

Abstract
Xenopus Laevis provides a robust model system to study cellular signaling and downstream processes during development both in vitro and in vivo. Intracellular signals must function within highly restricted spatial and temporal domains to activate specific downstream targets and cellular processes. Combining the versatility of developing Xenopus neurons with advances in fluorescent protein biosensors and imaging technologies has allowed many dynamic cellular processes to be visualized. This review will focus on the techniques we use to visualize and measure cell signaling, motility and adhesion by quantitative fluorescence microscopy in vitro and in vivo.
Introduction
Throughout development, embryonic cells use environmental factors to direct their migration and morphological differentiation. While many extracellular cues and their transmembrane receptors are known, the precise mechanisms that control directional cell movement remains poorly understood. This is particularly true in the developing nervous system, where hundreds of millions or billions of neurons (depending on species) must interconnect through trillions of synapses. Remarkably, these highly complex neural circuits assemble stereotypically using a relatively small set of substrata molecules, guidance cues and receptors. The highly diverse neuronal interconnections that form during development likely result from temporally controlled combinations of cues and receptors specific for individual neurons at precise times and locations during axon pathfinding to their targets. An additional level of control comes from distinct combinations of intracellular signaling pathways that are activated in neuronal growth cones downstream of receptor-generated signals. According to these diverse processes, a complete understanding of the molecular basis for how growth cones decipher environmental cues to assemble functional neural networks must come from multifaceted approaches using both reduced and intact systems.
Analysis of axon guidance mechanisms in vitro
Neurons cultured from Xenopus embryos have proven to be a highly productive in vitro model system amenable for testing the molecular basis of axon guidance. The rapid external development of their large embryos allows targeted ectopic protein expression and protein knock-down (Gomez et al., 2003). The first neurons are born in Xenopus just 15 hours post fertilization (hpf) and neurulation is complete by 22 hpf. Neurons are easily cultured from spinal cord, brain and retina, where they exhibit rapid neurite outgrowth on diverse substrata ranging from bare glass to purified cell adhesion molecules. Moreover, isolated neurons appear to continue to follow their normal programmed development, gaining and losing responsiveness to guidance cues over time (Stein and Tessier-Lavigne, 2001). The extent of ectopic protein expression and knock-down can be tightly controlled by varying the amount of mRNA or morpholino injected. It is important to note that expression level is critical, since all biosensors can be dominant negative if over-expressed. However, protein expression and knock-down by early blastomere injection does have the drawback that very early gene expression changes can alter early developmental processes. We use two different approaches to delay expression, neural-specific promoter or heat-shock promoter driven expression (Woo et al., 2009) and electroporation (Falk et al., 2007). Promoter specific expression in Xenopus requires genomic integration of transgenes. To accomplish this, we use the I-SceI meganuclease-mediated transgenesis method, which has been found to be effective in Laevis (Pan et al., 2006). This approach allows us to use either neural class-specific promoters, such as Rohon-Beard-specific neurogenin (Blader et al., 2003), or a heat shock promoter (HSP70). Specific tissues within Xenopus embryos can also be subjected to targeted electroporation of mRNA/DNA/morpholinos (Falk et al., 2007). For example. we have used electroporation to selectively express proteins in commissural interneurons (e.g. see Fig 4).
Figure 4. Labeling and measurement of CI axons.

A. Maximum projection of a confocal z-series of a ventral view of a 27 hpf neural tube immunolabeled with 3A10 (neurofilament in CIs) antibody (blue) and expressing GFP (green) in crossed CIs. Anterior is up in all ventral views. B. Traced trajectories of GFP-labeled CIs (from A) at various points before, during and after midline crossing. Red lines indicate the position of the ventral fascicles. Note that CIs directly cross the midline and turn to ascend in the contralateral spinal cord after midline crossing. C. Ventral view of a 27 hpf neural tube immunolabeled with 3A10 antibody. D. Same embryo as (C) showing GFP-FRNK (dominant-negative FAK) expression targeted to the left side. E. Merge of 3A10 (blue) and GFP-FRNK (green) fluorescence. F. Traced trajectories of FRNK-expressing axons in (D). One axon at the floor plate prematurely turned posteriorly (black asterisk), while two others aberrantly reoriented back towards the ipsilateral side (red asterisks). G, H. Confocal maximum projection of a ventral view of a 27 hpf neural tube that was electroporated at 21 hpf with mRNA encoding GFP and TMR-Dextran. Note that GFP expression occurs in the dorsal CIs that project their axons ventrally on one side (G), while TMR-D was electroporated into ventral cells on the opposite side of the spinal cord (H). This distribution is likely due to charge difference between mRNA (negative) and TMR-D (positive). I. β-tubulin (β-tub) labeling of the spinal cord from (G, H). Note that β-tub labels all neurons in the spinal cord, including the neurons that form the ventral fascicles (blue arrowheads). J. Merged image of the spinal cord from (G-I) showing GFP (green), TMR-D (red) and β-tub (blue). K. Optical cross sectional views of confocal image stacks of the spinal cord from (G-J) taken at the position indicated by the white arrowheads in (J). Scale bar, 50 μm (in A-J) and 40 μm (K).
The spinal cord of Xenopus embryos is a rich source of distinct neuronal classes. These include Rohon-Beard (RB) sensory neurons, motoneurons (MNs) and several classes of interneurons, including commissural interneurons (CIs). These neurons can be identified based on cell body position and their axonal projection pattern in vivo. However, it is often difficult to positively identify specific classes of neurons in vitro, especially in living cells. One approach we use to identify neuronal types in vitro is to culture spinal neurons from embryos pre-labeled for specific cell types by targeted blastomere injection using fate maps (Moody, 1987; Moody, 1987). Labeling specific regions of the spinal cord allows us to identify neurons derived from dorsal and ventral spinal cord in vitro (Fig 1). Using promoters to drive GFP in specific classes of neurons would provide even more specific labeling of distinct classes of neurons.
Figure 1. Identifying neuronal types in cell culture.
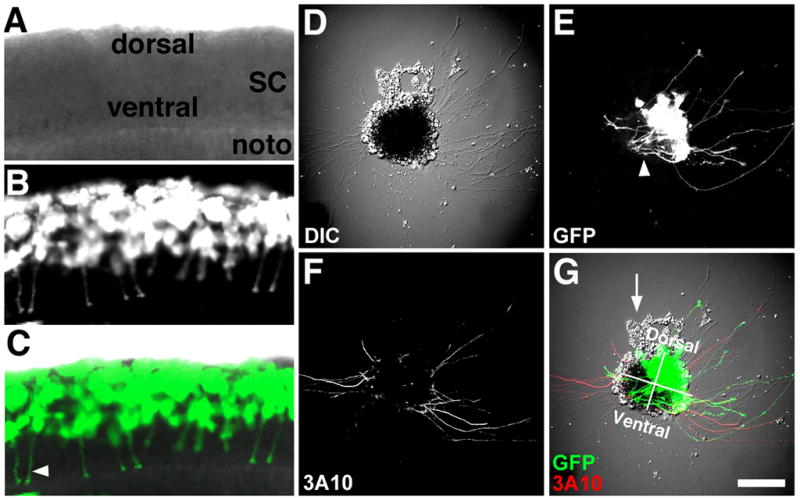
A–C. Confocal microscope images of a ~24 hpf live Xenopus embryo that was previously injected at the 8-cell stage with mRNA encoding GFP into a single blastomere fated to make dorsal spinal cord. Note many ventrally projecting CIs (commissural interneurons; arrowhead in C). D. DIC image of a spinal cord explant in cross-section from the same spinal cord as in (A–C) cultured onto PDL-LN coverslips for 24 hrs. E. GFP fluorescence shows many labeled RB (Rohon-Beard) and CIs extending from the dorsal-lateral region of this explant. Also note many presumed CIs that extend toward the floor plate and stay within the explant (arrowhead). F. Fixed explant that was immunofluorescently labelled with an anti-neurofilament antibody (3A10) that is specific for CIs and the peripheral process of RB neurons in vivo. Note dorsal lateral labeled axons emerge from both sides of spinal explant G. Merged image of fluorescent and DIC channels with dorsal-ventral and right-left quadrants indicated. Presumed neural crest cells (arrow) are often observed migrating from the dorsal aspect of spinal cord explants. Scale bar, 62 μm (in A-C) and 100 μm (in D–E).
Quantitative immunocytochemical (ICC) measurements
We use quantitative ICC with phospho-specific antibodies to assess cell signals associated with changes in growth cone motility downstream of axon guidance cues. Quantitative ICC offers several advantages over traditional biochemical measurements. Western blot analysis requires a sufficient sample size of cells from culture or tissues to detect proteins by blot, whereas fluorescent measurements can be made from individual immuno-labeled cells. Measurements made in neurons isolated in culture can be further restricted to specific neuronal types identified using neuron specific markers. This is in contrast to Western blotting from complex tissues such as spinal cord, which contain multiple neuronal types, as well as non-neuronal cells. Moreover, fluorescent ICC signals can be measured within specific sub-cellular regions, such as the tips of filopodia. Making fluorescent measurements from restricted regions of specific cell types can greatly increase the signal over background noise. This is particularly true for signaling proteins concentrated within growth cones that become phosphorylated in response to guidance cues. Isolating proteins from a heterogeneous population of cells from culture or tissues dilutes the relevant proteins with a vast over-abundance of protein originating from non-responsive cells or from parts of neurons that do not respond to guidance cues. As an example, we detect robust changes in the activation of the tyrosine kinase Src within growth cones, as determined by tyrosine phosphorylation of Y418-Src, after a brief treatment with brain derived neurotrophic factor (BDNF) by ICC (Robles et al., 2005). In contrast, we cannot detect any change in the phosphorylation of Y418-Src by Western blot from isolated pure Xenopus spinal cords treated with BDNF (not shown).
While we find that quantitative ICC is a highly sensitive tool to measure local changes in protein phosphorylation within growth cones, there are some caveats and shortcomings to this approach. First, stimulating cells with axon guidance cues can lead to dramatic changes in growth cone morphology. Growth promoting guidance cues, such as BDNF, often lead to growth cone expansion, which can dilute ICC labeling, whereas inhibitory guidance cues can have the opposite effect. To limit artifact due to morphological changes, we often stimulate neurons for short times (e.g. 2–5 min) before fixing and processing for ICC. This approach is effective since the signals that lead to morphological changes will precede those morphological changes. However, if morphological changes are unavoidable, we often normalize for cell volume by co-labeling with a total protein marker (Fig 2). Of course there are some well-recognized limitations to ICC. For example, some antibodies do not work well by ICC and give high background staining that makes detection of specific proteins difficult. Additionally, some proteins are expressed at levels too low to be detected or are lost during the fixation or extraction steps. Moreover, while ICC can be used to measure protein colocalization, this approach does not prove that two proteins physically interact in a complex. For this, co-immunoprecipitation followed by Western blot is the most common method. However, modern imaging methods that utilize fluorescence energy transfer now make it possible to measure protein-protein interactions within intact cells.
Figure 2. Quantification of active Src in growth cones by ICC.
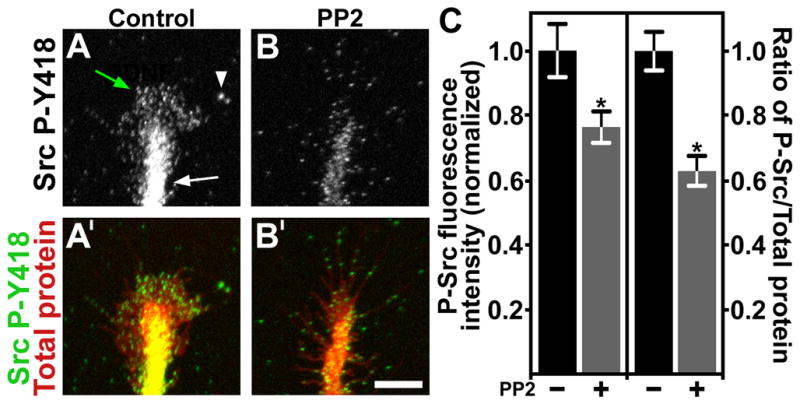
A. Control growth cone immunolabeled with antibodies against phospho-tyrosine 418 Src (P-Y418 Src). Note intense labeling in the axon shaft (white arrow), peripheral veil (green arrow) and some labeled filopodial tips (arrowhead). B. A growth cone treated with the Src inhibitor PP2 for 5 min before immunolabeling for P-Y418 Src. Note reduced labeling throughout this growth cone compared to control. A′, B′. The growth cones from (A, B) were co-labeled for total protein (pseudocolored red) with Alexa Fluor 647 carboxylic acid, succinimidyl ester. All images were collected with a confocal microscope. C. Normalized total fluorescent intensity measurements of P-Y418 Src immunolabeling, and the normalized P-Y418 Src/total protein ratio. PP2 treatment results in a significant decrease in P-Y418 Src by both measures. *P < 0.05. Scale bar, 10 μm.
Examining cells in real time with live imaging
While the analysis of fixed cells by ICC provides many insights into how growth cones respond to axon guidance cues, examining live cells under similar conditions offers several key advantages. First, the fixation, extraction and ICC labeling of neurons can alter fine cellular and molecular composition of growth cones. This is particularly true for extending filopodia, which contain numerous proteins that control actin polymerization at their growing tips (Svitkina et al., 2003; Robles et al., 2005). Second, live cell imaging allows researchers to track changes in growth cone motility over time and during stimulation with axon guidance cues. As there may be time-dependent changes in growth cones after stimulation with guidance cues, observing neurons with fast image acquisition over the short-term and less frequently over the long-term, can provide important insights into how guidance cues influence outgrowth. This is particularly true when imaging intracellular signaling events that often show dynamic changes over wide time scales. With the development of a tremendous number of fluorescent biosensors, many based on the fluorescent proteins (FPs), live cell imaging can now be used to track a wide variety of physiological and molecular processes within cells.
Versatile biosensors allow visualization cell signaling and structure
A wide variety of tools are available to visualize many aspects of cell signaling and structure in living cells. Synthetic dyes can be used to detect cell physiology, such as calcium (Ca2+) signals, expressed fluorescent fusion proteins can be localized to endogenous structures, and biosensors can report protein conformational changes and protein-protein interactions. In this section, we will detail a few of the biosensors we have used to study the role of key cell signals and subcellular structures in growth cones during extension and guidance.
Imaging intracellular signals
Ca2+ is a fundamental signaling ion in all cells. Changes in the intracellular Ca2+ concentration ([Ca2+]i) control a broad range of cellular functions throughout development (Webb and Miller, 2003) and into adulthood, including regulation of cell motility, muscle contraction and synaptic vesicle release (Augustine et al., 2003). According to these varied functions, Ca2+ acts through a number of Ca2+ binding proteins including, calmodulin, myosin regulatory light chain, protein kinase C and the protease calpain. Axon outgrowth and guidance is also strongly influenced by Ca2+ signals and many axon guidance cues modulate Ca2+ influx or release from stores (Gomez and Zheng, 2006).
Optical imaging of intracellular Ca2+ using fluorescent Ca2+ indicators has been widely used to study neuronal physiology. The use of synthetic Ca2+ sensors has made it possible to resolve spatially and temporally discrete Ca2+ signals in developing and adult neurons and their processes (Augustine et al., 2003). For example, using the highly sensitive Fluo dyes, we have observed distinct types of spontaneous Ca2+ transients in Xenopus spinal neuron growth cones. We have observed slow Ca2+ waves that spread throughout the growth cone both in vitro and in vivo (Gomez and Spitzer, 1999). Using faster imaging, we have also observed highly localized, brief Ca2+ transients that occur predominantly at the tips of filopodia (Gomez et al., 2001). These local Ca2+ signals require Ca2+ influx through a non-voltage gated channel(s) and appear to inhibit axon extension. Similar Ca2+ transients have been observed in dendritic filopodia (Lohmann et al., 2005). With the development of more sensitive genetically encoded Ca2+ indicators (GECIs), such as GCaMP3 (Tian et al., 2009), it will be interesting to test if similar signals can be resolved with targeted expression GCaMP3.
Fluorescent fusion proteins provide nearly limitless applications to track a wide variety of cellular signals. One approach is to exploit protein binding domains that are targeted to post-translational modifications of other proteins. For example, to investigate phosphotyrosine (PY) dynamics in growth cones we use a fluorescent PY reporter construct consisting of tandem PY-binding SH2 (Src homology-2) domains fused to GFP or mCherry (mCh-dSH2; Fig 3B; (Kirchner et al., 2003)). Using GFP-dSH2, we find that PY concentrates to the tips of extending filopodia (Robles et al., 2005), and within stable growth cone adhesions called point contacts (Robles and Gomez, 2006). Since the PY accumulations detected by GFP-dSH2 depend on the tyrosine kinase Src, this sensor can be used to determine whether guidance cues activate Src. Other protein-binding domain-based biosensors we use depend on the CRIB domain of Rho GTPase binding effector molecules to track the localization of active Rho GTPases (Bement et al., 2006). Rho GTPases act as molecular switches, active when bound to GTP and inactive when bound to GDP (Jaffe and Hall, 2005). In their active form, Rho family GTPases associate with specific effectors that propagate their signals. GFP fused to the binding domain (CRIB domain) of these effector proteins have proven an effective way to assess Rho GTPase activity in live cells, where an accumulation of fluorescence indicates a higher level of activity (Benink and Bement, 2005). Normalizing for cell volume with a second fluorescent dye, such as tetramethylrhodamine-dextran (TMR-D), allows the specific accumulations of Rho GTPase activity to be highlighted. We have used this approach with the CRIB domain from N-WASP (GFP-WBD; WASP binding domain) to track Cdc42 activity in growth cones and correlate changes in Cdc42 activity with cell protrusion and motility (Figure 3E-H). CRIB binding domains tagged with other fluorophores have been used in more complex imaging paradigms to simultaneously visualize two GTPases during lammelipodial protrusion and retraction (Machacek et al., 2009). Surprisingly, these investigators found that RhoA was elevated at the leading edge during protrusion, whereas active Rac1 and Cdc42 became localized with a short delay just behind leading edge. Similar studies in neuronal growth cones will be informative, as it is likely that attractive and repulsive axon guidance molecules differentially regulate Rho GTPase activities in a spatially and temporally distinct manner.
Figure 3. Analysis of cell signaling and structure with live cell imaging.

A, B. Total Internal Reflection Fluorescence (TIRF) microscopy images of paxillin-GFP and mCherry dual-Src homology 2 domain (mCh-dSH2) fluorescent images of a growth cone on PDL-LN. Note that paxillin and phosphotyrosine (PY), as revealed with mCh-dSH2, colocalize at adhesion sites (arrowheads in A, B), whereas the tip of a growing filopodium has PY, without paxillin (arrow in B). C. Merged image of the growth cone in (A, B) shows co-localization at several peripheral adhesions. Note that mCh-dSH2 puncta within the central domain are mobile vesicles (green arrow in B). D. Single line kymograph (see text for details) constructed from region between the arrowheads (C). Note a stable adhesion (arrow) that disassembles after a new protrusion extends forward, followed by the formation of a second adhesion (arrowhead), which stabilizes the receding protrusion. Scale bar, 10 μm in all images and as indicated in kymographs. E, F. GFP-WASP binding domain (GFP-WBD; active Cdc42 binding) and tetramethylrhodamine-dextran (TMR-D; volume marker) fluorescent images of a growth cone on LN captured with a confocal microscope. The average pixel intensities (8-bit scale) within regions at the growth cone periphery (red), central domain (blue) and axon (green) show that GFP-WBD is selectively enriched in the peripheral lamellipodia of this growth cone. G. A pseudo-colored ratio image of the growth cone in (E, F) shows the GFP-WBD/TMR-D ratio is elevated throughout peripheral lamellipodia and filopodia (arrows). H. Single line kymograph constructed from region indicated by the white line in (G). Note an elevated WBD/TMR-D ratio during lamellipodial protrusion. I, J. GFP-Utrophin (GFP-Utr; F-actin probe) and TMR-Kabiramide C (TMR-KabC; F-actin barbed end binding) fluorescent images of a growth cone on PDL-LN collected with a TIRF microscope. Note bright TMR-KabC fluorescence at the periphery (arrow in J), indicating a high concentration of F-actin barbed ends. K. Merged image of the growth cone in (I, J) shows strong co-localization of GFP-Utr and TMR-KabC in central domain foci, but primarily TMR-KabC in the peripheral domain. Weak labeling of peripheral actin with GFP-Utr is consistent with the slow association rate of GFP-Utr onto recently polymerized F-actin (Rybakova et al., 2006). L. Single line kymograph constructed from region indicated by the white line in (K). The slope of the diagonal bands of GFP-Utr and TMR-KabC fluorescence (arrows) indicates a retrograde flow rate of ~ 4 μm/min. Scale bars, 5 μm or as indicated.
While simply tagging individual protein-binding domains with fluorescent proteins can provide useful tools to track cell signaling through fluorescence localization, this approach does not measure the active state of signaling molecules. For this, techniques that require protein conformational changes have been designed. For example, biosensors that utilize Försters resonance energy transfer (FRET) or circular permutated GFP can quantitatively measure signals in live cells. FRET works by non-radiative energy transfer between a donor fluorophore and an acceptor fluorophore (Miyawaki, 2003). Energy transfer will only occur if donor and acceptor fluorophores are near (less than 10 nm) and several other conditions have been met. Since FRET efficiency depends on the distance between two fluorophores, biosensors can be designed that change energy transfer in response to intramolecular conformational changes or protein-protein interactions (intermolecular FRET). Many FRET constructs have been developed, including Ca2+ and cAMP indicators, Rho GTPases sensors, tyrosine kinase probes, as well as many others (Aye-Han et al., 2009). Other biosensors use a single fluorophore that changes in fluorescence intensity after a conformational change in the fluorophore. For example, the GCaMP family of GECIs use circular permuted GFP linked to proteins that change conformation upon Ca2+ binding leading to increased fluorescence from GFP (McCombs and Palmer, 2008).
Cytoskeletal imaging
Most cellular signals that modulate cell motility and axon outgrowth do so by directly or indirectly influencing the cytoskeleton. Cytoskeletal effector proteins regulate the polymerization, organization and movement of actin filaments (F-actin) and microtubules (MTs) (Dent and Gertler, 2003). Given their essential functions in the control of axon outgrowth and guidance, a number of tools have been developed to visualize cytoskeletal dynamics in live cells. Fluorescent fusion variants of G-actin and tubulin have been used for years to visualize F-actin and MT dynamics, but high background labeling from unpolymerized fluorescent monomers often makes it difficult to resolve filaments. More recently, fluorescent F-actin binding peptides and protein fragments have been used to specifically track F-actin in cells. For example, F-lifeact is a 17 amino acid F-actin binding peptide fused to GFP that labels F-actin in live cells (Riedl et al., 2008). As an alternative, we use the N-terminal actin binding domain of Utrophin (Utr) fused to GFP to monitor filamentous actin (F-actin) in live cells (Burkel et al., 2007). Expressing GFP-Utr in neurons allows us to monitor dynamic changes in the actin cytoskeleton of live growth cones (Fig 3I). GFP-Utr labels with high signal-to-noise similar structures observed with GFP-actin and actin isoforms by ICC, as well as with phalloidin staining (not shown). GFP-Utr localizes to linear actin bundles within filopodia and lamellipodia near the leading edge of growth cones, as well as to cortical actin surrounding the entire axon shaft and to actin puncta within the growth cone central domain. However, due to its slow on rate (Kd=18.6 μM; (Rybakova et al., 2006)), GFP-Utr does not label actin in the most distal extreme of newly emerging lamellipodia and filopodia (Fig 3I, K). To label the distal extent of peripheral veil and to monitor retrograde actin flow, we use the small molecule Kabiramide C (KabC; Fig 3J), which binds and caps the growing or barbed-end F-actin. A fluorescent conjugate of KabC has been used previously to monitor retrograde flow in cells (Petchprayoon et al., 2005). A low level of TMR-KabC loaded into neurons allows us to track retrograde flow (Fig 3L), without interfering with neurite outgrowth (not shown).
MT polymerization can also be tracked effectively in growth cones using +TIPS binding proteins. +TIPs proteins are a class of proteins that associate with and protect the growing end of MTs from depolymerization (Galjart, 2010). As these proteins are highly concentrated to the growing ends of MTs, which are relatively sparsely distributed in the peripheral domain of growth cones, simple fusion with fluorescent proteins provides an effective probe of MT polymerization. For example, GFP-EB3, GFP-CLIP170 or GFP-CLASP expressed in growth cones appear as bright, fast-moving “comets” of fluorescence that project throughout axons and dendrites and into growth cones and spines (Stepanova et al., 2003; Lee et al., 2004; Hu et al., 2008). With these probes, its possible to measure changes in the rate and spread of microtubule exploration in response to pharmacological agents or growth factors. When used in combination with other probes, such as F-actin markers, it is possible to correlate rates of microtubule polymerization with retrograde flow.
Cell adhesion imaging
Growth cones use specialized integrin-dependent adhesion sites called point contacts to stabilize new protrusions during axon extension on extracellular matrix (ECM) proteins (Gomez et al., 1996; Robles and Gomez, 2006). Point contacts are analogous to focal adhesions, which regulate motility and a number of other important cellular functions of non-neuronal cells (Zaidel-Bar et al., 2007). When cultured on ECM substrata, growth cone point contacts are visualized by imaging fluorescent fusion proteins of adhesion components, such as integrin receptors, paxillin, talin or vinculin (Worth and Parsons, 2008). To image cell adhesion markers in vitro, as well as many cellular signals, we use total internal reflection fluorescence (TIRF) microscopy. TIRF imaging uses an evanescent wave of light to excite fluorophores only approximately 100 nm into the cell, which greatly increases the image signal-to-noise. TIRF imaging of paxillin-GFP expressing growth cones reveals point contacts as stable puncta that assemble at the leading edge of nascent protrusions (Fig 3A). Point contacts are smaller and much shorter lived than focal adhesions, but some remain stable for minutes as the axon extends forward (Fig 3A–D). Despite differences in size and lifetime, point contacts and focal adhesions share important characteristics. For example, the assembly and lifetime of point contacts and focal adhesions are both regulated by focal adhesion kinase (FAK) and Src family kinases (Robles and Gomez, 2006; Woo et al., 2009). Interestingly, many axon guidance cues are known to signal through FAK and Src (Li et al., 2004; Liu et al., 2004; Ren et al., 2004; Robles and Gomez, 2006; Woo et al., 2009) and work from our lab suggests that axon guidance cues control axon growth on ECM by modulating point contact dynamics (Woo and Gomez, 2006; Woo et al., 2009). We found that the turnover of point contacts in Xenopus spinal neurons are regulated by balanced Rho GTPases signals and FAK in vitro. Moreover, in spinal neurons we found that Sema3A destabilized new and existing point contacts (Woo and Gomez, 2006). In contrast, we found that a sub-collapsing dose of EphrinA on retinal ganglion cell growth cones cause point contact stabilization (Woo et al., 2009). In both cases, disrupting FAK signaling altered point contact turnover in vitro and led to axon pathfinding defects in vivo.
Visualization and analysis of fluorescent signals in live cells
Imaging fluorescent fusion proteins in live cells allows investigators to correlate changes in protein distribution with cell motility and guidance. The dynamic distribution of proteins in growth cones varies depending on protein function. Moreover, stimulation of growth cones with guidance cues can alter the trafficking and localization of proteins. We use several approaches to measure the lifetime and movements of discrete protein puncta within live cells. For example, we use kymography to quantify leading edge protrusion, the rate of myosin-driven retrograde flow and the lifetime of substratum-associated point contacts (Fig 3D, H, L). Kymography is a method where a single line extending through a specified region of a cell is sampled over all frames and displayed over time in a two dimensional image. Typically time is displayed along the x-axis and distance along the y-axis. In this format, stable puncta appear as level streaks across the time axis, with the length of the line measuring puncta persistence. On the other hand, if the line is drawn along the length of speckled actin filaments, puncta flowing rearward appear as diagonal lines (Fig 3L). The slope of these diagonal lines indicates the rate of retrograde flow of actin. Probably the most common use of kymography is to quantify leading edge protrusion. Here the rate, frequency and duration of protrusions can be quantified (Robles and Gomez, 2006). In addition, for growth cones expressing fluorescent biosensors, leading edge protrusions can be associated with intracellular signals (Fig 3H).
One drawback with kymography is that it only samples individual, user-defined regions. This not only limits the areas measured, but also introduces subjectivity to the analysis since lines are user-selected. This can be a particular problem when studying adhesion site dynamics, as multiple point contacts form with varying lifetimes during even brief time-lapse sequences. One method we use to identify multiple point contacts across a growth cone is to merge three frames from a movie, which have been pseudo-colored red, green, and blue (Robles and Gomez, 2006). In this case, a point contact that is present in all three frames will appear white in the merged image, giving an indication of its lifetime. Our laboratory has recently developed an improvement of this technique that displays the summed fluorescence of every frame as a heat map, where the color of the puncta is a function of its lifetime. To do this, multiple image filters are applied in sequence using ImageJ open source plugins. The first filter is an image stabilization algorithm, to account for any drift in the stage that can occur over the time-lapse. The second filter is an unsharp mask, which sharpens the contrast of fluorescent puncta over the background. The third step is to threshold the fluorescence of the image so that only the puncta of interest are included. For growth cone point contacts, thresholding typically excludes any remaining background fluorescence and smaller, unstable puncta that can be present. Applying an 8-bit binary filter converts those thresholded puncta to a grey scale value of 255 and background to black. Finally, we sum the binary frames together giving a graded, 16-bit image, where the puncta intensity represents adhesion lifetime. Applying a color-look up table allows viewers to quickly assess adhesion life-time (Fig. 5).
Figure 5. Heat maps of point contact lifetime.

A, B. Heat maps indicating point contact lifetime generated by applying a series of filters (see the text for details). Images were created from of a Xenopus retinal ganglion cell growth cone expressing paxillin-GFP before (A) and after (B) the addition of 1 μg/ml EphrinA1. The arrows highlight point contacts. Note the stabilization of point contacts and partial collapse of the growth cone after addition of EphrinA1. Scale bar, 5 μm.
Analysis of axon guidance mechanisms in vivo
A great number of in vitro studies have provided a detailed molecular understanding into how growth cones use substratum and cell-associated ligands, as well as soluble cues to guide axons. However, since in vitro experimental conditions cannot precisely replicate true pathfinding in developing embryos, the molecular and cellular processes identified in vitro must be also tested in vivo. The use of developing Xenopus embryos has made it possible to quantitatively assess pathfinding by several clearly identifiable neuronal populations after 24–48 hrs post fertilization (hpf). Moreover, developing neurons can be assayed in both in fixed, immunolabeled tissues and in living neurons during pathfinding in vivo. Here we describe techniques that we and others have used to assess pathfinding in the developing spinal cord and in the retinotectal system. While these are the major systems we have examined, the development of other CNS tracks have been studied in Xenopus (Honore and Hemmati-Brivanlou, 1996; Wilson and Key, 2006).
Testing midline crossing by spinal commissural interneurons
One of the best understood pathfinding choices made by vertebrate neurons is midline crossing by spinal commissural interneurons (CIs). Multiple extracellular cues and receptors are involved in selectively attracting CI axons ventrally into the floor plate, then across the midline and finally into the contralateral spinal cord (Dickson and Zou, 2010). Additional factors are known to prevent CI axon recrossing and to promote growth cone turning to ascend longitudinally within the spinal cord. In fixed Xenopus embryos, the projection patterns of individual spinal axons can be visualized and quantified by immunohistochemical (IHC) staining and labeling of terminal growth cones with fluorescent-phalloidin (Moon and Gomez, 2005). Confocal z-stacks of fluorescently labeled neurons viewed within lateral and ventral whole mount preparations allow detailed examination of axon projection patterns and growth cone morphologies. At around 22 hpf, the first CIs in the anterior spinal cord begin extending their nascent axons toward the ventral midline. One to two hours later, the first CI growth cones are observed within the floor plate (Moon and Gomez, 2005). Normal axon crossing occurs in a highly regular manner, with an average crossing angle of nearly 90 degrees at the midline (Fig 4). Therefore, this stereotypical crossing can be used as a measure to test the molecular mechanisms governing guidance toward, across and away from the midline.
To test molecular mechanisms involved in axon guidance in the spinal cord and to express fluorescent fusion proteins in spinal neurons, we typically inject DNA or mRNA constructs or anti-sense morpholinos into targeted blastomeres at the eight-cell stage. While this approach can alter developmental processes before spinal cord development, protein expression levels can be tightly controlled by the amount of mRNA injected. This allows us to observe phenotypes of hypomorphic spinal neurons. However, if delayed expression becomes necessary, we have used both electroporation of neural plate embryos (Fig 4b), and heat-shock promoter driven expression (Woo et al., 2009). Typically, mutant proteins are either directly tagged with fluorescent proteins, or constructs are co-injected with fluorescent tracers. One advantage of fluorescently labeling spinal neurons is that axon guidance by CIs can be imaged in living embryos after spinal cord exposure by removing the overlying skin and somites (Moon and Gomez, 2005).
Our imaging studies in cell culture show that the rate of neurite outgrowth correlates with turnover of integrin-dependent adhesions (Robles and Gomez, 2006). Substrata that stimulate rapid adhesion turnover, promote the fastest rate of axon outgrowth. The non-receptor tyrosine kinases Src and FAK promote adhesion assembly and turnover. Interestingly, Src and FAK are necessary downstream of several axon guidance cues, including Netrin. Since netrin is an important chemotropic factor that attracts CIs into the floor plate, we used FAK loss of function embryos to test whether FAK is necessary for CI guidance toward the floor plate and across midline. By using the ventral preparation described above, we showed that FAK function is necessary for proper midline crossing by CIs (Fig 4A–F), but is not necessary for initial extension of axons toward the floor plate (Robles and Gomez, 2006). This result suggests that Netrin may more strongly influence crossing compared to guidance toward the floor plate. Additional mechanistic details, such as tyrosine phosphorylation status of FAK-related proteins, may also be measured within growth cones at various positions along their trajectory with this in vivo preparation.
Measuring axon extension and branching by peripheral Rohon-Beard axons
Rohon-Beard (RB) sensory neurons extend two central axons that fasciculate closely with other RB central processes to form the dorsal lateral fascicle within the spinal cord. In contrast to its central axons, RB peripheral processes project laterally along the skin basal lamina, where they arborize extensively to form an elaborate sensory plexus. The arborization pattern of RB peripheral processes is the result of both frequent axon branching and contact inhibition between neighboring RB processes (Moon and Gomez; Liu and Halloran, 2005). Using a neural tube-skin whole mount preparation, we have examined the molecular basis for RB peripheral axon growth and arborization. The peripheral processes of RB axons are selectively labeled by IHC using an antibody to HNK-1. Using this whole mount preparation we have found that gain and loss of function of intracellular signaling molecules such as, FAK and Vav2, a guanine nucleotide exchange factor, lead to stalled outgrowth and reduced axon arborization (Moon and Gomez; Robles and Gomez, 2006). While it remains uncertain what extracellular factors regulate peripheral axon branching and contact inhibition, one arborization factor that may influence peripheral RB branching is BDNF. Consistent with a role for BDNF is the high expression of TrkB in RB neurons (Islam et al., 1996). In addition, we find that RB neurons expressing dominant-negative TrkB exhibit reduced outgrowth and branching by their peripheral processes (TMG, unpublished observations).
Analysis of arborization by retinotectal axons
The retinotectal system is one of the most accessible CNS pathways that has been used extensively as a model of axon guidance and topographic mapping. In Xenopus, retinal ganglion cell (RGCs) axons first exit the eye at 32 hpf, cross the optic chiasm at 40 hpf and begin entering the tectum at 54 hpf, where they arborize extensively (Sakaguchi and Murphey, 1985). Multiple guidance cues direct RGC axons into the tectum, whereas Ephrin gradients influence the anterior-posterior and dorsal-ventral location of RGC arborization (Mann et al., 2004). The peripheral location of these CNS neurons allows many different types of experimental manipulations, including ectopic gene expression and knock-down, lipophilic dye tracing, live cell imaging, tissue transplantation and cell culture. Moreover, unlike the spinal neurons, RGCs are a relatively homogenous population of projection neurons that use many different guidance cues to reach the contralateral tectum and subsequently also the ipsilateral tectum (Mann et al., 2004). Once at the tectum, RGC axons arborize regionally within the tectum according to EphA/EphrinA forward and Eph/EphrinB reverse signaling rules (McLaughlin and O’Leary, 2005).
We recently tested whether Src/FAK signaling functions downstream of EphrinA in the regulation of retinotopic mapping. Since EphrinA acts as an inhibitory guidance cue toward temporal RGCs, we were surprised to find that both Src and FAK are activated downstream of EphrinA and were necessary for the inhibitory effects of EphrinAs (Woo et al., 2009). This suggests that activation of Src and FAK are necessary downstream of both growth promoting and inhibiting axon guidance cues. Several possible mechanisms could explain these contradictory results. For example, since FAK can directly or indirectly associate with a variety of different receptor types (Sieg et al., 2000; Ren et al., 2004; Moeller et al., 2006; Streuli and Akhtar, 2009), activation of different receptors may redistribute FAK, resulting in tyrosine phosphorylation of distinct target proteins. However, this does not explain how EphrinA can inhibit axon outgrowth by temporal RGCs, but promote outgrowth by nasal RGCs (Hansen et al., 2004; Woo et al., 2009). In this regard, it is interesting to note that we found that FAK expression levels were higher in temporal RGC growth cones compared to nasal RGC growth cones in vitro. Similarly, a low-to-high gradient of FAK expression was observed across the nasal-to-temporal axis within the RGC layer of the retina. Together, these results suggest that an optimum level of FAK activity may promote RGC axon outgrowth, with too little or too much being inhibitory to outgrowth. A mechanism like this would attract nasal RGC axons toward EphrinA in the posterior tectum, while stabilizing temporal RGC arbors in the anterior tectum. Importantly, we also tested if FAK function was required for proper mapping of RGCs within the tectum. Using several different methods to inhibit FAK function and two different model systems, we showed that loss of FAK function resulted in a posterior shift of temporal RGC arbors, consistent with a role for FAK in retinotopic mapping (Woo et al., 2009).
Visualizing signaling and structural dynamics in growth cones in vivo
Many of the molecular processes we study in cell culture can now be viewed within intact systems using confocal and two-photon microscopy. For example, local Ca2+ signals have been observed within growth cones in developing spinal cord and brain (Gomez and Spitzer, 1999; Hutchins et al., 2010), as well as in dendritic filopodia and spines in slice (Lohmann et al., 2005) and even in the intact brain of awake behaving animals (Lutcke et al., 2010). In addition, fluorescent protein-based biosensors have been used to measure cell structure and signaling in vivo and in slice. For example, dynamic actin correlates with dendritic remodeling downstream of synaptic potentiation in vivo (Krucker et al., 2000; Rex et al., 2010) and F-actin has been monitored within developing axons of developing zebrafish embryos (Andersen et al., 2010). Protein domain-based biosenors have also been used to measure phosphatidylinositol signaling at the leading edge of neutrophils undergoing chemotactic guidance in live zebrafish (Yoo et al.). Significantly, this study also used genetically encoded photoactivatable Rac to show that local activation of Rac was sufficient to orient neutrophil migration in vivo. Future studies using FRET-based biosensors to measure signaling in growth cones at choice points in vivo will be highly informative. By coupling more sensitive biosensors with advanced forms of microscopy, such as super-resolution microscopy, we should one day be able to visualize the precise cellular and molecular events associated with axon guidance behaviors at choice points in vivo.
Table 1.
Fluorescent probes used for imaging cell structure and signaling in motile cells and growth cones.
| Signal/cellular structure | Probes | Description/Function | Cell type | Reference |
|---|---|---|---|---|
| F-Actin |
|
|
Assorted non- neuronal, neurons in vivo and growth cones | (Dent and Kalil, 2001; Burkel et al., 2007; Riedl et al., 2008; Andersen et al., 2010; Jang et al., 2010; Marsick et al., 2010)(Figure 3) |
| G-Actin | Monomeric actin | Labels actin (G and F actin) | Assorted non- neuronal and growth cones | (Flynn et al., 2009) |
| F-Actin barbed end | KabiramideC | Binds and caps the plus end of F-actin. | Fibroblast, growth cone | (Petchprayoon et al., 2005; Keren et al., 2008) (Figure 3) |
| Microtubules |
|
|
Assorted non-neuronal and growth cones | (Dent and Kalil, 2001; Miller and Bement, 2009) |
| Microtubule plus ends | EB1, EB3, APC, clasp, etc. | Tracks polymerizing MTs | Assorted non- neuronal and growth cones | (Lee et al., 2004; Zhou et al., 2004; Koester et al., 2007) |
| Motor proteins | Dynein, Kinesin, Myosins | Transports cargo along MTs and F-actin | Assorted non- neuronal and growth cones | (Berg and Cheney, 2002; Brown and Bridgman, 2003; Ha et al., 2008) |
| Rho family GTPases |
|
|
Assorted non- neuronal, PC12, primary neurons and growth cones | (Aoki et al., 2004; Benink and Bement, 2005; Zhang et al., 2005; Machacek et al., 2009) (Figure 3) |
| Phosphatidyl-inositol | PH domain from PLC-δ1, Akt | Localizes to membrane rich with PIP2 and PIP3 | Assorted non- neuronal and growth cones | (Jacob et al., 2005; Ketschek and Gallo, 2010) |
| Tyrosine Kinases | Src, FAK (FRET) | Measures Src and FAK activity | Assorted non- neuronal cells | (Ting et al., 2001; Cai et al., 2008; Na et al., 2008) |
| Phosphotyrosine | dSH2 | Localizes to proteins tyrosine phosphorylated by Src | Assorted non- neuronal, growth cones | (Kirchner et al., 2003; Robles et al., 2005; Baker et al., 2008) (Figure 3) |
| Adhesions | Paxillin, Vinculin, Talin, Integrins | Targets focal adhesions and point contacts | Assorted non-neuronal and growth cones | (Smilenov et al., 1999; Robles and Gomez, 2006; Woo et al., 2009) (Figure 3) |
| cAMP | FlCRhR, PKA-, EPAC-based (FRET) | FRET sensors measure cytosolic cAMP levels | Neurons and growth cones | (Gorbunova and Spitzer, 2002; Dunn and Feller, 2008) |
| Calcium |
|
|
Assorted non neuronal, neurons in vivo and growth cones | (Gomez and Spitzer, 1999; Gomez et al., 2001; Horikawa et al., 2010; Hutchins et al., 2011) |
Acknowledgments
We thank Kate Kalil and members of the Gomez lab for comments on the manuscript. This work was supported by NIH NS41564 to T.M.G.
Bibliography
- Andersen E, Asuri N, Clay M, Halloran M. Live imaging of cell motility and actin cytoskeleton of individual neurons and neural crest cells in zebrafish embryos. J Vis Exp. 2010 doi: 10.3791/1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki K, Nakamura T, Matsuda M. Spatio-temporal regulation of Rac1 and Cdc42 activity during nerve growth factor-induced neurite outgrowth in PC12 cells. J Biol Chem. 2004;279:713–719. doi: 10.1074/jbc.M306382200. [DOI] [PubMed] [Google Scholar]
- Augustine GJ, Santamaria F, Tanaka K. Local calcium signaling in neurons. Neuron. 2003;40:331–346. doi: 10.1016/s0896-6273(03)00639-1. [DOI] [PubMed] [Google Scholar]
- Aye-Han NN, Ni Q, Zhang J. Fluorescent biosensors for real-time tracking of post-translational modification dynamics. Curr Opin Chem Biol. 2009;13:392–397. doi: 10.1016/j.cbpa.2009.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker MW, Peterson SM, Macagno ER. The receptor phosphatase HmLAR2 collaborates with focal adhesion proteins in filopodial tips to control growth cone morphology. Dev Biol. 2008;320:215–225. doi: 10.1016/j.ydbio.2008.05.522. [DOI] [PubMed] [Google Scholar]
- Bement WM, Miller AL, von Dassow G. Rho GTPase activity zones and transient contractile arrays. Bioessays. 2006;28:983–993. doi: 10.1002/bies.20477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benink HA, Bement WM. Concentric zones of active RhoA and Cdc42 around single cell wounds. J Cell Biol. 2005;168:429–439. doi: 10.1083/jcb.200411109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg JS, Cheney RE. Myosin-X is an unconventional myosin that undergoes intrafilopodial motility. Nat Cell Biol. 2002;4:246–250. doi: 10.1038/ncb762. [DOI] [PubMed] [Google Scholar]
- Blader P, Plessy C, Strahle U. Multiple regulatory elements with spatially and temporally distinct activities control neurogenin1 expression in primary neurons of the zebrafish embryo. Mech Dev. 2003;120:211–218. doi: 10.1016/s0925-4773(02)00413-6. [DOI] [PubMed] [Google Scholar]
- Brown ME, Bridgman PC. Retrograde flow rate is increased in growth cones from myosin IIB knockout mice. J Cell Sci. 2003;116:1087–1094. doi: 10.1242/jcs.00335. [DOI] [PubMed] [Google Scholar]
- Burkel BM, von Dassow G, Bement WM. Versatile fluorescent probes for actin filaments based on the actin-binding domain of utrophin. Cell Motil Cytoskeleton. 2007;64:822–832. doi: 10.1002/cm.20226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X, Lietha D, Ceccarelli DF, Karginov AV, Rajfur Z, Jacobson K, Hahn KM, Eck MJ, Schaller MD. Spatial and temporal regulation of focal adhesion kinase activity in living cells. Mol Cell Biol. 2008;28:201–214. doi: 10.1128/MCB.01324-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dent EW, Gertler FB. Cytoskeletal dynamics and transport in growth cone motility and axon guidance. Neuron. 2003;40:209–227. doi: 10.1016/s0896-6273(03)00633-0. [DOI] [PubMed] [Google Scholar]
- Dent EW, Kalil K. Axon branching requires interactions between dynamic microtubules and actin filaments. J Neurosci. 2001;21:9757–9769. doi: 10.1523/JNEUROSCI.21-24-09757.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson BJ, Zou Y. Navigating intermediate targets: the nervous system midline. Cold Spring Harb Perspect Biol. 2010;2:a002055. doi: 10.1101/cshperspect.a002055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn TA, Feller MB. Imaging second messenger dynamics in developing neural circuits. Dev Neurobiol. 2008;68:835–844. doi: 10.1002/dneu.20619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk J, Drinjakovic J, Leung KM, Dwivedy A, Regan AG, Piper M, Holt CE. Electroporation of cDNA/Morpholinos to targeted areas of embryonic CNS in Xenopus. BMC Dev Biol. 2007;7:107. doi: 10.1186/1471-213X-7-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn KC, Pak CW, Shaw AE, Bradke F, Bamburg JR. Growth cone-like waves transport actin and promote axonogenesis and neurite branching. Dev Neurobiol. 2009;69:761–779. doi: 10.1002/dneu.20734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galjart N. Plus-end-tracking proteins and their interactions at microtubule ends. Curr Biol. 2010;20:R528–537. doi: 10.1016/j.cub.2010.05.022. [DOI] [PubMed] [Google Scholar]
- Gomez TM, Harrigan D, Henley J, Robles E. Working with Xenopus spinal neurons in live cell culture. Methods Cell Biol. 2003;71:129–156. doi: 10.1016/s0091-679x(03)01008-2. [DOI] [PubMed] [Google Scholar]
- Gomez TM, Robles E, Poo M, Spitzer NC. Filopodial calcium transients promote substrate-dependent growth cone turning. Science. 2001;291:1983–1987. doi: 10.1126/science.1056490. [DOI] [PubMed] [Google Scholar]
- Gomez TM, Roche FK, Letourneau PC. Chick sensory neuronal growth cones distinguish fibronectin from laminin by making substratum contacts that resemble focal contacts. J Neurobiol. 1996;29:18–34. doi: 10.1002/(SICI)1097-4695(199601)29:1<18::AID-NEU2>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Gomez TM, Spitzer NC. In vivo regulation of axon extension and pathfinding by growth-cone calcium transients. Nature. 1999;397:350–355. doi: 10.1038/16927. [DOI] [PubMed] [Google Scholar]
- Gomez TM, Zheng JQ. The molecular basis for calcium-dependent axon pathfinding. Nat Rev Neurosci. 2006;7:115–125. doi: 10.1038/nrn1844. [DOI] [PubMed] [Google Scholar]
- Gorbunova YV, Spitzer NC. Dynamic interactions of cyclic AMP transients and spontaneous Ca(2+) spikes. Nature. 2002;418:93–96. doi: 10.1038/nature00835. [DOI] [PubMed] [Google Scholar]
- Ha J, Lo KW, Myers KR, Carr TM, Humsi MK, Rasoul BA, Segal RA, Pfister KK. A neuron-specific cytoplasmic dynein isoform preferentially transports TrkB signaling endosomes. J Cell Biol. 2008;181:1027–1039. doi: 10.1083/jcb.200803150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen MJ, Dallal GE, Flanagan JG. Retinal axon response to ephrin-as shows a graded, concentration-dependent transition from growth promotion to inhibition. Neuron. 2004;42:717–730. doi: 10.1016/j.neuron.2004.05.009. [DOI] [PubMed] [Google Scholar]
- Honore E, Hemmati-Brivanlou A. In vivo evidence for trigeminal nerve guidance by the cement gland in Xenopus. Dev Biol. 1996;178:363–374. doi: 10.1006/dbio.1996.0224. [DOI] [PubMed] [Google Scholar]
- Horikawa K, Yamada Y, Matsuda T, Kobayashi K, Hashimoto M, Matsu-ura T, Miyawaki A, Michikawa T, Mikoshiba K, Nagai T. Spontaneous network activity visualized by ultrasensitive Ca(2+) indicators, yellow Cameleon-Nano. Nat Methods. 2010;7:729–732. doi: 10.1038/nmeth.1488. [DOI] [PubMed] [Google Scholar]
- Hu X, Viesselmann C, Nam S, Merriam E, Dent EW. Activity-dependent dynamic microtubule invasion of dendritic spines. J Neurosci. 2008;28:13094–13105. doi: 10.1523/JNEUROSCI.3074-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchins BI, Li L, Kalil K. Wnt/calcium signaling mediates axon growth and guidance in the developing corpus callosum. Dev Neurobiol. 2010 doi: 10.1002/dneu.20846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchins BI, Li L, Kalil K. Wnt/calcium signaling mediates axon growth and guidance in the developing corpus callosum. Dev Neurobiol. 2011;71:269–283. doi: 10.1002/dneu.20846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam N, Gagnon F, Moss T. Catalytic and non-catalytic forms of the neurotrophin receptor xTrkB mRNA are expressed in a pseudo-segmental manner within the early Xenopus central nervous system. Int J Dev Biol. 1996;40:973–983. [PubMed] [Google Scholar]
- Jacob SN, Choe CU, Uhlen P, DeGray B, Yeckel MF, Ehrlich BE. Signaling microdomains regulate inositol 1,4,5-trisphosphate-mediated intracellular calcium transients in cultured neurons. J Neurosci. 2005;25:2853–2864. doi: 10.1523/JNEUROSCI.4313-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe AB, Hall A. Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol. 2005;21:247–269. doi: 10.1146/annurev.cellbio.21.020604.150721. [DOI] [PubMed] [Google Scholar]
- Jang KJ, Kim MS, Feltrin D, Jeon NL, Suh KY, Pertz O. Two distinct filopodia populations at the growth cone allow to sense nanotopographical extracellular matrix cues to guide neurite outgrowth. PLoS One. 2010;5:e15966. doi: 10.1371/journal.pone.0015966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren K, Pincus Z, Allen GM, Barnhart EL, Marriott G, Mogilner A, Theriot JA. Mechanism of shape determination in motile cells. Nature. 2008;453:475–480. doi: 10.1038/nature06952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketschek A, Gallo G. Nerve growth factor induces axonal filopodia through localized microdomains of phosphoinositide 3-kinase activity that drive the formation of cytoskeletal precursors to filopodia. J Neurosci. 2010;30:12185–12197. doi: 10.1523/JNEUROSCI.1740-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchner J, Kam Z, Tzur G, Bershadsky AD, Geiger B. Live-cell monitoring of tyrosine phosphorylation in focal adhesions following microtubule disruption. J Cell Sci. 2003;116:975–986. doi: 10.1242/jcs.00284. [DOI] [PubMed] [Google Scholar]
- Koester MP, Muller O, Pollerberg GE. Adenomatous polyposis coli is differentially distributed in growth cones and modulates their steering. J Neurosci. 2007;27:12590–12600. doi: 10.1523/JNEUROSCI.2250-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krucker T, Siggins GR, Halpain S. Dynamic actin filaments are required for stable long-term potentiation (LTP) in area CA1 of the hippocampus. Proc Natl Acad Sci U S A. 2000;97:6856–6861. doi: 10.1073/pnas.100139797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Engel U, Rusch J, Scherrer S, Sheard K, Van Vactor D. The microtubule plus end tracking protein Orbit/MAST/CLASP acts downstream of the tyrosine kinase Abl in mediating axon guidance. Neuron. 2004;42:913–926. doi: 10.1016/j.neuron.2004.05.020. [DOI] [PubMed] [Google Scholar]
- Li W, Lee J, Vikis HG, Lee SH, Liu G, Aurandt J, Shen TL, Fearon ER, Guan JL, Han M, Rao Y, Hong K, Guan KL. Activation of FAK and Src are receptor-proximal events required for netrin signaling. Nat Neurosci. 2004;7:1213–1221. doi: 10.1038/nn1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Beggs H, Jurgensen C, Park HT, Tang H, Gorski J, Jones KR, Reichardt LF, Wu J, Rao Y. Netrin requires focal adhesion kinase and Src family kinases for axon outgrowth and attraction. Nat Neurosci. 2004;7:1222–1232. doi: 10.1038/nn1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Halloran MC. Central and peripheral axon branches from one neuron are guided differentially by Semaphorin3D and transient axonal glycoprotein-1. J Neurosci. 2005;25:10556–10563. doi: 10.1523/JNEUROSCI.2710-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohmann C, Finski A, Bonhoeffer T. Local calcium transients regulate the spontaneous motility of dendritic filopodia. Nat Neurosci. 2005;8:305–312. doi: 10.1038/nn1406. [DOI] [PubMed] [Google Scholar]
- Lutcke H, Murayama M, Hahn T, Margolis DJ, Astori S, Zum Alten Borgloh SM, Gobel W, Yang Y, Tang W, Kugler S, Sprengel R, Nagai T, Miyawaki A, Larkum ME, Helmchen F, Hasan MT. Optical recording of neuronal activity with a genetically-encoded calcium indicator in anesthetized and freely moving mice. Front Neural Circuits. 2010;4:9. doi: 10.3389/fncir.2010.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machacek M, Hodgson L, Welch C, Elliott H, Pertz O, Nalbant P, Abell A, Johnson GL, Hahn KM, Danuser G. Coordination of Rho GTPase activities during cell protrusion. Nature. 2009;461:99–103. doi: 10.1038/nature08242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann F, Harris WA, Holt CE. New views on retinal axon development: a navigation guide. Int J Dev Biol. 2004;48:957–964. doi: 10.1387/ijdb.041899fm. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsick BM, Flynn KC, Santiago-Medina M, Bamburg JR, Letourneau PC. Activation of ADF/cofilin mediates attractive growth cone turning toward nerve growth factor and netrin-1. Dev Neurobiol. 2010;70:565–588. doi: 10.1002/dneu.20800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCombs JE, Palmer AE. Measuring calcium dynamics in living cells with genetically encodable calcium indicators. Methods. 2008;46:152–159. doi: 10.1016/j.ymeth.2008.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin T, O’Leary DD. Molecular gradients and development of retinotopic maps. Annu Rev Neurosci. 2005;28:327–355. doi: 10.1146/annurev.neuro.28.061604.135714. [DOI] [PubMed] [Google Scholar]
- Miller AL, Bement WM. Regulation of cytokinesis by Rho GTPase flux. Nat Cell Biol. 2009;11:71–77. doi: 10.1038/ncb1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyawaki A. Visualization of the spatial and temporal dynamics of intracellular signaling. Dev Cell. 2003;4:295–305. doi: 10.1016/s1534-5807(03)00060-1. [DOI] [PubMed] [Google Scholar]
- Moeller ML, Shi Y, Reichardt LF, Ethell IM. EphB receptors regulate dendritic spine morphogenesis through the recruitment/phosphorylation of focal adhesion kinase and RhoA activation. J Biol Chem. 2006;281:1587–1598. doi: 10.1074/jbc.M511756200. [DOI] [PubMed] [Google Scholar]
- Moody SA. Fates of the blastomeres of the 16-cell stage Xenopus embryo. Dev Biol. 1987;119:560–578. doi: 10.1016/0012-1606(87)90059-5. [DOI] [PubMed] [Google Scholar]
- Moody SA. Fates of the blastomeres of the 32-cell-stage Xenopus embryo. Dev Biol. 1987;122:300–319. doi: 10.1016/0012-1606(87)90296-x. [DOI] [PubMed] [Google Scholar]
- Moon MS, Gomez TM. Balanced Vav2 GEF activity regulates neurite outgrowth and branching in vitro and in vivo. Mol Cell Neurosci. 44:118–128. doi: 10.1016/j.mcn.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon MS, Gomez TM. Adjacent pioneer commissural interneuron growth cones switch from contact avoidance to axon fasciculation after midline crossing. Dev Biol. 2005;288:474–486. doi: 10.1016/j.ydbio.2005.09.049. [DOI] [PubMed] [Google Scholar]
- Na S, Collin O, Chowdhury F, Tay B, Ouyang M, Wang Y, Wang N. Rapid signal transduction in living cells is a unique feature of mechanotransduction. Proc Natl Acad Sci U S A. 2008;105:6626–6631. doi: 10.1073/pnas.0711704105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan FC, Chen Y, Loeber J, Henningfeld K, Pieler T. I-SceI meganuclease-mediated transgenesis in Xenopus. Dev Dyn. 2006;235:247–252. doi: 10.1002/dvdy.20608. [DOI] [PubMed] [Google Scholar]
- Petchprayoon C, Suwanborirux K, Tanaka J, Yan Y, Sakata T, Marriott G. Fluorescent kabiramides: new probes to quantify actin in vitro and in vivo. Bioconjug Chem. 2005;16:1382–1389. doi: 10.1021/bc050006j. [DOI] [PubMed] [Google Scholar]
- Ren XR, Ming GL, Xie Y, Hong Y, Sun DM, Zhao ZQ, Feng Z, Wang Q, Shim S, Chen ZF, Song HJ, Mei L, Xiong WC. Focal adhesion kinase in netrin-1 signaling. Nat Neurosci. 2004;7:1204–1212. doi: 10.1038/nn1330. [DOI] [PubMed] [Google Scholar]
- Rex CS, Gavin CF, Rubio MD, Kramar EA, Chen LY, Jia Y, Huganir RL, Muzyczka N, Gall CM, Miller CA, Lynch G, Rumbaugh G. Myosin IIb regulates actin dynamics during synaptic plasticity and memory formation. Neuron. 2010;67:603–617. doi: 10.1016/j.neuron.2010.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedl J, Crevenna AH, Kessenbrock K, Yu JH, Neukirchen D, Bista M, Bradke F, Jenne D, Holak TA, Werb Z, Sixt M, Wedlich-Soldner R. Lifeact: a versatile marker to visualize F-actin. Nat Methods. 2008;5:605–607. doi: 10.1038/nmeth.1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robles E, Gomez TM. Focal adhesion kinase signaling at sites of integrin-mediated adhesion controls axon pathfinding. Nat Neurosci. 2006;9:1274–1283. doi: 10.1038/nn1762. [DOI] [PubMed] [Google Scholar]
- Robles E, Woo S, Gomez TM. Src-dependent tyrosine phosphorylation at the tips of growth cone filopodia promotes extension. J Neurosci. 2005;25:7669–7681. doi: 10.1523/JNEUROSCI.2680-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybakova IN, Humston JL, Sonnemann KJ, Ervasti JM. Dystrophin and utrophin bind actin through distinct modes of contact. J Biol Chem. 2006;281:9996–10001. doi: 10.1074/jbc.M513121200. [DOI] [PubMed] [Google Scholar]
- Sakaguchi DS, Murphey RK. Map formation in the developing Xenopus retinotectal system: an examination of ganglion cell terminal arborizations. J Neurosci. 1985;5:3228–3245. doi: 10.1523/JNEUROSCI.05-12-03228.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieg DJ, Hauck CR, Ilic D, Klingbeil CK, Schaefer E, Damsky CH, Schlaepfer DD. FAK integrates growth-factor and integrin signals to promote cell migration. Nat Cell Biol. 2000;2:249–256. doi: 10.1038/35010517. [DOI] [PubMed] [Google Scholar]
- Smilenov LB, Mikhailov A, Pelham RJ, Marcantonio EE, Gundersen GG. Focal adhesion motility revealed in stationary fibroblasts. Science. 1999;286:1172–1174. doi: 10.1126/science.286.5442.1172. [DOI] [PubMed] [Google Scholar]
- Stein E, Tessier-Lavigne M. Hierarchical organization of guidance receptors: silencing of netrin attraction by slit through a Robo/DCC receptor complex. Science. 2001;291:1928–1938. doi: 10.1126/science.1058445. [DOI] [PubMed] [Google Scholar]
- Stepanova T, Slemmer J, Hoogenraad CC, Lansbergen G, Dortland B, De Zeeuw CI, Grosveld F, van Cappellen G, Akhmanova A, Galjart N. Visualization of microtubule growth in cultured neurons via the use of EB3-GFP (end-binding protein 3-green fluorescent protein) J Neurosci. 2003;23:2655–2664. doi: 10.1523/JNEUROSCI.23-07-02655.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streuli CH, Akhtar N. Signal co-operation between integrins and other receptor systems. Biochem J. 2009;418:491–506. doi: 10.1042/BJ20081948. [DOI] [PubMed] [Google Scholar]
- Svitkina TM, Bulanova EA, Chaga OY, Vignjevic DM, Kojima S, Vasiliev JM, Borisy GG. Mechanism of filopodia initiation by reorganization of a dendritic network. J Cell Biol. 2003;160:409–421. doi: 10.1083/jcb.200210174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian L, Hires SA, Mao T, Huber D, Chiappe ME, Chalasani SH, Petreanu L, Akerboom J, McKinney SA, Schreiter ER, Bargmann CI, Jayaraman V, Svoboda K, Looger LL. Imaging neural activity in worms, flies and mice with improved GCaMP calcium indicators. Nat Methods. 2009;6:875–881. doi: 10.1038/nmeth.1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting AY, Kain KH, Klemke RL, Tsien RY. Genetically encoded fluorescent reporters of protein tyrosine kinase activities in living cells. Proc Natl Acad Sci U S A. 2001;98:15003–15008. doi: 10.1073/pnas.211564598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb SE, Miller AL. Calcium signalling during embryonic development. Nat Rev Mol Cell Biol. 2003;4:539–551. doi: 10.1038/nrm1149. [DOI] [PubMed] [Google Scholar]
- Wilson NH, Key B. Neogenin interacts with RGMa and netrin-1 to guide axons within the embryonic vertebrate forebrain. Dev Biol. 2006;296:485–498. doi: 10.1016/j.ydbio.2006.06.018. [DOI] [PubMed] [Google Scholar]
- Woo S, Gomez TM. Rac1 and RhoA promote neurite outgrowth through formation and stabilization of growth cone point contacts. J Neurosci. 2006;26:1418–1428. doi: 10.1523/JNEUROSCI.4209-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo S, Rowan DJ, Gomez TM. Retinotopic mapping requires focal adhesion kinase-mediated regulation of growth cone adhesion. J Neurosci. 2009;29:13981–13991. doi: 10.1523/JNEUROSCI.4028-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worth DC, Parsons M. Adhesion dynamics: mechanisms and measurements. Int J Biochem Cell Biol. 2008;40:2397–2409. doi: 10.1016/j.biocel.2008.04.008. [DOI] [PubMed] [Google Scholar]
- Yoo SK, Deng Q, Cavnar PJ, Wu YI, Hahn KM, Huttenlocher A. Differential regulation of protrusion and polarity by PI3K during neutrophil motility in live zebrafish. Dev Cell. 18:226–236. doi: 10.1016/j.devcel.2009.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidel-Bar R, Itzkovitz S, Ma’ayan A, Iyengar R, Geiger B. Functional atlas of the integrin adhesome. Nat Cell Biol. 2007;9:858–867. doi: 10.1038/ncb0807-858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Webb DJ, Asmussen H, Niu S, Horwitz AF. A GIT1/PIX/Rac/PAK signaling module regulates spine morphogenesis and synapse formation through MLC. J Neurosci. 2005;25:3379–3388. doi: 10.1523/JNEUROSCI.3553-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou FQ, Zhou J, Dedhar S, Wu YH, Snider WD. NGF-induced axon growth is mediated by localized inactivation of GSK-3beta and functions of the microtubule plus end binding protein APC. Neuron. 2004;42:897–912. doi: 10.1016/j.neuron.2004.05.011. [DOI] [PubMed] [Google Scholar]