

Abstract
The smallest ribozyme that carries out a complex group transfer is the sequence GUGGC-3′, acting to aminoacylate GCCU-3′ (and host a manifold of further reactions) in the presence of substrate PheAMP. Here, I describe the enzymatic rate, the characterization of about 20 aminoacyl-RNA and peptidyl-RNA products and the pathways of these GUGGC/GCCU reactions. Finally, the topic is evolution, and the potential implications of these data for the advent of translation itself.
Keywords: RNA, aminoacylation, aminoacyl-RNA, peptidyl-RNA, translation
1. Introduction
A pentanucleotide ribozyme, discovered by a combination of selection and design, conducts one of the central group transfers of modern translation, linking an activated amino acid to the 2′,3′ terminus of a substrate oligoribonucleotide. The existence of such a simple catalyst raises many evolutionary questions, for example, might a similar means of aminoacyl-RNA production appear in the time of an RNA world, during the creation of a translation system?
2. A more precise selection was sought
At the initiation of these experiments, we had carried out many previous selections (the prototype is that of Illangasekare et al. [1]) for aminoacyl-RNA synthesis:
where the amino acid (aa) was activated by the biological leaving group 5′-adenylic acid (AMP). Selected RNAs both accelerated the transfer of the aminoacyl group and accepted the aminoacyl ester at a terminal 2′,3′ hydroxyl. The aminoacyl-RNA produced consequently connects the chemically activated amino acid (as its ribose ester) to an RNA sequence, which might serve as an anticodon, as also occurs in aatRNA during modern translation. By reacting with the unique α-amino groups of an RNA covalently linked to an amino acid, many different RNAs were selected for aaRNA synthesis [2,3]. Some were non-specific (i.e. accepted varied amino acid side chains), but others acted as rapidly and accurately as highly evolved protein aminoacyl-RNA synthetases perform a parallel aminoacylation. The transacylation active centre had been defined in an RNA as small as 29 nucleotides [4].
The impetus for yet another selection was the realization that earlier selections had been complicated by a well-described side reaction of RNA polymerases [5]. In the usual realization of a selection experiment [1], the selected molecule called RNA in the equation above is actually a runoff transcript made from a randomized-sequence synthetic DNA template. This had the consequence that the actual site of reaction, the 3′-terminal nucleotide ribose, was altered by the tendency of RNA polymerase to add untemplated A (and other nucleotides) to a templated 3′ terminus. Thus, the actual site of reaction, although its hydroxyl presumably must approach the substrate carbonyl to within atomic precision, was nonetheless on different nucleotides in different candidate molecules.
3. An arbitrary terminus sequence
And as if 3′ vagueness were not a sufficient hindrance, RNA for selection also had an arbitrary experimenter-specified sequence at its 3′ end. All molecules required unvarying terminal primer complement, required for reverse transcription and subsequent DNA amplification. These steps were essential to recapture a minority of active RNA sequences for further selection. Therefore, selection required reaction at the end of a sequence chosen for use in amplification without the knowledge of the needs of the transacylation reaction centre, necessarily close by. The success of these suboptimal selections (e.g. [6]) now appears to be a better argument than even we thought, in favour of the aptitude of compact RNA folds for aminoacyl-RNA synthesis.
However, both kinds of difficulty could be removed by ligating a short sequence that could be used to add the hepatitis delta virus (HDV) ribozyme to the 3′ of the already selected RNA [7]. This meant that subsequent transcripts (figure 1) would contain the HDV ribozyme at their ends, and accordingly cut themselves reproducibly, at the same point. Thus, an HDV-determined 3′ end could not grow. The HDV ribozyme also has the useful property of allowing almost any sequence upstream of the cut, including allowing any nucleotide (X in the figure 1) immediately 5′ of the cut, as the future reactive nucleotide.
Figure 1.

Transcript 3′ end designed for improved aaRNA selection. HDV ribozyme is shown at the 3′ terminus of an RNA transcript. It cleaves 3′-ward of X, leaving a terminus of any sequence at a fixed position, at the end of the randomized sequence (broad line), as the acceptor for later aminoacylation. HDV cleavage leaves an X 2′,3′ cyclic phosphate terminus, which is opened and dephosphorylated.
4. A contest between two forms of activated Phe
Another aspect of selection design was that two forms of activated phenylalanine were supplied at a similar concentration. If more frequent (simpler, smaller), RNA structures can transfer Phe from AMP, then selected RNAs will mostly employ PheAMP [1,8] (figure 2). Conversely, CoA as an activating group allows PheRNA derived from the coenzyme thioester. RNA aminoacylation from CoA has been observed [9]. Nonetheless, all detectable pool activities used PheAMP, the biological adenylate-activated amino acid, as the source of Phe. This finding is consistent with the idea that aaRNA synthesis on and by RNA could have mandated the initial choice of aaAMP for translation because it is more easily bound and oriented by an RNA catalyst.
Figure 2.

Simultaneous selection of aaRNA using two forms of activated Phe (PheAMP and PheCoA) in the same reactions allows determination of which substrate is more readily (simply) used by RNA.
5. A frequent structure satisfies selection
The majority of RNAs first detected [8] after only three steps of selection (by biotinylating the unique amino acid α-amino group, and binding to avidin) were two-helix junctions (74% could be described as in figure 3) in which an unpaired 3′-U and a junction-loop GU-3′ were conserved.
Figure 3.

Most common selected structure among self-aminoacylators with a uniform 3′ end. Independently derived RNAs had all four base pairs at the four leftward paired positions. Pairs to the right are constrained by constant sequence at the 5′ end, but vary in secondary structure and spacing from conserved nucleotides.
Strikingly, outside three conserved nucleotides, these folds varied widely—even though the helix to the right is partially constrained by a terminal constant sequence, it is at a varying location with respect to the three conserved site nucleotides because of ready variation in loop size (42% of loops are longer than the most prevalent trinucleotide length). Removal of the 5′ triphosphate made little difference, another hint that the 5′ RNA terminus might not be in the active site. All four standard base pairs appear at leftward helix positions. Tentatively, then, it appeared that conservation could be summarized very simply as: leftward pairing is required to draw the 3′ terminal site of aminoacylation to the active centre, but specifically required nucleotides may be three only: 3′-U (the aminoacyl acceptor) with GU-3′ opposite.
6. An active site hypothesis
The simplicity of this structure encouraged its study by computed energy minimization with solvent [8]. We found a recurrent stable structure in which PheAMP was stably poised in a cradle formed by the apparent conserved elements of the active site. This is schematized in figure 4.
Figure 4.
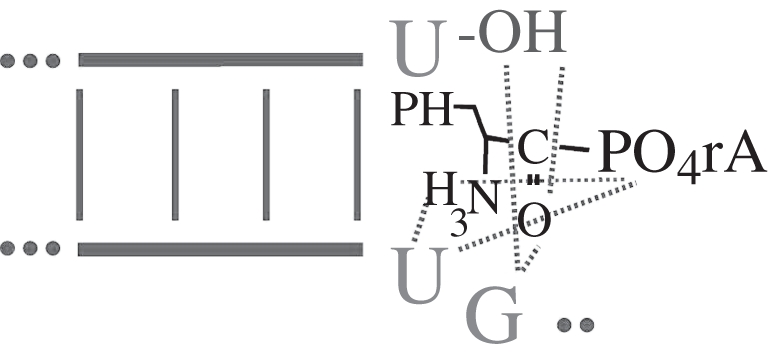
Schematic of PheAMP energy-minimized within the active site of C3 RNA [8]. Activated PheAMP substrate is localized by six hydrogen bonds that draw the carbonyl carbon to the attacking ribose oxygen. (PH, phenyl; r, ribose; A, adenine; thin-dotted lines, hydrogen bonds).
As predicted by the calculated low energy structure, neither the A of AMP nor the phenyl side chain of Phe is strongly engaged by the ribozymic centre. These and several other consequences of the model have been verified: both UMP–Phe and AMP–Met are functional substrates. In addition, the 2′ regiospecificity of aminoacylation, and strongly predicted stereoselectivity (d-PheAMP is not functional because of multiple interactions with α-amino) were confirmed. Because of these multiple successes, it seems likely that the computed model does capture essential aspects of this ribozyme's function.
7. Reduction to a minimal ribozyme complex
In drawing figure 4, I have quietly reduced the rest of the ribozyme structure to dots in order to suggest a striking possibility—that everything else might literally be dispensable. A functional aminoacyl transferase centre might, consistent with everything known at this point, be constituted from a few base pairs holding the receptive terminal ribose hydroxyl adjacent to the conserved GU sequence of the required loop, by poising all three nucleotides at the end of a short substrate:ribozyme helix. Even if the idea were correct, a minimal molecule need not have observable activity. However, a programme of deletions that successively reduced and tested the initial small RNAs [10] showed that a minimal active centre retained function. Indeed, a ribotetramer substrate, GCCU, was aminoacylated by the ribopentamer GUGGC. This is empirically minimal, because CCU is aminoacylated by GUGG at least 40-fold more slowly.
The minimal enzyme based on the originally selected RNAs is therefore as shown in figure 5. Although 87 per cent of the initially selected ribozyme has been discarded, the active centre still parallels many of the initially measured properties. It is
— only slightly dependent on divalents, functional with dilute monovalents;
— still yields highly regiospecific initial aminoacylation at U 2′-OH;
— faster at higher pH, suggesting that attack by 2′-OH is probably rate-limiting; and
— undiscriminating with regard to the aminoacyl group and activating nucleotide
Figure 5.
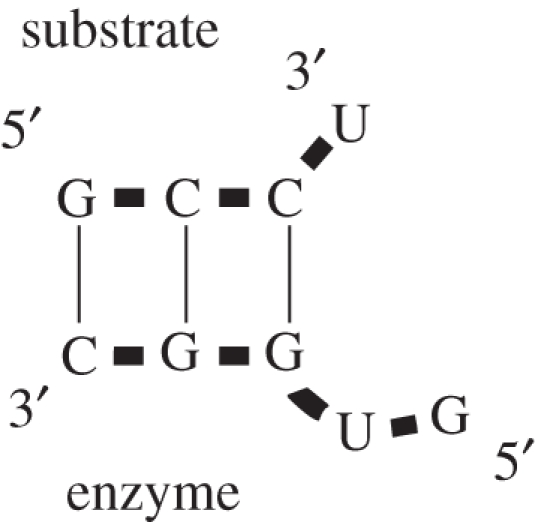
The minimal GCCU/GUGGC ribozyme system [10].
8. Kinetic analysis
Moreover, the kinetics of GUGGC/GCCU are describable as those of an ordered bisubstrate enzyme [11]. The chemistry seems necessarily ordered, because the substrate GCCU must assemble with ribozymic GUGGC before the active site for PheAMP binding exists. The small ribozyme readily turns over, with multiple aminoacylations per GUGGC ribozyme. It therefore qualifies as a true enzyme. Using essentially standard steady state enzyme methods with trace [32P]GCCU (so that GUGGC concentrations are known), we have measured Michaelis–Menten saturations and total rates (pH 7 in 5 mM Mg2+ at 4°C).
GUGGC/GCCU seems, on this basis, to comprise an ordinary small oligonucleotide strand association involving three GC pairs, followed by a loose association of PheAMP, as might be anticipated for a few hydrogen bonds in water (cf. figure 4). GUGGC/GCCU forms a Michaelis complex with PheAMP (it is not second order). It is also notable that all saturations follow normal Michaelis–Menten hyperbolae, confirming that despite a minimal size, each GUGGC/GCCU/PheAMP reacts as an independent enzymatic unit (the kinetic unit is not an aggregate). The most surprising aspect of the kinetics may be the relatively rapid first-order kcat for total transfer of the aminoacyl group, unexpectedly achieved within an active centre with only three lightly constrained nucleotides.
9. Versatility of a small active centre
The reader may think the phrase ‘total transfer’, used immediately above to characterize phenylalanylation, is superfluous: not so. GCCU/GUGGC has been characterized, since the earliest measurements, by multiple products. However, all products appear completely dependent on the first 2′ aminoacylation of U from PheAMP, and kinetic constants above refer to that event. However, the product manifold extends far beyond the initial aminoacylation. Figure 6 is a display of products [11] from (5′ 32P)GCCU at high (PheAMP), resolved by acid urea gel electrophoresis (low gel pH stabilizes esters and other less-stable products).
Figure 6.

Eight products (P1–P8) of vigorous aminoacylation of [32P]GCCU with PheAMP and GUGGC; e.g. at high (PheAMP), long incubation or high pH, with products fractionated by downward mild acid gel electrophoresis.
10. Analysis of products resolved on gels
The eight most intense product bands have been identified and inter-related by gel purification of individuals, then mass spectrometry, chemical reactivity, degradation by mild base and re-acylation with PheAMP [11]. This argument, positing 20 distinct products, is too lengthy to recreate here, but a plausible hypothesis for product inter-relations, consistent with all known data, is shown in figure 7.
Figure 7.

Origins and identities of eight bands of GCCU/GUGGC products [11] detected after acid gel electrophoresis. Stick diagrams of products depict the 3′ terminal ribose of GGCU with substituents at the 2′ atom (on the upper projection), and on the 3′ atom (on the lower projection). At the lower left are early reaction steps: strand association and PheAMP binding to GCCU/GUGGC. Short unidirectional arrows on the right are accelerated, but uncatalysed reactions of ester or peptidyl phenylalanine amino groups with PheAMP. F ≡ Phe.
11. More than 20 products
Figure 7 shows, at the lower left, the base pairing of the ribozyme GUGGC oligonucleotide with the substrate oligonucleotide GCCU (first line; refer to 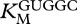 in table 1). Then, the assembled complex (terminal U now shown explicitly) interacts with PheAMP (arrow; refer to
in table 1). Then, the assembled complex (terminal U now shown explicitly) interacts with PheAMP (arrow; refer to 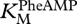 and kcat in table 1), specifically producing the left-hand element of product 1, GCCU-2′ Phe. Aminoacylation at 2′ is unique, based on the reactivity of synthetic GCC-3′dU and inactivity of GCC-2′dU.
and kcat in table 1), specifically producing the left-hand element of product 1, GCCU-2′ Phe. Aminoacylation at 2′ is unique, based on the reactivity of synthetic GCC-3′dU and inactivity of GCC-2′dU.
Table 1.
Kinetic constants (± s.e.) for GCCU/GUGGC.
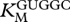 (µM) (µM) |
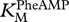 (mM) (mM) |
kcat (min−1) |
|---|---|---|
| 7.4 ± 4.3 | 6.4 ± 2.2 | 0.12 ± 0.03 |
Crucially, electrophoresis cannot distinguish the isomeric aminoacylated products GCCU-2′ Phe and GCCU-3′ Phe, which are interconnected by a fast (several per second) spontaneous transacylation [12]. Therefore, product 1 quickly becomes an unresolved mixture of these two species.
Transacylation of the initial Phe to 3′-OH opens up the 2′-OH for a second ribozymic 2′ aminoacylation (long, bent arrow), producing the diester, GCCU-2′, 3′ Phe2. This comprises most of the region resolved on the gel as P5. P2 through P4 cannot be re-aminoacylated to P6 through P8 (by GUGGC and PheAMP), so 3′ peptides (but not 3′ Phe) appear to inhibit re-aminoacylation at 2′, as shown (figure 7).
Transacylation is expected to be radically slowed after alteration of the α-amino by peptide formation, so that 2′ ↔ 3′ transaminoacylation, under our conditions, is most significant for the aminoacyl residues within P1. This will be increasingly true as the peptides lengthen, P2 (-FF, -Phe–Phe) through the minor P5 (-FFFFF, -Phe–Phe–Phe–Phe–Phe). Figure 7, as the simplest hypothesis, does not depict peptides as mobile.
All other arrows in figure 7 represent spontaneous peptide formation by successively encountered molecules of PheAMP, thereby building a manifold of peptides onto GCCU-2′ Phe and GCCU-3′ Phe (P1), but also forming the higher products on GCCU-2′,3′ Phe2. These reactions are not RNA-catalysed, but they are RNA-stimulated, because Phe esters of GCCU have altered pKas expected to greatly speed further attack on the activated Phe carbonyl in PheAMP. Esterification of amino acids lowers the pKa of the amino group by almost two orders [13], thereby similarly elevating the reactive form of the amino in the ester. Because peptide extension does not require specific catalysis, the point of reaction can move freely with respect to RNA structure, and peptides up to Phe5 are formed. These products are all normal peptides because exopeptidase treatment collapses all higher products to one band, corresponding to the ester P5, and similarly collapses all lower products to the esters P1. All these diester, ester/peptide and dipeptide products clearly come from the initial 2′ Phe because of the high specificity of GCCU/GUGGC for 2′ aminoacylation [10].
The mixed character of the gel region called P5, mentioned above, results from slow addition of Phe to GCCU-Phe4 to create some GCCU-Phe5 (easily seen in re-acylation experiments), which is not completely resolved from GCCU-2′,3′ Phe2 by electrophoresis. Accordingly, we expect three molecular species in the gel band called P5 (figures 6 and 7).
To summarize, in short incubations, initially specific initial phenylalanylation, facile transacylation and facilitated reaction with further molecules of PheAMP elaborate 20 or more molecular products on one active site nucleotide (figures 6 and 7).
12. Discussion
(a). The active site hypothesis, supported
Deletion and/or replacement of structural elements confirms the initial hypothesis supposing three critical active nucleotides [8]. The three nucleotides initially supposed to be in the active site cannot be effectively replaced. All other nucleotides and structures which appeared to be less stringently conserved are now seen to be dispensable (as for the right-hand helix in figure 3, or to have a function which can be served by many sequences (as for the left-hand helix in figure 3). Thus, limited nucleotide conservation among independently derived initially selected RNAs correctly predicted the active site [8,10].
(b). Variety of products
It seems likely that the relative indifference of the GCCU/GUGGC active centre to the amino acid side chain, and to the activating nucleotide arose via our emphasis on rapid selection, and therefore on simple mechanism [8]. In figure 4, the RNA directly interacts only with atoms within the first few bonds around reacting substrate atoms. This limits the active centre's scope, and presumably therefore simplifies its selection as well. However, it probably also makes it more likely that more distant groups, like the phenyl side chain and the adenine base, will have small effects. Thus, the ultimate stress on simplicity stresses the atoms directly linked to the chemical transfer, and by the same token, reduces the likelihood that more distant chemical qualities will be crucial. Accordingly, small active centres will be likely to catalyse reactions with materials with the same core chemical qualities, and to ignore peripheral, chemically less crucial qualities. In this sense, simple ribozymes will tend to be capable of a bigger array of products than larger, more complex catalysts.
This is relevant in another way to the large group of aminoacyl- and peptidyl-RNAs here. An open, unhindered active site allows the initially esterified amino acid to migrate to the adjacent ribose hydroxyl, and this allows a second specific acylation. Then, there are three aminoacyl-RNAs which can be extended into peptide, on encountering the abundant activated phenylalanine in the reaction. This readily happens to all three kinds of aminoacyl-RNAs, again probably owing to the open, unrestricted vicinity of the initial aminoacylations. Once again, now because of the lack of sophisticated structure, a simplified reaction centre has more products than a sophisticated one.
GCCU/GUGGC's activities therefore suggest that early enzymes may have served a different function than today's highly evolved, highly specific protein catalysts. Instead of furthering a pathway, or enforcing a highly specific fate for substrate, early catalysts may have facilitated many parallel reactions, and thereby allowed evolution to act on a variety of related products.
In particular, with regard to the evolution of a translation system: the vicinal diol of primordial aminoacylated ribose would have readily supported many forms of aminoacyl-RNA, aminoacyl-/peptidyl-RNA and dipeptidyl-RNAs. This structural and chemical variety could have easily participated in the genesis of an early coded peptide synthesis. For example, such a complex array of peptides and proximal, potentially catalytic groups (e.g. the terminal α-NH2s) on one ribose may create new entries for the list of evolutionarily significant reactions. It is notable that even in the modern ribosome, the ribose 2′-OH is arguably a highly significant catalytic contributor to peptidyl transfer [14].
(c). Origin of active RNAs
For some purposes, it is crucial to seek the smallest RNA structures that can perform biological reactions. This is true because it is probably difficult to make RNA-like molecules under primitive conditions, so the smallest RNA for activity is a critical parameter. The so-called ‘axiom of origin’ quantitates this notion [15]; it presumes that the threshold for the RNA world can be approximately computed as the point at which sufficient random-sequence RNA exists to allow significant RNA activities to be selected.
Our previous smallest aminoacylator had a total of 29 nucleotides [4], which reduced to 24 nucleotides at the beginning of the present selection [8]. Indeed, the repeated isolation of small numbers of aminoacyl transfer ribozymes from about 1014 initial, arbitrary-sequence molecules (by selection-amplification) suggests that active molecules have a frequency of ca 1 in 425 ≈ 1 in 1015 (there are more trials than molecules because selection potentially finds activity at several positions in each molecule). Or, said another way, it appears that aminoacylators have a frequency similar to a sequence constructed from 25 conserved nucleotides. This is very roughly consistent with the smallest previously observed self-acylators. Such findings enter the argument for an RNA world at a fundamental level, because only small sequences are plausible early on. Therefore, if RNA were not biochemically active at such sizes, then basic arithmetic would make an RNA world implausible [16].
However, it now appears that we previously used a biased selective task. Selection was more difficult than it need be, because of the positional instability and arbitrary sequence of the RNA terminus to be acylated. The difference between 25 and six specified nucleotides (GUNNN plus N′N′N′U; NN′ is one standard nucleotide pair, frequency = 1/4) is very significant, because for 1.66 essential nucleotides added, other considerations equal, one must search through 10-fold more RNA [15]. Thus, the difference between these experiments and earlier selections reduces the RNA that must be searched to find this essential translational group transfer by more than 11 orders of magnitude (i.e. 19 nt/1.66 nt order−1; cf. figure 8). One inevitably wonders if other fundamental RNA activities might similarly be vastly easier to find than we have usually assumed.
Figure 8.

Distribution of ribooligonucleotide lengths after undirected condensation of ribonucleotides to give dimer, on average. Total mole fractions (X) as a function of length, l: (X(l) = (1−f)fl−1 where f, fraction polymerized = 0.5, here)—diamonds [17]. Mole fraction of pentanucleotide sequences sampled ((l−4)X(l), l ≥5)—squares. Pentamers sample one 5-mer, hexamers sample two, heptamers three; all pentamers summed = 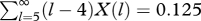 .
.
(d). The meaning of this functional RNA
I now want to re-examine the probable meaning of GCCU/GUGGC for the evolution of RNA-mediated aminoacyl transfer. I will do this first qualitatively, and then quantitatively, using a specific model for ribozyme origin.
Qualitatively: can anybody imagine a less complex ribozyme than this one? It seems that we are approaching a lower limit. The three base pairs used to acquire the GCCU substrate are minimal, in the sense that they cannot be reduced to two at usual temperatures. It is certainly possible that active centres comprising less than three nucleotides exist. Because these would be so frequent in random sequences, they should come to light readily, now that one knows they can occur. However, even if there are no simpler ones, then one needs only tetramers and pentamers to find this particular ribozyme.
One way to express the impact of such small size is to note that soluble, polar components from the Murchison meteorite are numerous even beyond a size of 2000 [18]. Both ribozyme GUGGC (m/z = 1583) and substrate GCCU (m/z = 1278) are therefore smaller than many products of meteorite chemistry. An essential reaction of protein biosynthesis, therefore, can be catalysed by agents less complex, by this criterion, than extraterrestrial organic chemicals.
More quantitatively: molecules similar to GUGGC/GCCU would become visible to selection as soon as the feeblest random polymerization of ribonucleotides existed on the Earth. Suppose a condensation [17] appears that yields random, untemplated oligoribonucleotides. For example, suppose ribonucleotides are activated by a small 5′ or 3′ leaving group that allows them to randomly polymerize in a primordial setting. Polymerization is assumed slight—consistent with primitive biochemistry and dilute concentrations, I posit that only half of activated nucleotides assemble into untemplated oligonucleotide products, leaving half still as monomers. Such a system has a mean length of two nucleotides, half as many final oligomers as initial activated nucleotides, and would exhibit the oligomer length distribution [17] shown in figure 8.
Figure 8 shows both total mole fractions (diamonds), and total randomized pentamers appearing (squares). I emphasize pentamers, the group to which the ribozyme belongs, because tetramers are invariably more frequent than pentamers, so a sufficiency of pentamer ribozymes in such a population, in most cases, ensures a sufficiency of (3′-terminal) tetramer substrates. How much RNA is needed? We need enough to ensure access to our ribozymes, so 5 × 45 = 5120 pentamers random at all positions (a mean of five copies of every possible pentamer sequence) seems enough. It would include not just GUNNN, but every sequence of five nucleotides (with probability (1−e−5) = 0.9933). Therefore, if there exist any other ribozyme systems of the size of GCCU/GUGGC, then they will also be present.
The total mole fraction of sampled pentamers summed over all oligonucleotides (figure 8) is 0.125 N0, where the latter is the initial nucleotide supply. Therefore, to possess our almost complete sample of 5120 pentamers, we must begin with
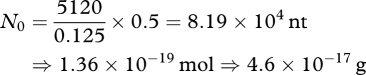 |
of nucleotide. As a result, 46 attograms of slightly activated, sluggishly oligomerizing nucleotides will sample every ribozyme like that treated here, almost certainly including GUGGC/GCCU itself. From the axiom of origin, the ancient thin edge of the RNA world might be defined, some rudiments of translation might have first appeared, when only 23 attograms of RNA world oligonucleotide were pooled (the calculated 46 attograms minus unpolymerized monomer). This is approximately the oligoribonucleotide in six poliovirus particles; a minute RNA sample by most standards.
While we have made some tacit approximations, they do not seem to be important for the conclusion. This is more remarkable because, for robustness, we sought all possible pentamers; calculating for one (e.g. GUGGC) or a few functional sequences would yield a yet more striking result. Accordingly, it is imprecise to require pools or piles of RNA, or locales encrusted with RNA to begin the RNA world—an amount so small that it is difficult to detect is probably enough [19].
(e). The existence of the RNA world, supported
The RNA world hypothesis is a productive one, having predicted many unknown RNA activities that have later been found. This is a convenience for experimentalists, but also has general implications.
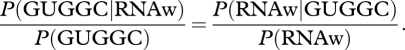 |
A form of Bayes' theorem is written above to point up a relation (the vertical bar means given that) between the probability (P) of existence of the pentamer ribozyme (GUGGC) and the existence of an RNA world (RNAw). It says, approximately, a pentanucleotide ribozyme seems surprising, but makes sense in terms of a role in an RNA world (left-hand side > 1); this necessarily increases the probability of an RNA world (right-hand side > 1). Such revision is not impressionistic, but a quantitative necessity. Other possible explanatory hypotheses substituted for RNAw (panspermia, pyrite, clay) do not make similar predictions. Moreover, increases for other independent RNA world discoveries can be multiplied [20]. However small the probability one initially assigned an RNA world, it has been raised in this way many times over.
(f). Later in the RNA world
GUGGC/GCCU-like molecules, as a consequence of their sizes, are relatively easily replicated. In fact, such molecules lie easily within the templated replicative capabilities (≤20 nt) of the RNA–RNA polymerase of Zaher & Unrau [21] and even more so within the ambit (≤95 nt) of the polymerase of Wochner et al. [22]. Thus, aminoacyl transfer activity could be captured and reproduced by RNA–RNA replicases already known (although these replicases are themselves presently larger RNAs). Replication remains possible even if the present molecule is enlarged by including both elements in a single GUGGC … GCCU sequence. Accordingly, §12(d) above emphasizes that GUGGC/GCCU-like ribozymes would easily be found, in fact, many examples would occur in weakly polymerizing random nucleotide mixtures comprising only 82 000 activated molecules. In addition, available replicase RNAs show that, once found useful, a GUGGC/GCCU-like enzyme could be captured and replicated to serve an evolving translation system in an RNA world. There is, therefore, a credible argument that this very system would first be found and later propagated. Consequently, it is realistic to posit that a congener of GUGGC/GCCU is a true progenitor, a molecule lying on the RNA world path actually taken by life on the Earth.
References
- 1.Illangasekare M., Sanchez G., Nickles T., Yarus M. 1995. Aminoacyl-RNA synthesis catalyzed by an RNA. Science 267, 643–647 10.1126/science.7530860 (doi:10.1126/science.7530860) [DOI] [PubMed] [Google Scholar]
- 2.Illangasekare M., Yarus M. 1999. Specific, rapid synthesis of Phe-RNA by RNA. Proc. Natl Acad. Sci. USA 96, 5470–5475 10.1073/pnas.96.10.5470 (doi:10.1073/pnas.96.10.5470) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee N., Bessho Y., Wei K., Szostak J. W., Suga H. 2000. Ribozyme-catalyzed tRNA aminoacylation. Nat. Struct. Biol. 7, 28–33 10.1038/71225 (doi:10.1038/71225) [DOI] [PubMed] [Google Scholar]
- 4.Illangasekare M., Yarus M. 1999. A tiny RNA that catalyzes both aminoacyl-RNA and peptidyl-RNA synthesis. RNA 5, 1482–1489 10.1017/S1355838299991264 (doi:10.1017/S1355838299991264) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Milligan J. F., Groebe D. R., Witherell G. W., Uhlenbeck O. C. 1987. Oligoribonucleotide synthesis using T7 RNA polymerase and synthetic DNA templates. Nucleic Acids Res. 15, 8783–8798 10.1093/nar/15.21.8783 (doi:10.1093/nar/15.21.8783) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Illangasekare M., Kovalchuke O., Yarus M. 1997. Essential structures of a self-aminoacylating RNA. J. Mol. Biol. 274, 519–529 10.1006/jmbi.1997.1414 (doi:10.1006/jmbi.1997.1414) [DOI] [PubMed] [Google Scholar]
- 7.Kieft J. S., Batey R. T. 2004. A general method for rapid and nondenaturing purification of RNAs. RNA 10, 988–995 10.1261/rna.7040604 (doi:10.1261/rna.7040604) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chumachenko N. V., Novikov Y., Yarus M. 2009. Rapid and simple ribozymic aminoacylation using three conserved nucleotides. J. Am. Chem. Soc. 131, 5257–5263 10.1021/ja809419f (doi:10.1021/ja809419f) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li N., Huang F. 2005. Ribozyme-catalyzed aminoacylation from CoA thioesters. Biochemistry 44, 4582–4590 10.1021/bi047576b (doi:10.1021/bi047576b) [DOI] [PubMed] [Google Scholar]
- 10.Turk R. M., Chumachenko N. V., Yarus M. 2010. Multiple translational products from a five-nucleotide ribozyme. Proc. Natl Acad. Sci. USA 107, 4585–4589 10.1073/pnas.0912895107 (doi:10.1073/pnas.0912895107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Turk R. M., Illangasekare M., Yarus M. 2011. Catalyzed and spontaneous reactions on ribozyme ribose. J. Am. Chem. Soc. 133, 6044–6050 10.1021/ja200275h (doi:10.1021/ja200275h) [DOI] [PubMed] [Google Scholar]
- 12.Taiji M., Yokoyama S., Miyazawa T. 1983. Transacylation rates of (aminoacyl)adenosine moiety at the 3′-terminus of aminoacyl transfer ribonucleic acid. Biochemistry 22, 3220–3225 10.1021/bi00282a028 (doi:10.1021/bi00282a028) [DOI] [PubMed] [Google Scholar]
- 13.Sober H. A. 1968. CRC Handbook of biochemistry: selected data for molecular biology. Cleveland, OH: Chemical Rubber Co [Google Scholar]
- 14.Schmeing T. M., Huang K. S., Kitchen D. E., Strobel S. A., Steitz T. A. 2005. Structural insights into the roles of water and the 2′ hydroxyl of the P site tRNA in the peptidyl transferase reaction. Mol. Cell 20, 437–448 10.1016/j.molcel.2005.09.006 (doi:10.1016/j.molcel.2005.09.006) [DOI] [PubMed] [Google Scholar]
- 15.Yarus M., Knight R. D. 2004. The scope of selection. In The genetic code and the origin of life (ed. Pouplana L. R.), pp. 75–91 Georgetown, TX: Landes Bioscience [Google Scholar]
- 16.Yarus M. 2010. Life from an RNA world. The ancestor within. MA, USA: Harvard University Press [Google Scholar]
- 17.Flory P. J. 1936. Molecular size distribution in linear condensation polymers. J. Am. Chem. Soc. 58, 1877–1885 10.1021/ja01301a016 (doi:10.1021/ja01301a016) [DOI] [Google Scholar]
- 18.Schmitt-Kopplin P., Gabelica Z., Gougeon R. D., Fekete A., Kanawati B., Harir M., Gebefuegi I., Eckel G., Hertkorn N. 2010. High molecular diversity of extraterrestrial organic matter in Murchison meteorite revealed 40 years after its fall. Proc. Natl Acad. Sci. USA 107, 2763–2768 10.1073/pnas.0912157107 (doi:10.1073/pnas.0912157107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kennedy R., Lladser M. E., Yarus M., Knight R. 2008. Information, probability, and the abundance of the simplest RNA active sites. Front. Biosci. 13, 6060–6071 10.2741/3137 (doi:10.2741/3137) [DOI] [PubMed] [Google Scholar]
- 20.Yarus M., Caporaso J. G., Knight R. 2005. Origins of the genetic code: the escaped triplet theory. Annu. Rev. Biochem. 74, 179–198 10.1146/annurev.biochem.74.082803.133119 (doi:10.1146/annurev.biochem.74.082803.133119) [DOI] [PubMed] [Google Scholar]
- 21.Zaher H. S., Unrau P. J. 2007. Selection of an improved RNA polymerase ribozyme with superior extension and fidelity. RNA 13, 1017–1026 10.1261/rna.548807 (doi:10.1261/rna.548807) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wochner A., Attwater J., Coulson A., Holliger P. 2011. Ribozyme-catalyzed transcription of a functional RNA gene. Science 332, 209–212 10.1126/science.1200752 (doi:10.1126/science.1200752) [DOI] [PubMed] [Google Scholar]