reaction [7]. Addition of 2-aminooxazole 3 to glyceraldehyde 5 then gives the pentose aminooxazolines 6 with ribo-6 and arabino-6 predominant [4,8]. Arabino-6 turns out to be the desired intermediate for pyrimidine ribonucleotide synthesis, but there is a stereochemical twist to the story. It is not the arabino-6 initially formed by this addition reaction that ends up as the pyrimidine ribonucleotides; if it were, then there would be no way to produce enantiomerically pure ribonucleotides unless a prebiotic route to enantiopure glyceraldehyde 5 existed. Routes to non-racemic glyceraldehyde can be envisaged [9], but obtaining this three-carbon sugar enantiopure looks very difficult. A means of enantioenrichment is, therefore, needed at some point in the synthetic route. Ribo-6 is highly crystalline [10–12] and the least soluble of the pentose aminooxazolines 6 [12]. Furthermore, when ribo-6 crystallizes from non-racemic solutions it becomes enantioenriched [8]. At a threshold enantiomeric excess (ee) of about 60 per cent in solution, the crystalline ribo-6 that forms is enantiopure. Thus, if 2-aminooxazole 3 adds to glyceraldehyde 5 with an ee of 60 per cent, enantiopure ribo-6 crystallizes out of the reaction mixture.
Now the twist—ribo-6 can be equilibrated with arabino-6 by incubation in phosphate buffer solution [13]; moreover, if enantiopure ribo-6 as purified by crystallization is incubated in such a solution, it is converted into a mixture of enantiopure arabino-6 and enantiopure ribo-6. The enantiopure arabino-6 in this mixture can then react with cyanoacetylene 7 to give the anhydronucleoside 8 in a reaction in which phosphate serves to keep the pH from rising above neutrality, and thereby prevents base-catalysed hydrolysis and other reactions of 8 [4]. The enantiopure ribo-6 in the mixture also reacts with cyanoacetylene 7 to give anhydronucleoside 9. Conditions whereby 9 might be converted into purine ribonucleotides are currently being sought. However, if they are not found then it transpires that subsequent phosphorylation and irradiation steps, which convert the isomer 8 into pyrimidine ribonucleotides, will otherwise destroy the material deriving from 9 because of a remarkable series of chemoselectivity effects [4,14].
The phosphorylation of 8 by inorganic phosphate, which is key to the synthesis, takes place either in urea 4 melts, or in formamide–urea 4 mixtures at elevated temperatures, due to nucleophilic catalysis by urea 4, as mentioned previously, a co-product of the first step of the reaction sequence. Chemoselective phosphorylation of the 3′-hydroxyl group of anhydronucleoside 8 followed by rearrangement of the resultant 3′-phosphate to ribo-cytidine-2′,3′-cyclic phosphate 10 (base = Cyt) is observed. 5′-Phosphorylation also takes place, though to a lesser degree if the ratio of 8 : phosphate is 1 : 1, resulting in the concomitant production of the diphosphate 11 [4]. Mechanistically (scheme 1, box), a carbonyl group lone-pair of 4 attacks a rare tautomer 12 of the phosphate mono-anion to displace water and generate the imidoyl phosphate 13. Although 12 is a minor species, it is highly reactive towards nucleophilic substitution because the phosphorus atom bears a good leaving group—water—and two negatively charged oxygen substituents, so that reaction can occur dissociatively via a metaphosphate-like transition state. Imidoyl phosphate 13 can then undergo attack at phosphorus either by water—in which case, the formation reaction is reversed—or by various alcohols. In the latter case, rare tautomers of alkyl monophosphate mono-anions are produced that then reversibly tautomerize to singly charged alkylphosphate mono-anions. The phosphorylation chemistry is slowly reversible, though the evaporation of water results in predominant alcohol phosphorylation.
This slow reversibility means that chemoselectivity can result from thermodynamic and kinetic effects. Kinetically, 3′-phosphorylation is favoured because an nO → π*CN stereoelectronic effect between the O5′ atom and C2 of the nucleobase in 8 reduces electron density on the O5′ atom and increases its steric bulk relative to the O3′ atom [14]. Thermodynamically, 3′-phosphorylation is favoured because the 3′-phosphate can rearrange to the more stable cyclic nucleotide 10 (base = Cyt) [15], whereas the 5′-phosphate cannot. It transpires that phosphate can also bring about the hydrolysis of the anhydronucleoside C2–O2′ bond via nucleophilic addition at C2 followed by dissociative nucleophilic attack at phosphorus by urea 4; another consequence of the nO → π*CN stereoelectronic effect in anhydronucleoside 8 is that this hydrolysis is slowed relative to that of isomer 9 because the electrophilicity of C2 of 8 is lower than that of C2 of 9 due to the partial donation of electron density from O5′ [14]. The resultant in situ hydrolysis of 9 to α-ribonucleoside 14 then allows phosphorylation of this free nucleoside to give a range of products all destined to be destroyed by UV irradiation in the next step, whereas 10 (base = Cyt) simply converts to 10 (base = Cyt, Ura) [4].
3. The need for membranes
The importance of membranes in the origin of life was noted by Haldane in the late 1920s [16]. The self-assembly of bilayer vesicles (including those encapsulating macromolecules) from lipids has been amply demonstrated [16] and various reactions have been shown to take place in, or be catalysed by, such structures [17]. Lipids are amphiphilic molecules normally composed of long chain aliphatic hydrocarbons with polar head groups. In a general sense, bilayer formation requires a lipid composition ratio of one polar head to two non-polar tails. Each polar head can have either one or two non-polar tails attached. For those polar heads attached to only one non-polar tail, a second non-polar tail molecule lacking a highly polar head is needed in roughly equimolar quantity. Single chain amphiphiles can form bilayers under conditions where an ionizable head group is only partly ionized—the unionized head group not being highly polar—otherwise such amphiphiles usually form micelles rather than bilayer structures such as vesicles [18,19].
In considering the possible prebiogenesis of lipids, we have realized that one likely synthetic sequence is linked to the production of phosphate on the early Earth (vide infra). Because of the central role of phosphate in our prebiotic synthesis of ribonucleotides, and the growing appreciation of the evolutionary advantages that accrue if (self-)replicating informational molecules are compartmentalized in (dividing) vesicles [20,21], we have sought prebiotic chemistry that leads to both ribonucleotides and lipids in a single system.
4. A linked source of phosphate and lipids: towards a higher order system
The most plausible prebiotic source of phosphate is from anoxic corrosion of schreibersite [22,23], a mineral found in iron–nickel meteorites. By a series of disproportionation reactions, this iron–nickel phosphide ((Fe,Ni)3P) produces ferrous and nickel phosphates as well as more reduced phosphorus-containing species. The phosphate can be mobilized in solution if ferrous and nickel ions are precipitated or sequestered in some way. Experimentally, sulphide addition causes precipitation of FeS and NiS, and this is probably prebiotically relevant as it is thought that significant H2S was available from volcanic emissions. Alternatively—or additionally—it is possible that hydrogen cyanide could sequester the ferrous iron as hexacyanoferrate(II) [24]. This is an attractive possibility as it would both liberate phosphate and concentrate hydrogen cyanide; subsequent irradiation could then release the hydrogen cyanide [24].
In keeping with the approach outlined in the introduction, this meteoritic source of phosphate suggests that geochemistry associated with iron/nickel meteorite impacts should be considered. Our modus operandi has it that the resultant sequence of geochemical conditions should be assessed on its own merits, but also with the requirements of our systems chemistry synthesis of ribonucleotides in mind, and triangulation our goal.
Large impactors would deliver considerable energy with meteorite and ejecta vapour persisting in the atmosphere after the impact. This would in turn cause significant ocean vapourization through downward thermal radiation, and production of atom and molecular fragment recombination products such as hydrogen cyanide and cyanoacetylene. Later, after partial cooling, hot iron/nickel particles in the water vapour- and CO2-rich atmosphere would catalyse first reduction of atmospheric CO2 to CO [25], and then Fischer–Tropsch (FT) chemistry (2CO + H2 → –CH2– + CO2) on a huge scale [26].
A prolonged hydrogen-rich atmosphere on the early Earth seems unlikely because this lightest of elements is unfettered by Earth's gravity. Accordingly, the particular type of FT chemistry that is most likely following iron/nickel meteorite impact is the Kölbel–Engelhardt (KE) variant (3CO + H2O → –CH2– + 2CO2) as this does not require an atmosphere rich in hydrogen per se. Instead, the hydrogen is synthesized in situ by a water gas shift reaction (CO + H2O → CO2 + H2) [27]. Under optimal laboratory conditions, the reaction proceeds with more than 80 per cent CO conversion. Hydrocarbon yields in the KE reaction are optimal when the CO : H2O ratio is ca 2.6—an excess of CO leads to carbon deposition, whereas an excess of H2O causes catalyst oxidation with concomitant production of H2 [27]. In the presence of a large amount of iron, these stoichiometric processes would be expected to shift the local CO : H2O ratio towards the optimal for hydrocarbon synthesis.
Significantly, the KE reaction produces a higher fraction of oxygenates than the FT reaction. Straight chain alkanols, ketones, aldehydes (including H2CO) and carboxylic acids are formed in addition to straight chain hydrocarbons [27]. This array of products is produced in the temperature range 180–280°C; at higher temperatures, CO conversions drop slowly but methane production becomes predominant (methanation). The percentage CO conversion is essentially constant over the pressure range 1–100 bar, but the oxygenate content rises with pressure (3% alcohols by weight at 3 bar, 30% by weight at 100 bar). Given these attributes of the KE reaction, the presence of mixed length straight chain alkanols on the early Earth seems highly likely.
The presence of NiS on the Earth's surface in a CO-containing atmosphere could have led to the synthesis of other FT-type products—including thioacids and the corresponding carboxylic acids—by the same sorts of chemistry that have been invoked by other authors for deep-sea hydrothermal vent scenarios [28,29]. However, the surface scenario we describe here has the added advantage that the other chemistries required to make nucleotides, inter alia, are possible in the same location, as conditions naturally change over time. In contrast, deep-sea vents do not allow drying down, dry state heating or UV irradiation. Thus, when conditions calmed down after an impact, phosphate and organic nucleotide precursors could have been present along with mixed alkanols. Gases such as cyanoacetylene and hydrogen cyanide would partition between the gaseous atmospheric phase and aqueous solution surface phase. As mentioned, hydrogen cyanide could be concentrated during phosphate mobilization, whereas cyanoacetylene would be expected to gradually dissolve in surface water and react with nucleophilic species dissolved therein. During such a period, the chemistry from 1, 2, 5 and 7 to anhydronucleosides 8 and 9 could have taken place. Then a temperature increase—perhaps associated with another impact—could cause aqueous surface phases to dry out and the residues to be heated; this is when the conversion of 8 and 9 to 10, 11 and other nucleotides could have taken place.
To reiterate: this sequence of conditions and the scenario causing it were not arbitrarily selected ahead of our experimental investigations into prebiotic nucleotide chemistry. The sequence of conditions is empirically realized by the chemistry we, and others, have discovered. The scenario is an attempt to match this sequence of conditions to a plausible sequence of geochemical events. The reason for attempting this is that such considerations point to the likely co-location of anhydronucleosides and mixed chain alkanols when phosphorylation conditions occurred.
5. Alkanol phosphorylation
In light of the foregoing, we investigated the phosphorylation of mixed length alkanols under the conditions previously established for anhydronucleoside phosphorylation and rearrangement [4] (scheme 2). Short chain alkanols tend to predominate over medium and long chain alkanols in the oxygenate fraction of products synthesized by the KE variant of the FT reaction [27], and so, as a representative KE oxygenate fraction for phosphorylation, we employed a mixture of ethanol, hexanol and decanol, with the C2 alkanol in a large molar excess over the longer chain species. Because ethyl phosphate is not amphiphilic, significant chemoselectivity would have to operate for membrane-forming amphiphiles to be produced by phosphorylation of such a mixture using sub-stoichiometric phosphate. In the event, we were delighted to find that when such an oxygenate fraction was mixed with ammonium phosphate and urea, dried down, and heated on a glass fibre support—to mimic spreading out on an (inert) mineral surface—no ethyl phosphate was apparent in the 1H-NMR spectrum of the products after washing of the solid support and lyophilization of the washings. Downfield, phosphorus-coupled methylene signals resonated as a quartet indicative of longer chain alkyl phosphate(s) (the methylene group of ethyl phosphate has a higher multiplicity). Furthermore, the next most downfield signal for the β-methylene protons was a quintet with no trace of the triplet expected for ethyl phosphate. Integration of all other methylene signals indicated that decyl phosphate and hexyl phosphate were both present in a ratio of 7 : 3 (no residual long chain alcohols were detectable—either the excess alkanol vapourized, or it is possible that these alcohols were not eluted from the solid phase during the washing protocol). Precise yield determination based on weight was not possible because of uncertainties concerning how much of the various reagents remained after the washing and lyophilization steps. However, related phosphorylations have been found to approach quantitative yields based on phosphate incorporation with only traces of inorganic phosphate, pyrophosphate and higher polyphosphates, if the alcohol to be phosphorylated is in (even slight) excess over phosphate.
Scheme 2.

Selective formation of long chain alkyl phosphates in the phosphorylation of mixed chain length alkanols.
The chemoselective formation of decyl phosphate over hexyl phosphate, and the lack of ethyl phosphate can be attributed to the reversibility of the phosphorylation, and the progressively lower volatility of ethanol, hexanol and decanol. It is highly significant that the temperature that was found experimentally to be ideal for the phosphorylation of the anhydronucleosides 8 and 9 also results in a chain length chemoselectivity for alkanol phosphorylation favouring the formation of amphiphilic medium and long chain alkyl phosphates. If such alkyl phosphates were to be mixed with similarly thermally fractionated alkanols, or if the phosphorylation reaction used less phosphate than mixed single chain amphiphile, long chain non-amphiphile combinations with the potential to form bilayer vesicles would result. This is currently being investigated experimentally. Furthermore, mixed alkanol/anhydronucleoside phosphorylations are being studied because of the potential for forming compartmentalized nucleotides as well as an additional aspect of chemoselectivity. As mentioned above, when 8 undergoes phosphorylation, significant quantities of the diphosphate 11 are produced in addition to the desired product 10. In fact, when pyrophosphate–urea mixtures are employed as the phosphorylating agent with a pyrophosphate:anhydronucleoside ratio of 5 : 2, 11 can be produced in yields of 70 per cent (J.D.S and M.W.P unpublished data).
It thus seems likely that phosphorylation of a mixture comprising anhydronucleoside 8, excess pyrophosphate, and an even greater excess of mixed chain length alkanols will give 10—rather than 11—in increased yield along with long chain alkyl phosphates and long chain alkanols. This expectation is based on the fact that an excess of pyrophosphate over 8 will drive phosphorylation of both the 5′- and 3′-hydroxyl groups, but phosphorylation of the former is reversible whereas phosphorylation of the latter is rendered irreversible by the rearrangement that generates the 2′,3′-cyclic phosphate. The reversible phosphorylation of the 5′-hydroxyl groups should then compete with reversible long chain alkanol phosphorylation, and if the alkanols are in an excess over the 5′-hydroxyl groups, the latter should not end up being extensively phosphorylated at equilibrium. When the analytical methods for fully characterizing such mixtures with respect to individual product yield and assessment of nucleotide-containing vesicle formation are established, we will carry out such experiments and report the results.
6. The way forward
The modus operandi for prebiotic chemistry adumbrated in this paper and our results thus far suggest a clear way forward. Through investigation of multi-component reactions and sequential one-pot multi-step synthetic sequences, productive subsystems will be further defined. After interrelation of these subsystems, it will hopefully prove possible to thereby adduce a sequence, set or range of geochemically plausible conditions that could have simultaneously and chemoselectively delivered a broad spectrum of biologically relevant molecules on the early Earth. Delivery of these molecules within the full system under such conditions can then be experimentally reproduced with high expectations of emergent properties.
7. Experimental section
(a). Synthesis of decyl and hexyl phosphate monoesters via the diallyl phosphotriesters
(i). Diallyl decyl phosphate
Decanol (36 µl, 0.19 mmol) was dissolved in anhydrous acetonitrile (1 ml) over 3 Å molecular sieves (30 mg, dried over P2O5 at 300°C) under a nitrogen atmosphere and the suspension was stirred for 30 min. N,N-Bis-iso-propyl-diallyl phosphoramidite (0.1 ml, 0.38 mmol) was added and the reaction was stirred for 30 min, before 1H-tetrazole solution (0.45 M in acetonitrile, 3.4 ml) was added and the mixture was stirred for 24 h. t-Butyl hydrogen peroxide solution (6.0 M in decane, 0.76 ml) was added and the mixture was stirred for 2 h. The reaction mixture was filtered through a pad of Celite (10 mm), concentrated in vacuo and the residue purified by silica gel flash column chromatography (eluting with cyclohexane : EtOAc 3 : 2) to give 45 mg (75%) of the title compound as a clear oil. Rf: 0.5 (cyclohexane : EtOAc 4 : 1). 1H-NMR (500 MHz, CDCl3) δH 5.86 ppm (ddtt, J = 17.2, 10.3, 5.5, 0.8 Hz, 2H, CH = CH2); 5.30 (dq, J = 17.2, 1.5 Hz, 2H, CH = CH2); 5.19 (dq, J = 10.3, 1.3 Hz, 2H, CH = CH2); 4.47 (ddt, J = 8.1, 5.5, 1.3 Hz, 4H, CH2CH = CH2); 3.98 (q, J = 6.8 Hz, 2H, POCH2CH2CH2); 1.64 (q, J = 2.5 Hz, 1H, POCH2CH2CH2); 1.62 (q, J = 6.6 Hz, 1H, POCH2CH2CH2); 1.12–1.37 (m, 14H, (CH2)7); 0.81 (t, J = 7.1 Hz, 3H, Me). 13C-NMR (101 MHz, CDCl3) δC 132.5 ppm (2C, CH = CH2); 118.1 (2C, CH = CH2); 68.1 (2C, CH = CH2CH2); 68.0 (POCH2CH2CH2); 31.9 (POCH2CH2CH2); 30.3 (POCH2CH2CH2); 30.2 (CH2); 29.5 (CH2); 29.3 (CH2); 29.1 (CH2); 25.4 (CH2); 22.7 (CH2); 14.1 (Me). 31P-NMR (162 MHz, CDCl3) δP –0.70 ppm (h, J = 7.8 Hz). 1H-decoupled 31P-NMR (162 MHz, CDCl3) δP –0.70 ppm. ES–MS (pos. m/z): 341 (100%, [M + Na+]+). HRMS (m/z): [M + Na+]+ calcd for C16H31O4NaP, 341.1852; found, 341.1855 (figure 1).
Figure 1.
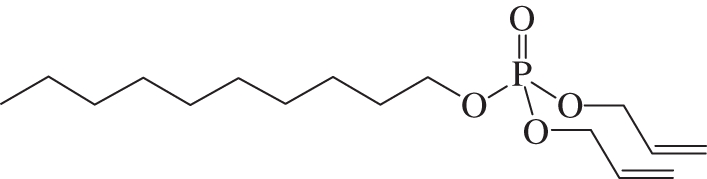
Diallyl decyl phosphate.
(ii). Sodium decyl phosphate
Diallyl decyl phosphate (30 mg, 0.10 mmol) was dissolved in CH2Cl2 (5 ml), trimethylsilyl bromide (64 µl, 0.5 mmol) was added and the solution was stirred under a nitrogen atmosphere for 15 h. The reaction mixture was then concentrated in vacuo. The residue was dissolved in H2O and the resultant solution was adjusted to pH = 9.0 with 1 M NaOH and lyophilized to give 20 mg (84%) of the title compound as a white semi-solid. 1H-NMR (400 MHz, D2O) δH 3.57 ppm (q, J = 7.0 Hz, 2H, POCH2CH2CH2); 1.43 (quin., J = 6.9 Hz, 2H, POCH2CH2CH2); 1.04–1.22 (m, 14H, (CH2)7); 0.71 (t, J = 7.1 Hz, 3H, Me). 31P-NMR (162 MHz, D2O) δP 3.86 ppm (t, J = 5.9 Hz). 1H-decoupled 31P-NMR (162 MHz, D2O) δP 3.86 ppm. ES–MS (neg. m/z): 237 (100%, [M–Na+]−). HRMS (m/z): [M–Na+]− calcd for C10H22O4P, 237.1261; found, 237.1263 (figure 2).
Figure 2.
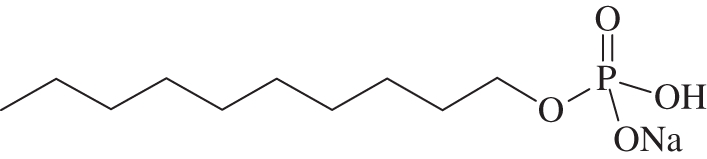
Sodium decyl phosphate.
(iii). Diallyl hexyl phosphate
Following the procedure described above for the preparation of diallyl decyl phosphate, hexanol (23 µl, 0.19 mmol) was converted to diallyl hexyl phosphate 41 mg (83%) as a clear oil. Rf: 0.5 (cyclohexane : EtOAc 4 : 1). 1H-NMR (500 MHz, CDCl3) δH 5.89 ppm (ddtt, J = 17.2, 11.1, 5.5, 1.0 Hz, 2H, CH = CH2); 5.30 (dq, J = 17.2, 1.5 Hz, 2H, CH = CH2); 5.19 (dq, J = 10.3, 1.3 Hz, 2H, CH = CH2); 4.44–4.50 (m, J = 8.1, 5.5, 1.3 Hz, 4H, POCH2CH = CH2); 3.99 (q, J = 6.8 Hz, 2H, POCH2CH2CH2); 1.61 (quin., J = 6.9 Hz, 2H, POCH2CH2CH2); 1.18–1.36 (m, 8H, (CH2)); 0.82 (t, J = 7.1 Hz, 3H, Me). 13C-NMR (101 MHz, CDCl3) δC 131.6 ppm (2C, CH = CH2); 117.1 (2C, CH = CH2); 67.5 (2C, CH = CH2CH2); 67.0 (POCH2CH2CH2); 30.3 (POCH2CH2CH2); 29.2 (POCH2CH2CH2); 24.1 (CH2CH2CH3); 21.5 (CH2CH3); 12.9 (Me). 31P-NMR (162 MHz, CDCl3) δP –0.70 ppm (h, J = 7.8 Hz). 1H-decoupled 31P-NMR (162 MHz, CDCl3) δP –0.70 ppm. ES–MS (pos. m/z): 285 (100%, [M + Na+]+). HRMS (m/z): [M + Na+]+ calcd for C12H23O4NaP, 285.1226; found, 285.1233 (figure 3).
Figure 3.
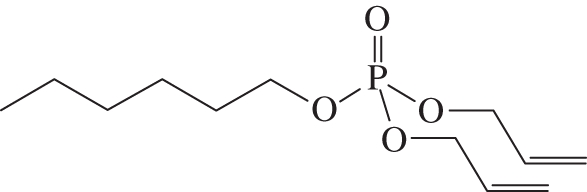
Diallyl hexyl phosphate.
(iv). Sodium hexyl phosphate
Following the procedure described above for the preparation of sodium decyl phosphate, diallyl hexyl phosphate (26 mg, 0.10 mmol) was converted to sodium hexyl phosphate 10 mg (54%) as a white solid. 1H-NMR (400 MHz, D2O) δH 3.57 ppm (q, J = 7.0 Hz, 2H, POCH2); 1.42 (quin., J = 7.1 Hz, 2H, POCH2CH2); 1.05–1.25 (m, 6H, (CH2)3); 0.71 (t, J = 6.7 Hz, 3H, CH2CH3). 31P-NMR (162 MHz, D2O) δP 2.60 ppm (t, J = 5.9 Hz). 1H-decoupled 31P-NMR (162 MHz, D2O) δP 2.60 ppm. ES–MS (neg. m/z): 181 (100%, [M–Na+]−). HRMS (m/z): [M–Na+]− calcd for C6H14O4P, 181.0635; found, 181.0631 (figure 4).
Figure 4.
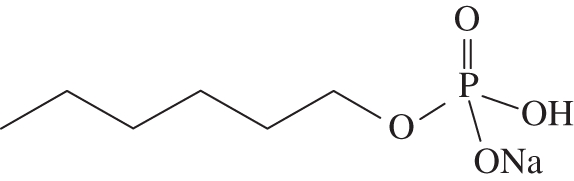
Sodium hexyl phosphate.
(b). Mixed chain length alkanol phosphorylation
Decanol (23.6 µl, 0.12 mmol), hexanol (15.5 µl, 0.12 mmol), ethanol (0.25 ml), ammonium phosphate (14.2 mg, 0.12 mmol) and urea (74 mg, 1.20 mmol) were dissolved in H2O (0.5 ml). The solution was applied evenly to two glass fibre discs (25 mm × 0.68 mm), the discs were dried at 40°C for 2 days and then heated at 100°C for 24 h. The discs were then washed with H2O (10 ml) and the washings were lyophilized. The lyophilisate was then dissolved in D2O (3 ml) and adjusted to pD = 9.0 to give complete solubility. The sample was analysed by 1H- and 31P-NMR and spiked with authentic standards of hexyl phosphate and decyl phosphate as prepared above. Relative integration of methylene, methyl and O-methylene protons showed no presence of ethyl phosphate, and a product ratio of decyl phosphate : hexyl phosphate 7 : 3 was observed. 1H-NMR (500 MHz, D2O) δH 3.71 ppm (q, J = 6.6 Hz, 2H, POCH2); 1.50 (quin., J = 6.6 Hz, 2H, POCH2CH2), 1.12–1.27 (m, 12H, (CH2)n); 0.75 (t, J = 6.8 Hz, 3H, Me).31P-NMR (162 MHz, D2O) δP 0.90 ppm (m). 1H-decoupled 31P-NMR (162 MHz, D2O) δP 0.90 ppm.
Acknowledgements
This work was supported by the Engineering and Physical Sciences Research Council through the provision of a studentship to M.W.P. We thank Samuel Bjork, Christopher Chan, Dr Colm Duffy and Dr Dougal Ritson for helpful suggestions.
References
- 1.Ugi I. 1962. The α-addition of immonium ions and anions to isonitriles accompanied by secondary reactions. Angew. Chem. Int. Ed. Engl. 1, 8–21 10.1002/anie.196200081 (doi:10.1002/anie.196200081) [DOI] [Google Scholar]
- 2.Lazcano A., Bada J. L. 2004. The 1953 Stanley L. Miller experiment: fifty years of prebiotic organic chemistry. Orig. Life Evol. Biosph. 33, 235–242 10.1023/A:1024807125069 (doi:10.1023/A:1024807125069) [DOI] [PubMed] [Google Scholar]
- 3.Pascal R., Taillades J., Commeyras A. 1978. Systemes de Strecker et apparentes—X: decomposition et hydratation en milieu aqueux basique des α-aminonitriles secondaires. Processus d'hydratation autocatalytique et catalysé par l'acetone. Tetrahedron 34, 2275–2281 10.1016/0040-4020(78)89038-3 (doi:10.1016/0040-4020(78)89038-3) [DOI] [Google Scholar]
- 4.Powner M. W., Gerland B., Sutherland J. D. 2009. Synthesis of activated pyrimidine ribonucleotides in prebiotically plausible conditions. Nature 459, 239–242 10.1038/nature08013 (doi:10.1038/nature08013) [DOI] [PubMed] [Google Scholar]
- 5.Mullen L. B., Sutherland J. D. 2007. Formation of potentially prebiotic amphiphiles by reaction of β-hydroxy-n-alkylamines with cyclotriphosphate. Angew. Chem. Int. Ed. Engl. 46, 4166–4168 10.1002/anie.200700394 (doi:10.1002/anie.200700394) [DOI] [PubMed] [Google Scholar]
- 6.Blackmond D. G. 2009. An examination of the role of autocatalytic cycles in the chemistry of proposed primordial reactions. Angew. Chem. Int. Ed. Engl. 48, 386–390 10.1002/anie.200804565 (doi:10.1002/anie.200804565) [DOI] [PubMed] [Google Scholar]
- 7.Lohrmann R., Orgel L. E. 1968. Prebiotic synthesis: phosphorylation in aqueous solution. Science 161, 64–66 10.1126/science.161.3836.64 (doi:10.1126/science.161.3836.64) [DOI] [PubMed] [Google Scholar]
- 8.Anastasi C., Crowe M. A., Powner M. W., Sutherland J. D. 2006. Direct assembly of nucleoside precursors from two- and three-carbon units. Angew. Chem. Int. Ed. Engl. 45, 6176–6179 10.1002/anie.200601267 (doi:10.1002/anie.200601267) [DOI] [PubMed] [Google Scholar]
- 9.Breslow R., Cheng L. 2010. L-Amino acids catalyze the formation of an excess of D-glyceraldehyde, and thus of other D sugars, under credible prebiotic conditions. Proc. Natl Acad. Sci. USA 107, 5723–5725 10.1073/pnas.1001639107 (doi:10.1073/pnas.1001639107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sanchez R. A., Orgel L. E. 1970. Studies in prebiotic synthesis V. Synthesis and photoanomerization of pyrimidine nucleosides. J. Mol. Biol. 47, 531–543 10.1016/0022-2836(70)90320-7 (doi:10.1016/0022-2836(70)90320-7) [DOI] [PubMed] [Google Scholar]
- 11.Borsenberger V., Crowe M. A., Lehbauer J., Raftery J., Helliwell M., Bhutia K., Cox T., Sutherland J. D. 2004. Exploratory studies to investigate a linked prebiotic origin of RNA and coded peptides. Chem. Biodivers. 1, 203–246 10.1002/cbdv.200490020 (doi:10.1002/cbdv.200490020) [DOI] [PubMed] [Google Scholar]
- 12.Springsteen G., Joyce G. F. 2004. Selective derivatization and sequestration of ribose from a prebiotic mix. J. Am. Chem. Soc. 126, 9578–9583 10.1021/ja0483692 (doi:10.1021/ja0483692) [DOI] [PubMed] [Google Scholar]
- 13.Powner M. W., Sutherland J. D. 2010. Phosphate-mediated interconversion of ribo- and arabino-configured prebiotic nucleotide intermediates. Angew. Chem. Int. Ed. Engl. 49, 4641–4643 10.1002/anie.201001662 (doi:10.1002/anie.201001662) [DOI] [PubMed] [Google Scholar]
- 14.Choudhary A., Kamer K. J., Powner M. W., Sutherland J. D., Raines R. T. 2010. A stereoelectronic effect in prebiotic nucleotide synthesis. ACS Chem. Biol. 5, 655–657 10.1021/cb100093g (doi:10.1021/cb100093g) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tapiero C. M., Nagyvary J. 1971. Prebiotic formation of cytidine nucleotides. Nature 231, 42–43 10.1038/231042a0 (doi:10.1038/231042a0) [DOI] [PubMed] [Google Scholar]
- 16.Deamer D. W. 1998. The molecular origins of life (ed. Brack A.), pp. 189–205 Cambridge, UK: Cambridge University Press [Google Scholar]
- 17.Segré D., Ben-Eli D., Deamer D. W., Lancet D. 2001. The lipid world. Orig. Life Evol. Biosph. 31, 119–145 10.1023/A:1006746807104 (doi:10.1023/A:1006746807104) [DOI] [PubMed] [Google Scholar]
- 18.Mansy S. S., Szostak J. W. 2008. Thermostability of model protocell membranes. Proc. Natl Acad. Sci. USA 105, 13 351–13 355 10.1073/pnas.0805086105 (doi:10.1073/pnas.0805086105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Apel C. L., Deamer D. W., Mautner M. N. 2002. Self-assembled vesicles of monocarboxylic acids and alcohols: conditions for stability and for the encapsulation of biopolymers. Biochim. Biophys. Acta 1559, 1–9 10.1016/S0005-2736(01)00400-X (doi:10.1016/S0005-2736(01)00400-X) [DOI] [PubMed] [Google Scholar]
- 20.Szostak J. W., Bartel D. P., Luisi P. L. 2001. Synthesizing life. Nature 409, 387–390 10.1038/35053176 (doi:10.1038/35053176) [DOI] [PubMed] [Google Scholar]
- 21.Mansy S. S., Schrum J. P., Krishnamurthy M., Tobé S., Treco D. A., Szostak J. W. 2008. Template-directed synthesis of a genetic polymer in a model protocell. Nature 454, 122–126 10.1038/nature07018 (doi:10.1038/nature07018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bryant D. E., Kee T. P. 2006. Direct evidence for the availability of reactive, water soluble phosphorus on the early Earth. H-Phosphinic acid from the Nantan meteorite. Chem. Commun. 2006, 2344–2346 10.1039/b602651f (doi:10.1039/b602651f) [DOI] [PubMed] [Google Scholar]
- 23.Pasek M. A., Lauretta D. S. 2006. Aqueous corrosion of phosphide minerals from iron meteorites: a highly reactive source of prebiotic phosphorus on the surface of the early Earth. Astrobiology 5, 515–535 10.1089/ast.2005.5.515 (doi:10.1089/ast.2005.5.515) [DOI] [PubMed] [Google Scholar]
- 24.Keefe A. D., Miller S. L. 1996. Was ferrocyanide a prebiotic reagent? Orig. Life Evol. Biosph. 26, 111–129 10.1007/BF01809851 (doi:10.1007/BF01809851) [DOI] [PubMed] [Google Scholar]
- 25.Kasting J. F. 1990. Bolide impacts and the oxidation state of carbon in the Earth's early atmosphere. Orig. Life Evol. Biosph. 20, 199–231 10.1007/BF01808105 (doi:10.1007/BF01808105) [DOI] [PubMed] [Google Scholar]
- 26.Nooner D. W., Oro J. 1987. Synthesis of fatty acids by a closed system Fischer–Tropsch process. Adv. Chem. 178, 159–171 10.1021/ba-1979-0178.ch012 (doi:10.1021/ba-1979-0178.ch012) [DOI] [Google Scholar]
- 27.Kölbel H., Ralek M. 1984. The Fischer–Tropsch synthesis (ed. Anderson R. B.), pp. 265–294 Orlando, FL: Academic Press [Google Scholar]
- 28.Loison A., Dubant S., Adam P., Albrecht P. 2010. Elucidation of an iterative process of carbon–carbon bond formation of prebiotic significance. Astrobiology 10, 973–988 10.1089/ast.2009.0441 (doi:10.1089/ast.2009.0441) [DOI] [PubMed] [Google Scholar]
- 29.Huber C., Wächterhäuser G. 1997. Activated acetic acid by carbon fixation on (Fe,Ni)S under primordial conditions. Science 276, 245–247 10.1126/science.276.5310.245 (doi:10.1126/science.276.5310.245) [DOI] [PubMed] [Google Scholar]