

Abstract
The ability of RNA to both store genetic information and catalyse chemical reactions has led to the hypothesis that it predates DNA and proteins. While there is no doubt that RNA is capable of storing the genetic information of a primitive organism, only two classes of reactions—phosphoryl transfer and peptide bond formation—have been observed to be catalysed by RNA in nature. However, these naturally occurring ribozymes use a wide range of catalytic strategies that could be applied to other reactions. Furthermore, RNA can bind several cofactors that are used by protein enzymes to facilitate a wide variety of chemical processes. Despite its limited functional groups, these observations indicate RNA is a versatile molecule that could, in principle, catalyse the myriad reactions necessary to sustain life.
Keywords: RNA world, ribozyme, nucleobase catalysis, two-metal ion mechanism, cofactor, peptide bond formation
1. Introduction
The information content of the cell is contained in DNA, which codes for proteins that carry out the majority of cellular functions. However, the discovery of catalytic RNA led to the RNA world hypothesis: at one point, RNA could have been both the information carrier and the functional molecule [1]. At first the main function would be self-replication, but as more complex RNAs evolved, they would need to synthesize their precursors, store and use energy, and isolate their reaction products from the environment. Eventually, RNA would have to transition towards modern biology, where proteins play most of the catalytic roles. This would involve the increasingly complex task of message-directed protein synthesis, including the specific activation of amino acids to be incorporated.
Is RNA up to the task? For the first two decades after the discovery of catalytic RNA, only one RNA-catalysed reaction was observed in nature—phosphoryl transfer. Furthermore, the functional groups in RNA are limited, with none having an unperturbed pKa near neutrality that could be easily implicated in catalysis (figure 1). Early speculation was that RNA functioned primarily by recruiting divalent metal ions to the active site. While metal ions are useful for catalysis and remain important for many protein enzymes, this strategy could not be wholly responsible for catalysing all the necessary reactions in an RNA world.
Figure 1.
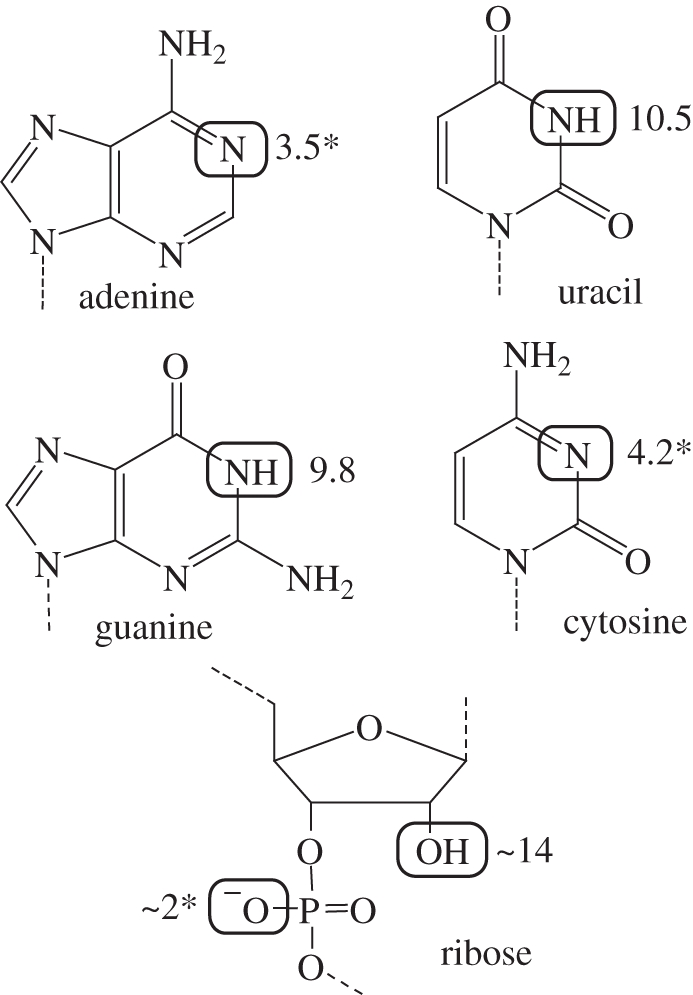
The functional groups of RNA. Primary sites of protonation and deprotonation are highlighted. pKas are shown; asterisk indicates the pKa of the conjugate acid.
It is now known that natural ribozymes use a variety of catalytic mechanisms, even for phosphoryl transfer. Furthermore, RNA regulatory elements, called riboswitches, have been shown to bind a variety of cofactors, small molecules which expand the repertoire of modern protein enzymes. In addition to phosphoryl transfer ribozymes, the RNA component of the ribosome is responsible for catalysing protein synthesis. This is the key reaction required to transition from RNA to protein as functional molecule. These observations indicate the versatility of RNA in catalysing chemical reactions, an essential characteristic for the RNA world.
2. Phosphoryl transfer reactions
The majority of natural ribozymes catalyse phosphoryl transfer: the cleavage and/or ligation of the RNA phosphodiester backbone (figure 2). The fastest ribozymes increase the rate of reaction by approximately 1012-fold when compared with the uncatalysed reaction, in the same range as protein enzymes such as ribonuclease (RNase) A [3]. An enzyme, whether made of RNA or protein, could catalyse this reaction in several ways. A general base could assist in deprotonation of the nucleophile, which is either water or a ribose hydroxyl. A general or Lewis acid could help protonate the 5′-oxygen leaving group. A positively charged moiety could stabilize the build-up of negative charge on the non-bridging oxygen atoms in the transition state. Finally, positioning of the nucleophile will also contribute to catalysis since the reaction occurs fastest when the nucleophile and leaving group are in an inline conformation.
Figure 2.

Catalytic mechanisms of phosphoryl transfer ribozymes. (a) General mechanisms of phosphoryl transfer catalysis. (b) The group I intron, which uses a two-metal ion mechanism. (c) The hammerhead ribozyme, which uses RNA functional groups for acid–base catalysis. (d) The glmS ribozyme, which uses a small molecule cofactor. (b,d) Adapted from Strobel & Cochrane [2].
RNA can form specific, high-affinity binding pockets for divalent metal ions, with ribose hydroxyls and negatively charged phosphate oxygens often serving as ligands. Since positively charged metal ions are suited for all of the catalytic roles above, it was postulated that all phosphoryl transfer ribozymes would be metalloenzymes. A direct role for RNA bases was disfavoured, as no functional group has a pKa near neutrality and RNA lacks positively charged moieties to stabilize the build-up of negative charge.
One example of a metalloribozyme is the group I intron, which catalyses two phosphoryl transfer reactions during the process of self-splicing. It was proposed that the intron would use a two-metal ion mechanism, analogous to protein phosphoryl transfer enzymes [4]. Metal-specificity switch experiments demonstrated that several functional groups near and including the scissile phosphate coordinate divalent metal ions [5]. Furthermore, structures of the complete intron show two magnesium ions bound 3.9 Å apart, consistent with the proposed two-metal ion mechanism (figure 2b) [6,7]. One metal ion is in position to deprotonate the nucleophile, the second to stabilize the negative charge on the leaving group and both can stabilize the negatively charged non-bridging oxygens.
Later, the structure of the group II intron showed that it also uses the two-metal ion mechanism [8]. The RNA components of other phosphoryl transfer enzymes—RNase P and the spliceosome—also bind divalent metal ions important for catalysis [9,10]. Clearly, RNA is suited for recruiting divalent metal ions, which can be used for a variety of catalytic roles.
The large ribozymes are all metalloenzymes, but surprisingly, the hairpin, Varkud Satellite (VS) and hammerhead ribozymes are active in the absence of divalent metal ions [11]. Each of these ribozymes, along with the hepatitis delta virus ribozyme (HDV), catalyse self-cleavage and ligation reactions as part of processing genome replicants.
The hammerhead ribozyme is perhaps the most extensively studied small ribozyme, and the wealth of biochemical and structural data support a catalytic mechanism dependent on RNA functional groups (figure 2c). Although early crystal structures of a minimal hammerhead construct conflicted with biochemical data, this has been reconciled by the structure of a full-length, fully active construct [12]. The N1 of G12 is positioned to abstract a proton from the 2′-oxygen nucleophile, and the 2′-OH of G8 is near the 5′-oxygen leaving group. Further evidence supports a role for both of these groups in acid–base catalysis. Mutation of G8 reduces catalysis 104-fold, but this effect is rescued if the bridging oxygen of the scissile phosphate is replaced with a sulphur leaving group, alleviating the need for a general acid [13]. In another study, a bromoacetamide group was attached to the 2′-oxygen nucleophile, which would produce a covalent adduct if a general base were in the vicinity [14]. Indeed, G12 becomes covalently linked to the nucleophile in this case. pH-dependence and mutational studies are also consistent with G8 being the general acid and G12 the general base [15].
This appears to be a common strategy for the small ribozymes. Similar experiments implicate nucleobases in acid–base catalysis for the hairpin, HDV and VS ribozymes [16–19]. This is unexpected because no RNA functional groups have unperturbed pKas near neutrality. It is possible that some of these bases are participating in hydrogen bonds instead of full acid–base catalysis. Alternatively, the bases could be functioning as alternate tautomers, especially for guanine, which has been repeatedly observed in position for acid–base catalysis [19–22]. Metal ions may also play an indirect role in altering the pKa of a nucleobase so it may act as a general acid or base [14]. Protein enzymes commonly have pKa shifts of several units; it appears that RNA is capable of the same.
A third strategy for catalysis is demonstrated by the glmS riboswitch. In response to high concentrations of glucosamine-6-phosphate, this riboswitch catalyses its own cleavage, which results in subsequent nuclease degradation of the mRNA (figure 2d). The pKas of glucosamine-6-phosphate and several analogues track closely to the observed reaction pKas of the ribozyme, suggesting a direct role in catalysis [23]. Glucosamine-6-phosphate binds in the active site, in position to donate an amino proton to the leaving group [20]. It is also possible that the positively charged amino group stabilizes the negatively charged transition state. In either case, recruitment of a small molecule cofactor expands the functional groups available for catalysis. Aided by this cofactor, glmS can reach rates as high as 5 s−1 [24].
3. Functional cofactors bound by riboswitches
Riboswitches are conserved gene elements generally found in the 5′-untranslated region of mRNAs. Each riboswitch binds a ligand and in response regulates expression of its associated mRNA. Riboswitches for more than 10 ligands have been identified by bioinformatic techniques [25]. Interestingly, several of these ligands are cofactors for protein enzymes (figure 3). Catalytic RNA could use these cofactors to increase the range of chemistries available, as protein enzymes do. The glmS riboswitch is a precedent for the use of a cofactor in RNA catalysis. Although it is the only riboswitch of this type discovered to date, it is reasonable to speculate that cofactors similar to those bound by other riboswitches could be used by ribozymes to aid in catalysis.
Figure 3.

Protein cofactors bound by riboswitches.
Riboswitches that bind S-adenosylmethionine (SAM), tetrahydrofolate (THF), flavin mononucleotide (FMN), thiamine pyrophosphate (TPP) and adenosylcobalamin (AdoCbl) have been identified. Each of these small molecules is ubiquitously employed by protein enzymes. What chemistry could ribozymes achieve with these cofactors?
Five classes of SAM riboswitches have been identified. These riboswitches are found upstream of genes involved in SAM, cysteine and methionine synthesis [26]. SAM is the second most common protein cofactor (after ATP) and the most common methyl donor [27]. In addition to its use as a regulatory signal, methylation often plays a functional role in RNA. Both the sugar and nucleobase of tRNA and rRNA are a frequent target of methylation aided by SAM [28]. Methylation of the 2′-oxygen in particular would stabilize an RNA against degradation, potentially increasing the lifetime of an RNA copy and allowing larger genomes. SAM is also a source of other synthetic building blocks, including amino, ribosyl and aminoalkyl groups [29]. It is also noteworthy that SAM is a relatively simple molecule, containing a single nucleotide and a single amino acid. Such a molecule could have been one of the first to be synthesized and bound by RNA.
The THF riboswitch is found upstream of genes encoding folate synthesis and uptake [30]. THF is a carrier of single-carbon units in a variety of oxidation states [31]. These units are used for the synthesis of purine nucleotides and the amino acid methionine. This cofactor would expand the ability for a primitive ribozyme to provide building blocks initially for nucleic acids and then proteins. THF also provides the 5-methyl group which differentiates the DNA nucleotide thymine from its RNA counterpart, uracil. It is strongly associated with two other cofactors bound by riboswitches: it provides the methyl group needed to recycle SAM, and is kept in an active form itself by a form of vitamin B12 (related to AdoCbl, below).
The FMN riboswitch is found upstream of genes involved in the biosynthesis and transport of riboflavin, a precursor to FMN [32]. Flavin is a redox active chromophore, with oxidized flavin able to be reduced by one or two electrons [33]. Flavin-dependent enzymes are involved in photosynthesis, pyruvate oxidation and nitrogen fixation [34,35]. A flavin-dependent ribozyme could carry out important reactions allowing it to harness energy from its environment, a key component of complex life. Interestingly, flavin-dependent enzymes are also involved in nucleic acid repair.
The TPP riboswitch, associated with genes for thiamine biosynthesis, phosphorylation and transport, is the only riboswitch to be identified in all three domains of life [36,37]. TPP-dependent enzymes catalyse the reversible cleavage of carbon–carbon bonds. These reactions are essential in the pentose phosphate pathway, Calvin cycle of photosynthesis and the tricarboxylic acid pathway [38]. These pathways store energy and create precursors to nucleic acids and aromatic amino acids. TPP is also involved in the production of ethanol by anaerobic fermentation in fungi. The ability of this cofactor to make and break carbon bonds would greatly expand the reactions that could be catalysed by RNA, both for biosynthesis and energy production.
The AdoCbl riboswitch regulates genes for the synthesis and import of AdoCbl, a form of vitamin B12 [39]. AdoCbl is a source of free radicals used to catalyse isomerization reactions [40]. These isomerizations would allow a primitive ribozyme to use alternative carbon sources, as some anaerobic bacteria do today. Ribonucleotide reductases also use AdoCbl, although they use an active site cysteine [41,42]. This functional group is lacking in RNA and would have to be provided by a second cofactor (possibly a free amino acid similar to cysteine). This reaction would be part of the pathway from RNA to DNA as carrier of genetic information.
4. Protein synthesis
It is necessary but not sufficient for the RNA world to be self-perpetuating: it must also have given rise to current biology featuring protein enzymes. The details of this transition are purely speculative, but it may have included the recruitment of amino acids or short peptides as enzymatic cofactors. Clearly, RNA is capable of specifically binding these ligands and could use them to assist in chemistry. Interestingly, many of the cofactors discussed above contain a nucleic acid element [43], suggesting that molecules like them may have been selected in part for their ability to bind to RNA. As peptides became more complex, the chances of encountering them randomly in solution would drop precipitously, and machinery for specifically synthesizing them would have to be developed. Eventually, this could become the mechanism for protein synthesis.
Since the machinery for making proteins would have to predate proteins, the RNA world hypothesis predicts such machinery would be made of RNA. In all extant organisms, protein synthesis is carried out by a large nucleoprotein complex, the ribosome. Strikingly, the active site for peptide bond formation is composed entirely of RNA [44,45]. This is possibly the single most significant piece of data supporting the RNA world hypothesis.
The ribosome manufactures proteins by linking individual amino acids through peptide bonds (figure 4). As in phosphoryl transfer, this reaction may be catalysed by deprotonating the nucleophile, protonating the leaving group and stabilizing build-up of charge in the transition state. Substrate positioning is expected to play a large role, as it does for all bimolecular reactions. This could be as general as bringing the two substrates in close proximity (essentially increasing the diffusion constant) or in orienting functional groups for productive chemistry.
Figure 4.
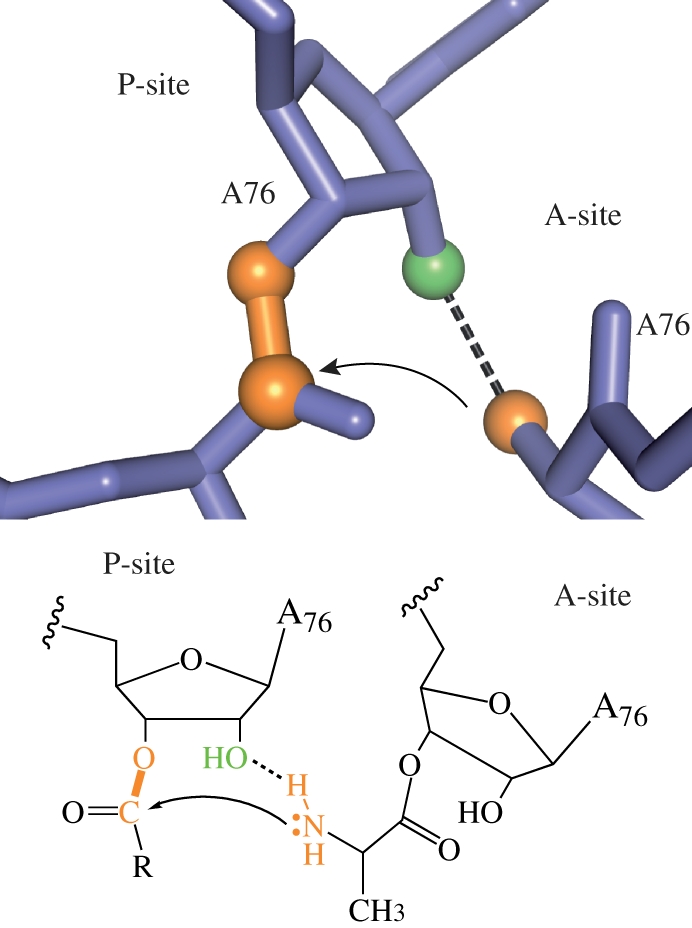
Peptide bond formation by the ribosome. The first step, nucleophilic attack by the aminoacyl-tRNA, is shown. The 2′-hydroxyl of peptidyl-tRNA may assist in shuttling the amino proton to the leaving group oxygen, carbonyl oxygen or an ordered water molecule (not shown). Adapted from Strobel & Cochrane [2].
Does the ribosome use similar strategies to the phosphoryl transfer ribozymes, or some novel strategy? Structural and biochemical data have mostly ruled out a significant role for metal ions or ribosomal functional groups [46]. No metal ions have been observed in any high-resolution crystal structures of the active site [47]. Furthermore, the only ribosomal functional group to have any effect on the rate when deleted is the 2′-hydroxyl of A2451 [48]. The effect is only 10-fold, consistent with it helping to position the nucleophile, but not making an important transition state hydrogen bond or assisting in proton transfer.
It could be that substrate positioning is then the only contribution of the ribosome to catalysis—it serves as an inert scaffold on which the substrates bind. It is easy to imagine such a molecule arising during the RNA world. However, it does appear that the ribosome plays a significant role in altering the reaction pathway, as many reaction parameters differ from similar uncatalysed reactions [49–51]. It may be that the ribosome uses a strategy made famous by protein enzymes—drastically altering the environment of the substrates by creating a fully enclosed active site largely devoid of water. This increases the entropy of the reaction (consistent with biochemical data [52]), and allows the reaction to proceed by pathways that are unlikely in bulk solvent.
The ribosome takes advantage not only of the versatility of RNA as a catalyst, but also its versatility as a substrate. For many phosphoryl transfer reactions, RNA provides its own nucleophile, the 2′-hydroxyl, greatly accelerating the reaction (hence the drastic difference in stability of DNA and RNA). For peptide bond formation, the ribosome may use the 2′-hydroxyl to aid in proton transfer or in strong transition state hydrogen bonds; deletion of this functional group significantly reduces activity [49]. This allows the reaction to progress even with limited water molecules available to accept and donate protons. In the case of the ribosome, this is a different role for the 2′-hydroxyl group from similar uncatalysed reactions, where the 2′-hydroxyl has a small effect [53].
It is unknown (and perhaps unknowable) what a more primitive ribosome looked like, and at what point aminoacyl adenylates were used as substrates. Although a small ribozyme has been developed by in vitro selection which can form a peptide bond between two aminoacyl adenylates [54], message-directed synthesis requires a more complex system with decoding abilities. Nevertheless, the modern ribosome is a vivid demonstration that RNA has the catalytic properties necessary to begin protein synthesis.
5. Concluding remarks
Only two reactions are observed to be catalysed by RNA in nature. However, the diversity of catalytic mechanisms for these reactions implies that RNA may be capable of much more. This is supported by the success of in vitro selection to develop ribozymes for RNA ligation [55], carbon–carbon bond formation [56], glycosidic bond formation [57] and other activities. Furthermore, in vitro-selected ribozymes can use catalytic cofactors, demonstrated by an alcohol dehydrogenase [58].
If the RNA world hypothesis is correct, then almost all ribozymes have been replaced by protein counterparts. Why then do some ribozymes persist in the presence of proteins, while others do not? First, the lower reaction rate could be compensated for by the increased effective concentration, since most ribozymes exist in nature as cis-acting elements, and not true trans-acting enzymes. In fact, the deficiency of many ribozymes compared with protein enzymes is not in the catalytic rate, but in binding the engineered substrate. Further, some ribozymes are mobile genetic elements, which benefit from requiring as little exogenous protein as possible (and, in fact, introns often contain their own open reading frames) [59]. Many ribozymes are found in viruses, where they may confer a selective advantage by requiring fewer resources—compare the genetic cost of the hammerhead (approx. 50 nucleotides) with even a small protein, such as RNase A (approx. 400 nucleotides, plus the machinery to translate mRNA to protein).
On the other hand, the ribosome is an extremely complex machine, consisting of three RNA molecules and 34 proteins in prokaryotes with a total mass around 2.5 MDa. In addition, it requires numerous tRNAs and synthetases to provide activated amino acids, and another set of proteins for initiation and elongation. It may be that by the time protein enzymes were fully developed, the functional role of RNA in the ribosome was too interwoven and complex to be replaced.
It is unlikely that any other massive, complex ribozymes such as the ribosome will be discovered. Instead, unidentified ribozymes are likely to be low-cost alternatives to proteins, while still maintaining high catalytic activity. The versatility of RNA indicates a wider range of ribozymes fitting this description may be possible.
Acknowledgements
Work described in this review was supported by an NIH post-doctoral fellowship to D.A.H. and NIH grant 054839 to S.A.S.
References
- 1.Gilbert W. 1986. Origin of life: the RNA world. Nature 319, 618. 10.1038/319618a0 (doi:10.1038/319618a0) [DOI] [Google Scholar]
- 2.Strobel S. A., Cochrane J. C. 2007. RNA catalysis: ribozymes, ribosomes, and riboswitches. Curr. Opin. Chem. Biol. 11, 636–643 10.1016/j.cbpa.2007.09.010 (doi:10.1016/j.cbpa.2007.09.010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Doudna J. A., Cech T. R. 2002. The chemical repertoire of natural ribozymes. Nature 418, 222–228 10.1038/418222a (doi:10.1038/418222a) [DOI] [PubMed] [Google Scholar]
- 4.Steitz T. A., Steitz J. A. 1993. A general two-metal-ion mechanism for catalytic RNA. Proc. Natl Acad. Sci. USA 90, 6498–6502 10.1073/pnas.90.14.6498 (doi:10.1073/pnas.90.14.6498) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yoshida A., Sun S., Piccirilli J. 1999. A new metal ion interaction in the Tetrahymena ribozyme reaction revealed by double sulfur substitution. Nat. Struct. Biol. 6, 318–321 10.1038/7551 (doi:10.1038/7551) [DOI] [PubMed] [Google Scholar]
- 6.Adams P. L., Stahley M. R., Kosek A. B., Wang J., Strobel S. A. 2004. Crystal structure of a self-splicing group I intron with both exons. Nature 430, 45–50 10.1038/nature02642 (doi:10.1038/nature02642) [DOI] [PubMed] [Google Scholar]
- 7.Stahley M. R., Strobel S. A. 2005. Structural evidence for a two-metal-ion mechanism of group I intron splicing. Science 309, 1587–1590 10.1126/science.1114994 (doi:10.1126/science.1114994) [DOI] [PubMed] [Google Scholar]
- 8.Toor N., Keating K. S., Taylor S. D., Pyle A. M. 2008. Crystal structure of a self-spliced group II intron. Science 320, 77–82 10.1126/science.1153803 (doi:10.1126/science.1153803) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Warnecke J. M., Held R., Busch S., Hartmann R. K. 1999. Role of metal ions in the hydrolysis reaction catalyzed by RNase P RNA from Bacillus subtilis. J. Mol. Biol. 290, 433–445 10.1006/jmbi.1999.2890 (doi:10.1006/jmbi.1999.2890) [DOI] [PubMed] [Google Scholar]
- 10.Yean S., Wuenschell G., Termini J., Lin R. 2000. Metal-ion coordination by U6 small nuclear RNA contributes to catalysis in the spliceosome. Nature 408, 881–884 10.1038/35048617 (doi:10.1038/35048617) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murray J. B., Seyhan A. A., Walter N. G., Burke J. M., Scott W. G. 1998. The hammerhead, hairpin and VS ribozymes are catalytically proficient in monovalent cations alone. Chem. Biol. 5, 587–595 10.1016/S1074-5521(98)90116-8 (doi:10.1016/S1074-5521(98)90116-8) [DOI] [PubMed] [Google Scholar]
- 12.Martick M., Scott W. G. 2006. Tertiary contacts distant from the active site prime a ribozyme for catalysis. Cell 126, 309–320 10.1016/j.cell.2006.06.036 (doi:10.1016/j.cell.2006.06.036) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thomas J. M., Perrin D. M. 2009. Probing general acid catalysis in the hammerhead ribozyme. J. Am. Chem. Soc. 131, 1135–1143 10.1021/ja807790e (doi:10.1021/ja807790e) [DOI] [PubMed] [Google Scholar]
- 14.Thomas J. M., Perrin D. M. 2008. Probing general base catalysis in the hammerhead ribozyme. J. Am. Chem. Soc. 130, 15 467–15 475 10.1021/ja804496z (doi:10.1021/ja804496z) [DOI] [PubMed] [Google Scholar]
- 15.Han J., Burke J. M. 2005. Model for general acid–base catalysis by the hammerhead ribozyme: pH–activity relationships of G8 and G12 variants at the putative active site. Biochemistry 44, 7864–7870 10.1021/bi047941z (doi:10.1021/bi047941z) [DOI] [PubMed] [Google Scholar]
- 16.Das S. R., Piccirilli J. A. 2005. General acid catalysis by the hepatitis delta virus ribozyme. Nat. Chem. Biol. 1, 45–52 10.1038/nchembio703 (doi:10.1038/nchembio703) [DOI] [PubMed] [Google Scholar]
- 17.Suydam I. T., Strobel S. A. 2008. Fluorine substituted adenosines as probes of nucleobase protonation in functional RNAs. J. Am. Chem. Soc. 130, 13 639–13 648 10.1021/ja803336y (doi:10.1021/ja803336y) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suydam I. T., Levandoski S. D., Strobel S. A. 2010. Catalytic importance of a protonated adenosine in the hairpin ribozyme active site. Biochemistry 49, 3723–3732 10.1021/bi100234v (doi:10.1021/bi100234v) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wilson T. J., McLeod A. C., Lilley D. M. J. 2007. A guanine nucleobase important for catalysis by the VS ribozyme. EMBO J. 26, 2489–2500 10.1038/sj.emboj.7601698 (doi:10.1038/sj.emboj.7601698) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cochrane J. C., Lipchock S. V., Strobel S. A. 2007. Structural investigation of the GlmS ribozyme bound to its catalytic cofactor. Chem. Biol. 14, 97–105 10.1016/j.chembiol.2006.12.005 (doi:10.1016/j.chembiol.2006.12.005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rupert P. B., Massey A. P., Sigurdsson S. T., Ferré-D'Amaré A. R. 2002. Transition state stabilization by a catalytic RNA. Science 298, 1421–1424 10.1126/science.1076093 (doi:10.1126/science.1076093) [DOI] [PubMed] [Google Scholar]
- 22.Klein D. J., Ferré-D'Amaré A. R. 2006. Structural basis of glmS ribozyme activation by glucosamine-6-phosphate. Science 313, 1752–1756 10.1126/science.1129666 (doi:10.1126/science.1129666) [DOI] [PubMed] [Google Scholar]
- 23.Winkler W. C., Nahvi A., Roth A., Collins J. A., Breaker R. R. 2004. Control of gene expression by a natural metabolite-responsive ribozyme. Nature 428, 281–286 10.1038/nature02362 (doi:10.1038/nature02362) [DOI] [PubMed] [Google Scholar]
- 24.Cochrane J. C., Lipchock S. V., Smith K. D., Strobel S. A. 2009. Structural and chemical basis for glucosamine 6-phosphate binding and activation of the glmS ribozyme. Biochemistry 48, 3239–3246 10.1021/bi802069p (doi:10.1021/bi802069p) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roth A., Breaker R. R. 2009. The structural and functional diversity of metabolite-binding riboswitches. Annu. Rev. Biochem. 78, 305–334 10.1146/annurev.biochem.78.070507.135656 (doi:10.1146/annurev.biochem.78.070507.135656) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Winkler W. C., Nahvi A., Sudarsan N., Barrick J. E., Breaker R. R. 2003. An mRNA structure that controls gene expression by binding S-adenosylmethionine. Nat. Struct. Biol. 10, 701–707 10.1038/nsb967 (doi:10.1038/nsb967) [DOI] [PubMed] [Google Scholar]
- 27.Cantoni G. L. 1975. Biological methylation: selected aspects. Annu. Rev. Biochem. 44, 435–451 10.1146/annurev.bi.44.070175.002251 (doi:10.1146/annurev.bi.44.070175.002251) [DOI] [PubMed] [Google Scholar]
- 28.Loenen W. A. M. 2006. S-adenosylmethionine: jack of all trades and master of everything? Biochem. Soc. Trans. 34, 330–333 10.1042/BST20060330 (doi:10.1042/BST20060330) [DOI] [PubMed] [Google Scholar]
- 29.Roje S. 2006. S-Adenosyl-l-methionine: beyond the universal methyl group donor. Phytochemistry 67, 1686–1698 10.1016/j.phytochem.2006.04.019 (doi:10.1016/j.phytochem.2006.04.019) [DOI] [PubMed] [Google Scholar]
- 30.Ames T. D., Rodionov D. A., Weinberg Z., Breaker R. R. 2010. A eubacterial riboswitch class that senses the coenzyme tetrahydrofolate. Chem. Biol. 17, 681–685 10.1016/j.chembiol.2010.05.020 (doi:10.1016/j.chembiol.2010.05.020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wagner C. 2001. Biochemical role of folate in cellular metabolism. Clin. Res. Regul. Aff. 18, 161–180 10.1081/CRP-100108171 (doi:10.1081/CRP-100108171) [DOI] [Google Scholar]
- 32.Winkler W. C., Cohen-Chalamish S., Breaker R. R. 2002. An mRNA structure that controls gene expression by binding FMN. Proc. Natl Acad. Sci. USA 99, 15 908–15 913 10.1073/pnas.212628899 (doi:10.1073/pnas.212628899) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Massey V. 1995. Introduction: flavoprotein structure and mechanism. FASEB J. 9, 473–475 [DOI] [PubMed] [Google Scholar]
- 34.Swartz T. E., Corchnoy S. B., Christie J. M., Lewis J. W., Szundi I., Briggs W. R., Bogomolni R. A. 2001. The photocycle of a flavin-binding domain of the blue light photoreceptor phototropin. J. Biol. Chem. 276, 36 493–36 500 10.1074/jbc.M103114200 (doi:10.1074/jbc.M103114200) [DOI] [PubMed] [Google Scholar]
- 35.Sancho J. 2006. Flavodoxins: sequence, folding, binding, function and beyond. Cell. Mol. Life Sci. 63, 855–864 10.1007/s00018-005-5514-4 (doi:10.1007/s00018-005-5514-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Winkler W., Nahvi A., Breaker R. R. 2002. Thiamine derivatives bind messenger RNAs directly to regulate bacterial gene expression. Nature 419, 952–956 10.1038/nature01145 (doi:10.1038/nature01145) [DOI] [PubMed] [Google Scholar]
- 37.Cheah M. T., Wachter A., Sudarsan N., Breaker R. R. 2007. Control of alternative RNA splicing and gene expression by eukaryotic riboswitches. Nature 447, 497–500 10.1038/nature05769 (doi:10.1038/nature05769) [DOI] [PubMed] [Google Scholar]
- 38.Frank R. A. W., Leeper F. J., Luisi B. F. 2007. Structure, mechanism and catalytic duality of thiamine-dependent enzymes. Cell. Mol. Life Sci. 64, 892–905 10.1007/s00018-007-6423-5 (doi:10.1007/s00018-007-6423-5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nahvi A., Sudarsan N., Ebert M. S., Zou X., Brown K. L., Breaker R. R. 2002. Genetic control by a metabolite binding mRNA. Chem. Biol. 9, 1043–1049 10.1016/S1074-5521(02)00224-7 (doi:10.1016/S1074-5521(02)00224-7) [DOI] [PubMed] [Google Scholar]
- 40.Marsh E. N. 1999. Coenzyme B12 (cobalamin)-dependent enzymes. Essays Biochem. 34, 139–154 [DOI] [PubMed] [Google Scholar]
- 41.Reichard P. 1993. From RNA to DNA, why so many ribonucleotide reductases? Science 260, 1773–1777 10.1126/science.8511586 (doi:10.1126/science.8511586) [DOI] [PubMed] [Google Scholar]
- 42.Booker S., Licht S., Broderick J., Stubbe J. 1994. Coenzyme B12-dependent ribonucleotide reductase: evidence for the participation of five cysteine residues in ribonucleotide reduction. Biochemistry 33, 12 676–12 685 10.1021/bi00208a019 (doi:10.1021/bi00208a019) [DOI] [PubMed] [Google Scholar]
- 43.White H. B. 1976. Coenzymes as fossils of an earlier metabolic state. J. Mol. Evol. 7, 101–104 10.1007/BF01732468 (doi:10.1007/BF01732468) [DOI] [PubMed] [Google Scholar]
- 44.Ban N., Nissen P., Hansen J., Moore P. B., Steitz T. A. 2000. The complete atomic structure of the large ribosomal subunit at 2.4 Å resolution. Science 289, 905–920 10.1126/science.289.5481.905 (doi:10.1126/science.289.5481.905) [DOI] [PubMed] [Google Scholar]
- 45.Nissen P., Hansen J., Ban N., Moore P. B., Steitz T. A. 2000. The structural basis of ribosome activity in peptide bond synthesis. Science 289, 920–930 10.1126/science.289.5481.920 (doi:10.1126/science.289.5481.920) [DOI] [PubMed] [Google Scholar]
- 46.Youngman E. M., Brunelle J. L., Kochaniak A. B., Green R. 2004. The active site of the ribosome is composed of two layers of conserved nucleotides with distinct roles in peptide bond formation and peptide release. Cell 117, 589–599 10.1016/S0092-8674(04)00411-8 (doi:10.1016/S0092-8674(04)00411-8) [DOI] [PubMed] [Google Scholar]
- 47.Schmeing T. M., Huang K. S., Kitchen D. E., Strobel S. A., Steitz T. A. 2005. Structural insights into the roles of water and the 2′ hydroxyl of the P site tRNA in the peptidyl transferase reaction. Mol. Cell 20, 437–448 10.1016/j.molcel.2005.09.006 (doi:10.1016/j.molcel.2005.09.006) [DOI] [PubMed] [Google Scholar]
- 48.Erlacher M. D., Lang K., Shankaran N., Wotzel B., Huttenhofer A., Micura R., Mankin A. S., Polacek N. 2005. Chemical engineering of the peptidyl transferase center reveals an important role of the 2′-hydroxyl group of A2451. Nucl. Acids Res. 33, 1618–1627 10.1093/nar/gki308 (doi:10.1093/nar/gki308) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zaher H. S., Shaw J. J., Strobel S. A., Green R. 2011. The 2′-OH group of the peptidyl tRNA stabilizes an active conformation of the ribosomal PTC. EMBO J. 30, 2445–2453 10.1038/emboj.2011.142 (doi:10.1038/emboj.2011.142) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Seila A. C., Okuda K., Nunez S., Seila A. F., Strobel S. A. 2005. Kinetic isotope effect analysis of the ribosomal peptidyl transferase reaction. Biochemistry 44, 4018–4027 10.1021/bi047742f (doi:10.1021/bi047742f) [DOI] [PubMed] [Google Scholar]
- 51.Kingery D. A., Pfund E., Voorhees R. M., Okuda K., Wohlgemuth I., Kitchen D. E., Rodnina M. V., Strobel S. A. 2008. An uncharged amine in the transition state of the ribosomal peptidyl transfer reaction. Chem. Biol. 15, 493–500 10.1016/j.chembiol.2008.04.005 (doi:10.1016/j.chembiol.2008.04.005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sievers A., Beringer M., Rodnina M. V., Wolfenden R. 2004. The ribosome as an entropy trap. Proc. Natl Acad. Sci. USA 101, 7897–7901 10.1073/pnas.0402488101 (doi:10.1073/pnas.0402488101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wolfenden R. 1963. The mechanism of hydrolysis of amino acyl RNA*. Biochemistry 2, 1090–1092 10.1021/bi00905a031 (doi:10.1021/bi00905a031) [DOI] [PubMed] [Google Scholar]
- 54.Zhang B., Cech T. R. 1997. Peptide bond formation by in vitro selected ribozymes. Nature 390, 96–100 10.1038/36375 (doi:10.1038/36375) [DOI] [PubMed] [Google Scholar]
- 55.Bartel D., Szostak J. 1993. Isolation of new ribozymes from a large pool of random sequences (see comment). Science 261, 1411–1418 10.1126/science.7690155 (doi:10.1126/science.7690155) [DOI] [PubMed] [Google Scholar]
- 56.Tarasow T. M., Tarasow S. L., Eaton B. E. 1997. RNA-catalysed carbon–carbon bond formation. Nature 389, 54–57 10.1038/37950 (doi:10.1038/37950) [DOI] [PubMed] [Google Scholar]
- 57.Unrau P. J., Bartel D. P. 1998. RNA-catalysed nucleotide synthesis. Nature 395, 260–263 10.1038/26193 (doi:10.1038/26193) [DOI] [PubMed] [Google Scholar]
- 58.Tsukiji S., Pattnaik S. B., Suga H. 2003. An alcohol dehydrogenase ribozyme. Nat. Struct. Biol. 10, 713–717 10.1038/nsb964 (doi:10.1038/nsb964) [DOI] [PubMed] [Google Scholar]
- 59.Lambowitz A. M., Belfort M. 1993. Introns as mobile genetic elements. Annu. Rev. Biochem. 62, 587–622 10.1146/annurev.bi.62.070193.003103 (doi:10.1146/annurev.bi.62.070193.003103) [DOI] [PubMed] [Google Scholar]